DOI:
10.1039/B508166A
(Paper)
Analyst, 2006,
131, 55-61
Immobilization of RNase S-Peptide on a single-stranded DNA-fixed gold surface and effective masking of its surface by an acridinyl poly(ethylene glycol)†
Received
9th June 2005
, Accepted 8th November 2005
First published on 23rd November 2005
Abstract
Oligonucleotide–peptide conjugate 1 was synthesized by coupling of RNase S-peptide to a 24-mer single-stranded DNA (ssDNA) oligonucleotide to be immobilized on its complementary ssDNA oligonucleotide-fixed gold surface of sensor chip or electrode. Immobilization of 1 on the ssDNA-fixed gold surface through DNA duplex formation was confirmed by quartz crystal microbalance (QCM) and electrochemical measurements. After treating with a synthetic acridinyl poly(ethylene glycol) (APEG), specific interaction of S-protein with the S-peptide immobilized on the gold surface was demonstrated by QCM without nonspecific adsorption of unrelated proteins such as BSA and RNase A at the surfaces. This result suggested that the acridine parts of APEG could bind to the DNA duplex on the gold surface and the poly(ethylene glycol) parts were fastened on the surface to resist the adsorption of proteins. Thus, the combination of oligonucleotide–peptide conjugate, ssDNA-fixed chip and APEG with effective masking property provides a new tool for the analysis of specific peptide–protein interactions without disturbance by other unrelated proteins.
1. Introduction
Since the first oligonucleotide–peptide conjugate was synthesized by Haralambidis et al.,1 many researchers have been exploring more effective synthetic methods from the viewpoint of the use of the functionalized nucleotides as new antigene and antisense therapeutic reagents with improved permeability to cell membranes.2 Oligonucleotide–peptide conjugate has also potential utilities for the construction of peptide chips which have attracted widespread interest as analytical tools for biomolecular recognition events.3 When the conjugate is allowed to hybridize with its complementary single-stranded DNA (ssDNA) on the DNA chip, the peptide can be immobilized on the proper position of the DNA chip, and the DNA chip can be converted to a peptide chip. However, the preparation of peptide chips by using the conjugate has not yet been reported to date. On the other hand, the application of oligonucleotide–protein conjugate for the construction of protein chips from the DNA chips was reported by Niemeyer and co-workers.4,5 Recently, Schultz and co-workers reported the preparation of the peptide chip by using peptide nucleic acid–peptide conjugate.6 This system was effective for the orientated and addressed immobilization of peptides on the chip regardless of the sequence and length limitation of the peptide nucleic acid. Therefore, the conjugate is expected to provide a more suitable immobilization method of peptides to form the peptide chip because of its economical advantages to use a variety of sequences and lengths of the oligonucleotide compared with those of the peptide nucleic acid. The concept of this strategy is shown in Fig. 1.
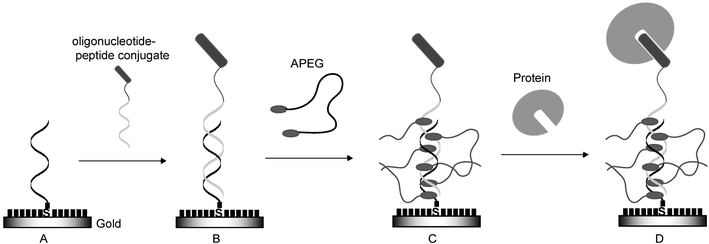 |
| | Fig. 1 Concept of the construction of peptide chip by using oligonucleotide–peptide conjugate coupled with ssDNA-fixed chip through DNA duplex formation. The position and orientation of peptide on the chip can be controlled by selecting the appropriate DNA oligonucleotide sequence to form the specific DNA duplex. ssDNA is fixed on gold substrate (A) and the conjugate is allowed to hybridize on this substrate (B). After treatment with acridinyl poly(ethylene glycol) (APEG), the acridine moiety of APEG intercalates with the DNA duplex region and the poly(ethylene glycol) moiety gives effective masking to prevent non-specific adsorption (C). This peptide chip permits the specific interaction between peptide and target protein (D). Short black bars on gold indicate mercaptohexanol. | |
Furthermore, we intended to use a simple acridinyl poly(ethylene glycol) (APEG) for anchoring and masking on a bare gold surface. Since a DNA duplex region exists in the peptide chip prepared by the immobilization of the conjugate on the DNA chip, the acridine moiety of APEG can bind to this region as an anchor and the poly(ethylene glycol) moiety can mask the bare gold surface. This masking can avoid nonspecific adsorption of proteins on the surface. The masking on the surface of the protein chip is also very important to resist the nonspecific adsorption of proteins and, in fact, poly(ethylene glycol) or a carbohydrate-derived component has been used for this purpose in self-assembled monolayer systems.7–11
In order to demonstrate the feasibility of the above approach, we selected popular RNase S-peptide and S-protein interaction systems with low dissociation constants.12 Firstly, the conjugate (1) of RNase S-peptide and a 24-mer oligonucleotide (5′-CGA CGT TGT AAA ACG ACG GCC-3′) was synthesized and immobilized on the gold surface previously fixed with its complementary oligonucleotide whose sequence is derived from the multicloning site of bacteriophage M13mp18.13 Secondly, APEG was added to this gold surface to evaluate its masking effect. Finally, specific interaction of S-protein with the S-peptide immobilized on this gold surface was studied. Each step of the process (Fig. 1) was evaluated by using quartz crystal microbalance (QCM) or electrochemical experiments.
2. Experimental
2.1 Instrumentation
UV-vis absorption spectra were measured by a Hitachi U-3310 spectrophotometer equipped with a Hitachi SPR-10 temperature controller (Hitachi Ltd., Japan). Fluorescence spectra were measured by an LS50 luminescence spectrometer (Perkin Elmer). Matrix-assisted laser desorption ionization-TOF MS (MALDI-TOF MS) was measured by a Voyager Linear-SA mass spectrometer (PerSeptive Biosystems). RP-HPLC analysis and purification were performed by a Hitachi HPLC system composed of an L-4200 UV-VIS detector, an L-6200 intelligent pump, an L-6000 pump and a D-2600 chromato-integrator using a Mightysil RP-18 column (4.6 × 250 mm) (Kanto Chemical Co., Inc., Japan) or an Inertsil ODS-3 column (4.6 × 250 mm) (GL Science Inc., Japan). Quartz crystal microbalance (QCM) experiments were conducted with an AffinixQ system (Initium Co., Japan). Electrochemical experiments were performed by an ALS model 600 electrochemical analyzer (CH Instrument Inc., USA). The peptides were synthesized on a Model 433A peptide synthesizer (Applied Biosystems) using the Fmoc/tert-butyl strategy.14
Oligonucleotide–peptide conjugate 1 (Fig. 2A) was prepared essentially following the method described by Haralambidis and co-workers, where peptides and oligonucleotide were synthesized separately and then each of them was coupled by a chemoselective conjugation reaction.15
2.2.1 Synthesis of maleimidopeptide.
Firstly, the RNase S-peptide (1-19)16 was assembled on the Model 433A peptide synthesizer on a 0.25 mmol scale using Fmoc-Ala-Alko-resin (0.7 mmol g−1, Watanabe Chemical Industries, Ltd., Japan) and the FastMoc protocol as described previously.17 Fmoc-8-amino-3,6-dioxaoctanoic acid (Peptide International Inc., USA) as a linker chain was coupled twice to the N-terminus of the peptide-resin. The peptide-resin (167 mg, 35.6 µmol) removed from the reaction vessel was suspended in 1.5 mL of N-methylpyrrolidone (NMP) and stirred for 30 min at room temperature to be swelled. The maleimido reagent, N-(4-maleimidobutyryloxy)succinimide ester (50 mg, 178 µmol, 5 equiv.; Pierce Biotechnology Inc., USA), and triethylamine (50 µL, 10 equiv.) were added to this suspension and stirred overnight at room temperature. After filtration, the peptide-resin was washed with NMP and dichloromethane and dried under reduced pressure. The peptide was cleaved from the resin with concomitant removal of the protecting groups by treatment with a mixture of trifluoroacetic acid (TFA)/m-cresol/thioanisole/ethanedithiol (92.5 ∶ 2.5 ∶ 2.5 ∶ 2.5 v/v/v/v) for 2 h at room temperature. The crude peptide was purified by the Hitachi HPLC system using the Inertsil ODS-3 column and the following HPLC conditions: a linear gradient of acetonitrile in 0.1% aq. TFA from 7 to 38.5% over 30 min at a flow rate of 1.0 mL min−1 to provide the desired maleimidopeptide. Its structure was verified by MALDI-TOF MS using α-cyano-4-hydroxycinnamic acid as a matrix: calcd. [M + H]+ = 2550.9, found [M + H]+ = 2550.24.
2.2.2 Coupling of maleimidopeptide to 5′-thiolated oligonucleotide.
The maleimidopeptide (2 mg) obtained above was dissolved in 0.1 M sodium phosphate buffer (pH 7.0) containing 40 nmol of 5′-mercaptohexyloligonucleotide (5′-HS(CH2)6-CGA CGT TGT AAA ACG ACG GCC AGT custom-synthesized by Genenet Co., Japan) and stirred for 24 h at room temperature. After addition of 0.1 M triethylamine acetate (TEAA) buffer (pH 7.0) to the reaction mixture up to 1 mL of total volume, the crude oligonucleotide–peptide conjugate was purified by a NAP-10 column (Amersham Biosciences) and then by the Hitachi HPLC system using the Mightysil RP-18 column and the following HPLC conditions: a linear gradient of acetonitrile in 0.1 M TEAA buffer (pH 7.0) from 10 to 40% over 30 min at a flow rate of 1 mL min−1 to give the target conjugate 1. Its structure was confirmed by MALDI-TOF MS using 3-hydroxypicolinic acid as a matrix: calcd. [M − H]− = 10131.53, found [M − H]− = 10136.92.
2.3 Synthesis of APEG
Poly(ethylene glycol) carrying amino moieties at its terminus (5 g, 1.3 mmol, average Mr = 4000 ) obtained from Sanyo Chemical Industries, Ltd., Japan and phenoxyacridine (1.08 g, 3.9 mmol) were dissolved in phenol and stirred for 4 h at 120 °C as described previously.18 After being cooled, the solid was dissolved in a small amount of acetone and added to excess amount of hexane under cooling in an ice bath. The precipitate obtained was dissolved in a small amount of acetone and poured into an excess amount of hexane under cooling in an ice bath. This procedure was repeated twice. The precipitate thus obtained was dried under reduced pressure to afford the desired APEG (5.19 g, 90%). MALDI-TOF MS spectrum of this APEG (Fig. 2B) is shown in Fig. 3, indicating that the chain length distribution ranges from about n = 66 to 111 ethylene glycol units with a maximum at n = 90. Molecular weights determined from MALDI-TOF MS: Mn (number average) = 4524.05, Mw (weight average) = 4556.87, Mw/Mn = 1.01. The concentration of an APEG stock solution was determined by using the molar absorptivity of its quinacrine.19
 |
| | Fig. 3 MALDI-TOF MS spectrum of APEG which exhibits bell-shaped distribution of 44 m/z-spaced signals. The chain length distribution ranges from about n = 66 to 111 ethylene glycol units with maximum at n = 90. Sinapic acid was used as a matrix. | |
2.4 Estimation of the binding constant of APEG with double-stranded DNA
UV-vis absorption spectra of APEG became unchanged within 1 min after addition of sonicated calf thymus DNA (double-stranded DNA) even though APEG is a high molecular weight polymer. Absorption changes of APEG were monitored upon addition of various amounts of sonicated calf thymus DNA. Scatchard plots were calculated using this change and the binding constant of APEG with the double-stranded DNA (dsDNA) was estimated by fitting the McGhee and von Hippel equation:20r/L = K(1 −
nr){(1 −
nr)/[1 − (n
− 1)r]}n
− 1, where r, L, K, and n refer to mole of the bound APEG per base pair, the concentration of unbound APEG, the binding constant, base pair coverage when APEG bound to dsDNA, respectively.
2.5 Preparation of ssDNA-fixed QCM chip
A 27 MHz, AT-cut QCM sensor chip (Initium Co., Japan)17,21,22 covered with a gold surface (diameter: 2.5 mm; area: 4.9 mm2) was used in the QCM experiment. CapOligo, 5′-thiolated oligonucleotide (5′-HS-(CH2)6-ACT GGC CGT CGT TTT ACA ACG TCG) custom-synthesized by Genenet Co., Japan, could be immobilized on the gold surface of the QCM chip with good reproducibility in the following procedure (Fig. 1A). The gold surface of this chip was treated with a piranha solution (conc. H2SO4/30% H2O2; 7 ∶ 3 v/v) for 2 × 5 min. After washing with MilliQ water, 10 µL of 20 µM CapOligo was placed on this surface and kept overnight at room temperature under high humidity conditions. After washing with MilliQ water, 10 µL of 1 mM mercaptohexanol was placed on this surface and kept for 2 h at room temperature. Caution: the piranha solution reacts violently with many organic materials and should be handled with great care.
2.6 Electrochemical hybridization assay by ferrocenylnaphthalene diimide
This assay was conducted following the method reported previously.23 A gold electrode with an area of 2 mm2 (BAS Inc., Japan) was used in this assay. Differential pulse voltammetry (DPV) was measured in an electrolyte containing 0.1 M potassium acetate buffer (pH 5.6), 0.1 M KCl and 0.05 mM ferrocenylnaphthalene diimide (FND) with normal three-electrode configurations of an oligonucleotide-fixed electrode as a working electrode, a Pt counter electrode, and an Ag/AgCl reference electrode. The fixation of CapOligo was performed as follows. The gold electrode was pretreated with boiling 2 M NaOH for 1 h and conc. HNO3 for 30 min at room temperature. After washing with MilliQ water, 1 µL of 2 µM CapOligo was placed on the surface of this gold electrode and kept for 1 h. The hybridization event was evaluated by the peak current of io and i obtained before and after hybridization with 1 or the oligonucleotide carrying the same sequence as that of 1.24 The same experiments were conducted three times and each datum has reproducibility within 13% of relative standard deviations.
3. Results and discussion
3.1 Interaction of APEG with dsDNA
UV-vis absorption spectrum of APEG showed hypochromic effect and red shift after addition of sonicated calf thymus DNA in 10 mM sodium 2-(N-morpholino)ethanesulfonate buffer (pH 6.5) containing 1 mM EDTA and 0.1 M NaCl (Fig. 4A). The absorption change after addition of calf thymus DNA became unchanged within 1 min. The behavior was similar to that of quinacrine which is a classical intercalator, suggesting that APEG can intercalate with dsDNA. Scatchard analysis of the interaction of APEG and dsDNA was achieved using this absorbance change (Fig. 4B), giving the binding constant of 3.3 × 104 M−1 with a site size of n = 9 in the presence of 0.1 M NaCl. Since aminoacridine has a binding constant of 6.1 × 104 M−1 with n = 5,25 it is suggested that the poly(ethylene glycol) chain of APEG does not disturb the dsDNA binding. Scatchard analyses of the interaction of APEG and dsDNA were also performed under different salt concentrations and log K was plotted against −log [Na+]. The slope of this plot was 0.4 ± 0.1, suggesting that the behavior of APEG was not affected by the salt concentration.19 APEG behaved as a monocationic intercalator judging from its pKa = 8.0 under the experimental conditions and this corresponds to the obtained slope. APEG might be a good candidate for use as a masking reagent because it does not interfere with DNA binding under various salt concentrations.
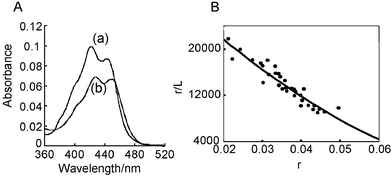 |
| | Fig. 4 (A) UV-vis absorption spectra of 2 µM APEG in the absence (a) and presence (b) of 60 µM sonicated calf thymus DNA in 10 mM sodium 2-(N-morpholino)ethanesulfonate buffer (pH 6.5) containing 1 mM EDTA and 0.1 M NaCl. (B) Scatchard plots of the interaction of APEG with calf thymus DNA obtained from the data of the absorption change in A. | |
3.2 Binding of 1 with CapOligo-fixed gold surface
The duplex formation of 1 with the CapOligo-fixed gold surface shown in Fig. 1B was evaluated by an electrochemical hybridization assay and a QCM experiment. The electrochemical hybridization assay with FND can monitor the hybridization event from the current intensity of FND before and after hybridization.23 This is based on the redox reaction of the concentrated FND to ss or ds DNA-immobilized electrode.24 After DNA duplex formation, the peak current is increased in DPV measurement.24 Thus the DPV measurement was conducted on the CapOligo-fixed gold electrode before and after hybridization with 1 or the oligonucleotide carrying the same sequence as that of 1. The increased DPV current was observed in both cases suggesting DNA duplex formation on the electrode (Fig. 5A and 5B). However, increased current of DPV was not observed after treatment with S-peptide (Fig. 5C). These results suggested that 1 did not non-specifically bind to the mercaptohexanol monolayer and could be immobilized on the surface through DNA duplex formation.
 |
| | Fig. 5 Background-corrected DPV of CapOligo-fixed gold electrode before (⋯) and after (—) hybridization with 1 (A) or oligonucleotide carrying the same sequence as that of 1 (B) in the electrolyte containing 0.1 M potassium acetate buffer (pH 5.6), 0.1 M KCl and 0.05 mM FND. (C) DPV was also measured before (⋯) and after (—) treatment with S-peptide. | |
Quantitative analysis was achieved by QCM measurement. The CapOligo-fixed gold surface of the QCM chip was immersed into 8 mL of 10 × SSC (0.15 M sodium citrate, 1.5M NaCl) buffer and frequency changes were monitored to reach a plateau. After that, 50 µL of 1 µM 1 was added into the buffer solution and the frequency changes were monitored to reach a plateau. The frequency change was saturated at −88 Hz as shown in Fig. 6A whose value corresponds to 53.7 ng cm−2 of 1,21,22 suggesting that 0.26 pmol of 1 was allowed to hybridize with the CapOligo on this chip. When DNA duplex is regarded as a column with a diameter of 2 nm, the cross-section area of the DNA column is 3.14 × 10−12 mm2, and the coverage area by the total DNA duplex formed by 1 (0.26 pmol or 1.56 × 1011 molecules) was calculated to be 0.49 mm2. As the area of the gold surface of the sensor chip was 4.9 mm2, 10% of the surface was expected to be covered by 1 in this case. The same QCM experiments were conducted at least twice and each datum is within error.
 |
| | Fig. 6 (A) Frequency changes of CapOligo-fixed QCM gold sensor chip upon addition of 1 in 10 × SSC buffer and after further addition of APEG in 1 × PBS buffer (pH 7.4). (B) Frequency changes of CapOligo (—) or mercaptohexanol (⋯) fixed QCM chip upon addition of APEG in 1 × PBS buffer (pH 7.4). Position of ▼ indicates the addition of 50 µl of 1 µM 1 and those of ▽ indicate the addition of 10 µl of 1 µM APEG. | |
3.3 Binding behavior of APEG with DNA duplex-immobilized gold surface
After hybridization of 1 with the CapOligo-fixed gold surface of the QCM chip, the chip was immersed into 8 mL of PBS (137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4; pH 7.4) buffer and the frequency change was monitored upon addition of 10 µL of 1 mM APEG. As shown in Fig. 6A, the settled frequency decrease was 160 Hz, suggesting the immobilization of 98.0 ng cm−2 or 1.10 pmol of APEG on the dsDNA-immobilized surface. Similar experiments were performed using CapOligo- or mercaptohexanol-fixed gold surface. Large frequency decrease was observed in the case of the CapOligo-fixed gold surface, whereas a very little frequency decrease was observed in the case of mercaptohexanol-fixed gold surface (Fig. 6B). These results suggest that APEG could bind to dsDNA by the intercalation and also to ssDNA possibly through an electrostatic or stacking interaction with ssDNA. Considering the use of APEG as a masking reagent, this characteristic property is suitable for effective masking of the ssDNA area.
3.4 Evaluation of masking effect of APEG by QCM experiment
The QCM chip carrying 0.28 pmol of 1 with RNase S-peptide was immersed into 8 mL of PBS buffer, and kept until stable frequency was obtained. After addition of 10 µL of 10 µM BSA into this buffer solution, frequency changes were monitored. Large frequency change was observed as shown in Fig. 7. Further addition of RNase S-protein (20–124)16 showed a very little frequency decrease. This result suggested that BSA was adsorbed on the gold surface nonspecifically and the adsorbed BSA disturbed specific binding of S-protein. On the other hand, the QCM chip carrying 0.26 pmol of 1 was pretreated with APEG and frequency changes were monitored upon addition of 10 µL of 10 µM BSA and 10 µL of 10 µM S-protein in this order. Fig. 8 shows that no frequency change was observed upon addition of BSA, but a large frequency decrease was observed upon addition of S-protein. Alternative addition of BSA and S-protein in order showed a frequency decrease only in the case of S-protein. This result suggested that APEG could cover the gold surface effectively and prevented the nonspecific adsorption of BSA but allowed the specific interaction of S-protein. Finally, a settled frequency decrease of 65 Hz was obtained upon addition of S-protein, indicating 40 ng cm−2 or 0.17 ± 0.01 pmol of S-protein binding. Considering the immobilization of 0.26 ± 0.02 pmol of S-peptide on the QCM chip surface, almost 1 ∶ 1 ratio binding between S-peptide and S-protein could be observed on the gold surface. Furthermore, the effect of RNase A which is a parent protein of S-peptide and S-protein12 was checked but no frequency decrease was observed under similar conditions. However, a frequency decrease was observed on further addition of S-protein (Fig. 9). Thus, APEG could mask the gold surface to avoid the nonspecific binding of such an acidic protein, BSA (pI = 4.7), and a basic protein, RNase A (pI = 9.3)26 allowing the specific S-protein–S-peptide interaction. This masking ability of APEG was very effective even in 10% coverage of dsDNA on the gold surface.
 |
| | Fig. 7 Frequency changes of S-peptide-immobilized QCM chip after addition of 10 µl of 10 µM BSA (▽) and 10 µl of 10 µM S-protein (▼) in 1 × PBS buffer (pH 7.4). | |
 |
| | Fig. 8 Frequency changes of S-peptide-immobilized QCM chip after APEG treatment upon addition of 10 µl of 10 µM BSA (▽) and 10 µl of 10 µM S-protein (▼) in 1 × PBS buffer (pH 7.4). | |
 |
| | Fig. 9 Frequency changes of S-peptide-immobilized QCM chip after APEG treatment upon addition of 10 µl of 10 µM RNase A (▽) and 10 µl of 10 µM S-protein (▼) in 1 × PBS buffer (pH 7.4). | |
3.5 Evaluation of specific interaction of S-protein with S-peptide-immobilized gold surface
To evaluate the specificity of this interaction, the interaction of S-protein with S-peptide-immobilized QCM chip was studied by QCM measurement in the presence of several kinds of peptides (Fig. 10). Firstly, the CapOligo-fixed QCM chip was allowed to hybridize with 1, followed by APEG treatment to afford the S-peptide-immobilized QCM chip. Competitive experiment was performed in the presence of S-peptide analogs, LB2,16 A5W,16 S-peptide or sunflower trypsin inhibitor (SFTI-1).17 The S-peptide analogs, LB2 and A5W, have been shown to bind to S-protein with high affinity by the surface plasmon resonance experiment and their enzymatic activities.16 SFTI-1 was used as a negative control with a non-binding property to S-protein. To confirm that LB2 and A5W can bind to the S-peptide binding site of S-protein, competitive assay in a homogenous aqueous solution was carried out by a similar fluorimetric method to that reported previously.27 Fluorescence intensity of A5W in 1 × PBS buffer (pH 7.4) at 355 nm after excitation at 295 nm was increased upon addition of S-protein (Fig. 11A). This fluorescence increase might be caused by the presence of Trp residue in A5W. It is suggested that the Trp residue of A5W is located in a more hydrophobic area after binding to S-protein.16 Then, S-peptide was added to this complex solution and the fluorescence decrease was observed as shown in Fig. 11B. This also suggested competitive binding of S-peptide and A5W to the same binding site of S-protein. This behavior was also monitored by the QCM experiment (Fig. 10). S-protein was added to the solution where the S-peptide-immobilized QCM chip was dipped, and a frequency decrease of 90 Hz or 55 ng cm−2 was observed. Similar experiments were performed in the simultaneous presence of S-protein and 12.5 µM LB2, A5W, S-peptide or SFTI-1. SFTI-1 had no influence on the frequency decrease. However, LB2, A5W and S-peptide were shown to inhibit the frequency decrease. This suggested that LB2, A5W or S-peptide acted as a competitor for the binding of the S-peptide immobilized on the QCM chip to S-protein. The inhibition order was in good agreement with the binding affinity of these peptides.16
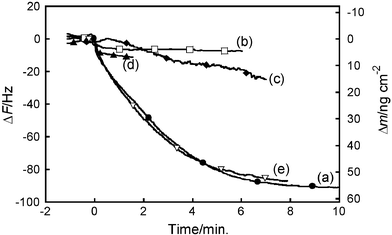 |
| | Fig. 10 Frequency changes of S-peptide-immobilized QCM chip after addition of 10 µl of 10 µM S-protein in the absence (a) or presence of 12.5 µM LB2 (b), A5W (c), S-peptide (d) or SFTI-1 peptide (e) in 1 × PBS buffer (pH 7.4). | |
4. Conclusion
In this study, we have shown the usefulness of the combination of oligonucleotide–peptide conjugate, ssDNA-fixed gold sensor chip and a newly synthesized APEG for the analysis of specific peptide–protein interactions. As an example of the utilization of this system, firstly the immobilization of oligonucleotide–peptide conjugate 1 containing RNase S-peptide on the CapOligo-fixed gold surface through duplex formation was demonstrated. Secondly, we developed APEG as a novel masking reagent for the gold surface and applied it for masking of the surface immobilized with 1 on ssDNA-fixed gold surface through the binding of the acridine unit of APEG to the formed DNA duplex. Finally, this masking was demonstrated to be very effective for the analysis of the specific interaction of S-protein with the S-peptide immobilized on the gold surface without nonspecific adsorption of unrelated proteins such as BSA and RNase A. Competitive assay using several peptides also showed the specificity of this interaction. Thus, oligonucleotide–peptide conjugate and ssDNA-fixed gold sensor chip should be useful for peptide chip construction with addressable immobilization of the peptide and simple treatment with APEG should provide effective masking on this chip, allowing quantification of specific peptide–protein interactions.‡
Acknowledgements
This work was partly supported by a grant from the “University Start-Ups Creation Support System” of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The authors would like to thank the Foundation “Hattori-Hokokai” for the financial support.
References
- J. Haralambidis, L. Duncan and G. W. Tregear, Tetrahedron Lett., 1987, 28, 5199 CrossRef CAS.
- C.-H. Tung, Bioconjugate Chem., 2000, 11, 605 CrossRef CAS.
- D-H. Min and M. Mrksich, Curr. Opin. Chem. Biol., 2004, 8, 554 CrossRef CAS.
- C. M. Niemeyer, L. Boldt, B. Ceyhan and D. Blohm, Anal. Biochem., 1999, 268, 54 CrossRef CAS.
- M. Lovrinovic, R. Seidel, R. Wacker, H. Schroeder, O. Seitz, M. Engelhard, R. S. Goody and C. M. Niemeyer, Chem. Commun., 2003, 2003, 822 RSC.
- N. Winssinger, J. L. Harris, B. J. Backes and P. G. Schultz, Angew. Chem., Int. Ed., 2001, 40, 3152 CrossRef CAS.
- S. Herrwerth, T. Rosendahl, C. Feng, J. Fick, W. Eck, M. Himmelhaus, R. Dahint and M. Grunze, Langmuir, 2003, 19, 1880 CrossRef CAS.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, B15, 3 CrossRef.
- M. Metzke, J. Z. Bai and Z. Guan, J. Am. Chem. Soc., 2003, 125, 7760 CrossRef CAS.
- D. F. Siqueira Petri, S. W. Choi, H. Beyer, T. Schimmel, M. Bruns and G. Wenz, Polymer, 1999, 40, 1593 CrossRef CAS.
- J. Ji, L. Feng, Y. Qiu and X. Yu, Polymer, 2000, 41, 3713 CrossRef CAS.
- R. T. Raines, Chem. Rev., 1998, 98, 1045 CrossRef CAS.
- C. Yanisch-Perron, J. Vieira and J. Messing, Gene, 1985, 33, 103 CrossRef CAS.
-
W. C. Chan and P. D. White, in Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York, 2000 Search PubMed.
- N. J. Ede, G. W. Tregear and J. Haralambidis, Bioconjugate Chem., 1994, 5, 373 CrossRef CAS.
- J. J. Dwyer, M. A. Dwyer and A. A. Kossialoff, Biochemistry, 2001, 40, 13491 CrossRef CAS.
- K. Ohtsuka, R. Kajiki, M. Waki, T. Nojima and S. Takenaka, Analyst, 2004, 129, 888 RSC.
- S. Takenaka, N. Shigemoto and H. Kondo, Supramol. Chem., 1998, 9, 47 CrossRef CAS.
- W. D. Wilson and I. G. Lopp, Biopolymers, 1979, 18, 3025 CrossRef CAS.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469 CrossRef CAS.
- S. Fukusho, H. Furusawa and Y. Okahata, Chem. Commun., 2002, 2002, 88 RSC.
- H. Matsuno, K. Niikura and Y. Okahata, Chem.–Eur. J., 2001, 7, 3305 CrossRef CAS.
- S. Takenaka, K. Yamashita, M. Takagi, Y. Uto and H. Kondo, Anal. Chem., 2000, 72, 1334 CrossRef CAS.
- S. Takenaka, Polym. J., 2004, 36, 503 CAS.
- D. D. McPherson and J. M. Pezzuto, J. Chromatogr., 1983, 281, 348 CrossRef CAS.
- N. Ui, Biochim. Biophys. Acta, 1971, 229, 567 CAS.
- Z. Getahun, C.-Y. Huang, T. Wang, B. D. Leon, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2003, 125, 405 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Three times repeating hybridization experiments with the same 5′-thiolated oligonucleotide-fixed QCM gold sensor chip (Fig. S1). See DOI: 10.1039/b508166a |
| ‡ Abbreviations: APEG, acridinyl poly(ethylene glycol); QCM, quartz crystal microbalance; NMP, N-methylpyrrolidone; TEAA buffer, triethylamine aceteate buffer; DPV, differential pulse voltammetry; BSA, bovine serum albumin; PBS, phosphate buffered saline; ss, single stranded; ds, double stranded; MALDI, matrix-assisted laser desorption ionization; TFA, trifluoroacetic acid; FND, ferrocenylnaphthalene diimde; SFTI-1, sunflower trypsin inhibitor. |
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here. 
