
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis
Megan Amy
Bryden
and
Eli
Zysman-Colman
 *
*
Organic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: http://www.zysman-colman.com/
First published on 18th May 2021
Abstract
Organic compounds that show Thermally Activated Delayed Fluorescence (TADF) have become wildly popular as next-generation emitters in organic light emitting diodes (OLEDs). Since 2016, a subset of these have found increasing use as photocatalysts. This review comprehensively highlights their potential by documenting the diversity of the reactions where an organic TADF photocatalyst can be used in lieu of a noble metal complex photocatalyst. Beyond the small number of TADF photocatalysts that have been used to date, the analysis conducted within this review reveals the wider potential of organic donor–acceptor TADF compounds as photocatalysts. A discussion of the benefits of compounds showing TADF for photocatalysis is presented, which paints a picture of a very promising future for organic photocatalyst development.
 Megan Amy Bryden | Megan Amy Bryden obtained her MChem from the University of Durham in 2018, with her master's project being undertaken at the Université Grenoble Alpes focusing on luminescent lanthanide redox probes. She is now completing her PhD project at the University of St Andrews and is interested in exploring new photocatalytic reactions with TADF compounds as the photocatalyst. |
 Eli Zysman-Colman | Eli Zysman-Colman obtained his PhD in Chemistry from McGill University in 2003 with Prof. David N. Harpp. He then completed two postdoctoral fellowships, one in supramolecular chemistry with Prof. Jay Siegel at the University of Zurich and the other in inorganic materials with Prof. Stefan Bernhard at Princeton University. He joined the Université de Sherbrooke as an assistant professor in 2007. In 2013, he moved to the University of St Andrews where he is presently Professor of Optoelectronic Materials, Fellow of the RSC and is the holder of a Royal Society Leverhulme Trust Senior Research Fellowship. |
1. Introduction
In the most basic definition, photocatalysis involves using light as an energy input in concert with a light-absorbing material to increase reaction rates. Since 2007, there has been a resurgence of activity in the domain of photocatalysis following the pioneering demonstrations by the likes of MacMillan, Yoon and Stephenson, who showed that photoactive organometallic complexes can mediate useful organic reaction transformations.1–3 The prospect of the widescale development of greener, more economical reactions that proceed under mild reaction conditions has propelled photocatalysis from a niche concept to a widely accepted and highly useful synthetic strategy. Photocatalysis presents a significant advantage over traditional synthetic methodologies in that it can allow for generation of products which are inaccessible thermally. Moreover, the reactive intermediates are generated under mild conditions, which opens the door to new transformations and to reaction conversions at higher efficiencies.Upon absorption of light, the photocatalyst (PC) accesses its excited state, PC*. From here, photoinduced energy transfer (PEnT) and/or photoinduced electron transfer (PET) can occur between the PC* and the substrate, with the latter requiring an additional single electron transfer (SET) step to close the photocatalytic cycle, reflected in Fig. 1. When PET is in operation, this is termed photoredox catalysis.
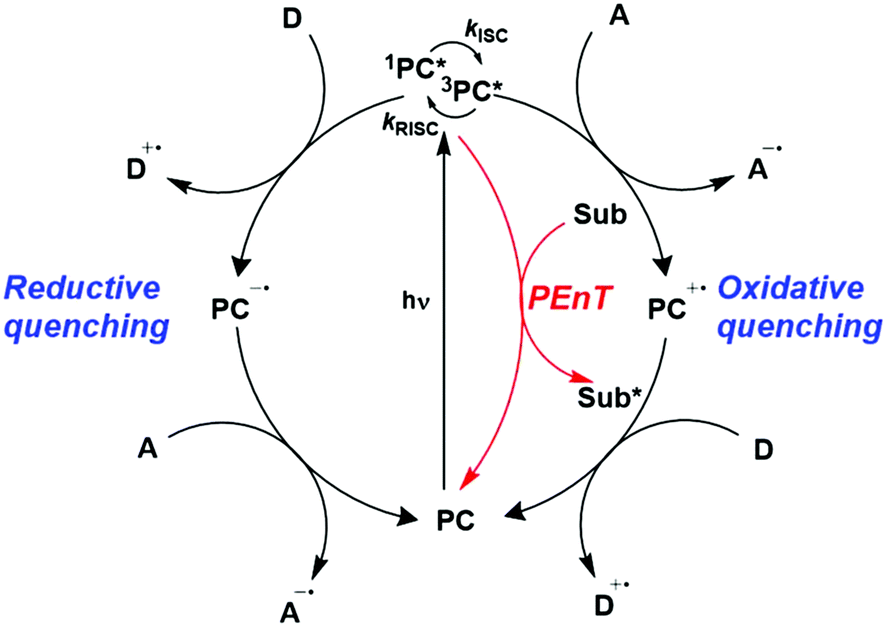 | ||
| Fig. 1 Energy transfer, oxidative quenching and reductive quenching photocatalytic cycles of a generic TADF PC with donor (D) and acceptor (A) substrates (Sub). | ||
Regardless of the mechanism in operation, it is important to use a PC that can be photoexcited selectively in the presence of the organic substrate or the product formed. Thus, PCs with absorption bands in the visible (or just within the UV) spectrum are desirable as the absorption spectra of many organic compounds are transparent in this regime. As the energy used to photoexcite the photocatalyst increases, direct homolytic cleavage of bonds within both the PC and the organic substrates becomes more prevalent, resulting in undesired side reactions and, thus, limiting the usefulness of the process.
The yield of the formation of the excited state should be as high as possible, which relates to both the quantum yield of its formation, as well as the absorption cross section of the PC (as measured by the concentration and the molar extinction coefficient, ε).4 Ideally, the quantum yield, Φ(λ), for a photocatalytic reaction, as defined in eqn (1), should be as close to 1 as possible, for reactions not proceeding via a radical chain mechanism.
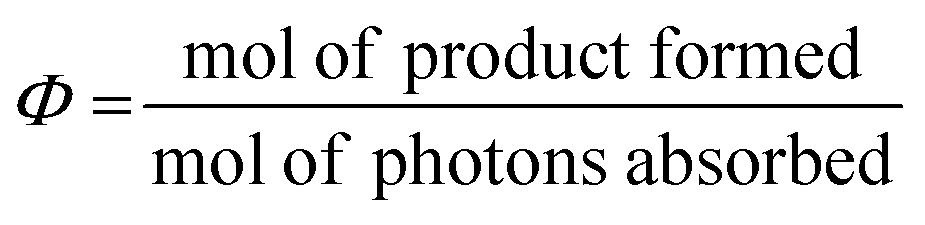 | (1) |
The intensity of the excitation source, the molar extinction coefficient of the PC at the excitation wavelength and the optical path length each affect the reaction rate.5 Additionally, the excited state of the PC must be sufficiently long-lived, on the order of at least a few nanoseconds, in order to overcome diffusion processes that are required to generate an encounter complex in these bimolecular reactions.6–10 The diffusion rates to generate the required encounter complex define a rate-limiting step in photocatalysis.
Traditionally, visible light PCs have been based around 4d- and 5d-transition metal complexes that possess low-lying charge transfer excited states. The most popular PCs remain those based on iridium(III) and ruthenium(II) complexes (cf.Fig. 9 and 10).11 These phosphorescent complexes possess long-lived excited states. For instance, [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) has a lifetime, τPL, of 1100 ns in acetonitrile12 while fac-Ir(ppy)3 (ppyH = 2-phenylpyridine) has a τPL of 1322 ns in acetonitrile (Table 1).13,14 However, despite their inherently attractive photophysical and electrochemical properties, the scarcity of the metals in these commonly used PCs, their expensive cost (1287 GBP/Oz and 215 GBP/Oz for iridium and ruthenium, respectively),15 and the unfavourable toxicity profiles are all detracting features that present barriers to wider and larger scale adoption of photocatalysis.16–18
| Photocatalyst | λ abs/nm | λ PL/nm | E 0,0/eV (kJ mol−1) | E ox/V | E red/V | E ox*/V | E red */V | τ/ns | Ref. |
|---|---|---|---|---|---|---|---|---|---|
a All potentials are given in V versus SCE. Eox* = Eox − E0,0 and Ered* = Ered + E0,0. 1 eV = (1.602 × 10−22 kJ) × NA where NA = Avogradro's constant. λabs refers to the absorption maximum of the lowest energy absorption band. λPL refers to the photoluminescence maximum. E0,0 values given correspond to the optical gap to the S1 state unless otherwise noted. τpf, τdf, τS and τT are the prompt fluorescence lifetime, the delayed fluorescence lifetime, the singlet excited state lifetime and the triplet excited state lifetime, respectively. Data reported in MeCN at room temperature unless otherwise noted.
b Determined in CH3CN:H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
c Determined in methanol.
d Determined in H2O.
e Determined in 10−4 M NaOH in an air saturated sample.
f Corresponds to the optical gap to the T1 state. :1).
c Determined in methanol.
d Determined in H2O.
e Determined in 10−4 M NaOH in an air saturated sample.
f Corresponds to the optical gap to the T1 state.
|
|||||||||
| 4CzIPN | 435 | 535 | 2.67 (258) | 1.52 | −1.21 | −1.04 | 1.35 | 18.7 τpf | 60, 114 and 119–121 |
| 1390 τdf | |||||||||
| fac-Ir(ppy)3 | 375 | 518 | 2.75f (265) | 0.77 | −2.19 | −1.73 | 0.31 | 1322 | 14, 122 and 126 |
| [Ir(ppy)2(dtbbpy)]PF6 | 410 | 581 | 1.21 | −1.51 | −0.96 | 0.66 | 557 | 122, 123, 127 and 128 | |
| [Ir(dF(CF3)ppy)2(bpy)]PF6 | 405 | 473 | 2.20f (212) | 1.69 | −1.37 | −1.00 | 1.32 | 2280 | 11 and 122 |
| [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 | 380 | 470 | 2.77f (267) | 1.69 | −1.37 | −0.89 | 1.21 | 2300 | 11 and 122–124 |
| [Ir(dF(Me)ppy)2(dtbbpy)]PF6 | 360 | 512 | 2.97f (287) | 1.49 | −1.44 | −0.97 | 0.92 | 1187 | 123 and 128 |
| [Ir(F(Me)ppy)2(dtbbpy)]PF6 | 543 | 2.44f (235) | 1.33 | −1.50 | −0.77 | 0.94 | 1100 | 123 | |
| [Ru(bpy)3](PF6)2 | 452 | 615 | 2.10f (203) | 1.29 | −1.33 | −0.81 | 0.77 | 1100 | 12 and 53 |
| [Acr-Mes]ClO4 | 430 | 570 | 2.65 (256) | −0.57 | 2.08 | 6.4 τS | 23, 107 and 129 | ||
| 30 000 τT | |||||||||
| Eosin Y | 520c | 537d | 2.31c (223) | 0.78b | −1.06b | −1.11b | 0.83b | 2.10cτS | 118 and 130–134 |
| 160 000dτT | |||||||||
| Rose Bengal | 558 | 576 | 2.17c (209) | 0.84c | −0.99c | −0.96c | 0.81c | 2.38 τS | 132 and 135–137 |
| 2400eτT | |||||||||
To address these problems, PCs based on either Earth-abundant metal complexes, previously reviewed by us and others,19,20 or organic fluorescent compounds6 have been developed. Organic PCs are generally inexpensive, possess lower toxicity profiles than transition metal complexes and are widely available.21 A broad range of organic PCs have now been studied. Notable examples include acridinium salts22,23 and organic dyes such as eosin Y,24,25 fluorescein26,27 and rose bengal28,29 (cf.Fig. 9 and 10), which all have moderate redox windows making them suitable in photoredox catalysis (Table 1).28 In multiple cases, these organic PCs have been shown to outperform their transition metal-based counterparts,30–34 although their limitations must still be considered. Lengthy and often complex syntheses may be required to produce the PC, which can reduce its desirability to industry. Moreover, there have also been reported problems with separation and low recyclability when using organic PCs.35 One of the main disadvantages with organic PCs is the limited flexibility in terms of tuning their redox potentials in comparison to transition metal complexes, whereby modification of the ligands generally permits a wide tuning of the redox properties.
Although these phosphorescent and fluorescent compounds have been well documented in the literature as PCs, beyond their ground and excited state redox potentials, their intrinsic photophysical properties have often been neglected when gauging their effectiveness as PCs. These compounds differ greatly in terms of available excited states, and thus their ability as PCs is quite different. Thus, a fundamental understanding of their photophysics must first be considered.
General photophysics of luminescent compounds
In photocatalysis, the first fundamental step involves formation of the excited PC, whereby a photoinduced transition from the ground state to an excited state must occur. The rate and feasibility of radiative transitions are dependent on the spin, vibrational and electronic wavefunctions of the initial and finial electronic states. The probability of transitions to/from these states are reflected in the oscillator strength, f, which takes into account orbital, vibrational and spin configuration factors and varies between 0 and 1.36For spin considerations, only transitions between states of equivalent spin are formally allowed (spin selection rule), although this can be relaxed through spin orbit coupling (SOC).
The dependence of the vibrational wavefunctions is in accordance with the Frank–Condon factor, F, defined in eqn (2), where Ψvib,f and Ψvib,i are the final and initial vibrational wavefunctions, respectively. This gives the transition probability, which for absorption reflects the transition from the zeroth vibrational level of the ground state to the mth vibrational level of the excited state. A greater overlap of the vibrational wavefunctions will lead to a greater probability that the transition occurs.
| F = |〈Ψvib,f|Ψvib,i〉|2 | (2) |
The electronic factor dictates the overall intensity of the transition and is proportional to the overlap of the initial and final state wavefunctions, as well as the transition dipole moment.
Therefore, to maximize the probability of a transition, the spin must be conserved, the vibrational and electronic wavefunctions of the initial and final states should each overlap strongly and a change in symmetry must occur. The more probable an absorption transition, the greater the associated molar absorptivity, ε, a beneficial factor to the efficiency of a photocatalytic process.
Fig. 2 shows a Jablonski diagram outlining many of the different photophysical and photochemical processes available to the excited PC. The probable pathways for each of fluorescent, phosphorescent and TADF PCs depend on the relative magnitudes of the rate constants for each class of PC. The photophysical processes shown in Fig. 2 are mononuclear; however, the photocatalysis implicates bimolecular reactions. Promotion of an electron from the closed shell singlet ground state, S0, to a higher-level singlet excited state, Sn, occurs upon photoexcitation, with the initially accessed excited state based on the energy of the excitation source. Rapid vibrational relaxation and internal conversion (IC) occur typically within picoseconds, causing the excited electron to populate the lowest vibrational level within the lowest singlet electronic excited state, S1, in accordance with Kasha's rule.37 Radiative decay, in the form of fluorescence, or non-radiative decay can ensue from S1 to regenerate the ground state, which are both spin-allowed processes, hence the lifetime of molecules in the S1 state is short, on the order of a few nanoseconds. Intersystem crossing (ISC) can also occur from S1 to the triplet manifold, although this a formally spin-forbidden process (see below for a detailed discussion of ISC rates). Subsequent IC results in population of the lowest triplet excited state, T1, which can decay radiatively (via phosphorescence), or non-radiatively to the ground state, both of which are spin-forbidden processes. On account of the formally spin-forbidden nature of these decay process from the T1 state, the triplet excited state is typically much longer-lived, with a lifetime on the order of microseconds.
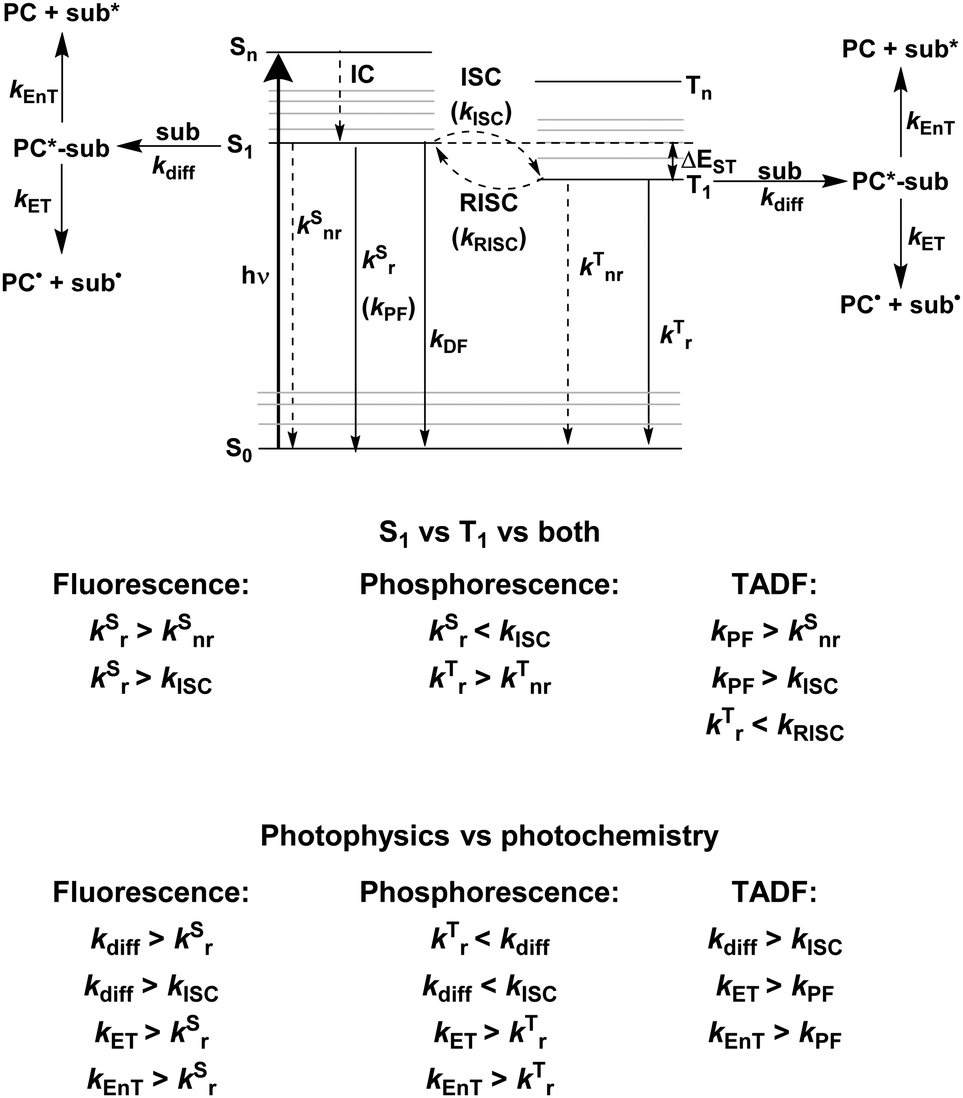 | ||
| Fig. 2 Jablonski diagram for luminescent compounds alongside possible photochemistry routes of the excited states. The prototypical rate constants for defining the dominant photoactive excited state for fluorescent, phosphorescent and organic TADF compounds are given alongside the required rate constants for productive photocatalysis, where kEnT and kET are the rate constants for energy and electron transfer, respectively, kdiff is the rate constant for diffusion, kSnr and kSr are the rate constants for non-radiative and radiative decay, respectively, from the singlet state, kTnr and kTr are the non-radiative and radiative decay, respectively, from the triplet state and kPF and kDF are the rate constants for prompt and delayed fluorescence, respectively. | ||
These are the available photophysical processes of all compounds, with the dominating processes depending upon the intrinsic properties and structure of the molecule in question and will be discussed in detail in the following sections. The rate constant for diffusion, kdiff, to form an encounter complex, corresponds to the first step in a photochemical reaction, and as previously mentioned, is a bimolecular process on the order of 109 M−1 s−1.38 An encounter complex can be formed between the substrate and either 1PC* or 3PC*. Which encounter complex is most likely, is dependent upon the magnitude of the intersystem crossing rate constant, kISC. When kISC < kdiff, this implies the singlet excited state of the PC is involved in encounter complex formation, while when kISC > kdiff, the triplet excited state of the PC participates. It is also possible that kISC ∼ kdiff, in which case both 1PC* or 3PC* are available to participate in the photochemistry. Upon formation of the encounter complex, subsequent electron and energy transfer can occur, with relative rate constants of kET and kEnT, respectively. These must occur sufficiently fast to outcompete alternative radiative and non-radiative decay pathways from the excited PC.
To ensure that photochemistry occurs (PET and/or PEnT), the associated rates for these processes must outcompete those of alternative photophysical pathways. To simplify the analysis of competing pathways, it can be helpful to use a PC with a high photoluminescence quantum yield, ΦPL, (eqn (3)), where in this limiting case only the radiative decay rate constant, kSr or kTr, need be considered as a competing pathway as either will outcompete knr. This simplifies the analysis as the only competitive pathway for PET and PEnT that must be considered is radiative decay, which can be experimentally determined through emission lifetime measurements. The rate of radiative decay must still be slow compared to kET and kEnT. However, a high PLQY is not a necessity for a compound to be a useful PC. Ultimately, as long as kET and kEnT are faster than all other mononuclear decay processes, radiative or non-radiative, then photocatalysis is kinetically feasible.
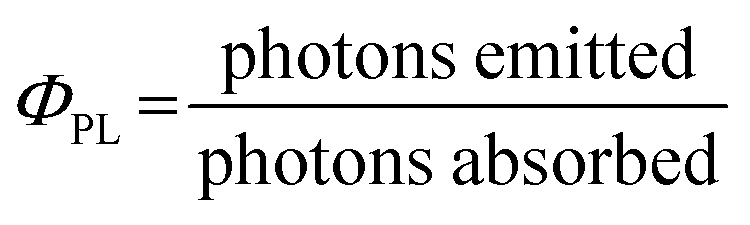 | (3) |
The rate constant kISC is key to determining whether 1PC* or 3PC* is involved in the subsequent photocatalysis, the importance of these states will be discussed in the photocatalysis mechanism section. As ISC is a formally spin-forbidden process, it is important to understand the factors that moderate the kinetics of this pathway.
Spin-forbidden transitions are only possible when state mixing occurs. This transpires with the aid of spin orbit coupling (SOC), which is a relativistic effect.39 SOC occurs through coupling of the total spin angular momentum, ![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) , and total orbital angular momentum,
, and total orbital angular momentum, ![[L with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004c_20d1.gif) , to yield a total angular momentum,
, to yield a total angular momentum, ![[J with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004a_20d1.gif) , defined by the vector sum in eqn (4).40 Since the total angular momentum must be conserved during a transition, a change in can be compensated for through a corresponding change in , to allow for to remain constant. This allows for the spin flip of an electron, required for processes such as ISC, RISC or phosphorescence to be feasible. Hence, a formally spin-forbidden process becomes possible through the perturbation of singlet and triplet state wavefunctions, which is made possible by SOC.
, defined by the vector sum in eqn (4).40 Since the total angular momentum must be conserved during a transition, a change in can be compensated for through a corresponding change in , to allow for to remain constant. This allows for the spin flip of an electron, required for processes such as ISC, RISC or phosphorescence to be feasible. Hence, a formally spin-forbidden process becomes possible through the perturbation of singlet and triplet state wavefunctions, which is made possible by SOC.
| = + | (4) |
The importance of SOC is manifested in terms of El Sayed's rule,41 which imposes a symmetry change between the Sn and Tn state in order to facilitate spin-forbidden ISC. This rule was initially developed to describe ISC in carbonyl compounds and states that there must be a change in orbital angular momentum in concert with the change in spin. This implies that rates of spin-forbidden processes, such as ISC, become faster when there is a greater difference in orbital symmetry of the excited states. For example, 1(π–π*) → 3(n–π*) is fast while 1(π–π*) → 3(π–π*) is formally forbidden. However, it should be noted that this rule assumes the involvement of pure electronic states in a static nuclear configuration.
Within the El Sayed framework, there is the intrinsic spin–orbit coupling constant associated with each atom involved in the transition. As the mass of the atom increases, so does the magnitude of the SOC; this is known as the heavy atom effect. The degree of state mixing is also dependent on the energy difference between the two states, as shown in eqn (5). The first order mixing coefficient between the S1 and T1 states, λ, is proportional to both the direct spin–orbit coupling (HSO) (which is itself proportional to the fourth power of the atomic number (Z) of atoms involved in the S1 and T1 transitions divided by the third power of the principal quantum number (n)), and on the aforementioned difference in orbital symmetry between the two states, and inversely proportional to the energy gap between the S1 and T1 states, ΔEST.
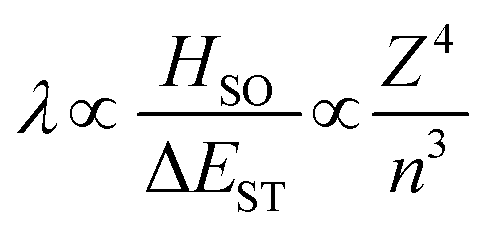 | (5) |
This description of state mixing is suitable for static nuclear configurations. When vibrational motion is taken into account, both spin-vibronic coupling and SOC must be considered. This can be done using second-order perturbation theory, which involves both spin–orbit and non-adiabatic coupling, with additional intermediate state(s) mediating the coupling.42,43 This provides a route for El Sayed-forbidden ISC to occur between S1 and T1. These factors play a role, to varying degrees, to explain the importance of ISC as a potentially competing pathway in Fig. 2 for fluorescent, phosphorescent and thermally activated delayed fluorescence (TADF) compounds.
An additional consideration to be made is the nature of the excited state. For organic compounds, there are two predominant types of excited states: locally excited (LE) or charge transfer (CT). In the former, the electron density distribution is retained in the same area of the molecule during the transition, which usually involves the promotion of an electron from a π-orbital to a π*-orbital in a symmetry-allowed transition. In the latter, the electron density distribution is displaced from one part of the molecule, the donor moiety, to another, the acceptor moiety. Here, the overlap of the orbitals involved in the symmetry-allowed transition is smaller and so the probability of the transition is lower, reflected in the smaller molar absorptivity values observed in the absorption spectrum of the compound.
Transitions to/from LE states usually occur with a higher oscillator strength in comparison to the CT state, since transitions to/from LE states exhibit a greater amount of orbital overlap. Accordingly, LE absorptions will have a greater ε, and from this, may seem like the preferential state to access in terms of photocatalysis. However, these LE states are typically shorter-lived than CT states owing to their larger radiative rate constants, kr, according to Fermi's golden rule.44,45
There are of course some exceptions to the relative behaviour of LE and CT states typically observed, which presuppose that both transitions are symmetry-allowed. Pyrene is a notable example where transitions to/from the LE state (a S0 → S1 transition) of this rigid organic molecule are weak despite strong spatial overlap of the relevant orbitals. This is due to the similar symmetries of the HOMO and LUMO wavefunctions, which results in a symmetry-forbidden transition that is reflective of the low oscillator strength (f = 0.0016 in n-heptane).46 Consequently, a weak fluorescence is observed with a much longer radiative decay rate (kr ≈ 106 s−1).47 A long-lived excited state is preferable in terms of photocatalysis, as if the LE (or CT) state decays quicker than the rate at which an encounter complex can form, productive photocatalysis cannot occur. Therefore, although generally the longer-lived CT states would be more advantageous than the shorter-lived LE states for photocatalysis, the example of pyrene illustrates that LE states are not always short-lived. Thus, emission lifetimes should be explicitly determined to assess whether the excited state of the compound is sufficiently long-lived for it to act as a photocatalyst.
Further, the energy of the CT state (but not the LE state) can be significantly influenced by the polarity of the medium. The size of the Stokes shift in a particular solvent, and hence the available energy to initiate the photocatalysis, becomes dependent on whether the excited state is LE or CT in nature. For most of the PCs discussed, the CT excited states have a larger associated transition dipole moment (TDM) in comparison to the permanent dipole moment associated with the ground state. In these cases, increasing polarity of the medium is associated with a more stabilized CT excited state, reflected in a red-shifted emission (positive solvatochromism).48 The energy of LE states is relatively independent of medium as there is no large TDM, hence the Stokes shift will be small. In the context of photocatalysis, a smaller Stokes shift would be preferred as this indicates that most of the energy absorbed is available for the photochemistry.
This behaviour is present in not only organic compounds, but also organometallic complexes with excited states with CT character, such as metal to ligand charge transfer (MLCT) or a ligand to metal charge transfer (LMCT) states.
Consideration of these fundamental photophysical concepts is important when discussing the pros and cons of each subcategory of compounds used in the literature as PCs.
Fluorescent compounds as photocatalysts
The vast majority of fluorescent compounds are organic molecules, with photophysical processes occurring as described by Fig. 2. Fluorescence, in almost all cases, occurs from the S1 state (adhering to Kasha's rule), hence, is independent of the excitation wavelength. For fluorescence to be detected, the non-radiative decay rate originating from S1 must not be more than three orders of magnitude greater than kr; with knr two orders of magnitude faster than kr, emission will appear very faint.49The rate constants that typically define a successful fluorescent PC are shown in Fig. 2. Fluorescent compounds possess slow kISC and so there is little to no population of the T1 state. This is a generalised statement, typically organic fluorescent compounds have kISC in the region of 106 s−1.50–52 Assuming the rate constant for fluorescence is faster than kISC, fluorescence is the only radiative decay process possible.
For productive photocatalysis, diffusion of the excited PC to form the encounter complex must ensue and be faster than kSr (and kISC and kSnr) to allow for bimolecular reactions to compete with monomolecular photophysical decay pathways of the excited state. Typically, if the lifetime of the singlet excited state is less than 1 ns, the compound will not be useful as a PC as the compound will fluoresce before diffusion to the substrate can occur.6 Upon formation of the encounter complex, kET and kEnT must then be sufficiently fast to occur prior to dissociation of the encounter complex and to outcompete alternative decay pathways for the excited PC in terms of radiative and non-radiative decay.
Phosphorescent compounds as photocatalysts
Phosphorescent compounds are typically transition metal complexes. The presence of the central metal allows for these compounds to readily access the triplet excited state on account of the heavy atom effect. This increases the magnitude of the SOC, in accordance with eqn (5), and relaxes the spin-forbidden nature of the ISC. As a result, rapid population of the T1 state occurs, which is long-lived with τPL typically on the order of microseconds.In most low oxidation state transition metal complexes surrounded by π-accepting ligands, absorption of a photon typically results in promotion of an electron from a metal d-orbital to a ligand π* orbital,53,54 which describes a MLCT transition and is symmetry-allowed. Rapid ISC from the singlet to the triplet MLCT state requires these states to have different symmetry for a direct transition to be allowed, for example when the π* orbital is the same, but the d-orbital is different.55 When the two MLCT states have the same symmetry, an intermediate state of different symmetry must instead mediate the ISC. Typically, ISC occurs from the S1 state with 1dπ* character, to a higher lying triplet state, Tn, as S1 and T1 have analogous electronic transitions, hence the ISC is symmetry-forbidden.55 Since ISC is usually very fast, (∼1010–1012 s−1 regime for complexes with strong MLCT character in the excited state),55,56kISC therefore outcompetes kSr by several orders of magnitude. The presence of the heavy metal contributes to near unity intersystem crossing quantum yield, ΦISC, being frequently observed.57 Thus, the rapidly generated, long-lived 3MLCT state is the only accessible excited state when considering the subsequent photochemistry. This implies that kISC > kdiff; however, kdiff must be faster than kTr and kTnr to allow for formation of the encounter complex. As these latter two rate constants are typically much slower, this is not an issue. Subsequent kET and kEnT must also be fast processes with respect to other decay pathways of the excited PC.
TADF compounds as photocatalysts
A third class of homogeneous PCs that can be used are TADF compounds. Although examples of organic, organometallic and inorganic TADF compounds have each demonstrated photocatalytic behaviour,58,59 this review focuses solely on the use of the organic analogues.The S1 state is rapidly populated upon photoexcitation. Radiative decay can then occur by direct fluorescence, known as prompt fluorescence (PF), with a lifetime of τpf on the order of nanoseconds, analogous to that observed for normal fluorescent compounds. Alternative competing pathways include non-radiative decay back to the ground state or ISC to the triplet manifold followed by IC such that the electron populates the lowest triplet excited state, T1. Phosphorescence from this state remains a spin-forbidden process and so is typically not observed at ambient temperatures in these molecules (due to large ΔES0T1, cf.eqn (5)); however, reverse intersystem crossing (RISC) from T1 back to S1 is possible at ambient temperatures due to the small singlet–triplet energy gap, ΔEST, between the lowest lying singlet and triplet excited states that can be overcome thermally, resulting in delayed fluorescence (DF), which occurs on a time scale of similar order to phosphorescent complexes, typically in the microsecond regime.60 RISC can also be mediated by spin-vibronic coupling, as discussed earlier (Fig. 3). As shown in Fig. 2, the rate constant for prompt fluorescence, kPF, is greater than kISC (and kSnr), which explains why prompt fluorescence is observed. Delayed fluorescence occurs as kRISC is faster than both kTr and kTnr.
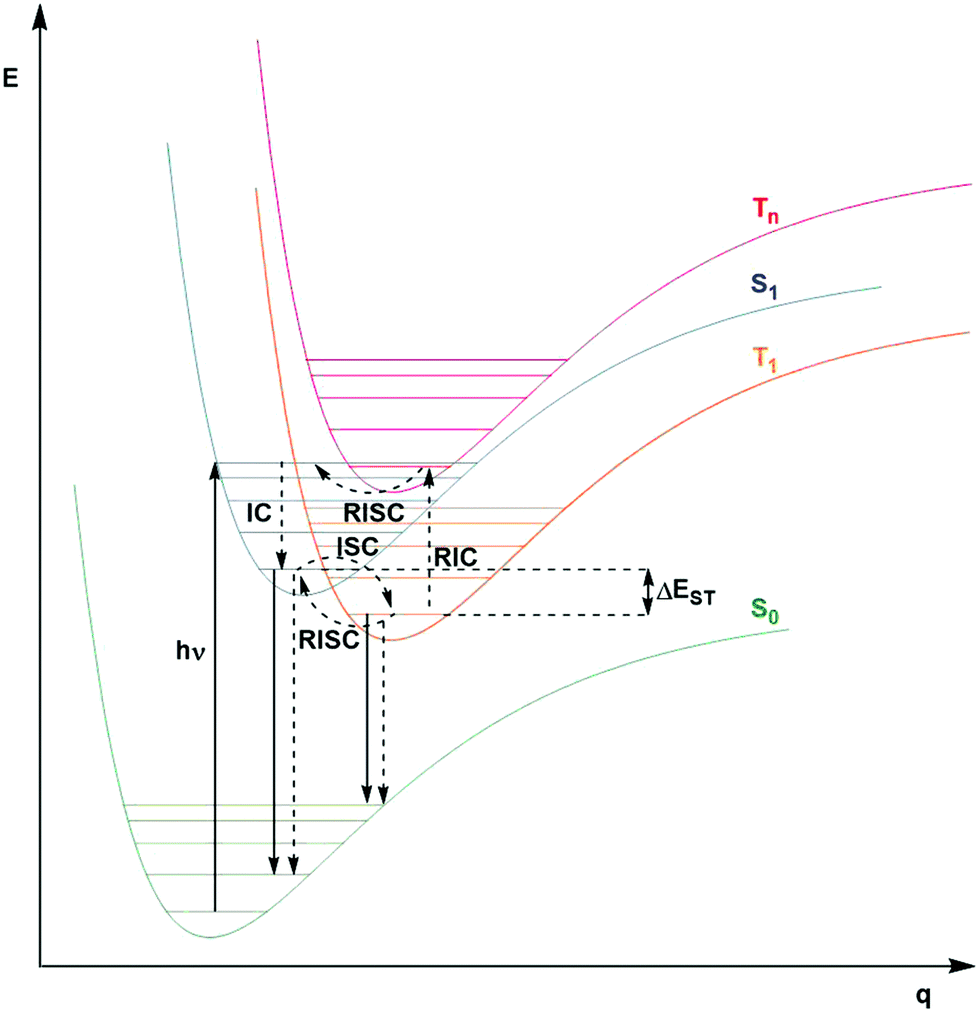 | ||
| Fig. 3 Modified Jablonski type diagram with Morse potentials, showing the photophysical processes of TADF molecules including photon absorption, radiative (solid arrow) and non-radiative (dashed arrow) decay, internal and reverse internal conversion (IC and RIC, respectively), intersystem and reverse intersystem crossing (ISC and RISC, respectively) along nuclear coordinate, q. | ||
For organic TADF compounds kISC is typically <107 s−1,56 although this value can vary widely depending on the individual compound. For this class of compounds kISC < kdiff, hence PET and PEnT are most likely to occur from the S1 state. For TADF compounds in which faster kISC rates are observed, this opens the availability of the triplet manifold to participate in photocatalysis. When this is the case, the relative populations of the singlet and triplet excited states, which are dependent on the quantum yields of prompt, ΦPF, and delayed fluorescence, ΦDF, respectively, become significant in identifying the probablity of PET and PEnT from each state.
The excited state from which the photocatalysis occurs is directly dependent on both kISC and kRISC in TADF PCs. The rates associated with ISC/RISC are, to a first approximation, understood through the relation disclosed in eqn (5), where the rate constants are dependent upon both HSO and ΔEST.
Organic TADF compounds distinguish themselves by possessing a small ΔEST, typically less than 0.3 eV,61 which is the primary factor that governs the degree of the mixing of states.62 The ΔEST itself is proportional to the exchange integral, J, between the excited singlet and triplet states, according to eqn (6).61,63 The exchange integral is dependent on the orbital overlap of the frontier molecular orbitals involved in the transitions to S1/T1 (eqn (7)), where Φ and Ψ are typically the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) wavefunctions, respectively, as these are involved in the S0–S1 and S0–T1 transitions, and e is the electron charge.
| ΔEST = ES − ET = 2J | (6) |
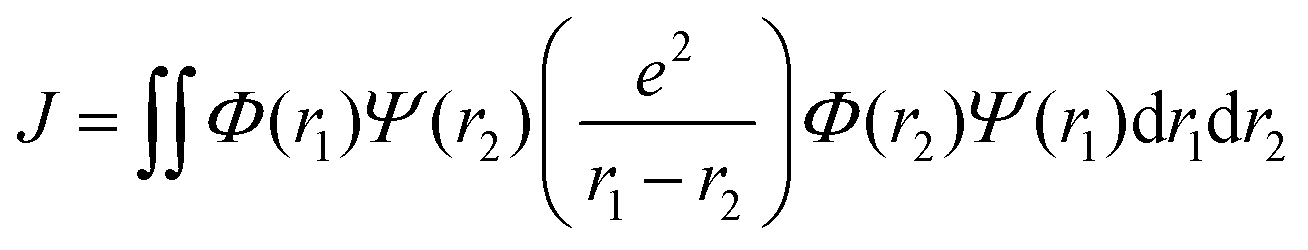 | (7) |
From eqn (7), it becomes clear that by minimising the overlap of the electron density distribution between the HOMO and LUMO, a reduction in J will be observed and hence a small ΔEST will be obtained.61 Spatially separated and electronically decoupled donor–acceptor compounds in which there are large torsions between the donor and acceptor groups are thus the most commonly employed molecular design strategies invoked in TADF emitters, in which the HOMO and LUMO are localised on the donor and acceptor units, respectively.64
Since the excited singlet state of TADF compounds is CT in nature, positive solvatochromism will be exhibited. Therefore, solvent choice should be considered when utilising these compounds as PCs, as it will affect not only the excited state redox potentials, but also the relative competition between kISC/kRISC and therefore from which state the PET or PEnT originates.
Photocatalysis mechanism
All three subcategories of luminescent compounds previously discussed; fluorescent, phosphorescent and TADF, have been successfully applied as photocatalysts in both PEnT and PET reactions. An understanding of their intrinsic photophysical properties is required in order to correlate the performance of the photocatalysts to their structure and to distinguish under which mechanism the photocatalysis takes place, a point which is all too often not adequately discussed in the photocatalysis literature.PEnT photocatalysis
In the photoinduced energy transfer (PEnT) mechanism, the excited PC sensitises the substrate by direct energy transfer (Fig. 1). Upon acceptance of the embedded energy, the substrate is in an electronically excited state, allowing for the subsequent reaction to occur.65 This energy transfer process can proceed via Förster or Dexter quenching mechanisms, which are both non-radiative decay processes.66 The former involves dipole–dipole coupling interactions mediated through space, whereby the transition dipole moment on the donor (the excited PC) and the acceptor (the substrate) couple non-radiatively, generating the required electronic transition through relaxation of the donor and simultaneous excitation of the acceptor.67 Since this mechanism is dipolar in nature, orbital overlap of the two species is not required, allowing for Förster resonance energy transfer (FRET) to occur over long distances (1–10 nm).68 Spectral overlap of the emission of the donor and absorption of the acceptor species is instead crucial for this mechanism, as quantified in the overlap integral, JF, in eqn (8), where fD is the normalised donor emission spectrum, [(fD(σ) is the photon exitance of the donor at wavenumber σ], εA(σ) is the molar extinction coefficient of the acceptor at wavenumber σ, and σ is the wavenumber.69,70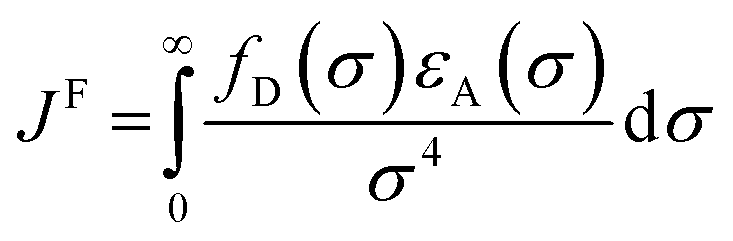 | (8) |
The distance dependence associated with FRET in indicated in eqn (9), which shows the dependence of the rate constant of FRET, kFRET, on both the spectral overlap integral and the donor–acceptor distance, RDA.67 The (RDA)−6 distance dependence of FRET allows for this energy transfer to still be possible at longer distances. In eqn (9), κ2 is the orientation parameter, n is the refractive index of the medium, NA is Avogadro's constant and τ0r(D) is the lifetime of the donor in the absence of the acceptor.
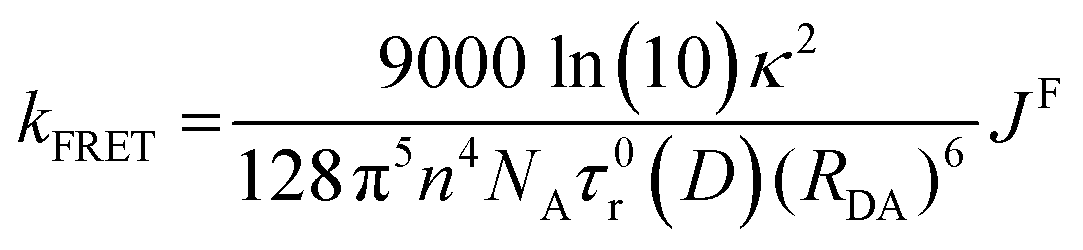 | (9) |
By contrast, the Dexter energy transfer (DET) mechanism typically involves simultaneous intermolecular electron exchange between the excited state of the donor, the excited PC, and the ground state of the acceptor, the substrate.71 The DET mechanism is electric-dipole forbidden on account of the change in the local spin state of the excited donor and acceptor, which disables the FRET mechanism.72
For DET, orbital overlap is essential, which is achieved through collisional interaction for bimolecular PEnT mechanisms. To aid this orbital overlap, easily accessible spin density on the excited PC is beneficial, which is linked to the shape of the PC. Spectral overlap of the emission of the donor (phosphorescence) and absorption of the acceptor (singlet to triplet absorption) is likewise important, which is calculated according to eqn (10) for DET, where fD is the normalised donor emission spectrum and εA is the normalised acceptor absorption spectrum.66,70 Unlike Förster PEnT, which requires spectral overlap of spin-allowed transitions, for Dexter PEnT the spectral overlap required is for spin-forbidden transitions and so the overlap integral is inherently smaller in this case.
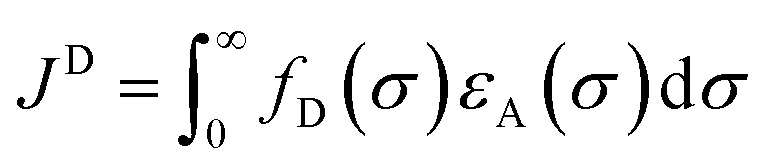 | (10) |
The absence of information on spin-forbidden transitions for many organic substrates means that assessing whether spectral overlap occurs becomes challenging. In light of this, the energy of the triplet state, ET, of the donor (the photocatalyst) relative to the acceptor (the substrate) is instead used to provide an estimation of the thermodynamic feasibility of the energy transfer mechanism.66 The criterion for an exergonic energy transfer thus is ET (D) > ET (A). In this instance, it is assumed that JD is large, which correlates to a faster rate of DET according to eqn (11), where RDA is the donor–acceptor distance, K is the parameter for specific donor and acceptor orbital interactions and L is the sum of the van der Waals radii of the donor and acceptor.71
If, however, ET (D) < ET (A), then energy transfer from donor to acceptor is only possible if the donor, in its vibrationally/rotationally excited state of its electronically excited state, is higher in energy than the acceptor in its electronic excited state. Spectral overlap between the emission of the donor and the absorption of the acceptor remains a criterion for energy transfer.
The main drawback of the Dexter mechanism is that it is limited to shorter distances of typically no more than 10 Å due to the exponential dependence of the energy transfer rate with donor–acceptor distance, eqn (11).73 Comparing the two energy transfer mechanisms, both FRET and DET require spectral overlap for the energy transfer to occur; however, the latter also requires orbital overlap as it is a double electron exchange mechanism. For bimolecular mechanisms, since DET requires collisional interactions, higher reactant concentrations are beneficial for this mechanism, while FRET can operate efficiently at lower concentrations.69 At all but the shortest donor–acceptor distances, FRET will be faster than DET, due to the RDA−6 distance dependence of FRET (eqn (9)) in comparison to the exponential distance dependence of DET (eqn (11)).
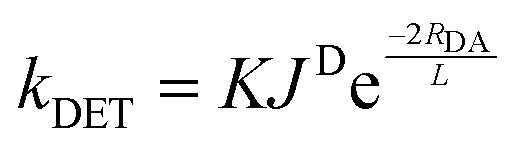 | (11) |
At larger distances, FRET occurs at a faster rate and is consequently the dominant PET mechanism while DET only becomes the dominant pathway when FRET is not allowed. Regardless of which PEnT mechanism is in operation, a sufficiently fast rate of energy transfer is required in order to outcompete other radiative and non-radiative decay pathways available to the excited PC. Similarly, for exergonic energy transfer, both mechanisms require the excited state energy of the donor to be greater than that of the acceptor.
The nature of the FRET and DET mechanisms mean compounds with a predominately photoactive singlet state, namely fluorescent compounds, undergo energy transfer through a FRET mechanism while phosphorescent compounds, whose photophysics relies on triplet excited states, undergo DET, since the FRET mechanism would result in breaking of the spin conservation rule of Wigner.66,74
Some energy transfer photochemistry proceeds via the singlet excited state of the substrate while other photochemistry requires accessing the triplet excited state of the substrate, and the precise nature of the photochemical reaction must be considered when designing a PC that can participate in PEnT photocatalysis. For example, in the Paternò–Buchi [2+2] cycloaddition reaction of 2,3-dihydrofuran with different aromatic carbonyls (Fig. 4), the nature of the excited state of the carbonyl impacts upon the diastereoselectivity of the reaction. Naphthaldehydes react through excited singlet states while benzaldehydes (such as mesitylaldehyde), react through excited triplet states.75
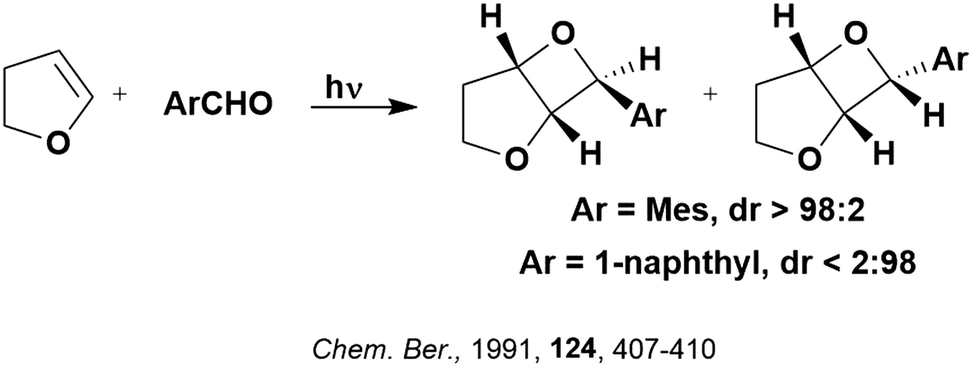 | ||
| Fig. 4 Reaction scheme for the Paternò–Buchi [2+2] cycloaddition reaction of 2,3-dihydrofuran with different aromatic aldehydes. | ||
Another example of the importance of singlet state photochemistry is the Norrish–Yang photocyclization.76 When using an alanine derivative, and in the presence of a triplet quencher, the cyclization proceeds through a helically chiral singlet biradical, conveying a “memory of chirality” effect to the product.77 If the reaction proceeds in the absence of a triplet quencher, the longer-lived triplet biradical forms and this leads to racemization, destroying the enantioconservation.
Triplet state sensitisation occurs through the DET mechanism, typically forming triplet biradical intermediates. These tend to have lifetimes of the order of nanoseconds to microseconds, which is enough time to allow for single bond rotations to occur.78 By contrast, singlet biradical intermediates, formed through a FRET mechanism, are much more short lived,79 hence the configuration found in the starting material is retained in the product. High enantioconservation is thus typically observed with singlet photochemistry.80–82 This is also possible with triplet biradical intermediates which only slowly interconvert.83,84
The benefit of TADF compounds is that they have the potential to access both FRET and DET pathways. However, since kISC is typically slower than kdiff, DET from the triplet state is inefficient for organic TADF compounds, much more so than for phosphorescent complexes. The capacity for organic TADF compounds to participate in triplet DET is dependent on the intersystem crossing quantum yield, ΦICS, which describes the population of PCs in their triplet excited states.85 Under the constant irradiation conditions employed in photocatalysis and given the long delayed lifetimes of most organic TADF PCs, there is sufficient population of triplet excitons to permit triplet sensitization reactions by organic TADF PCs. Additionally, it should be noted that DET is not limited to PEnT from triplet states, and can indeed occur from the singlet excited state.86 Since TADF compounds have singlet excited states that are accessible for subsequent photochemistry, this opens the pathway for DET energy transfer to occur from this state.
Undoubtedly, TADF compounds can access the FRET pathway, a mechanism exploited in the sensitization of fluorophores by TADF compounds, coined Hyperfluorescence™, that has been exploited in OLED developement.87 Accessing the FRET mechanism is beneficial in photocatalysis as it allows the reaction to occur over much longer distances, implicating lower required concentrations. However, as stated above, this relies on the singlet radical intermediate formed being productive in forming the required product.
PET photocatalysis
Photoredox catalysis involves in nearly all cases single electron transfer (SET) processes. Photocatalysis proceeding by PET is more prevalent than by PEnT and the majority of the examples in this review proceed via photoredox catalysis. It is important to first understand both the thermodynamic and kinetic factors at play when discussing PET.Upon absorption of light, the excited PC becomes both a stronger oxidising and reducing agent than in the ground state. It can then either be oxidatively or reductively quenched via SET (Fig. 1), depending on the relative energies of the molecular orbitals of the substrate compared to the PC. The PC, in its oxidized or reduced form, respectively, can then participate in a second SET that will close the photocatalytic cycle and regenerate the ground state of the photocatalyst. Typically, a sacrificial oxidant or reductant is employed to complete or initiate the photocatalytic cycle; however, if none is required then the process is considered redox neutral. Photoredox catalysis typically produces radical ion intermediates as a result of the SET, which are usually difficult to access using conventional chemical synthetic procedures.88
The redox properties of a PC define its oxidising and reducing power. The ground state reduction and oxidation potentials are defined as Ered = (PC/PC˙−) and Eox = E(PC˙+/PC), respectively, while the excited state reduction and oxidation potentials are given by Ered*(PC*/PC˙−) = Ered(PC/PC˙−) + E0,0 and Eox*(PC˙+/PC*) = Eox(PC˙+/PC) − E0,0,6,9,89,90 where E0,0 is the lowest energy electronic transition between the lowest vibrational states of the ground and first excited state, otherwise known as the optical gap. In fluorescent compounds, this is defined as the intersection point between the normalized absorption and emission spectra of the PC, and typically involves a transition between the S0 and S1 states.69 By contrast, in phosphorescent compounds, the E0,0 is correlated with the triplet state energy, ET, and is estimated from the onset of the phosphorescence spectrum.
According to Marcus theory, the redox potentials of the PC must be sufficiently oxidising or reducing to allow the required SET to occur. To permit the maximum reactivity it is therefore desirable that the PC have a wide redox window, both in the ground and the excited states. When proceeding via a reductive quenching cycle, the PC is described as a photooxidant. To ensure that oxidation of the substrate is thermodynamically feasible, v must be more positive than Eox of the substrate. By contrast, for an oxidative quenching cycle, the PC behaves as a photoreductant. In this case, Eox* must be more negative than Ered of the substrate for the reduction to take place.6,9,19,89
These criteria are encapsulated within the Gibbs energy of photoinduced energy transfer (eqn (12)),9,89,91 which relates the change in the Gibbs free energy, ΔG0, and hence the thermodynamically feasibility of the reaction, with the ground state redox potentials and the optical gap. In eqn (12), e is the electronic charge, E0(D˙+/D) is the oxidation potential of the donor, E0(A/A˙−) is the reduction potential of the acceptor, ε0 is the vacuum permittivity, ε is the dielectric constant of the solvent, r is the electron donor–acceptor distance and E0,0 is the optical gap of the photoexcited species.
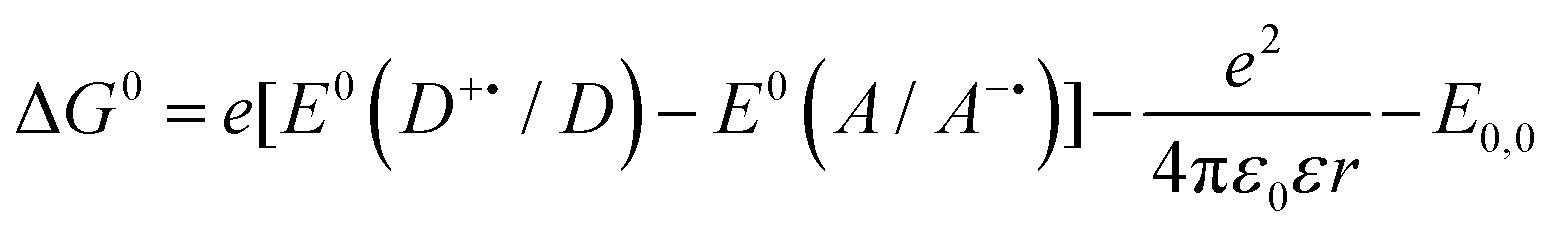 | (12) |
For the electron transfer to be thermodynamically allowed, ΔG0 must be negative. Since the excited triplet state is lower in energy than the excited singlet state, the absolute ΔG0 value for electron transfer from the triplet state is smaller than that from the singlet state. Taking this into consideration, this information can be applied to the kinetics of electron transfer, which is explained using the Marcus equation (eqn (13)).10,92 Here, the rate of electron transfer, kET, is linked to the electronic coupling between the initial and final states, Hif, the reorganisation energy, λ, and ΔG0.
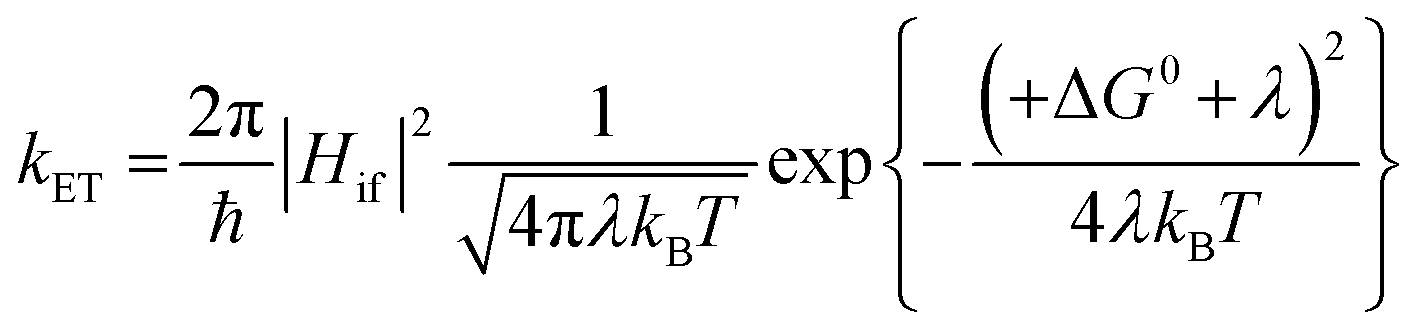 | (13) |
When ΔG0 is equal to λ, this represents a situation in which the activation energy, ΔG‡, is equal to zero and represents the maximum rate of the electron transfer. In the normal Marcus region, increasing the absolute value of ΔG0 causes a decrease in ΔG‡ and an increase in kET. However, in the inverted Marcus region, the opposite relationship is observed, whereby increasing the thermodynamic driving force, ΔG0, actually causes a decrease in kET (and an increase in ΔG‡). In PET from an excited compound to another reactant, namely the forward electron transfer, the normal Marcus region is in operation;9,89 however, in back electron transfer the inverted Marcus region has been observed.93–102 In the forward PET regime, in the highly exergonic region, the absence of the inverted region is thought to be related to λ, which increases in value on going from a contact radical ion pair to a solvent separated radical ion pair since the latter involves a greater distance between the radical ions.103
Considering only forward electron transfer first, since the absolute value of ΔG0 is smaller when electron transfer occurs from the triplet state, the rate of electron transfer, according to eqn (13), is slower when arising from the triplet state rather than the singlet.104,105 Recall that kET must be sufficiently fast to outpace unproductive decay pathways, both radiative and non-radiative. With these thermodynamic and kinetic factors in mind, the nuances in PET mechanisms of fluorescent, phosphorescent and TADF compounds can be understood. Since fluorescent compounds are usually dominated by singlet excited states, PET occurs from this state. For phosphorescent compounds, the triplet excited state is the assumed state from which electron transfer can occur. Despite kET being slower from triplet excited states, this does not present much of a problem in phosphorescent compounds since the T1 states are so long-lived, reflecting slower radiative and non-radiative rates.
For TADF compounds, the situation is less clearly ascertained. Since TADF compounds are characterised by small ΔEST, ΔG0 and consequently kET will be similar from both the singlet and triplet states, suggesting that there is negligible difference in electron transfer rates from these states when using TADF compounds as PCs.
Since kET is rate-limited by kdiff, it is therefore less than or equal to 109 s−1. As this is faster than the typical kISC observed for organic TADF compounds (<107 s−156), the excited state involved in SET is likely to be the S1 state. If kISC and kRISC are fast in comparison to kET, then a steady-state approximation may be assumed, and the relative populations of singlet and triplet excited states will be governed by a Boltzmann distribution. The majority of excitons will thus exist in the triplet state, on account of its lower energy.106 The exact populations of these states are determined by the respective quantum yields of the prompt and delayed fluorescence. SET under this scenario is thus more likely to occur from the 3PC*.
Therefore, on account of the small ΔEST of TADF compounds, the reactivity and kinetics of PET from both singlet and triplet excited states are similar, hence from this point of view, both states would be equally productive in photoredox catalysis.
Back electron transfer (BET) can be a detrimental path in photocatalysis. As previously mentioned, BET has been shown to occur through the inverted Marcus region, whereby the rate of electron transfer increases with decreasing thermodynamic driving force. This is exemplified in the selective oxygenation of p-xylene to p-tolualdehyde, using 10-methyl-9-phenylacridinium perchlorate ([AcrPh]ClO4), as the PC (Fig. 5).107 The proposed mechanism involves PET from p-xylene to the singlet excited state of the PC, as supported by previous work from the same group.108 Deprotonation of the resultant p-xylene radical cation occurs in competition with BET. When the former outcompetes the latter, a p-xylenyl radical is formed, which is trapped by oxygen, reduced by the reduced PC and decomposed to yield p-tolualdehyde selectively.
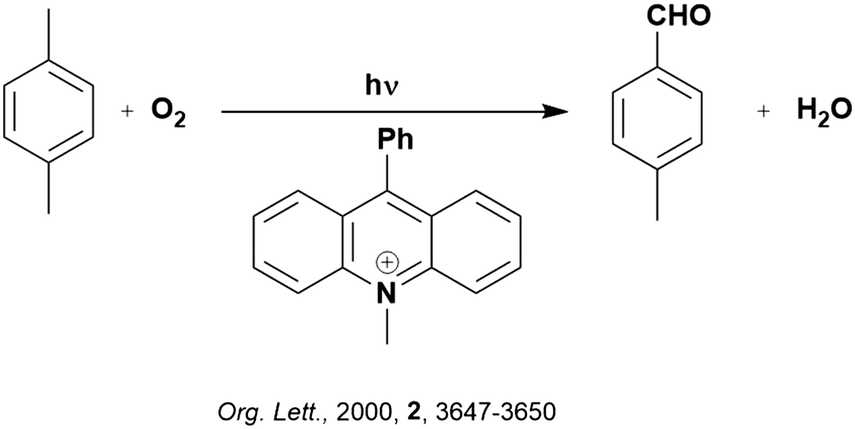 | ||
| Fig. 5 Reaction scheme for the oxygenation of p-xylene to p-tolualdehyde. | ||
In changing the solvent for this reaction from MeCN to chloroform, the yield of product increases despite a slower Stern–Volmer quenching rate constant observed (kq = 8.6 × 109 M−1 s−1 and 4.2 × 109 M−1 s−1 for MeCN and chloroform, respectively). Since the reorganization energies (0.34 eV and 0.27 eV for MeCN and chloroform, respectively) are much smaller than the driving force of the back electron transfer (ΔG0 = −2.36 eV when using [AcrH]ClO4 as the PC), the back electron transfer falls within the Marcus inverted region. With decreasing solvent polarity, a slower back electron transfer rate is observed, and thus an increase in final product yield. This example highlights the impact of solvent selection on reaction yield when conducting photocatalytic reactions. Of course, the choice of PC is also important, if using [AcrPh]ClO4 instead of [AcrH]ClO4, the thermodynamic driving force increases (2.48 eV and 2.34 eV, respectively), resulting in slower BET and consequently higher yields.
Whether PET occurs from the S1 or T1 state may also have an impact on whether BET becomes an important non-productive pathway (Fig. 6). Upon SET transfer from a donor (D) to the excited PC in a reductive quenching mechanism, a radical ion pair (RIP) is formed (Fig. 6a). For successful photocatalysis, this RIP will then dissociate to form the free ions/radicals, which can then react productively to complete the reaction. An alternative unproductive mechanism for this RIP involves BET from the PC˙− back to the D˙+, regenerating the ground state PC and the donor. A spin flip must occur for the electron during BET from the PC˙− to the D˙+ when the PC has a triplet excited state, whereas this is not the case if the PC were in the singlet excited state. As a spin-forbidden process is required for BET when involving the 3PC*, this makes the BET a much slower process, therefore making dissociation to the free ions the more favourable pathway.109,110 For 1PC*, the BET is a spin-allowed process, hence is much faster, and can provide significant competition to the productive dissociation pathway. Therefore, BET is likely to be least competitive when PET occurs from the triplet excited state of the PC. Moreover, PET from 3PC* does not have to overcome the associated pairing energy penalty present when the PC is in the singlet excited state. A similar situation is shown in Fig. 6b, where an oxidative quenching mechanism is in operation. Assuming the rate determining BET occurs as shown in Fig. 6, then this signifies that BET becomes more problematic when the PC is in its singlet excited state rather; however, it should be noted that kBET may not be a significant competing pathway to productive photocatalysis.
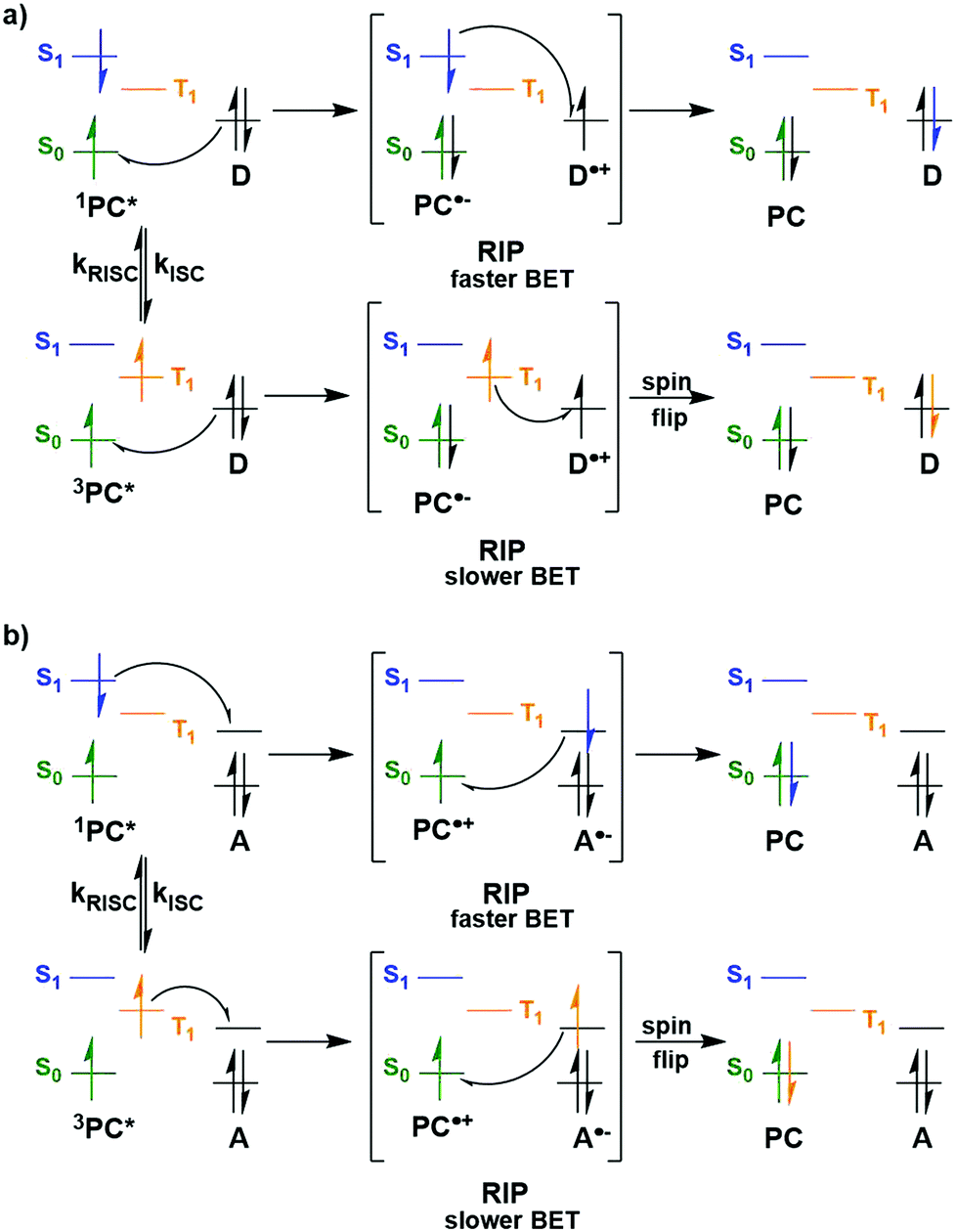 | ||
| Fig. 6 Back electron transfer mechanisms for a) reductive quenching and b) oxidative quenching of the PC with donor (D) and acceptor (A) substrates. | ||
Thus, both the energetics of the states and the nature of the excited state are important determinants for kBET.
In diffusion-controlled photocatalysis processes, the triplet states are perhaps more attractive owing to their longer-lived excited states in comparison to singlet states. However, when assessing the potential thermodynamic driving force of a PC, the energy loss from the initial photon through processes such as IC and ISC within the PC must be considered. For example, for compounds with fast and efficient ISC, such as Ir(III) photocatalysts, an energy loss equivalent to ΔEST occurs to solvent (0.65–1.08 eV, 63–105 kJ mol−1), resulting in a less potent PC.111 Since the singlet state retains a larger component of the initial photon energy in comparison to the triplet state, it is a priori more desirable to access this state for photoredox catalysis. A recent study by Lupton et al. concluded that when the photocatalysis mechanism involves pre-association of the PC and the substrate, the long excited state lifetime of the PC no longer becomes an important feature since diffusion is not a limiting factor.112 In this case, the authors propose electron transfer from the singlet state to be much more beneficial as a result of the lower available energy in triplet states in comparison to singlets.
A combination of all the aforementioned factors must be taken into consideration when evaluating whether electron transfer from the singlet or triplet excited states is preferable. Electron transfer from the singlet state is more exergonic according to the Gibbs free energy and therefore will have faster electron transfer rates. However, utilisation of the triplet excited state means slower BET as well as having the advantage of a longer excited state lifetime, making electron transfer a more competitive pathway to radiative decay as kr will be slower. Quantifying the relative rates of ISC and electron transfer is crucial to understanding which state is predominantly active in the SET. Notably, measurements of these kinetic parameters are rarely reported in photocatalysis investigations.
In-depth mechanistic consideration of the photoactive state of organic photocatalysts has seldom been reported, with this being non-existent for TADF PCs. A study from Kwon et al. focused on using TADF compounds as PCs in polymerisation reactions (see Section 7), implicating the triplet state as responsible for electron transfer,113 although no evidence was provided to support this conjecture. As TADF compounds have accessible singlet and triplet excited states, SET from each is viable. At this point, it is unclear whether the nature of TADF compounds is particularly beneficial for facilitating photoredox reaction. Typically, the success of TADF (and indeed most) PCs are rationalised using only thermodynamic considerations by acknowledging their promising redox potentials in comparison to alternative photocatalysts.114 Recently, the success of these compounds as PCs has been linked to their accessible CT states which, as previously mentioned, are significant in photoredox catalysis. This is seen in the hypothesis of Diebel et al. who suggested the presence of chlorine in 3CzClIPN increases the charge transfer character of the excited states in comparison to 4CzIPN, which may contribute to the former outperforming the latter in a recently demonstrated oxidation reaction.115 While this observation may be true, there is no explanation provided as to how an increase in the CT character can translate into improved thermodynamics or kinetics for aiding the photocatalysis process.
Although the specific impact the TADF character of the PC has on its photocatalytic potential is unclear at present, what is evident is that TADF compounds have long excited state lifetimes, comparable to those of iridium and ruthenium PCs. Redox potentials of TADF compounds in both the ground and excited state are of comparable magnitude to those of heavy metal PCs, and the organic compounds have been shown to have in many cases enhanced molar absorptivity for the CT band compared to transition metal complexes. All of these are contributing factors to their success as PCs.
The significance of 4CzIPN in photocatalysis
The first TADF compound to be tested as a PC was eosin Y,116 an organic dye that was one of the first organic compounds identified as showing TADF.117 In fact, for many years TADF was known as E-type fluorescence, acknowledging eosin Y's historic link to this radiative decay process. The moderate redox potentials (Eox = 0.78 V, Ered = −1.06 V, Eox* = −1.11 V and Ered* = 0.83 V in MeCN:H2O 1:1) as well as absorption in the visible region (λabs = 520 nm in MeOH), have contributed to the popularity of this commercially available and inexpensive dye being widely used as a PC.25 However, to the best of our knowledge, no mention of its TADF character has ever been discussed in the context of its use in photocatalysis. Interestingly, it has been widely accepted that SET occurs from the triplet state of eosin Y,6 owing to its large ΦISC as well as its extremely short singlet excited state lifetime (2 ns in MeOH),118 which reduces the probability of interaction of the PC in this excited state with the substrate. The ability of eosin Y to act as a PC has been well documented previously and so will not be included within this review.
Of the articles published using TADF compounds as PCs, one TADF material, 2,4,5,6-tetra(carbazol-9-yl)benzene-1,3-dicarbonitrile, 4CzIPN, first reported by Adachi et al. in 2012,60 has been predominant (Fig. 7a). 4CzIPN is composed of four carbazole electron-donor moieties and an isophthalonitrile that acts as the electron-acceptor component. The torsions of the carbazolyl groups from the plane of the dicyanobenzene are large due to steric interactions between adjacent carbazoles. This twisted conformation results in the HOMO and LUMO being localised on the donor and acceptor moieties, respectively, thus reducing the exchange integral between the two and ensures a sufficiently small ΔEST of 0.08 eV in toluene,60 allowing for the necessary RISC to occur to make this compound TADF.
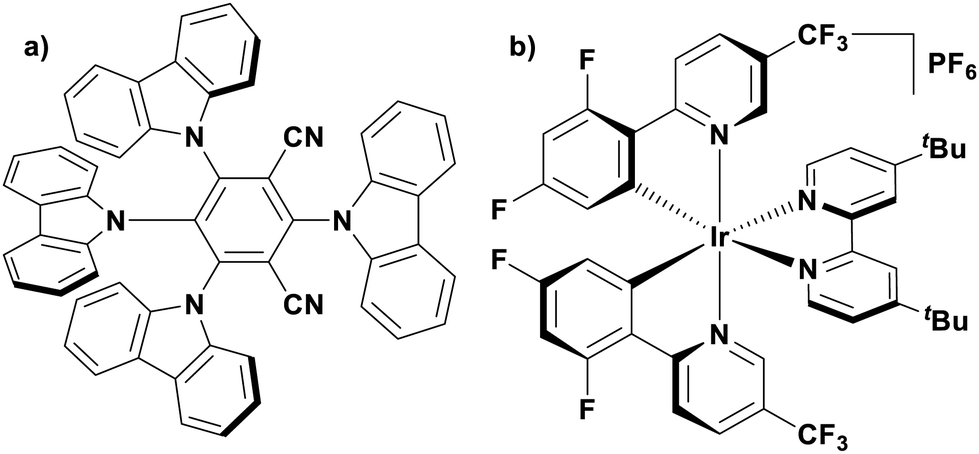 | ||
| Fig. 7 Structures of (a) 4CzIPN and (b) [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
In polar solvents such as MeCN, the ΦISC is extremely low at 0.041, with kISC being 2.2 × 106 s−1.119 These data indicate minimal formation of triplet excitons and implies that the singlet excited state is the state from which subsequent photochemistry is likely to occur in this solvent. In apolar solvents like toluene, the value of ΦISC increases dramatically to 0.73. This clear difference in intersystem crossing quantum yields upon changes in medium highlights the significant role that solvent can play in modulating the photophysical and potentially the photochemical properties of a PC.
The photophysical and electrochemical properties of 4CzIPN, summarised in Table 1, mimic quite closely those of the commonly used iridium complex [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (Fig. 7b) [(dF(CF3)ppy) = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridinato and dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine], as can be seen clearly in Fig. 8, and, as will become evident below, can serve as a useful replacement for the latter. Both compounds exhibit CT excited states. In 4CzIPN this is manifested in a radical cation delocalised over the donor component and a radical anion associated with the isophthalonitrile acceptor unit. Similarly, in the mixed metal-to-ligand/ligand-to-ligand charge transfer excited state of the iridium complex, the radical cation has dπ(dF(CF3)ppy) character and the central metal can be considered to be oxidised to Ir(IV), while the dtbbpy ligand is reduced to its radical anion.
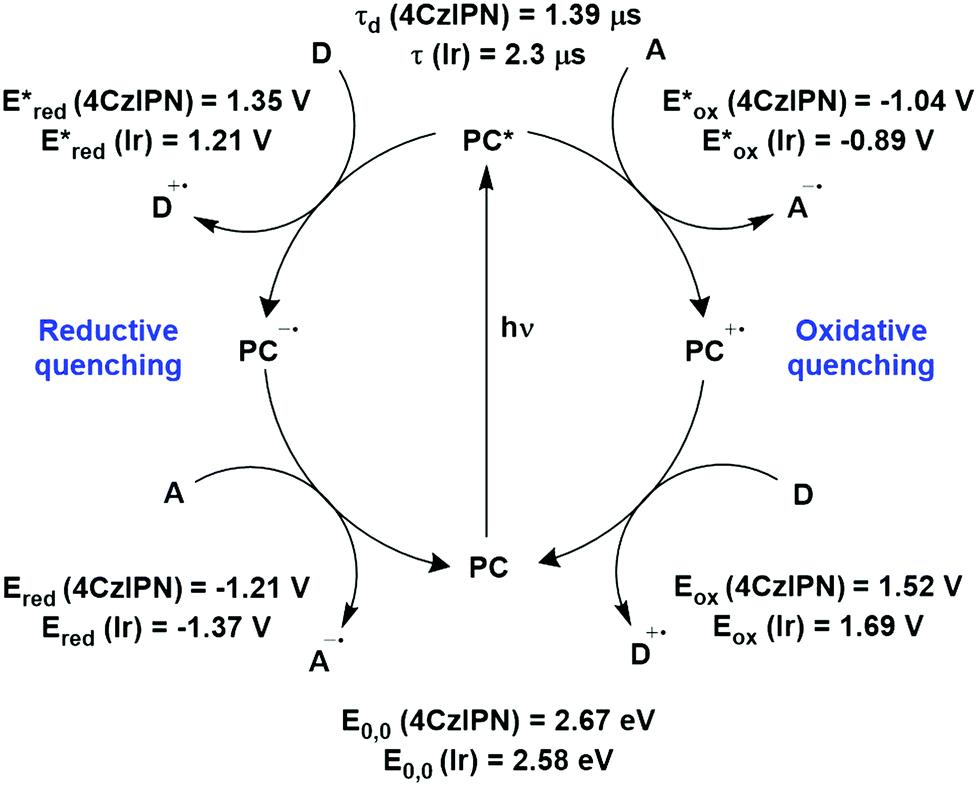 | ||
| Fig. 8 Photocatalytic cyclic comparing selected photophysical and redox properties of 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 in MeCN.11,60,114,119–124 | ||
The excited-state redox properties of 4CzIPN are particularly significant, making it a very strongly photooxidising and photoreducing photocatalyst in comparison to other commonly used visible light photocatalysts, as shown in Fig. 9 and 10.
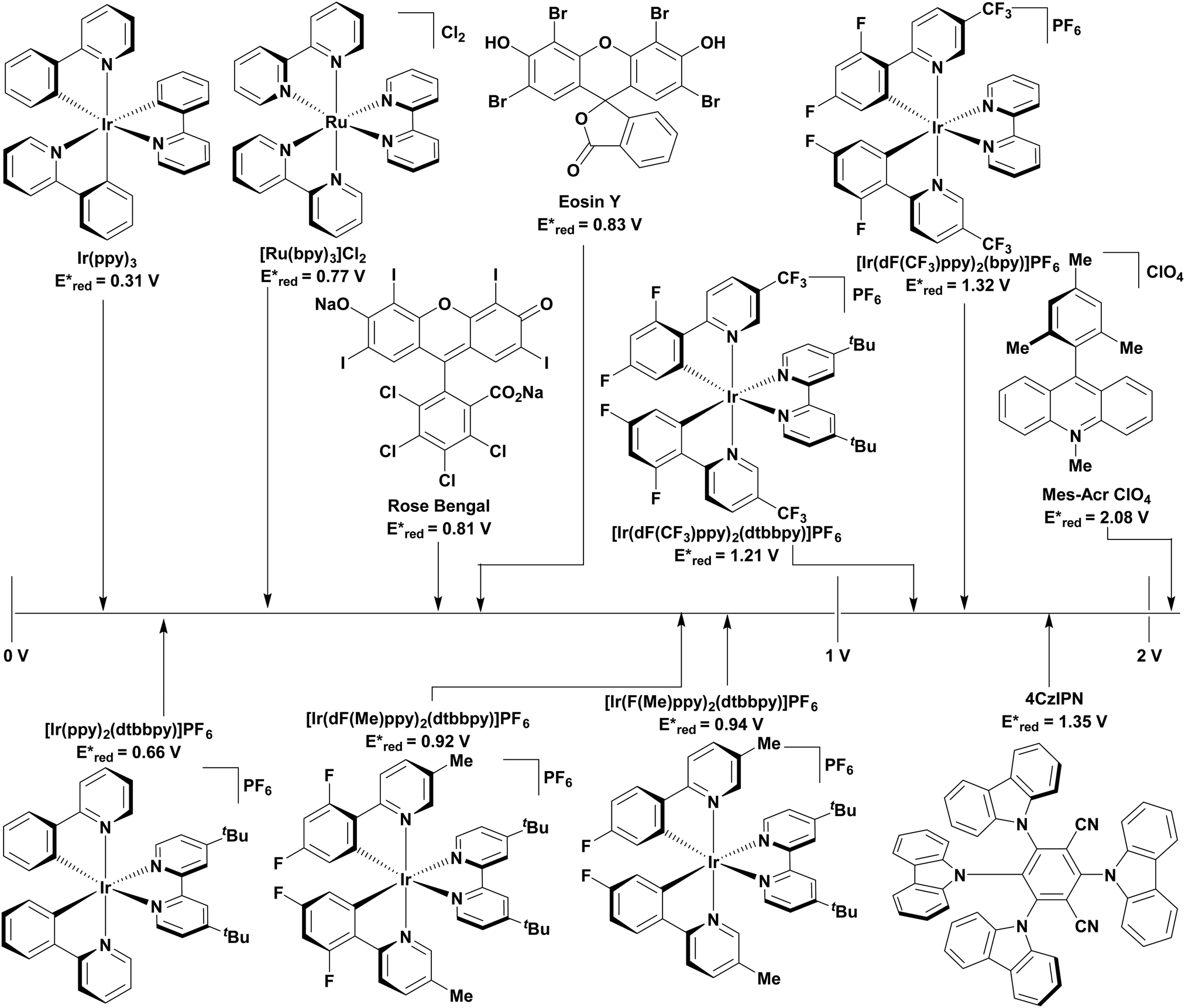 | ||
| Fig. 9 Excited state reduction potentials of common, visible light PCs. | ||
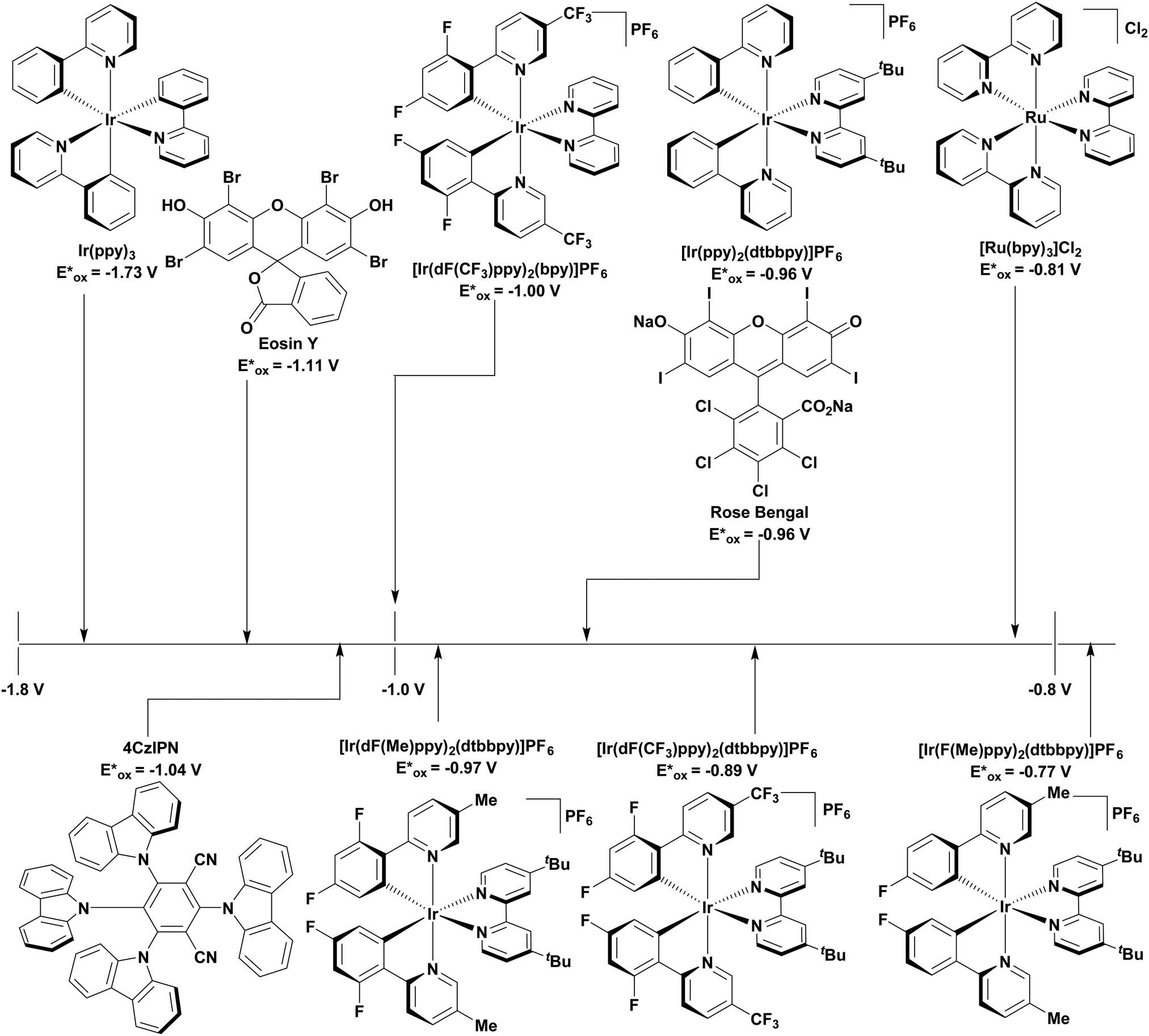 | ||
| Fig. 10 Excited state oxidation potentials of common, visible light PCs. | ||
One of the primary advantages of 4CzIPN is its relatively low synthesis cost. The one-pot synthesis of 4CzIPN from the corresponding dicyanotetrafluorobenzene via an SNAr reaction translates to an approximate cost of $5 per mmol for 4CzIPN in comparison to $935 per mmol for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (prices are in US$ in 2020).125 These values come from Wendlandt et al. in 2020,125 who determined this cost based on the price of the reagents, solvent and energy as well as the product yield. Cost considerations, in addition to the lower toxicity profile, wide redox potentials, appreciable visible-light absorption (ε = 6.2 × 103 M−1 cm−1 at 380 nm for [Ir(dF(CF3)ppy)(dtbbpy)]PF6 in MeCN123 and ∼1.7 × 105 M−1 cm−1 at 377 nm for 4CzIPN60) and long excited-state lifetime (τpf = 18.7 ns and τdf = 1390 ns in MeCN)119 are all important factors that contribute to the popularity of 4CzIPN as a PC.
Throughout this review, we will highlight the comparative performance of TADF photocatalysts with alternative PCs. However, there are some caveats to these comparisons and caution must be taken when drawing conclusions as to performance. Firstly, in almost all articles referenced, a photocatalyst is considered the best when it provides the highest yield of product. It is not always the case that the reaction has been identified as going to full conversion and so differences in yield may reflect a combination of differences in reaction kinetics and differences in reactivity. As such, we define a threshold for yield comparison whereby a variation of less than 5% between reaction yields obtained using different photocatalysts will be classed as not statistically significantly different. Further, product yields are sometimes based on isolated yields where there is a greater chance for variance between experiments, NMR yields by contrast, provide more accurate quantification of product formation. For photocatalysts, a more quantitative method is the quantum yield of reaction, Φ(λ), (eqn (1)); however, these are seldom reported.138 A maximum quantum yield of one is obtained for situations in which one mol of photons absorbed produces one mol of product. Typically, quantum yields are only reported as mechanistic evidence for a radical chain mechanism or a lack thereof, whereby one mol of photons forms n mols of product, producing a quantum yield >1. Usually, a quantum yield less than one signifies that a radical chain mechanism is not operational; however, this is a largely oversimplified interpretation of the reaction quantum yield. It is possible that the reaction proceeds through a radical chain mechanism even with a quantum yield less than 1, but with a highly inefficient initiation step. Estimations of the chain length are possible if the quenching fraction is taken into consideration,139 although such an analysis is rarely undertaken in much of the photocatalysis literature. As such, we are largely limited to reporting the quantum yield determined by the authors and the conclusions that have been drawn from such a value.
Cross-comparison of multiple studies becomes fraught when the excitation source changes, affecting both the photon flux and the excitation wavelength used to photoexcited the PC.138,140 Throughout the development of photocatalysis, a wide variety of excitation sources have been employed from simple household compact fluorescent lightbulbs (CFLs) with moderate light intensity and a broad excitation spectrum to high-powered light emitting diode (LED) lamps, which have controllable intensity and narrow excitation spectra.141 Further, photoreactors that are capable of capturing and reflecting all of the photons emanating from the excitation source and directing them into the reaction vessel will lead to more efficient reaction transformations.
Furthermore, throughout this review mechanisms are shown for each reaction mentioned; however, it should be noted that for the most part, these are putative with frequently little evidence provided to corroborate the proposed mechanism. Typically, only a comparison of the thermodynamic parameters of the PCs and the substrates is used to support the mechanistic proposals. This type of comparison and its correlation to reaction yields provides a very crude assessment of PC potency and ignores the reaction kinetics. For example, a study by Wolf et al. investigated the impact of the lifetime of the photoexcited state of the PC on the product yield in the trifluoromethylation of quinoline.142 It was found that a longer excited-state lifetime resulted in faster SET involving the excited PC; however, there was no significant impact on the yield, indicating the rate determining step is not the initial SET for this reaction; indeed, further investigations suggested the second SET was the rate limiting step. The importance of reaction kinetics was highlighted also in the work of Scaiano et al.143 It was concluded that while appropriate redox potentials are necessary for the PC to initiate the required SET, the kinetics of the process may be the deciding factor as to how successful the photocatalyst is. Unfortunately, kinetics studies like these are rarely completed.
Despite of these lacunes, this review aims to summarize the reactivity and document the potential of TADF compounds, such as 4CzIPN, as photocatalysts.
2. 4CzIPN as an independent photocatalyst
Thus far, 4CzIPN is the most widely studied TADF-based photocatalyst. The majority of examples in this section proceed via PET and are therefore photoredox catalysis reactions although a very small handful to be discussed occur via PEnT. Where applicable, the relative success of 4CzIPN is compared with other photocatalysts.Photoinduced decarboxylation of carboxylic acids in the formation of C–C and C–X bonds in a reductive quenching mechanism
Many examples employing 4CzIPN as a photocatalyst involve decarboxylation of the substrate, whereby a reductive quenching mechanism is typically in operation. Utilisation of carboxylic acids to form C(sp3) radicals via photoinduced oxidative decarboxylation has opened a variety of new synthetic pathways for the formation of C–C or C–X bonds.144 The carboxylic acid or carboxylic acid derivative is activated through SET to the excited photocatalyst, forming an alkyl or haloalkyl radical. This requires the photocatalyst to be a relatively strong photooxidant in order to oxidise the carboxylate (Eox of carboxylates typically ranges from 1.2–1.5 V).145 Through either a radical addition or radical coupling mechanism, a C–C or C–X bond (where X = O or N) is formed. This step is usually facilitated through a SET reduction from the reduced photocatalyst, concomitantly closing the photocatalytic cycle and regenerating the photocatalyst. A generic mechanism for this process, involving a radical addition mechanism can be seen in Fig. 11.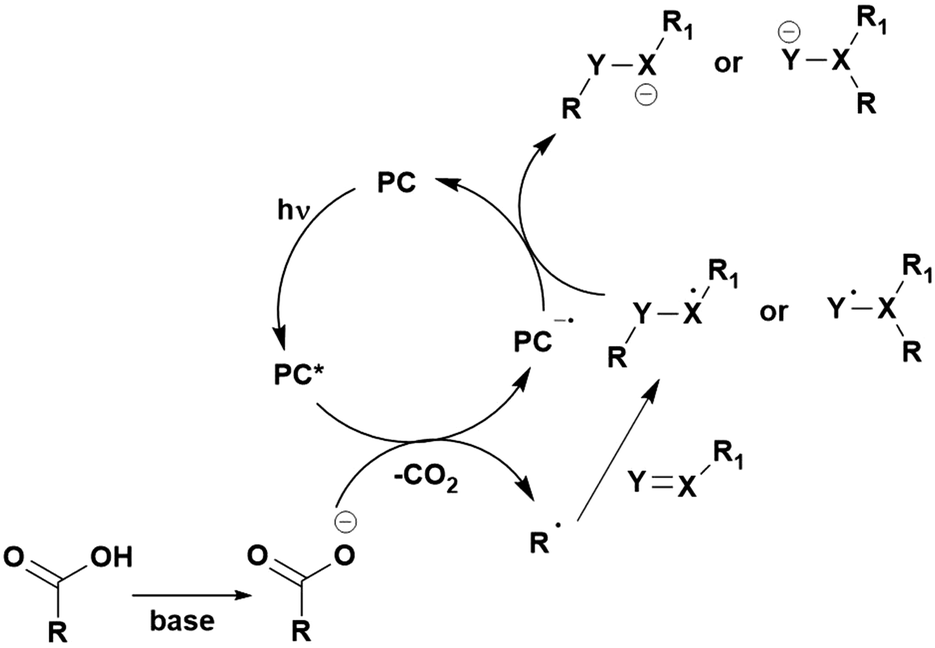 | ||
| Fig. 11 Photoinduced oxidative decarboxylation catalytic cycle using a radical addition mechanism where X = C or N, and Y = C or O. | ||
Reactions that have exploited this decarboxylation mechanism in combination with a radical addition include the formation of: formyl radical equivalents (from 2,2-diethoxyacetic acid)146 which can be used in hydroformylation reactions (Fig. 12a), alkyl radicals which can subsequently react with nitrosoarenes to synthesise hydroxylamines (Fig. 12b),147 α-amino radicals which can be added to vinyl boronic esters producing γ-amino boronic esters (Fig. 12c),148 amidyl radicals which can be added to alkenes (Fig. 12d),149 alkyl radicals, which can be added to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of amino acids or peptides (Fig. 12e)150 and silyl radicals, which can be added to alkenes (Fig. 12f).151
C bonds of amino acids or peptides (Fig. 12e)150 and silyl radicals, which can be added to alkenes (Fig. 12f).151
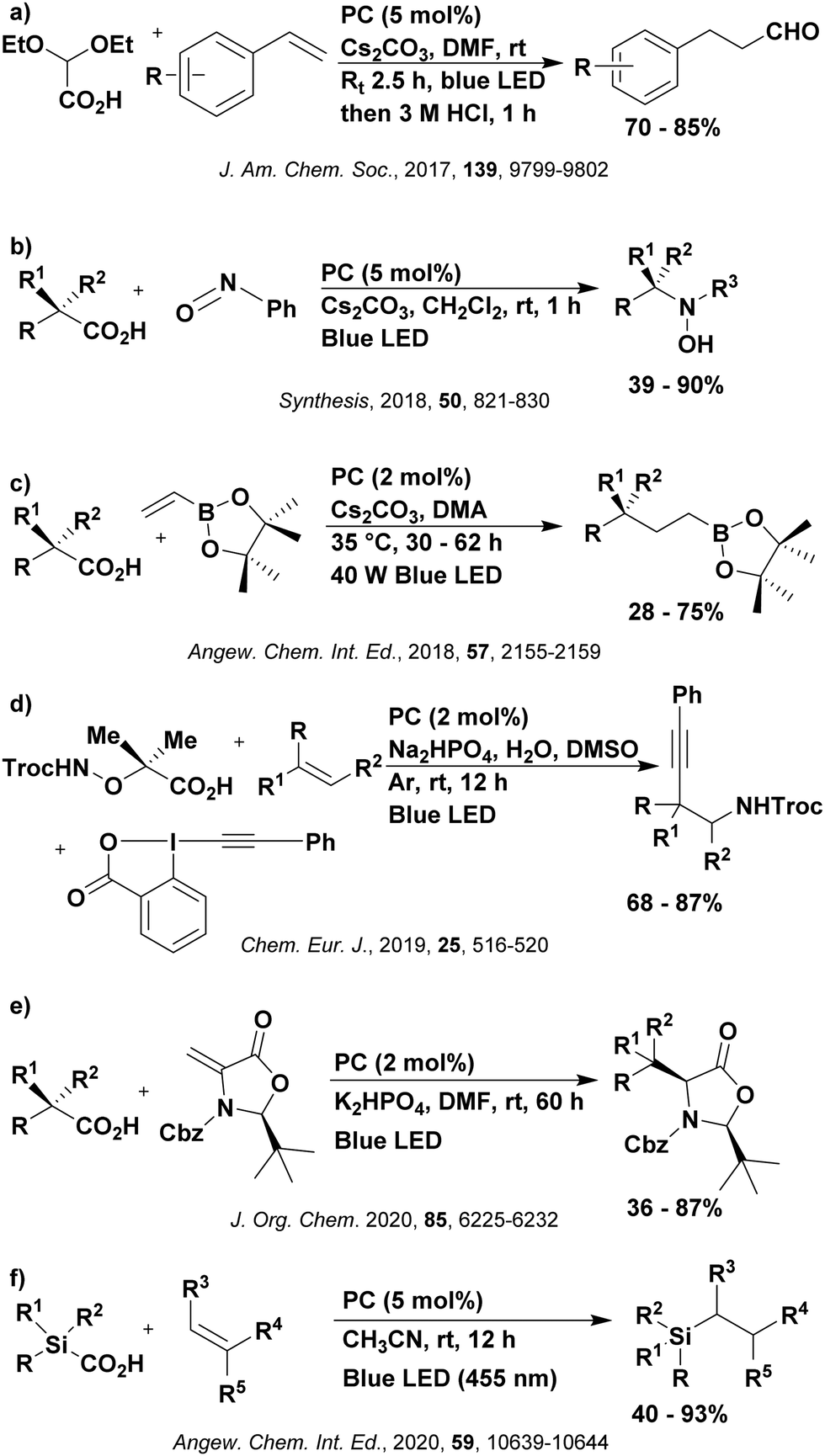 | ||
| Fig. 12 Reaction schemes involving photocatalytic decarboxylation; (a) hydroformylation of aromatic vinyl substrates, (b) synthesis of hydroxylamines, (c) synthesis of γ-amino boronic esters, (d) 1,2-amidoalkynylation of alkenes, (e) derivatisation of amino acids and (f) hydrosilylation of alkenes. Rt = residence time in the flow reactor. | ||
These reactions all proceed via a photoinduced decarboxylation followed by a radical addition mechanism (Fig. 11). For the hydroformylation reaction (Fig. 12a), 4CzIPN is the most efficient photocatalyst, obtaining 70% yield of product, as well as 100% conversion, in the batch reaction while all other organic photocatalysts tested, including eosin Y and [Mes-Acr]ClO4, obtained no product.146 Similarly, in the 1,2-amindoalkynlation of alkenes (Fig. 12d), 4CzIPN produced a 77% yield of product when using 2-methylpent-1-ene as the alkene, while other organic photocatalysts yielded between 7–33% of product, with eosin Y being the next best photocatalyst at 33% yield.149 These differences in yield correlate with the ground state reduction potential of the photocatalysts (Ered = −1.21 V, −1.06 V and −0.57 V for 4CzIPN, eosin Y and [Mes-Acr]ClO4, respectively), suggesting this thermodynamic parameter may have crucial impact on the success of the photocatalyst. In the hydrosilylation of alkenes (Fig. 12f),1514CzIPN considerably outcompeted the iridium and ruthenium complexes, yielding 82% while the next best PC, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, could manage only 45%. This is likely on account of the superior photooxidising ability of 4CzIPN (Ered* = 1.35 V and 1.21 V for 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6, respectively), which is implicated in the oxidation of the silacarboxylate (Eox(Ph2MeSiCO2˙/Ph2MeSiCO2−) = 1.32 V vs. SCE). For the derivatisation of amino acids (Fig. 12e),1504CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6 provided the same yields (90% and 89%, respectively), with 4CzIPN being selected as the PC for the subsequent substrate scope.
Unfortunately, 4CzIPN proved unsuccessful in synthesising the hydroxylamine product (Fig. 12b), as did the iridium photocatalyst tested, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6.147 Both [Mes-Acr]ClO4 and [Mes-Acr]BF4 worked as photocatalysts for this reaction, which is likely due to their far superior oxidising capability in the excited state (Ered* = 2.06 V for Mes-Acr+ compared to 1.35 V for 4CzIPN and 1.21 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6). For γ-amino boronic ester synthesis (Fig. 12c), four photocatalysts were considered: [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ru(phen)3]Cl2 and 4CzIPN. [Ir(ppy)2(dtbbpy)]PF6 was the most efficient photocatalyst with the substrate Boc-Pro-OH, giving a final product yield of 88% in the presence of the additive Cs2CO3, although 4CzIPN also provided a moderate yield of 66% in the absence of this additive. No yield was given for the use of 4CzIPN with the additive Cs2CO3, making a direct comparison somewhat challenging. Nevertheless, a possible reason for [Ir(ppy)2(dtbbpy)]PF6 outperforming 4CzIPN is their relative ground state reduction potentials (Ered = −1.51 V and −1.21 V, respectively). The more negative Ered value for the iridium photocatalyst facilitates the SET between the reduced photocatalyst and the intermediate α-boryl radical and thus the turnover of the photocatalytic cycle.
A mechanistically related example can be seen in the remote functionalisation of amides and amines using electrophilic nitrogen radicals (Fig. 13).152 This reaction proceeds via decarboxylation of the carboxylic acid derivatives to generate an electrophilic amidyl radical, which can then undergo a 1,5-hydrogen atom transfer (HAT) to form a distal alkyl radical (Fig. 14). Homolytic atom/group transfer with a polarised SOMOphile (SOMO = singly occupied molecular orbital) affords amines or amides that are remotely substituted. An extensive SOMOphile substrate scope study was undertaken, in the comparison of 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN gave a higher product yield in γ-chlorination (76% vs. 64%), thioetherification (71% vs. 59%), cyanation (64% vs. 21%) and alkynylation (80% for 4CzIPN, iridium PC not tested). Unusually, for γ-fluorination, 4CzIPN was not tested, so in this case [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was the best photocatalyst. Quantum yields were obtained for the reactions involving the five SOMOphile substrates with their respective superior photocatalyst and varied from 0.02–0.08. Unfortunately, quantum yields were not obtained across the PCs tested and so a direct comparison of the photocatalysts is not possible using this metric.
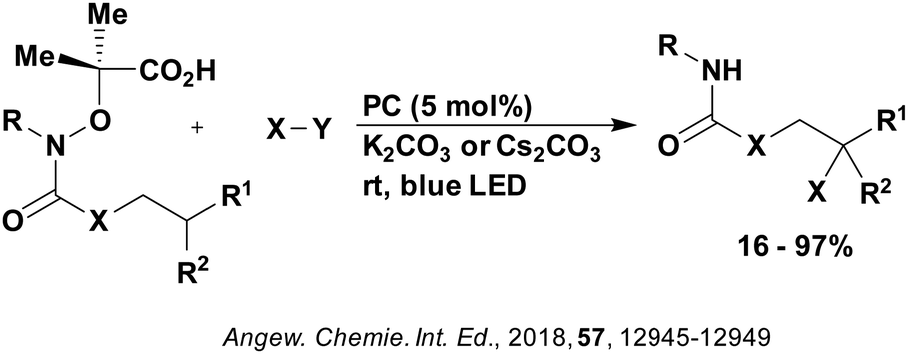 | ||
| Fig. 13 Reaction scheme for the remote functionalisation of amides and amines using electrophilic nitrogen radicals. | ||
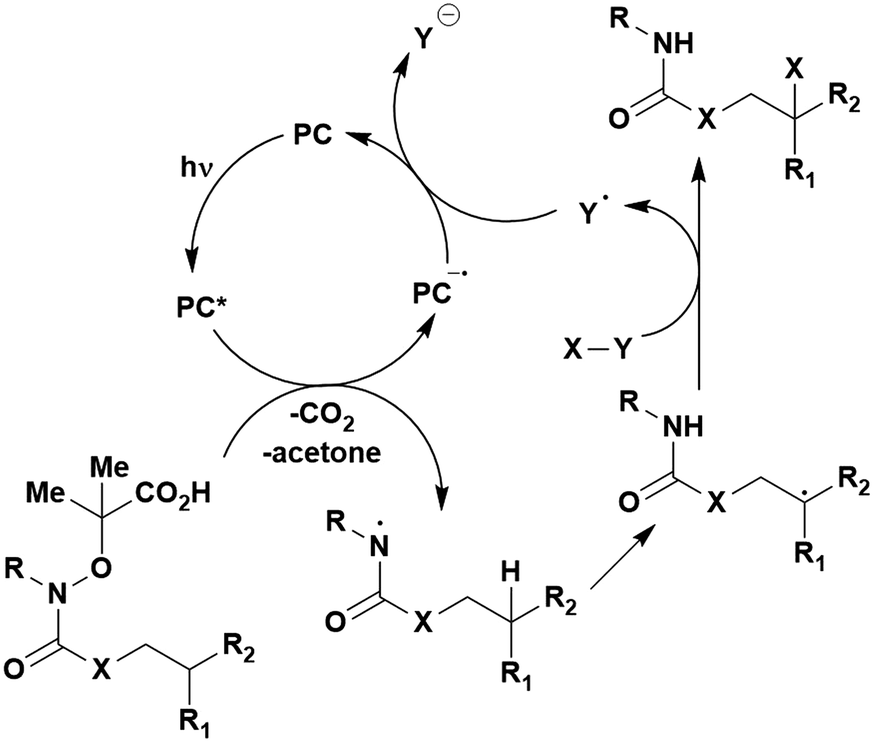 | ||
| Fig. 14 Proposed photocatalytic cycle for decarboxylation and group transfer in C–H functionalisation.152 | ||
Carboxylic acids may be used as radical precursors in C(sp3)–C(sp) type coupling reactions, for example the acetalation of alkynyl bromides (Fig. 15).153 Decarboxylation occurs to generate an acetal radical, which then adds to alkynyl bromide, forming a bromoalkenyl radical (Fig. 16). Elimination of a bromyl radical, which is reduced by the reduced photocatalyst, results in the formation of product. Radical trapping experiments confirmed the presence of the acetal radical while radical inhibition experiments provided further confirmation this is a radical-based process. Only 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 provided any product (36% and 5% yield, respectively), with photocatalysts such as [Ru(bpy)3](PF6)2 and eosin Y giving no product. Wang et al. postulated that this is a result of the photooxidising capability of the PC. The caesium salt of 2,2-diethoxyacetic acid has an oxidation potential of Eox = 0.95 V,21 hence the excited state reduction potential of the photocatalyst must be more positive than this for the transformation to be thermodynamically feasible. Both 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, are sufficiently photooxidizing (Ered* = 1.35 V and 1.21 V, respectively) and their relative strength as photooxidants may explain why the former outperforms the latter. For [Ru(bpy)3](PF6)2 and eosin Y, however, this SET is not thermodynamically possible (Ered* = 0.77 V and 0.83 V, respectively).
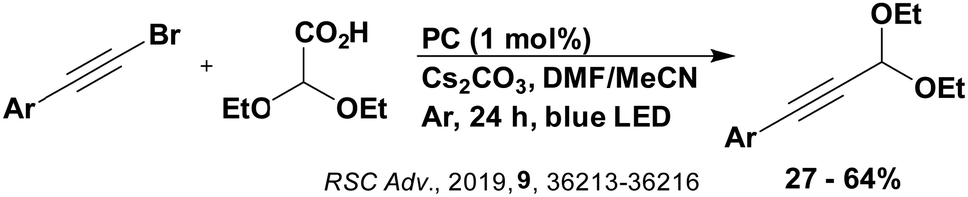 | ||
| Fig. 15 Reaction scheme for the acetalation of alkynyl bromides. | ||
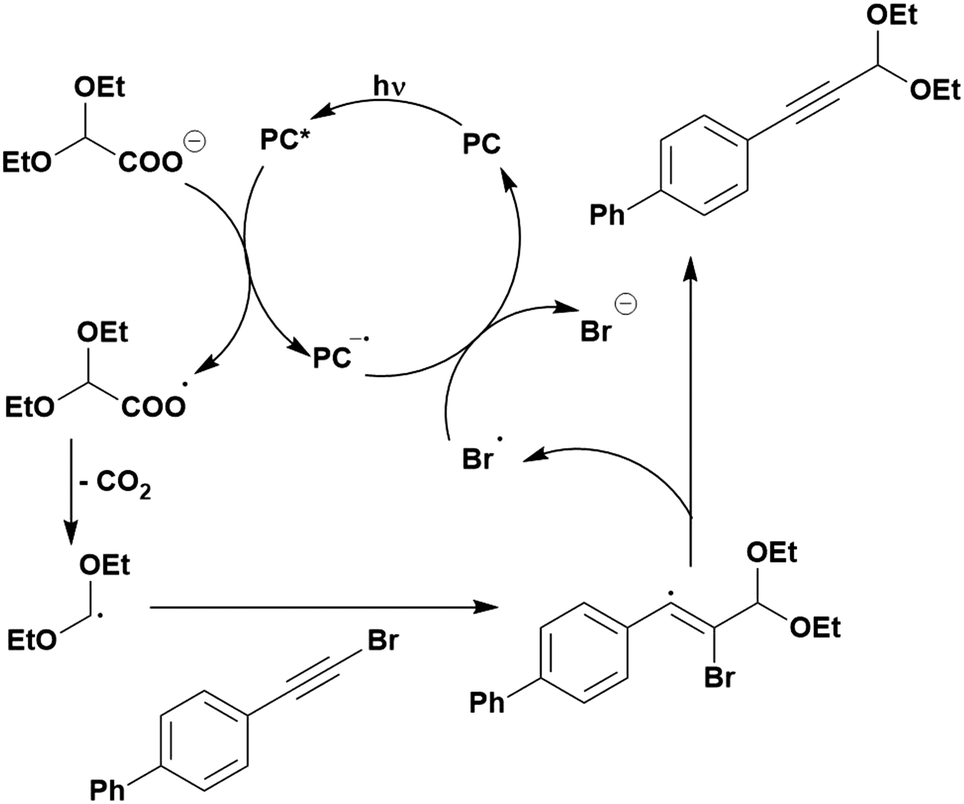 | ||
| Fig. 16 Putative mechanism for the photocatalytic acetalation of alkynyl bromides.153 | ||
Cyclisation reactions following an oxidative decarboxylation have also been reported. This can be observed in the synthesis of functionalised cyclopropanes (Fig. 17a),154 intramolecular arene alkylation (Fig. 17b)155 and the synthesis of 2-substituted piperazines (Fig. 17c).156 The former involves a radical addition-polar cyclisation cascade mechanism, generating functionalised cyclopropanes through a polar 3-exo-tet cyclisation (Fig. 18a).154 This mechanism was supported by quantum yield measurements for the reaction between Boc-Pro-OH and (4-chlorobut-1-en-2-yl)boronic acid pinacol ester, giving a quantum yield of 0.65. Since the quantum yield is below 1, this helps to support the proposal of the closed photocatalytic mechanism, rather than any alternative radical chain mechanisms. Under the optimised conditions, 4CzIPN was highlighted as the best photocatalyst, providing a 99% yield of product with a slightly lower yield of 90% obtained with [Ir(ppy)2(dtbbpy)]PF6 under the same reaction conditions. The wide ground state redox window of 4CzIPN allows the substrate scope to go beyond just homoallyl chlorides bearing electron-withdrawing substituents. For example, 4CzIPN (Ered = −1.21 V) could undergo SET with α-carboxylate ester radicals (Fig. 18b) (Ered(R˙/R−) ≈ −0.6 V)157 as well as electron-rich benzylic radicals (Fig. 18c) (Ered(R˙/R−) < −1.4 V).158
 | ||
| Fig. 17 Reaction schemes for cyclisation reactions involving a photoinduced decarboxylation mechanism for the formation of (a) functionalised cyclopropanes, (b) intramolecularly alkylated arenes and (c) 2-substituted piperazines. | ||
 | ||
| Fig. 18 (a) Proposed mechanism for the photoinduced cyclopropanation using carboxylic acids as radical precursors (EWG = electron withdrawing group), (b) carboxylate radical intermediate and (c) electron rich benzylic radical intermediate. | ||
The intramolecular arene alkylation reaction (Fig. 17b) also involves a radical addition mechanism in combination with decarboxylation (Fig. 19).155 Stern–Volmer quenching experiments were conducted in order to support the proposed mechanism as well as quantum yield determination (0.022 for 1,3-dioxoisoindolin-2-yl 5-oxo-5-phenylpentanoate). For this reaction, 23 photocatalysts were screened initially with 4CzIPN and iridium photocatalysts [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(bpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(dF(Me)ppy)2(dtbbpy)]PF6, Ir(dFppy)3, Ir(dF(tBu)ppy)3 providing the highest yields (70–75%). Since 4CzIPN provided high yields at a lower cost, it was chosen as the PC for the scope study.
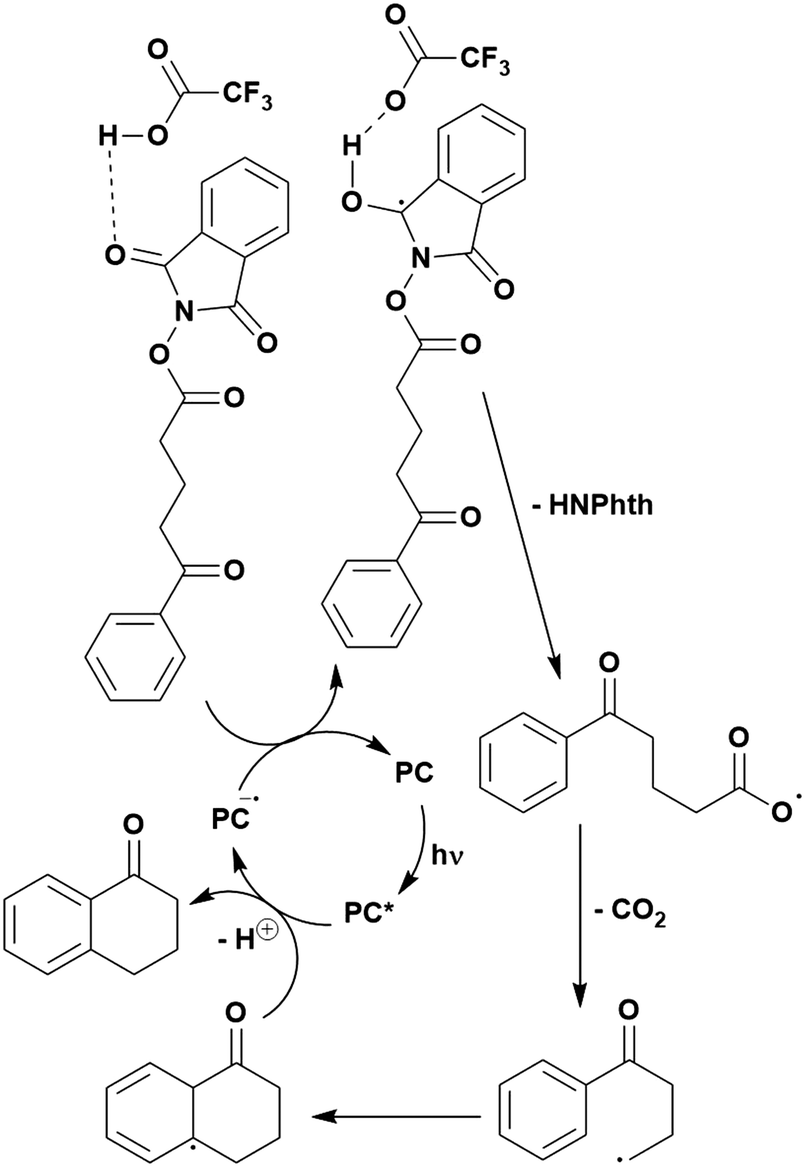 | ||
| Fig. 19 Possible mechanism for the photoinduced intramolecular arene alkylation of N-(acyloxy)phthalimides (NAPs). | ||
The synthesis of 2-substituted piperazines (Fig. 17c)156 proceeds through condensation of the aldehyde and amine to generate an imine prior to irradiation. Oxidative decarboxylation of the imine by the excited PC leads to the α-amino radical, which intramolecularly adds to the imine. Closure of the photocatalytic cycle is proposed to occur through either reduction of the resultant N-centred radical, which after protonation yields the final piperazine product, or the N-centred radical abstracts a proton from the solvent, MeCN, to afford the piperazine directly, and the cyanomethyl radical is reduced by the reduced PC. Both 4CzIPN and [Ir(ppy)2(dtbbpy)](PF6) provided essentially the same yields under the same conditions (70% and 75%, respectively), with [Ru(bpy)3](PF6)2 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 both managing only 33%. There seems to be no obvious link between the thermodynamic parameters of these PCs and the yields obtained.
Additionally, the C(sp3) radical produced from oxidative decarboxylation of carboxylic acids or their derivatives can be used to generate new C–C or C–O bonds via a radical coupling mechanism. In the reductive quenching photocatalytic reaction conditions, two different radical species from two different substrates may be formed that can then couple together. For example, a benzylic radical and an α-amino radical anion were demonstrated to couple together to produce β-arylethylamines (Fig. 20a)159 The proposed mechanism (Fig. 21) was supported by Stern–Volmer quenching studies that indicated that the excited photocatalyst is not quenched by the imine, but rather by the carboxylic acid in the presence of K2HPO4. For PC optimisation, both 4CzIPN and [Ir(dF(CF3)ppy)2(bpy)]PF6 yielded a comparable amount of product (45% and 50%, respectively). Perhaps the stronger ground state reduction capacity of the iridium PC (Ered = −1.21 V and −1.37 V for 4CzIPN and [Ir(dF(CF3)ppy)2(bpy)]PF6, respectively) is the reason it performs slightly better, especially since imines can have challenging reduction potentials (up to Ered = −2 V).160,161
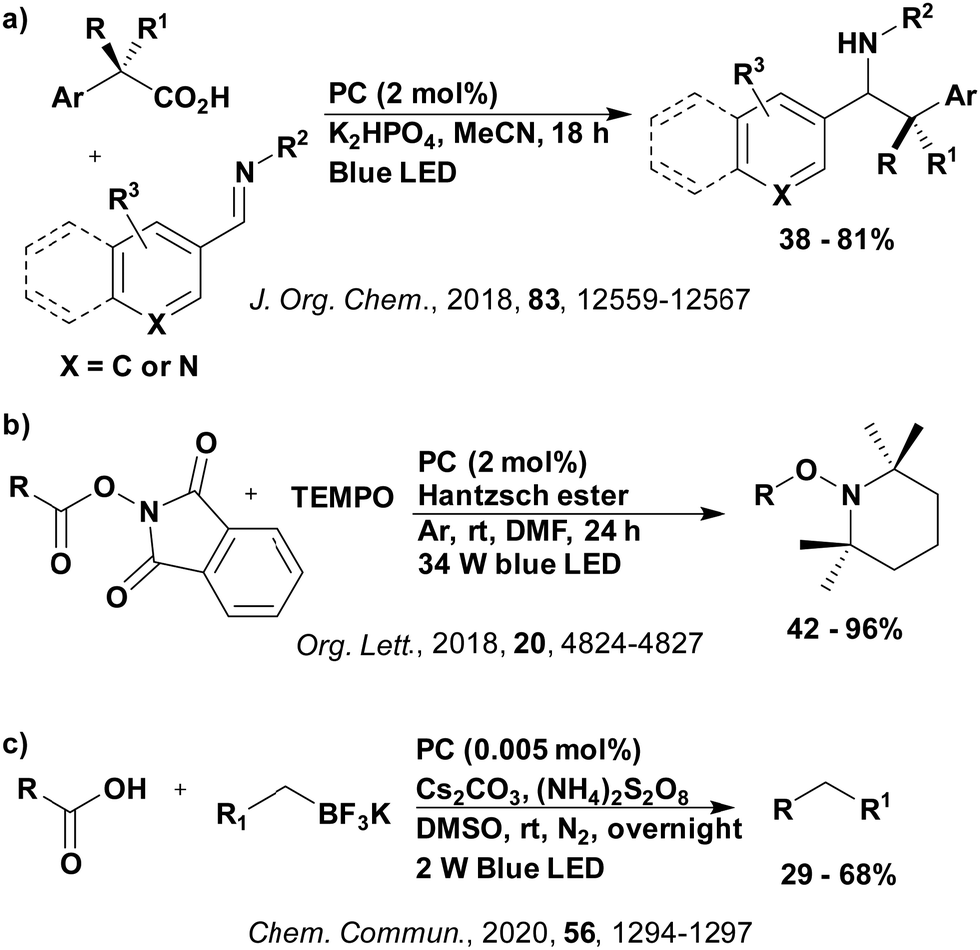 | ||
| Fig. 20 Reaction schemes for (a) benzylation of imines, (b) oxygenation of aliphatic carboxylic acids and (c) coupling of aliphatic acids with trifluoroborate salts. | ||
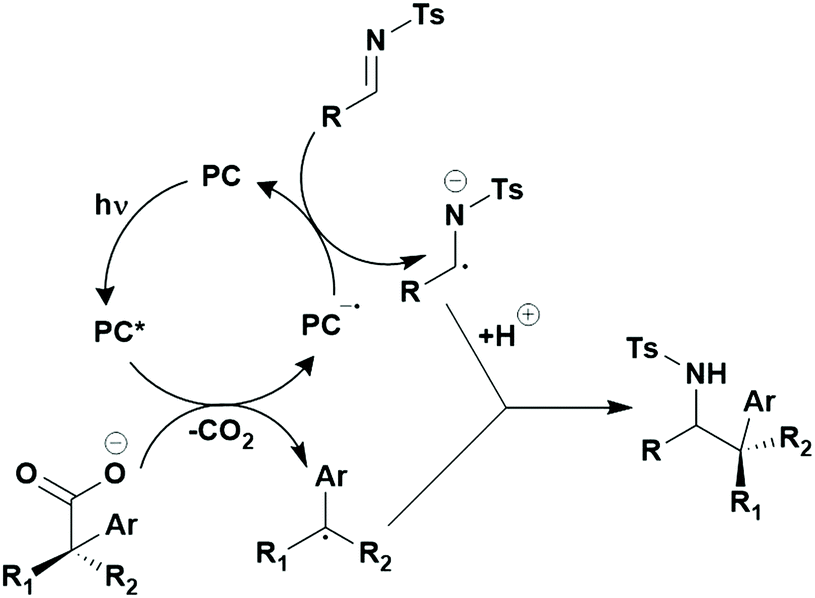 | ||
| Fig. 21 Photoinduced oxidative decarboxylation catalytic cycle used for a radical–radical coupling mechanism.159 | ||
An additional example involves an alkyl radical and (2,2,6,6-tetramethylpiperidin1-yl)oxyl (TEMPO), which couples to form an alkoxyamine (Fig. 20b).162 In this case, 4CzIPN is outperformed as a PC by [Ru(bpy)3]Cl2.6H2O (yields of 80% and 95%, respectively), which again could be related to the regeneration of the PC. The PC needs to be moderately reducing in the ground state in order to reduce the N-(acyloxy)phthalimides, which have a reduction potential of Ered = −1.26 to −1.37 V.162 Since the ruthenium PC is more reducing in the ground state (Ered = −1.33 V), this may explain the difference in yields. A quantum yield of 6.7 was obtained with the ruthenium(II) photocatalyst. It is unlikely that a radical chain process is involved since the TEMPO trapping of the alkyl radical is a termination process, hence Chen et al. propose an energy transfer process is likely to be occurring simultaneously to rationalize the high quantum yield, although no further investigations were undertaken to substantiate this claim.
Similarly, radical–radical coupling can be seen in the cross-coupling of aliphatic acids with trifluoroborates salts (Fig. 20c).163 In this case, the excited PC is reductively quenched by both the carboxylic acid and the trifluoroborate salt to generate two alkyl radicals, suggesting two photocatalytic cycles are in operation. These alkyl radicals can then couple together to form the product. The photocatalyst is regenerated in both cycles by oxidation from persulfate. This proposed mechanism is supported by the observation that both carboxylic acids and trifluoroborate salts quench the photocatalyst emission, while persulfate did not; radical trapping experiments are corroborative. Only 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6 were considered as photocatalysts, with both providing similar yields (50% and 44%, respectively). The slightly higher yield obtained by 4CzIPN is likely related to its superior photooxidising behaviour (Ered* = 1.35 V and 0.92 V for 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6, respectively) which is needed for the oxidation of both carboxylic acids and trifluoroborate salts.
Photoinduced decarboxylation of carboxylic acids in the formation of C–C and C–X bonds in an oxidative quenching mechanism
Carboxylic acids have also been used as carbon-centred radical precursors in C–C or C–X bond forming reactions when then photocatalyst is operating in an oxidative quenching mechanism, for example in formation of urethanes and ureas (Fig. 22a).164 Reduction of oxamic acids by the excited photocatalyst precipitates a decarboxylation to generate a carbamoyl radical. When this reaction was first developed, the carbamoyl radical was oxidised to form an isocyanate, regenerating the photocatalyst (Fig. 23). Radical trapping experiments with TEMPO were conducted to corroborate the mechanism proposed. 4CzIPN was chosen as the photocatalyst, despite slower reaction kinetics, requiring 24 hours to reach a 91% product yield in comparison to [Ru(bpy)3]Cl2, which gave a 94% product yield in 15 hours. This is due to the authors’ aim of identifying a “green” route to the synthesis of urethanes and ureas, which 4CzIPN provides in comparison to transition metal photocatalysts. A follow-up study demonstrated that the carbamoyl radical could be reacted with heteroarenes (Fig. 22b).165 Of the photocatalysts screened, 4CzIPN gave the highest yield of 95%, with [Mes-Acr]ClO4 managing 70%, eosin Y obtaining only a trace of product and rose bengal being completely unreactive. This is likely to be related the superior ground state oxidation potential of 4CzIPN (Eox = 1.52 V for 4CzIPN compared with 0.78 V for eosin Y and 0.84 V for rose bengal).28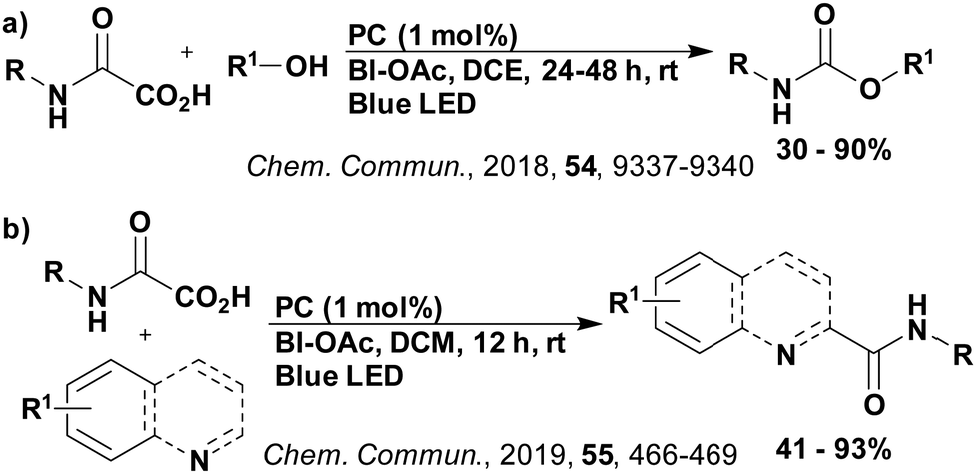 | ||
| Fig. 22 Reaction scheme for oxidative decarboxylation of oxamic acids with (a) alcohols and (b) heteroarenes. | ||
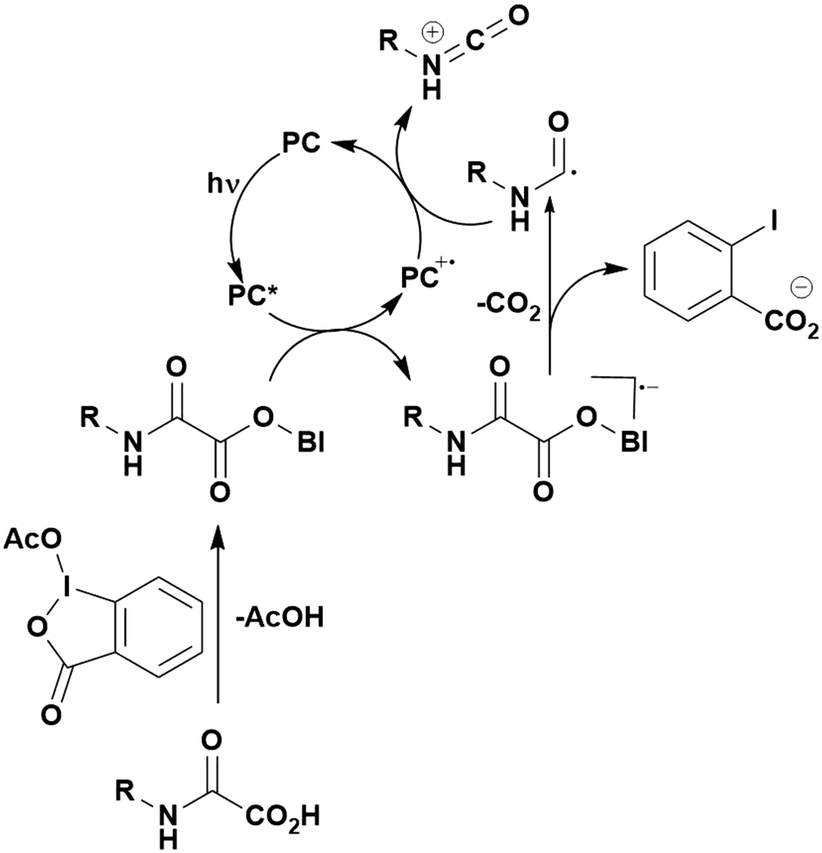 | ||
| Fig. 23 Possible mechanism for the oxidative decarboxylation of oxamic acids using a photocatalyst.164 | ||
Analogously, oxidative quenching of the excited photocatalyst by N-(acyloxy)phthalimides (NAPs) generates an alkyl radical,166 which can then undergo radical addition to a heteroarene to afford the functionalised product (Fig. 24). Three photocatalysts achieved comparable yields: 4CzIPN, [Ir(dF(CF3)ppy2)(dtbbpy)]PF6 and fac-Ir(ppy)3; however, 4CzIPN was chosen as the photocatalyst since the authors sought an organocatalysed reaction to prevent elemental impurity toxicity concerns presented by iridium photocatalysts.
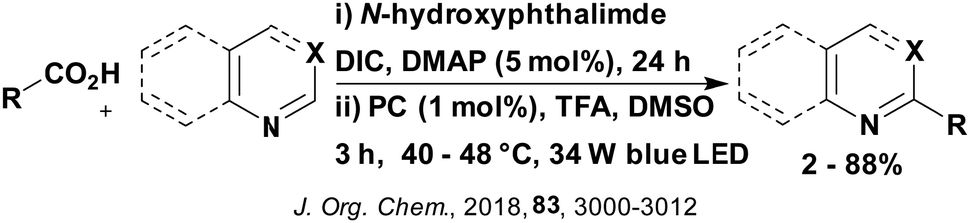 | ||
| Fig. 24 Reaction scheme for a Minisci reactions using carboxylic acids via N-acyloxyphthalimides. | ||
A similar mechanism is involved in the formation of C–F bonds using N-hydroxyphthalimide esters as the radical precursor (Fig. 25).167 The excited PC is proposed to be oxidatively quenched by this redox-active ester, forming an alkyl radical (Fig. 26). Rather than this radical adding to a double bond or coupling with another radical, as has been the case so far, the alkyl radical is oxidised by the oxidised PC to form a carbocation, closing the photocatalytic cycle. The carbocation is trapped by the fluoride ion to yield the alkyl fluoride. This proposed mechanism is consistent with the measured quantum yield of 0.37, suggesting this is not a chain mechanism. Moreover, Stern–Volmer quenching experiments revealed that the excited PC is indeed quenched efficiently by the phthalimide ester. Addition of TEMPO inhibits formation of the desired product, instead favouring a TEMPO adduct, which confirms the formation of the alkyl radical. The presence of the alkyl radical is also demonstrated by addition of other radical trapping reagents, including methyl acrylate and styrene. Finally, the formation of the carbocation was corroborated through trapping experiments with a range of nucleophiles, including alcohols and 1,3,5-trimethoxybenzene. 4CzIPN was the only organic PC considered for this reaction, with the desired product formed in 91% yield, which is comparable to the yields obtained using Ir(dFppy)3 (99%), fac-Ir(ppy)3 (96%) and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (94%).
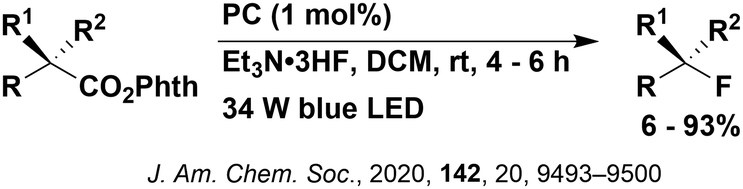 | ||
| Fig. 25 Reaction scheme for the nucleophilic fluorination of redox active esters. | ||
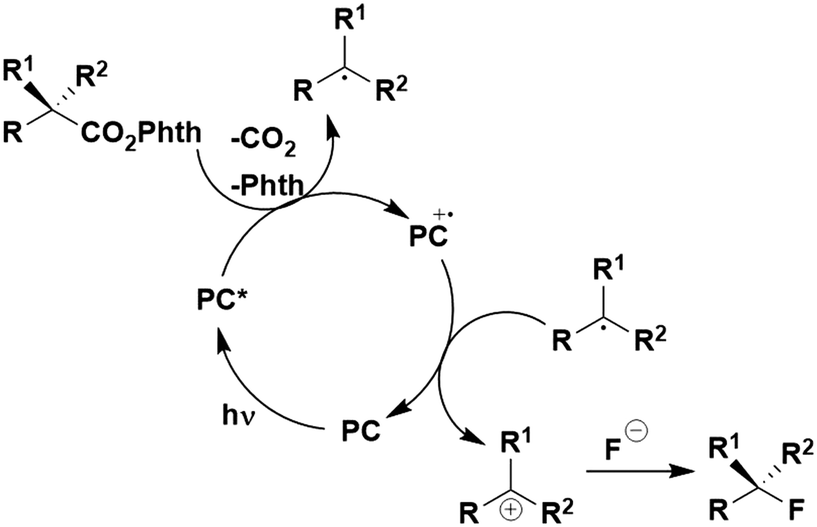 | ||
| Fig. 26 Suggested mechanism for the nucleophilic fluorination of redox active esters. | ||
Thus far, the excited photocatalyst has been oxidatively quenched by the carboxylic acid derivative to generate the required alkyl radical; however, it is possible for the oxidised photocatalyst to be responsible for generation of the alkyl radical. This can be observed in the photocatalytic synthesis of aldehydes and ketones from arylacetic acids (Fig. 27a),168 a δ-C–H mono and dihalogentation reaction (Fig. 27b),169 the intermolecular hydroalkylative dearomatization of electron-deficient indole derivatives (Fig. 27c),170 the intermolecular dearomatization of naphthalene derivatives by 1,2-hydroalkylation (Fig. 27d)171 and the decarboxylative radical addition bifunctionalization cascade for the production of 1,4-amino alcohols (Fig. 27e).172
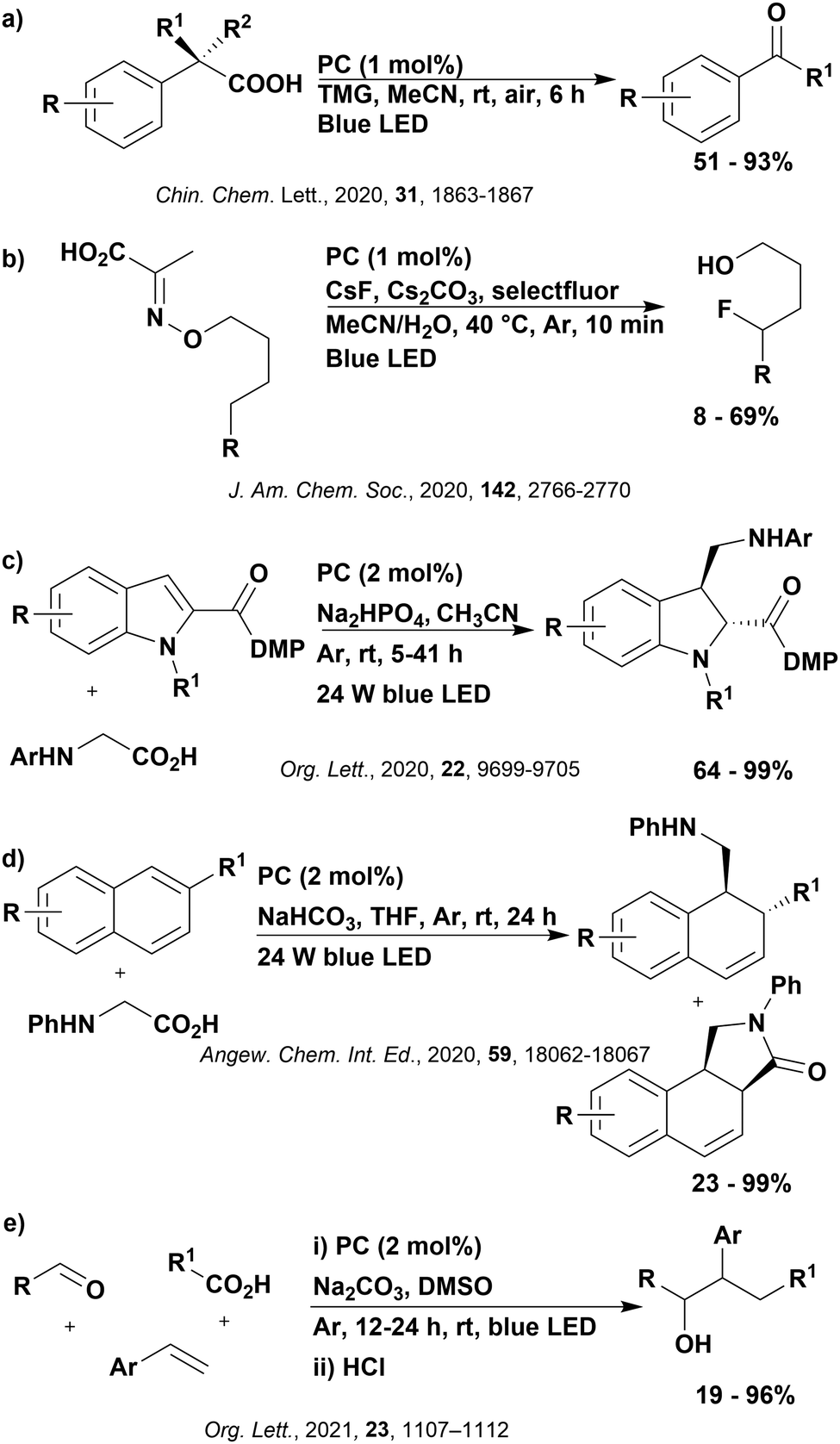 | ||
| Fig. 27 Reaction scheme for the (a) photocatalytic generation of aldehydes and ketones from carboxylic acids, (b) δ-fluorination using carboxylic acid derivatives, (c) intermolecular hydroalkylative dearomatization of electron deficient indole derivatives, (d) intermolecular dearomatization of naphthalene derivatives by 1,2-hydroalkylation and (e) decarboxylative radical addition bifunctionalization cascade to produce 1,4-amino alcohols. | ||
In the former, radical coupling of the alkyl radical with a superoxide radical (generated by SET from the excited PC), followed by protonation and dehydration yields the final product (Fig. 28). Stern–Volmer quenching experiments indicated that significant PC luminescence quenching was only observed with oxygen. Moreover, when 4-benzoquinone was added to the reaction, which acts as a superoxide radical scavenger, the yield decreased markedly from 91% to 18%, suggesting that the superoxide radical does play an active role in the mechanism. Electron paramagnetic spectroscopy (EPR) provided further evidence of the presence of the superoxide radical. Addition of TEMPO arrested the reaction, providing further evidence that this reaction involves radical processes. Only organic photocatalysts were considered, with 4CzIPN giving an impressive yield of 91% while others, such as eosin Y or rose bengal provided only a trace amount of product. This is likely related to the ground state oxidation potential of the photocatalyst, with eosin Y and rose bengal (Eox = 0.78 V and 0.84 V, respectively) not capable of oxidising the carboxylate (Eox = 1 to 1.25 V),173 while 4CzIPN is more than capable of doing this transformation (Eox = 1.52 V). Decreasing the photocatalyst loading from 5 mol% to 1 mol% resulted in a slightly higher product yield (91% to 98%, respectively).
 | ||
| Fig. 28 Plausible reaction mechanism for the photocatalytic generation of aldehydes and ketones from carboxylic acids. | ||
In the δ-C–H mono and dihalogentation reaction (Fig. 27b),169 the oxidised photocatalyst is used to oxidise the carboxylic acid derivative, which loses CO2 and MeCN, and undergoes 1,5-HAT to yield an alkoxy radical (Fig. 29). This radical then abstracts a F atom from selectfluor to generate the fluorinated product, with the selectfluor radical cation being reduced by the excited PC. This mechanism was supported by Stern–Volmer quenching experiments. A very short reaction time of 10 mins was needed to accomplish the fluorination reaction (with 1 mol% of PC) while for chlorination, a reaction time of 18 h was necessary (with 3 mol% of PC), hence quantum yield measurements were undertaken to confirm suspicion of a radical chain mechanism for the fluorination reaction. However, quantum yields of 0.070 and 0.016 were obtained for the fluorination and chlorination, respectively, suggesting radical chain mechanisms are not in operation. This result instead seemed to indicate that the fluorination reaction is more efficient, even with lower photocatalyst loading. Indeed, the rate of quenching of the alkoxy radical was found to be 53 times higher in the fluorination than chlorination reaction through radical clock experiments. Only 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 were considered as photocatalysts, providing the same yields (67% isolated yield with 4CzIPN and 71% LCMS yield with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6) with the former being chosen as the photocatalyst of choice.
 | ||
| Fig. 29 Proposed mechanism for the fluorination of carboxylic acid derivatives.169 | ||
For the reaction involving intermolecular hydroalkylative dearomatization of electron-deficient indole derivatives (Fig. 27c),170 a similar mechanism is invoked. The excited PC is oxidatively quenched by the indole to generate a radical species, which after protonation, couples with the α-amino radical formed from the oxidative decarboxylation of the carboxylic acid derivative (induced by the oxidised PC), to yield the final product. The proposed mechanism is supported by Stern–Volmer quenching experiments, which indicated that the excited PC is quenched by the indole much faster than by glycine (Stern–Volmer constant, KSV = 1460 and 173, respectively). Moreover, addition of a radical trapping agent proved the presence of the indole-based radical. 4CzIPN provided an essentially quantitative yield of 99%, while high yields were obtained by the transition metal PCs [Ir(ppy)2(dtbbpy)]PF6 (84%), fac-Ir(ppy)3 (73%) and [Ru(bpy)3]Cl2 (56%). The relative yields correlate generally with the photoreducing ability of the PC (Eox* = −1.04 V, −0.96 V and −0.81 V for 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 and [Ru(bpy)3]Cl2, respectively); however, the data do not align for fac-Ir(ppy)3 (Eox* = −1.73 V). Instead, the lower yields obtained for fac-Ir(ppy)3 may be related to its less positive ground state oxidation potential (Eox = 1.52 V and 0.77 V for 4CzIPN and fac-Ir(ppy)3, respectively).
Penultimately, the intermolecular dearomatization of naphthalene derivatives by 1,2-hydroalkylation (Fig. 27d)171 occurs analogously to the previous example. Stern–Volmer experiments indicated the naphthalene derivative oxidatively quenches the excited PC at a significantly faster rate than the carboxylic acid (Stern–Volmer constant, KSV, = 3870 and 253, respectively, in MeCN and 3068 and 232, respectively, in THF). 4CzIPN performed considerably better in this reaction than alternative PCs, yielding 90% of the product (2:1 ratio of uncyclized and cyclised product), while the next best competitor, [Ir(ppy)2(dtbbpy)]PF6 managed only 60% (3.7:1 ratio of products). It is unclear why this difference in yield was obtained, especially given that thermodynamically, neither should be capable of reducing the naphthalene derivative (Ered = −1.45 V vs. SCE for (3,5-dimethyl-1H-pyrazol-1-yl)-2-naphthalenylmethanone) and Eox* = −1.04 V and −0.96 V for 4CzIPN and [Ir(ppy)2(dtbbpy)]PF6, respectively).
Finally, in the decarboxylative radical addition bifunctionalization cascade for the production of 1,4-amino alcohols (Fig. 27e)172 the excited PC is oxidatively quenched by the aldehyde to form a ketyl radical. The oxidised PC generates the alkyl radical from the carboxylate analogously to the previous two examples. The alkyl radical adds to the olefin, with the resultant radical coupling with the ketyl radical. Subsequent protonation yields the final product. Addition of a radical scavenger TEMPO and deuterium labelling experiments substantiated the proposed mechanism. Of the eight PCs screened, only 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 yielded any product (90% and 42%, respectively). This may be on account of their photoreducing ability (Eox* = −1.04 V and −0.89 V, respectively, compared to −0.81 V for [Ru(bpy)3]Cl2 which obtained no product). Alternatively, the authors imply a reductive quenching mechanism may also be in operation, the lack of mechanistic clarity makes it difficult to ascertain why 4CzIPN performed so excellently in comparison to the other PCs.
Alternative radical precursors in forming C–C or C–X bonds
In lieu of carboxylic acids, a number of other precursors have been used to generate carbon-centred radicals via a reductive quenching pathway. These include alkyltrifluoroborates,174,175 alkyl bis(catecholato)silicates (or other alkyl silicates),161,175–180 4-alkyl-1,4-dihydropyridine derivatives (DHPs)177,181 and α-trimethylsilylamines175 (Fig. 30), all of which are oxidised via SET by the excited PC. These subsequent radicals were then reported to react with a structurally diverse number of substrates such as alkenes (Fig. 31a),175 heteroarenes (Fig. 31b, 31c and 31i),174,177,180 imines (Fig. 31d),161 CO (Fig. 31e and 31h),176,179 SO2 (Fig. 31f)181 and N-acylhydrazones (Fig. 31g).178 | ||
| Fig. 30 Radical precursors which are used in photoredox reactions: (a) trifluoroborate salts, (b) 4-alkyl-1,4-dihydropyridine derivatives (DHPs), (c) silicates, (d) carboxylic acids and (e) α-trimethylsilylamines. | ||
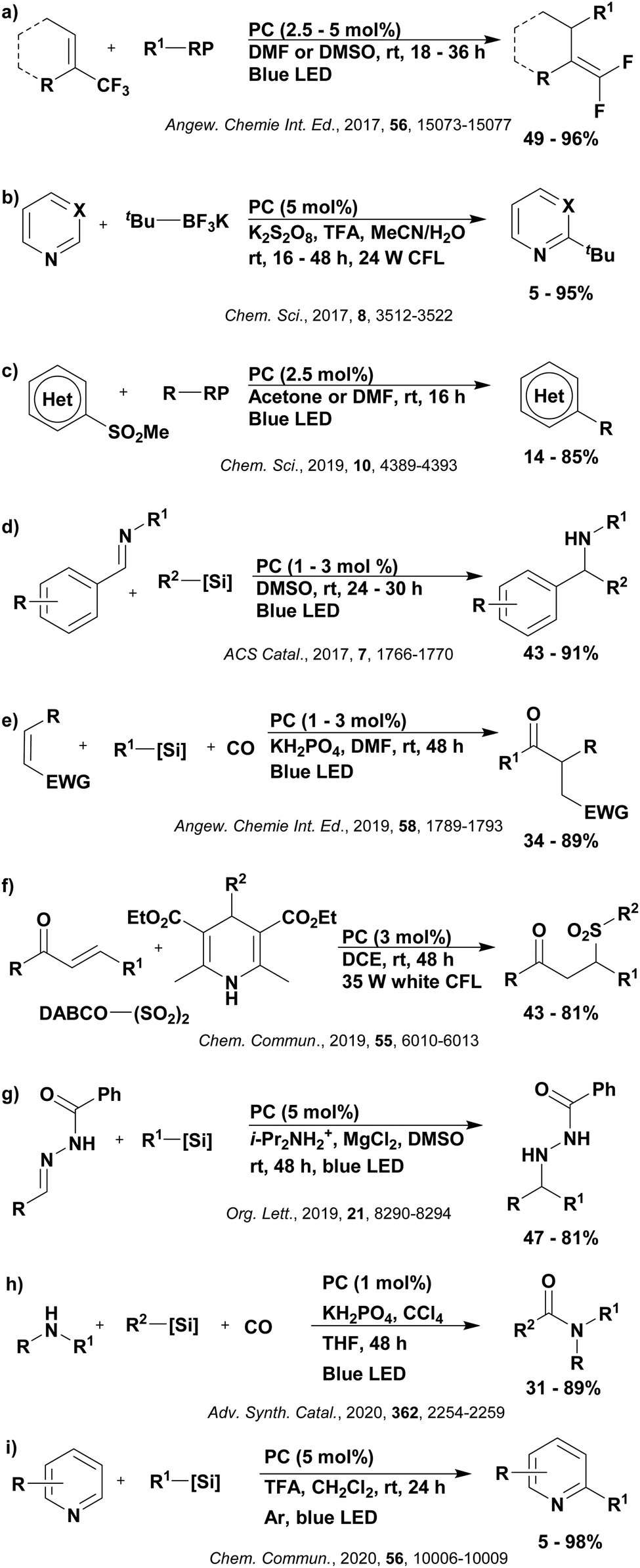 | ||
| Fig. 31 Reaction schemes for the use of common radical precursors other than carboxylic acid derivatives in C–C and C–X bond forming reactions. | ||
For the alkylation of imines (Fig. 31d),161 the carbonylation of alkenes (Fig. 31e),176 hydrosulfonylation of alkenes (Fig. 31f)181 and synthesis of aliphatic amides (Fig. 31h),1794CzIPN was demonstrated to be the best photocatalyst as it provided the highest product yields. In the alkylation of N-acylhydrazones (Fig. 31g), 4CzIPN was the only photocatalyst considered, reaching very high yields of 88% during the optimisation experiments.178
However, in the alkylation of heteroarenes (Fig. 31b, c and i), the identification of the superior photocatalyst is far more substrate dependent. For example, primary alkyltrifluoroborates have a significantly higher oxidation potential (Eox = 1.90 V vs. SCE)182 than their secondary and tertiary counterparts, hence require a much stronger photooxidant. Since [Mes-Acr]* has a considerably greater excited state oxidising capacity compared with 4CzIPN (Ered* = 2.06 V and 1.35 V, respectively), it is not surprising that it performed better; 4CzIPN, however, was a promising alternative for tertiary alkylation, outperforming [Mes-Acr]+ (96% yield for 4CzIPN and 57% for the acridinium salt).174 For the heteroarene alkylation shown in Fig. 31i, 4CzIPN was again outcompeted by [Mes-Acr]+ (73% and 87%, respectively).1804CzIPN was shown to give high product yields when DHPs (Eox ≈ 1.05 V vs. SCE)183 were used as the radical precursor (although no other photocatalysts were tested), but unfortunately gave low yields when silicates were used (Eox ≈ 0.75 V vs. SCE),177,184 achieving only 14% of product in comparison to 78% obtained with [Ru(bpy)3](PF6)2. The inferiority of 4CzIPN to [Ru(bpy)3](PF6)2 was also evident in the defluorinative alkylation protocol (Fig. 31a), whereby the ruthenium photocatalyst resulted in a 0.28 ratio of product:internal standard, while 4CzIPN managed only 0.23.175 Perhaps this is related to the regeneration of the photocatalyst, since 4CzIPN is less reducing in the ground state (Ered = −1.21 V for 4CzIPN compared to −1.33 V for [Ru(bpy)3](PF6)2).
Another important class of radical precursor is amines, which undergo SET to the excited PC, generating α-amino radicals after a subsequent deprotonation. This reactivity is seen in the dicarbofunctionalization of styrenes with amines and CO2 to generate γ-amino acids (Fig. 32).185 The α-amino radical adds to the styrene derivative, with the resultant γ-amino benzylic radical being reduced by the reduced PC to form a carbanion. Nucleophilic addition of this carbanion to CO2, followed by protonation, yields the final product. The quantum yield of the reaction was determined to be 0.04, implying a radical chain mechanism is unlikely; radical scavengers such as TEMPO inhibited product formation, confirming that the reaction is a radical process. Deuterium labelling studies implicated the presence of the γ-amino benzylic anion as an intermediate.
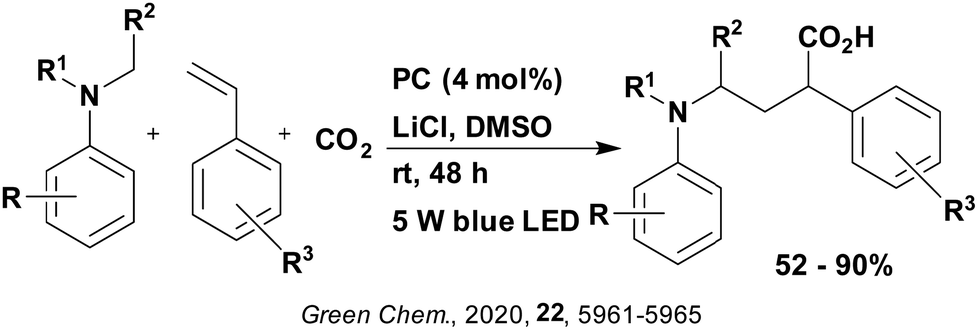 | ||
| Fig. 32 Reaction scheme for the dicarbofunctionalization of styrenes with amines and CO2 to generate γ-amino acids. | ||
4CzIPN performed excellently, yielding 95% of product. Cationic iridium and ruthenium complexes also performed very well ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2 obtained 76%, 84% and 83%, respectively). fac-Ir(ppy)3 was the only PC in the study that performed poorly, yielding only 12% of product. The success of 4CzIPN may be on account of its stronger photooxidising ability (Ered* = 1.35 V, 1.21 V, 0.66 V, 0.77 V and 0.31 V for 4CzIPN, ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(ppy)2(dtbbpy)]PF6, [Ru(bpy)3](PF6)2 and fac-Ir(ppy)3, respectively).
Interestingly, the α-amino radicals formed may not necessarily be present in the final product, but may instead be used as a halogen atom transfer (XAT) agents in the alkylation (Fig. 33a) and allylation (Fig. 33b) of alkyl and aryl halides.186 A proposed mechanism is shown in Fig. 34 for the alkylation reaction, with a similar mechanism being suggested also for the allylation. A quantum yield of less than 1 for both reactions is suggestive that this is not a radical chain process. Only 4CzIPN was utilised as a PC in both reactions; however, in the study, Leonori et al. do justify this choice by conducting a PC optimisation for the dehalogenation reaction involving a HAT catalyst, finding 4CzIPN to be the highest yielding and employing this PC in the subsequent reactions. For further information on this study and the PC optimisation, refer to the HAT dual catalysis Section 3.
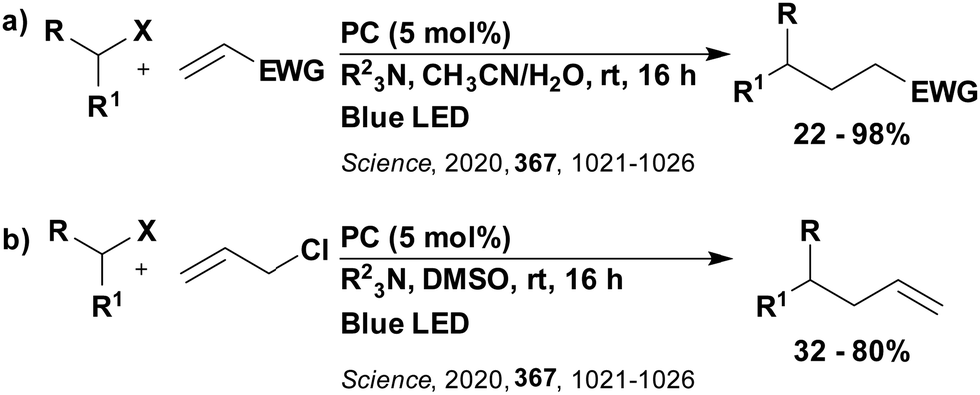 | ||
| Fig. 33 Reaction scheme for the (a) alkylation and (b) allylation of alkyl and aryl halides. | ||
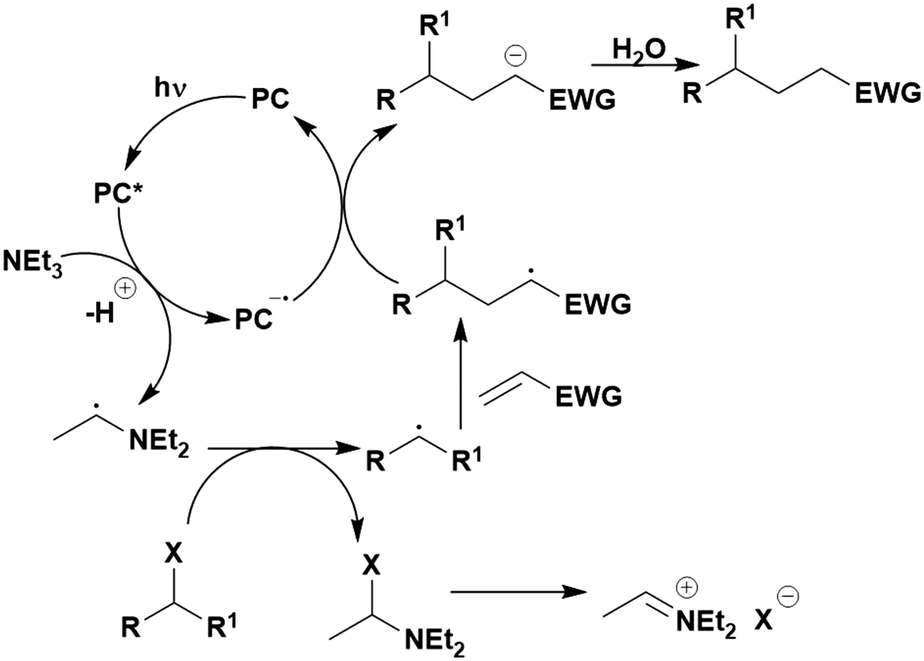 | ||
| Fig. 34 Proposed mechanism for the alkylation of alkyl halides. | ||
Alkylboranes may be used as the radical precursors, which themselves are obtained from functionalised 1,2-bis(boronic esters) (Fig. 35).187 A boronate complex is first formed through reaction of the 1,2-bis(boronic esters) with an aryl lithium species, which is oxidised by the excited PC, generating a primary radical (Fig. 36). A 1,2-boron shift results in the more thermodynamically favourable secondary radical, which undergoes radical addition to an alkene. Reduction of this species by the reduced photocatalyst closes the photocatalytic cycle and allows for the formation for the final functionalised product. This mechanism, particularly the 1,2-boron shift, was supported by mechanistic studies, DFT modelling and boron isotope labelling. Only 4CzIPN and fluorescein were considered as PCs in this study, with 4CzIPN vastly outperforming the latter (100% and 11% yield, respectively). These results were expected based on their relative excited state reduction potentials (Ered* = 1.35 V and 0.77 V, respectively).
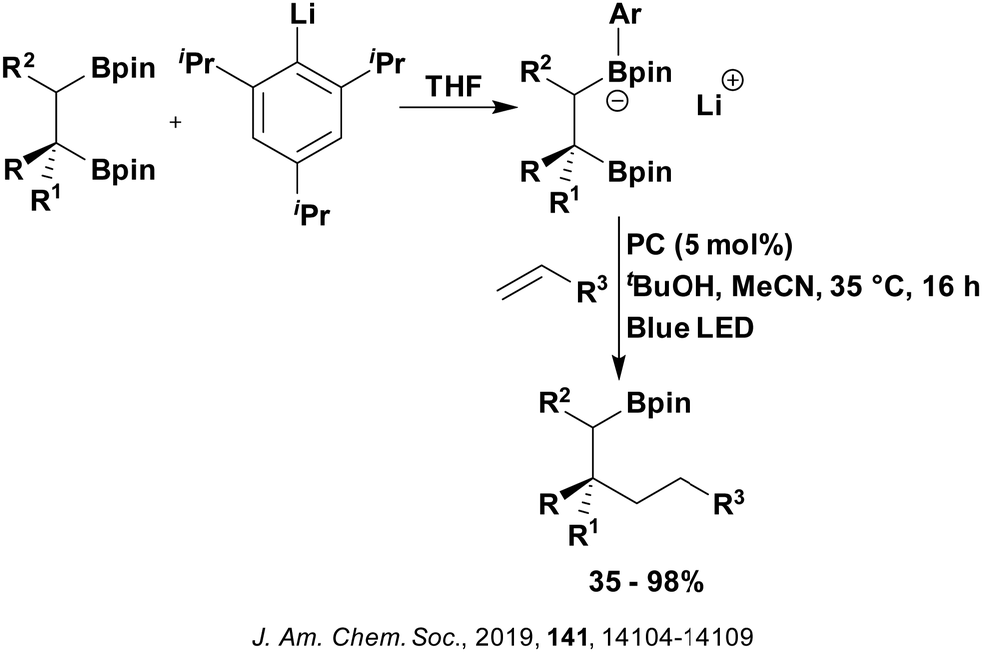 | ||
| Fig. 35 Reaction scheme for the functionalisation of 1,2-bis(boronic esters). | ||
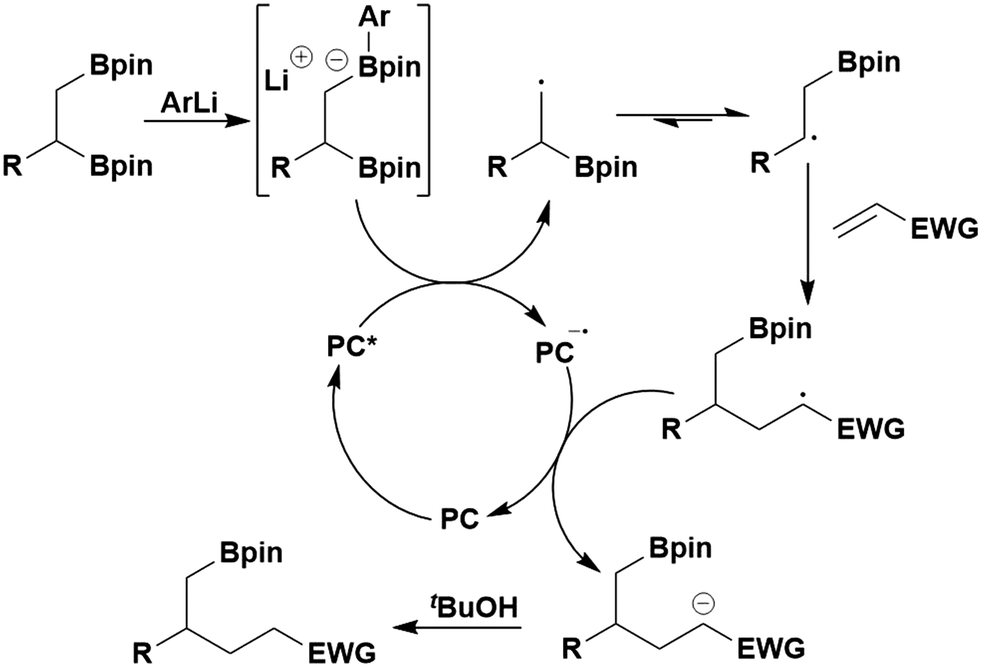 | ||
| Fig. 36 Proposed mechanism for the functionalisation of 1,2-bis(boronic esters). | ||
Borylation of aryl groups is additionally possible in a sp3-sp2 type coupling to form C-B bonds (Fig. 37).188 All six PCs considered in this study performed well, ranging between 68–95%, with 4CzIPN performing the best. Since the mechanism is unknown as of yet, it is difficult to ascertain why 4CzIPN yielded the largest amount of product.
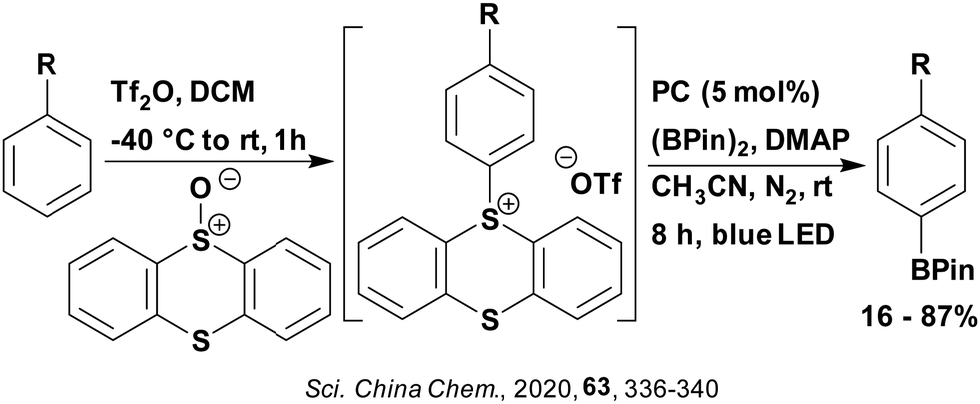 | ||
| Fig. 37 Reaction scheme for the borylation of aryl groups. R = alkyl, aryl, F, OR or NR2. | ||
Carbotrifluoromethylation via photocatalytic decomposition of reagents such as CF3SO2Na to generate CF3 radicals has been reported to proceed by a reductive quenching pathway.189–192 This electrophilic trifluoromethyl radical can then add to electron-rich substrates such as alkenes in an anti-Markovonikov fashion to afford trifluoromethylated products. When the substrate is an α,α-diaryl allylic alcohol, β-trifluoromethyl-α-aryl ketones are obtained (Fig. 38a), proceeding via a radical 1,2-aryl migration.189 When the substrate is an N-aryl acrylamide, an oxindole is generated (Fig. 38b),190 proceeding via a radical cyclisation (Fig. 39) while if the additional reactants involve quinoxalin-2(1H)-ones and alkenes, 3-trifluoroalkylated quinoxalin-2(1H)-ones are formed (Fig. 38c).191 Finally, in the combination of styrene and carbonyls, β-trifluoromethyl-α-substituted alcohols are obtained (Fig. 38d).192
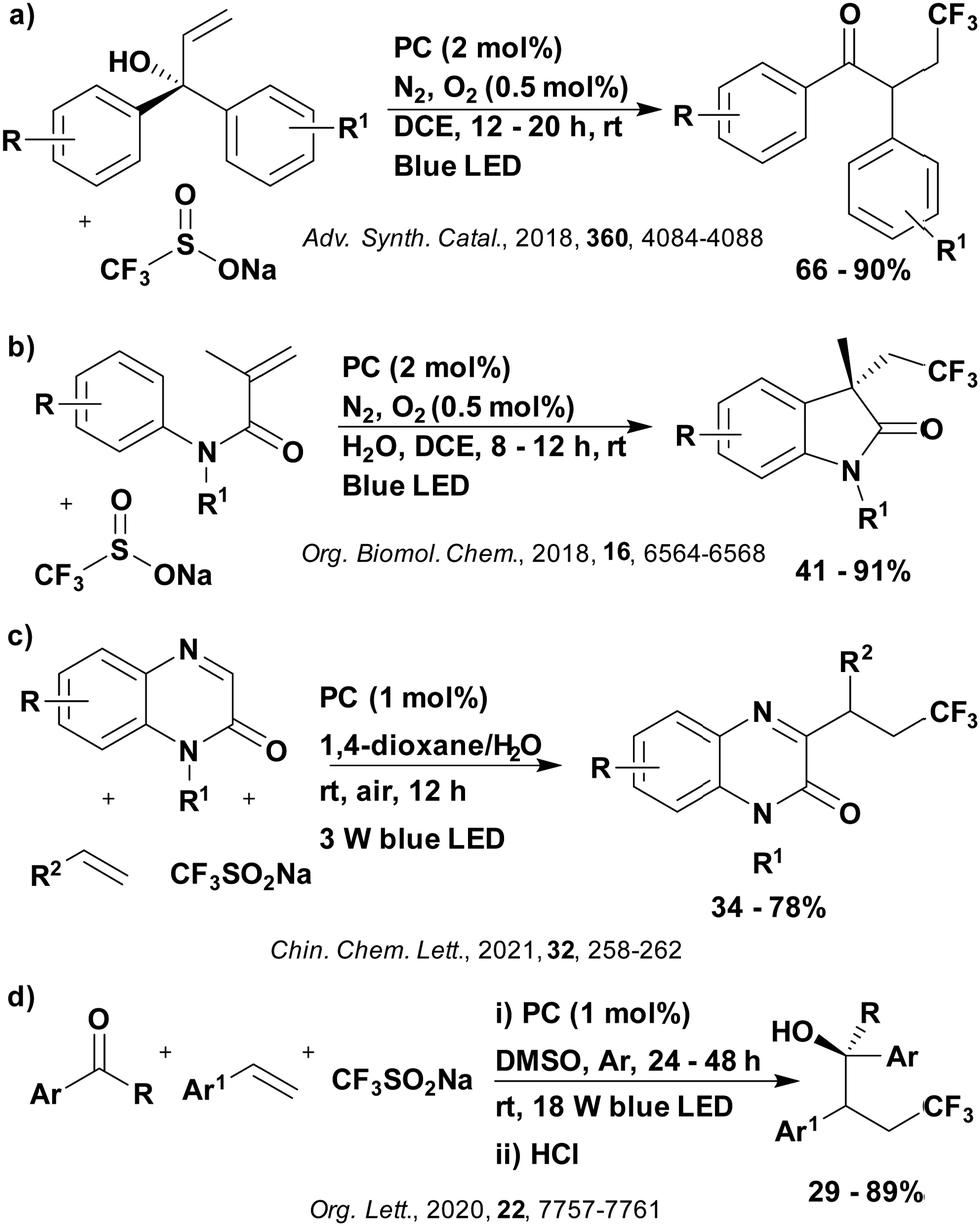 | ||
| Fig. 38 Reaction scheme showing carbotrifluoromethylation of (a) allylic alcohols, (b) N-aryl acrylamides, Nc) quinoxalin-2(1H)-ones and alkenes and (d) styrenes and carbonyls. | ||
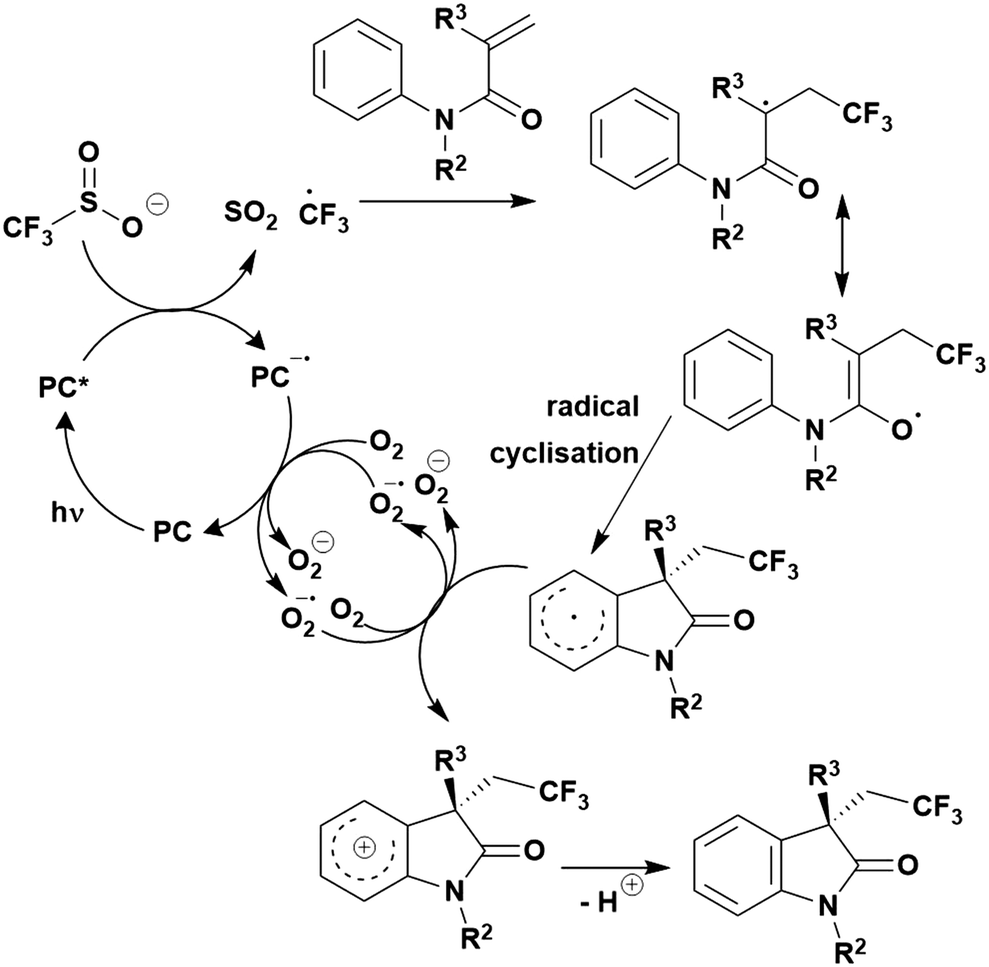 | ||
| Fig. 39 Proposed mechanism for the photocatalytic carbotrifluoromethylation of N-aryl acrylamides. | ||
The proposed mechanism for carbotrifluoromethylation involving N-aryl acrylamides was supported by radical trapping experiments and the results from control reactions. A quantum yield of 3.33 was obtained for the cyclisation reaction of N-methyl-N-(p-tolyl)-methacrylamide using 4CzIPN, suggesting strongly that a radical chain process is involved.190
In the reactions shown in Fig. 38a and b, 4CzIPN gave the highest product yields, with [Ir(ppy)2(dtbbpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 being the closest competitors (86%, 45% and 68% yields, respectively in the reaction shown in Fig. 38a, 83%, 64% and 71% yield, respectively in the reaction in Fig. 38b). For the reaction shown in Fig. 38d, only 4CzIPN and [Ir(dF(CF3)ppy)2(bpy)]PF6 provided any product (82% and 48%, respectively). The superiority of 4CzIPN could be corelated to its excited state reduction potential (Ered* = 1.35 V), which is considerably more positive than those of the other PCs considered (Ered* = 0.66 V, 1.21 V and 1.32 V for [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy2(dtbbpy)]PF6 and [Ir(dF(CF3)ppy)2(bpy)]PF6, respectively). Given the oxidation potential of CF3SO2Na (Eox = 1.05 V),193 there is a greater thermodynamic driving force for the generation of the CF3 with 4CzIPN, which may be why it attains the highest yield.
Meanwhile, in the synthesis of 3-trifluoroalkylated quinoxalin-2(1H)-ones (Fig. 38c), the mechanism differs, as shown in Fig. 40.191 In this case, the excited PC is reductively quenching by the qunoxalin-2(1H)one, affording a nitrogen centred radical cation. The CF3 radical, generated by oxidation and fragmentation of CF3SO2Na using molecular oxygen, then adds to the alkene. The carbon-centred radical intermediate couples with the aforementioned nitrogen-centred radical cation, which after deprotonation, yields the final product. The photocatalytic cycle is closed by oxidation of the reduced PC induced by molecular oxygen. Addition of the radical scavenger TEMPO inhibited the reaction, forming instead an adduct with CF3, confirming the presence of the CF3 radical and that this is a radical process. Stern–Volmer quenching experiments conducted by Wei et al. indicated that only quinoxaline-2(1H)-one could quench the excited PC while CF3SO2Na did not, which contradicts the mechanism shown in Fig. 39, as proposed by Huang, Cai and Liang.189,190,192 Only organic PCs were considered for this reaction, with 4CzIPN outperforming its competitors, obtaining 56% yield while the next best PC, rose bengal, afforded only 30%. This is likely due to the superior photooxidising ability of 4CzIPN (Ered* = 1.35 V and 0.81 V for 4CzIPN and rose bengal, respectively).
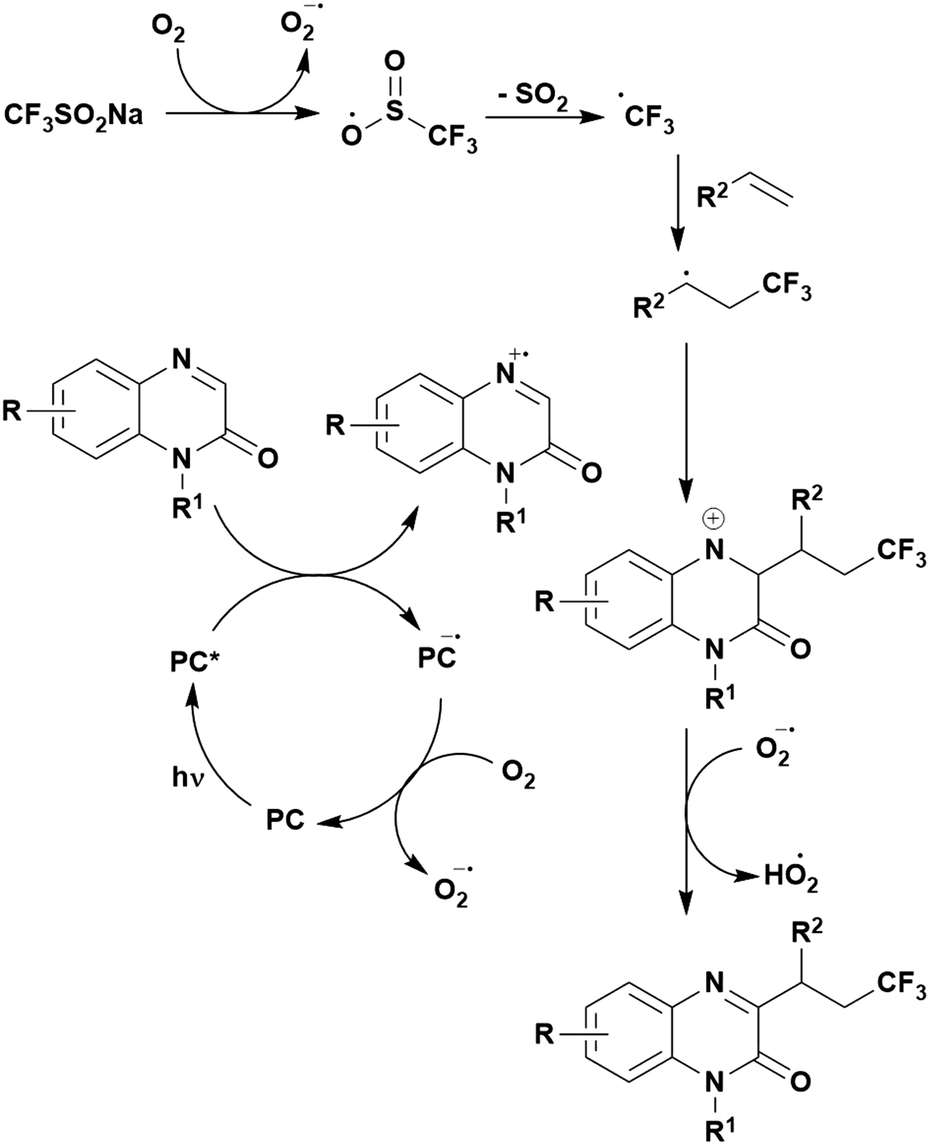 | ||
| Fig. 40 Putative mechanism for the synthesis of 3-trifluoroalkylated quinoxalin-2(1H)-ones. | ||
Carbotrifluoromethylation can also proceed via an oxidative quenching cycle using CF3SO2Cl or Togni's reagent [1,3-dihydro-3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole] as the trifluoromethylating agent (Fig. 41).193 The CF3 radical, generated through reduction by the excited PC, can undergo radical addition to benzyl-protected homoallylic alcohol and amine derivatives, which upon 1,5-HAT, produce a radical that is oxidized by the oxidized PC, closing the photocatalytic cycle (Fig. 42); the corresponding benzylic carbocation is quenched by the alcohol solvent to form an acetal, with an aqueous work-up resulting in the formation of the final product. Of the PCs investigated, eosin Y produced only trace amounts of the desired product while [Ir(ppy)2(dtbbpy)]PF6 yielded 57% and 4CzIPN 69%. The more positive oxidation potential for 4CzIPN than the other PCs (Eox = 1.52 V compared with 0.78 V for eosin Y and 1.21 V for the iridium PC), may account for its improved performance. Replacement of the CF3 source, in this case Togni's reagent or CF3SO2Cl, with CF3SO2Na altered the mechanism, favouring a reductive quenching cycle instead, as had been demonstrated by Huang et al.189
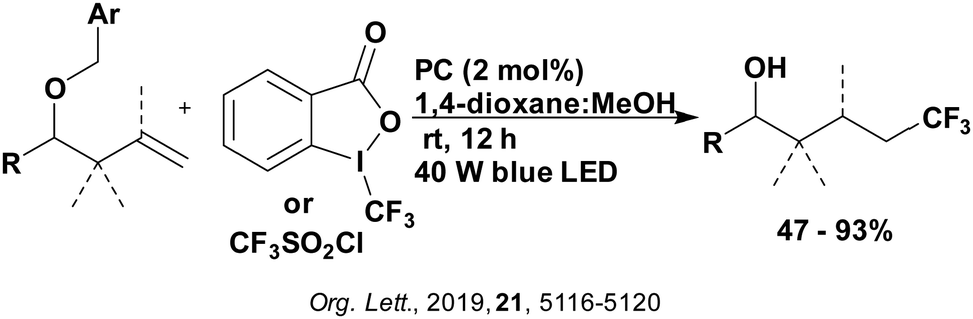 | ||
| Fig. 41 Carbotrifluoromethylation reaction scheme for the synthesis of δ-fluoromethylated alcohols. | ||
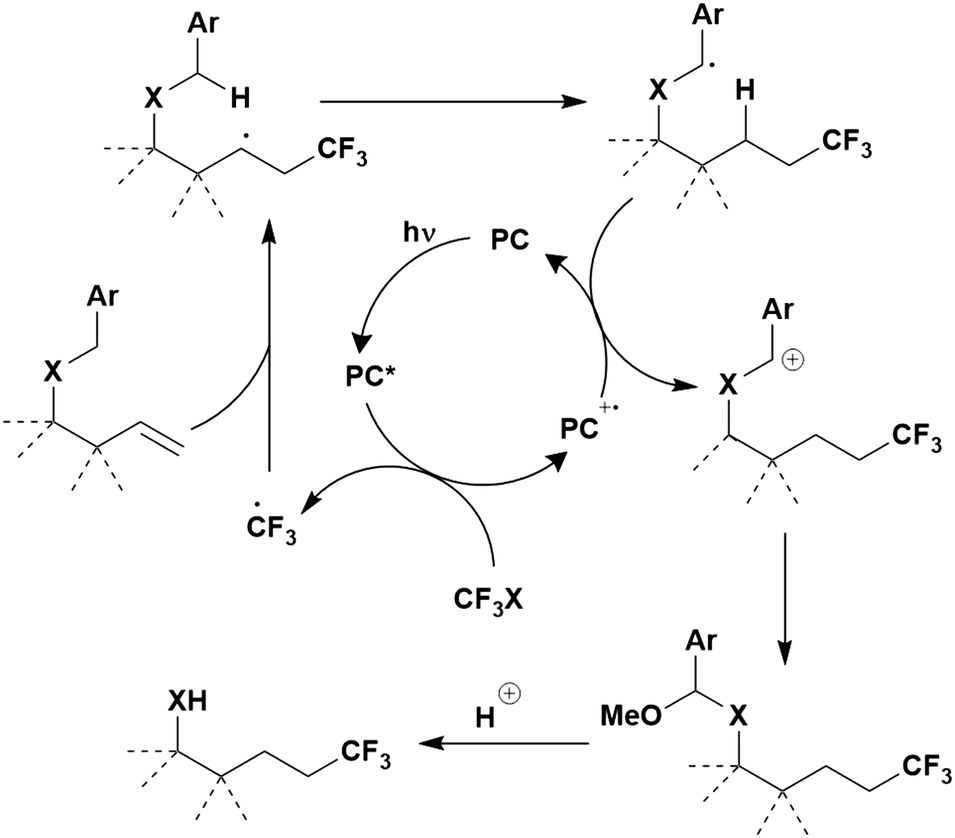 | ||
| Fig. 42 Proposed mechanism for organic photoredox catalysed synthesis of δ-trifluoromethylated alcohols and amines where X = O or NAr. | ||
Coupling of a CRF2 group is likewise possible using photocatalysis (Fig. 43). The excited photocatalyst is first reductively quenched by an in situ formed hydrazone, resulting in a radical intermediate (Fig. 44). The reduced photocatalyst then generates the difluoroalkyl radical from BrCF2R by SET, closing the photocatalytic cycle. The two radical intermediates subsequently couple together to form α,α-difluoroketone hydrazones.194 Only 4CzIPN was considered for this reaction and investigation into the catalyst loading revealed that increasing the loading from 2 to 4 mol% did little to impact the product yield (increasing from 72 to 75%). The suggested mechanism (Fig. 44) was supported by radical trapping experiments with TEMPO as well as Stern–Volmer quenching studies.
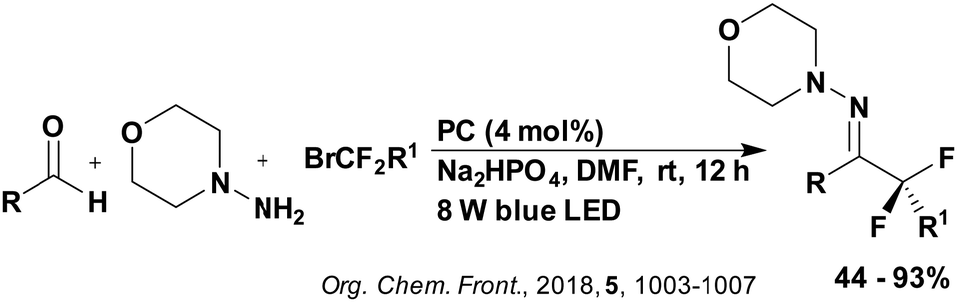 | ||
| Fig. 43 Reaction scheme for the coupling of aldehydes with hydrazines and bromodifluorinated reagents. | ||
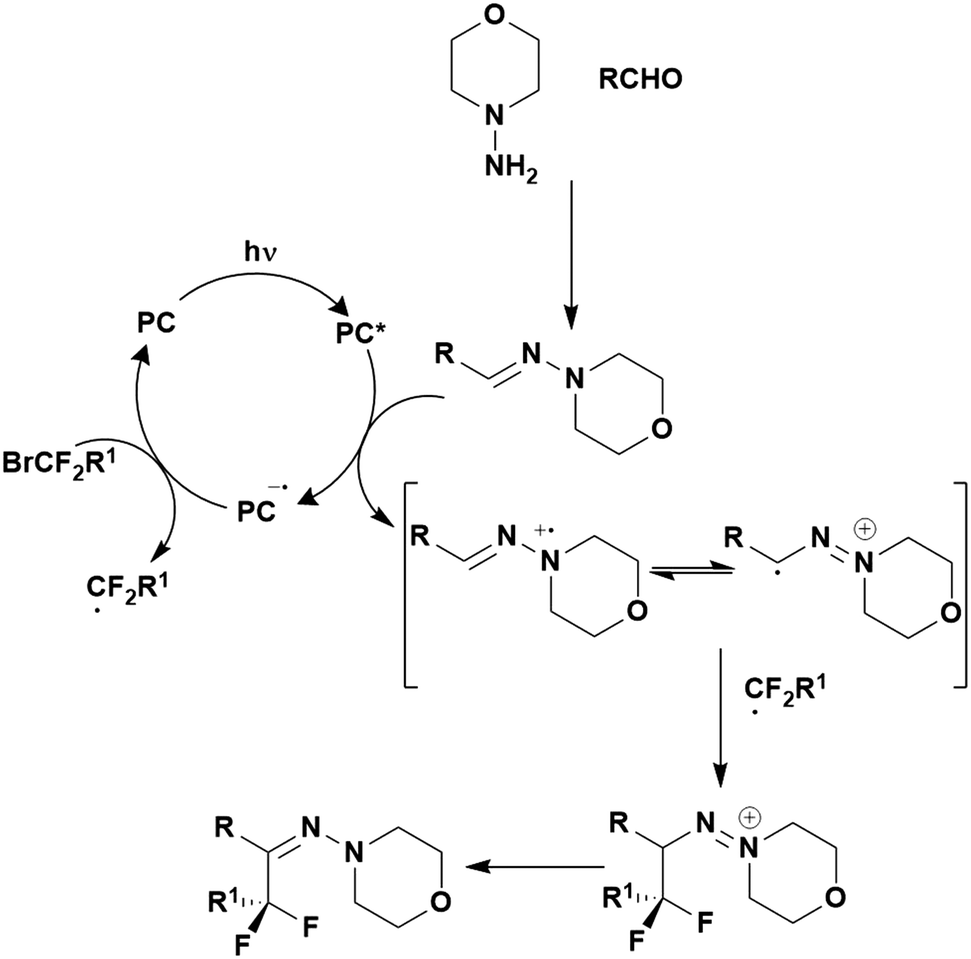 | ||
| Fig. 44 Potential mechanism for the formation of α,α-difluoroketone hydrazones using photocatalysis. | ||
A similar example involves the fluoroalkylation of arylidene and alkylidene amidrazones (Fig. 45).195 The putative mechanism involves first the reductive quenching of the excited PC by DABCO. The reduced PC is then proposed to reduce the fluorinated iodide reagent to yield the fluorinated radical. This radical then adds to the amidrazone and after oxidation (from oxidized DABCO) and deprotonation, the final product is formed. No evidence is provided to support this mechanism. 4CzIPN performed very well in this reaction (89% yield), closely followed by [Ru(bpy)3](PF6)2 (78% yield), while [Ir(ppy)2(dtbbpy)]PF6 and eosin Y acted poorly (38% and 10%, respectively). The trends in reaction yield may be linked to the photooxidising ability of the PC (Ered* = 1.35 V, 0.77 V, 0.66 V and 0.83 V for 4CzIPN, [Ru(bpy)3](PF6)2, [Ir(ppy)2(dtbbpy)]PF6 and eosin Y, respectively), which is linked to the capacity to oxidise DABCO (Eox = 0.69 V vs. SCE).
 | ||
| Fig. 45 Reaction scheme for the fluoroalkylation of arylidene and alkylidene amidrazones. | ||
Alkyl formates have been utilised as precursors for alkoxycarbonyl radicals, which can then react with alkenes to form a variety of substituted alkanoates, depending on the conditions employed (Fig. 46).196 The proposed mechanism involves oxidative quenching of the excited PC by the N-alkyoxyazinium salt (Fig. 47) to yield an isopropoxy radical, which first abstracts a hydrogen atom from methyl formate and then adds to the alkene. The resultant radical is oxidised by the oxidised PC. From here, products can form depending on the reaction conditions, as shown in Fig. 47. For example, when excess formic acid is present, this acts as a nucleophile and a β-formyloxy ester is produced (Fig. 46c) while when MeOH is present as the nucleophile, a β-methoxy ester is formed (Fig. 46a). Support for this mechanism was provided in the form of Stern–Volmer quenching experiments, which revealed that the N-alkyoxyazinium salt was responsible for quenching the excited PC. Complementary mechanistic studies involved isotope labelling, addition of radical scavengers and control experiments to investigate the role of the additives used. The 1,2-methoxy methoxycarbonylation of α-methyl styrene was investigated as the model reaction in order to determine the optimal reaction conditions. Only 3 PCs were tested, 4CzIPN (80%), [Ru(bpy)3]Cl2 (72%) and [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (62%). This difference in yield seems to be a reflection of the photoreducing ability of the PC (Eox* = −1.04 V, −0.89 V and −0.81 V). Consequently, 4CzIPN was chosen for the substrate scope.
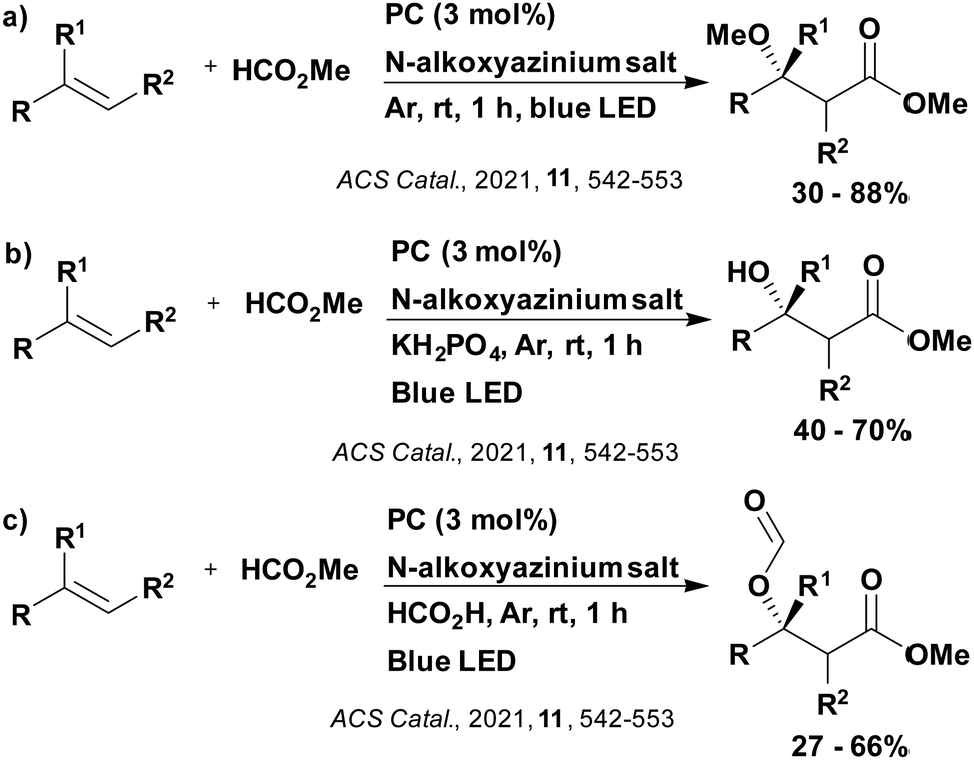 | ||
| Fig. 46 Reaction schemes for the difunctionalisation of alkenes. | ||
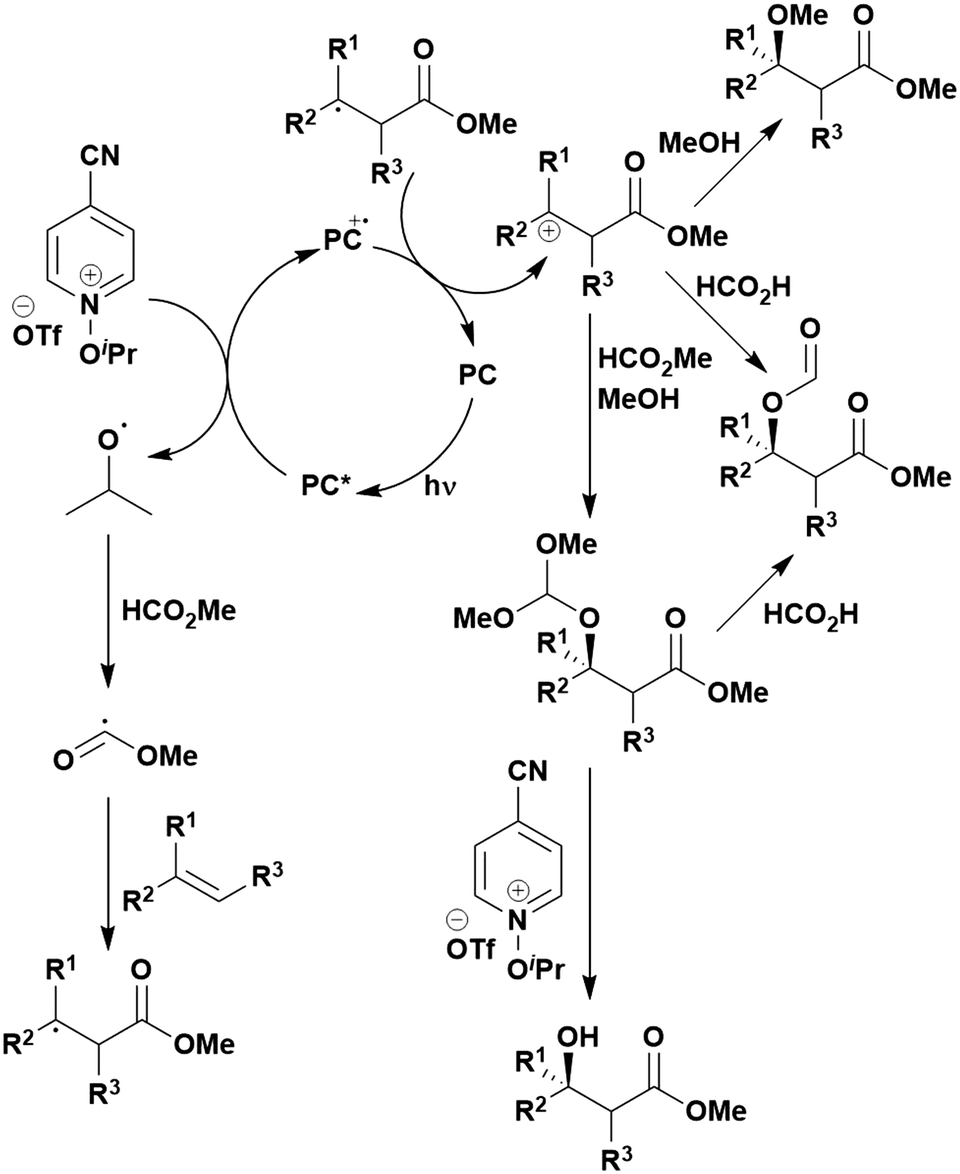 | ||
| Fig. 47 Proposed mechanism for the dinfunctionalisation of alkenes using alkyl formates. | ||
All radical precursors shown thus far can be used to generate the corresponding radical species relatively easily through SET to or from the excited or ground state PC; however, the situation becomes more complex when the reagents used are difficult to oxidise or reduce directly. This is exemplified through the use of aldehydes as radical precursors. Oxygen is required as an additive to allow for the formyl radical to be accessed, which upon dicarbonylation form alkyl radicals that can be added to heteroarenes (Fig. 48).197 In this case, the excited PC is oxidatively quenched by oxygen, generating a superoxide radical anion, which can abstract a proton from the aldehyde, forming the acyl radical. Decarbonylation of this acyl radical followed by addition to the heteroarene and deprotonation, results in a tertiary radical species that is then oxidised by the oxidised PC (Fig. 49). This mechanism is supported by radical trapping experiments using TEMPO, whereby an adduct was formed between TEMPO and the acyl radical of the aldehyde reagent. Stern–Volmer quenching experiments identified oxygen as the only significant quencher involved in the reaction. Both 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 provided the same yields (70% and 66%, respectively). These high yields are likely due to the strong ground state oxidation potential of these PCs (Eox = 1.52 V and 1.69 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively) in comparison to that of eosin Y (Eox = 0.78 V), which afforded only 33% of product.
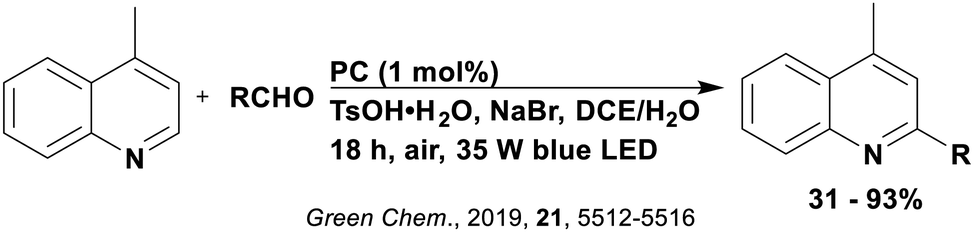 | ||
| Fig. 48 Reaction scheme for the alkylation of heteroarenes using aldehydes. | ||
 | ||
| Fig. 49 Possible mechanism for the photocatalytic alkylation of heteroarenes using aldehydes. | ||
Alternatively, LiBr has been used as an additive for accessing a formyl radical species from aldehydes as shown in the hydroxyalkylation of quinolines with aryl aldehydes (Fig. 50a)198 and alkyl aldehydes (Fig. 50b).199 A reductive quench of the PC is proposed to occur by the bromide anion, forming a bromyl radical, which abstracts a proton from the aldehyde via a deprotonated electron transfer (DPET) step (Fig. 51). The resultant acyl radical can add to the quinolines, yielding the hydroalkyl radical after subsequent deprotonation and a spin-centre shift. This radical is reduced by the reduced PC to form the product and close the photocatalytic cycle. Control deuterium labelling experiments indicated that a ketyl radical pathway was not occurring and formation of the acyl radical was likely the dominant pathway. This was further confirmed by the trapping of an acyl adduct with TEMPO, with product formation inhibited. Stern–Volmer quenching experiments showed that only the bromide ion acted to significantly quench the luminescence of the PC.
 | ||
| Fig. 50 Reaction scheme for the hydroxyalkylation of quinolines with (a) aryl aldehydes and (b) alkyl aldehydes. | ||
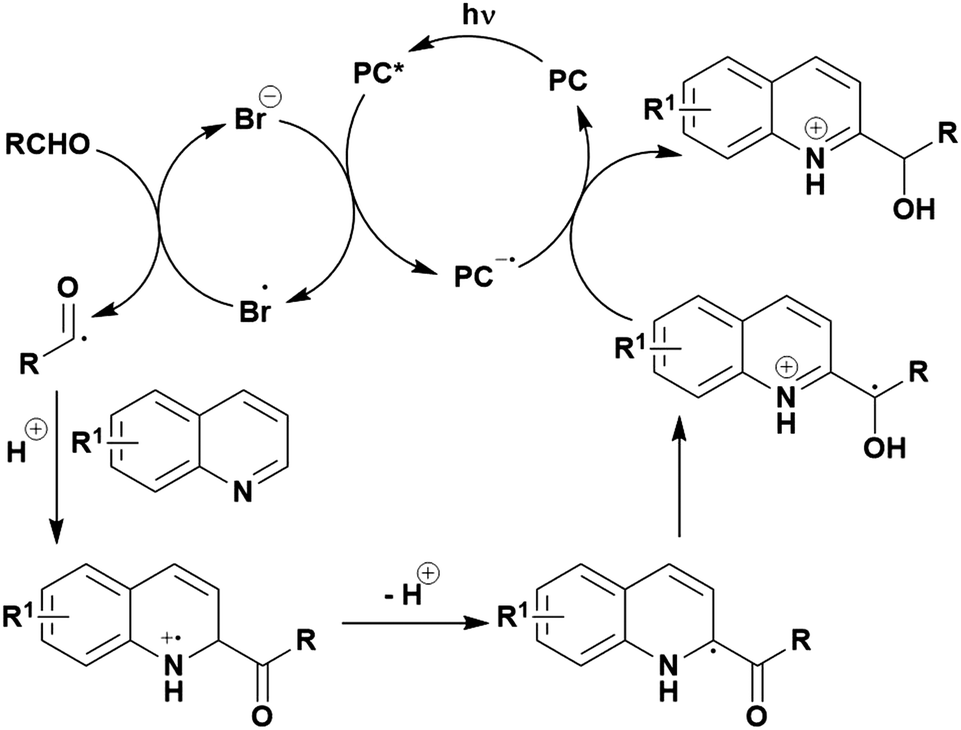 | ||
| Fig. 51 Putative mechanism for the hydroxyalkylation of quinolines with aldehydes. | ||
In the coupling with alkyl aldehydes, an additional additive Et3SiH was included, which was proposed to cause the mechanism to differ. The putative mechanism still involves the reductive quenching of the excited PC by the bromide ion, but the resultant bromyl radical then undergoes HAT with Et3SiH. The silyl radical adds to the aldehyde to form a nucleophilic silylether radical, which then adds to the protonated heteroarene. The mechanism then follows similarly to that shown in Fig. 51, where deprotonation followed by spin-centre shift (and elimination of triethylsilanol) forms a carbon-centred radical that is then reduced by the reduced PC to yield the final product.
Only 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 were investigated as PCs in the coupling with aryl aldehdyes,198 providing yields of 77% and 66%, respectively. The relative yields correlate with the photooxidising ability of the PC (Ered* = 1.35 V and 1.21 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), which must be capable of oxidising the bromide anion. Since single electron oxidation of halides cannot accurately be determined by cyclic voltammetry, recent estimations from Marcus theory suggest the single electron oxidation of bromide in MeCN is Eox = 1.11 V vs. SCE.200 The greater thermodynamic driving force for 4CzIPN for the oxidation of bromide may explain the higher product yield for this PC.
In the follow up study with alkyl aldehydes, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 shown to be superior to 4CzIPN (60% and 41%, respectively), while the other PCs employed, such as eosin Y or [Ru(bpy)3](PF6)2, could not complete the transformation.199 These other PCs are likely to have failed on account of their poor photooxidising ability (Ered* = 0.83 V and 0.77 V for eosin Y and [Ru(bpy)3](PF6)2, respectively). It is unclear why the iridium PC outperformed 4CzIPN in this case.
The use of bromide ions to facilitate the formation of radical species can also be seen in the coupling of alkyl bromides and heteroarenes using silyl-mediated visible light photocatalysis (Fig. 52).201 The excited PC oxidises the bromide ion, generating an electrophilic bromyl radical (Fig. 53). This radical then abstracts a hydrogen atom from (Me3Si)3SiH, producing a silyl radical species, which in turn abstracts the halogen atom from the alkyl bromide. The resultant alkyl radical then undergoes a radical addition mechanism with the protonated heteroarene, forming the alkylated product. The photocatalytic cycle is closed in the presence of a sacrificial perfsulfate oxidant. Both 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 were evaluated as the photocatalyst for this reaction, with 4CzIPN providing slightly superior yields (81% and 70%, respectively), which may be linked to its greater photooxidizing power (Ered* = 1.35 V for 4CzIPNvs. 1.21 V for the iridium photocatalyst).
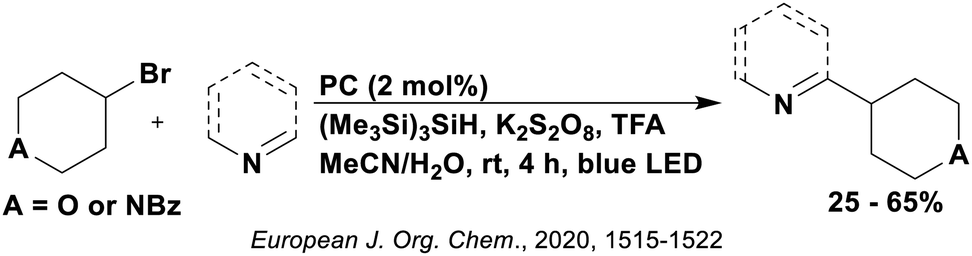 | ||
| Fig. 52 Reaction scheme for the coupling of alkyl bromides and heteroarenes, where A = O or NBz. | ||
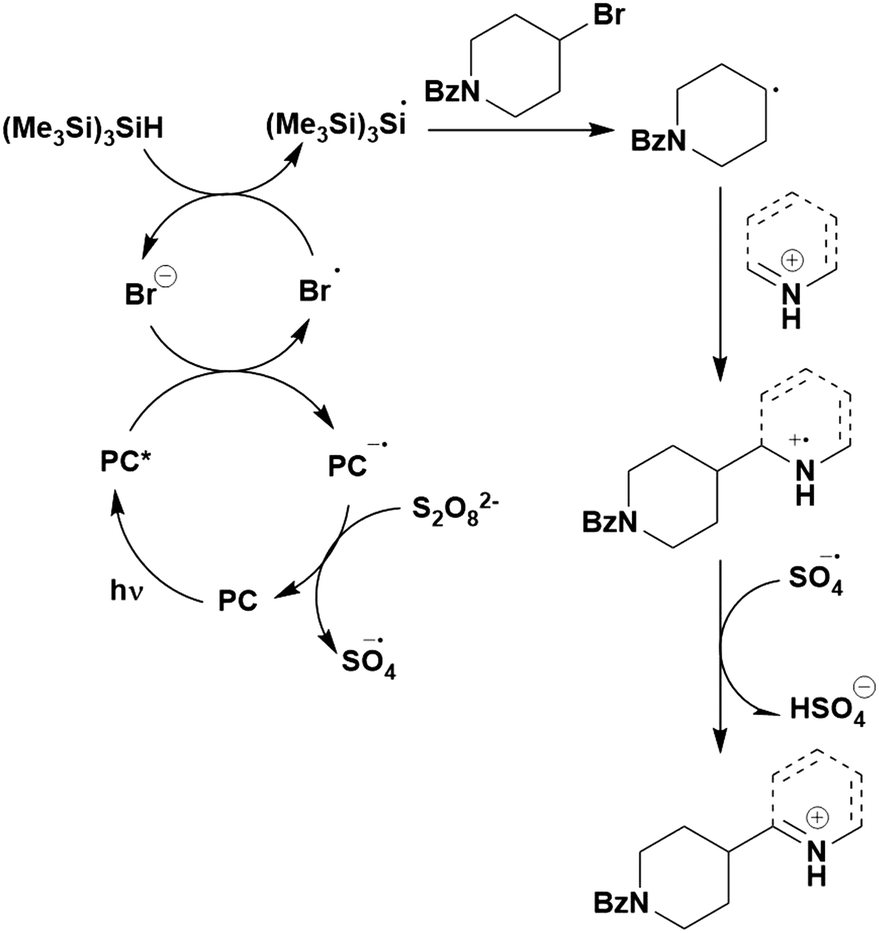 | ||
| Fig. 53 Proposed mechanism for the coupling of unactivated alkyl bromides with heterocycles. | ||
The aid of the hypervalent iodine oxidant trifluoroacetoxyiodobenzene (PIFA), which is reduced in the presence of the excited photocatalyst, helps to promote the formation of radicals that may be difficult to generate through direct oxidation/reduction from the PC. The resultant iodanyl radical is then used to generate the alkyl radical via H-atom abstraction from the substrate. Three mechanistically related examples involving PIFA have been reported: the oxidative alkylation of unactivated alkenes with DMSO (Fig. 54a),202 of N-aryl/benzoyl acrylamides with acetonitrile (Fig. 54b)203 and of alkenes with carbonyl compounds (Fig. 54c).204 In the former, α-sulfinyl radicals are generated from DMSO, which then add to a α,α-diaryl allylic alcohol, producing α-aryl-γ-methylsulfinyl ketones (Fig. 54a).202 Stern-Vomer quenching experiments support the proposed mechanism (Fig. 55) since quenching of the PC is only observed with the hypervalent iodide compound, not the alcohol. Of the PCs tested, 4CzIPN provided a considerably greater yield of 89% in comparison to [Ru(bpy)3](PF6)2 (23%) and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (47%). The higher yield is likely due to the greater reducing power of 4CzIPN in the excited state (Eox* = −1.04 V for 4CzIPN, −0.81 V for [Ru(bpy)3](PF6)2 and −0.89 V for [Ir(dF(CF3)ppy)(dtbbpy)]PF6.
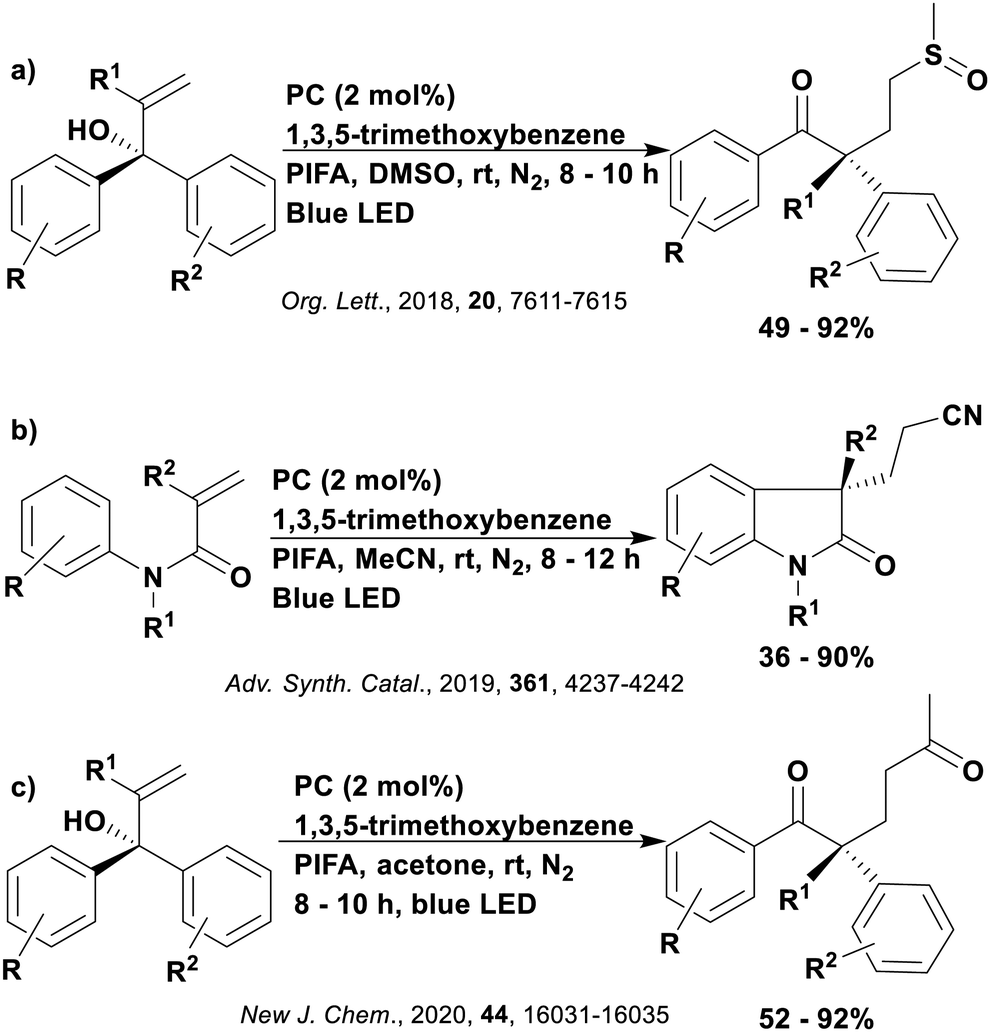 | ||
| Fig. 54 Reaction schemes for (a) oxidative alkylation of unactivated alkenes with DMSO, (b) oxidative alkylarylation of N-aryl/benzoyl acrylamides and (c) oxidative alkylation of alkenes with carbonyl compounds. | ||
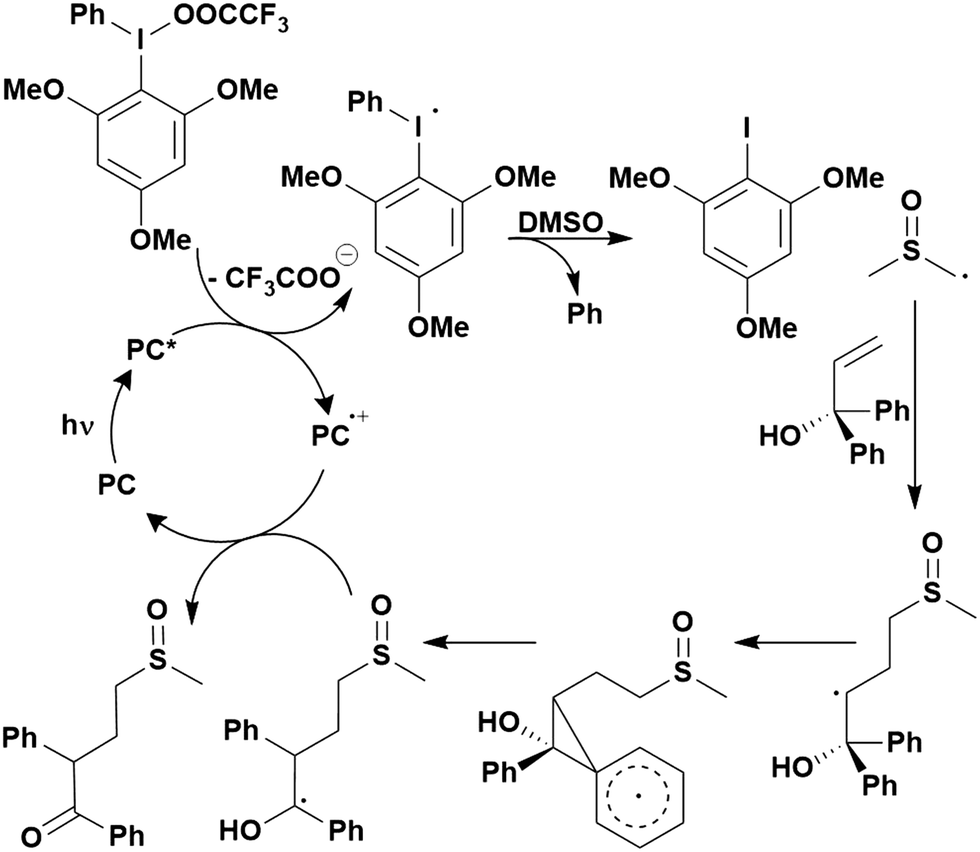 | ||
| Fig. 55 Suggested mechanism for the oxidative alkylation of alkenes using DMSO. | ||
A similar mechanism was invoked for the oxidative alkylarylation of N-aryl/benzoyl acrylamides (Fig. 54b), whereby the iodanyl radical was used to abstract a H atom from acetonitrile, generating the key alkyl radical, which in turn adds to the N-aryl acrylamide.203 Subsequent cyclisation, oxidation and deprotonation results in the required product. This mechanism was supported by radical trapping experiments with TEMPO, as well as Stern–Volmer quenching studies. Kinetic isotope effect studies (KH/KD = 3.2) implied that the C–H bond cleavage is likely to be the rate-determining step. 4CzIPN performed similarly to both [Ru(bpy)3]Cl2 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, (82%, 76% and 79% product yields, respectively), and was chosen as the PC for the remainder of the study.
Fig. 54c documents the oxidative alkylation of alkenes with carbonyl compounds.204 In this case, the iodanyl radical abstracts a H atom from acetone to generate the α-carbonyl alkyl radical, which then adds to the alkene. Addition of TEMPO inhibits the reaction and a kinetic isotope effect of KH/KD = 5.6 again implies that the C–H bond cleavage constitutes the rate determining step. In the PC screen, 4CzIPN yielded 78% of product, similar to that of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 at 72%. This modest statistically different yield may relate to the slightly stronger photoreducing ability of 4CzIPN.
As previously discussed, alkyl or haloalkyl radicals generated from radical precursors can then add to alkenes to form alkylated products. An alternative reaction pathway can involve reduction of the radical intermediate to the anion, which then can undergo a cyclisation reaction (Fig. 56a–c) in the presence of a remote leaving group. For example, cyclopropanation can occur through anionic 3-exo-tet ring closure (Fig. 56a with the mechanism shown in Fig. 57).205,206 Additionally, 4-exo-tet cyclisation can be observed to form functionalised cyclobutanes (Fig. 56b), and further functionalised cycloalkanes can also be synthesised depending on the length of the alkyl chain linking the halide leaving group to the alkene.207 The proposed mechanism was supported by Stern–Volmer quenching studies, and a quantum yield value of 0.066 indicates the likely absence of a radical chain process.206
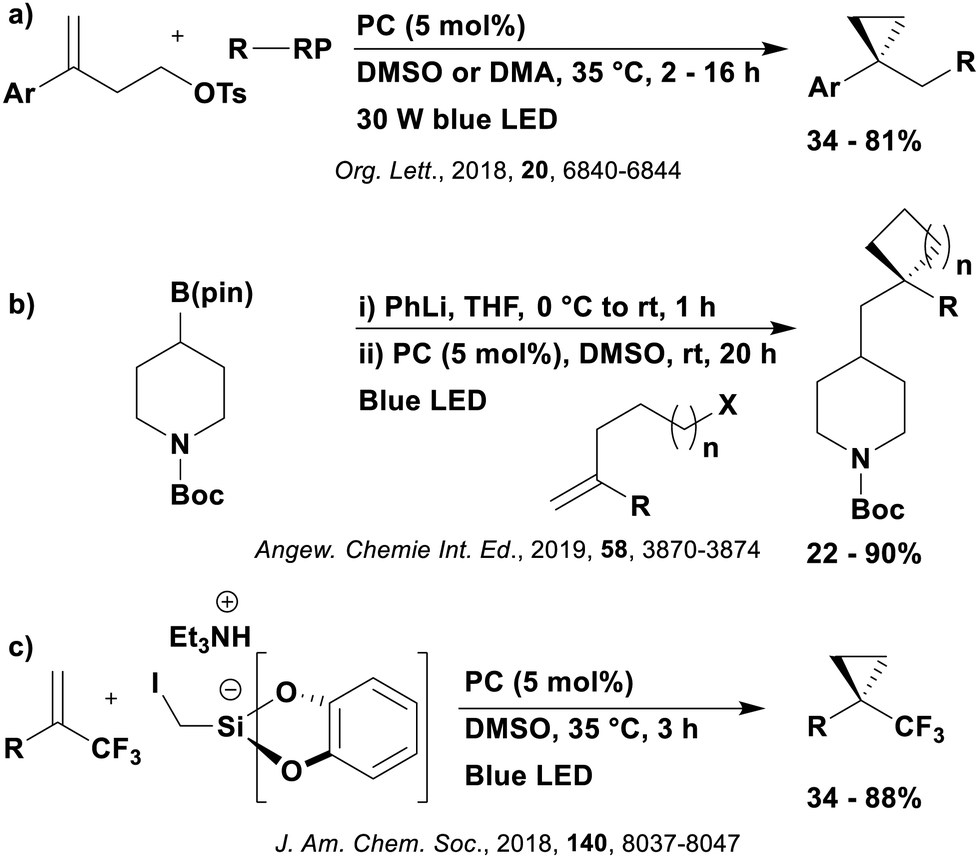 | ||
| Fig. 56 Reaction schemes for using radical precursors (RP) in cyclisation reactions; (a and c) cyclopropanes and (b) cyclobutanes. | ||
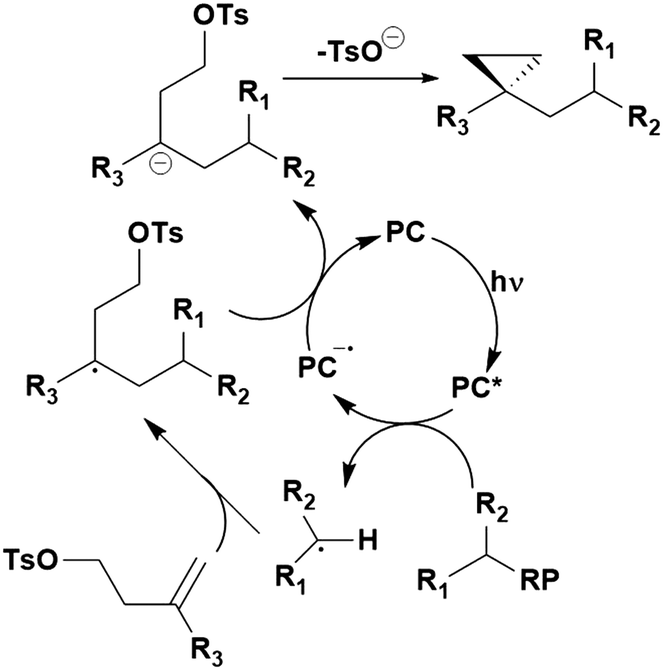 | ||
| Fig. 57 Viable mechanism for photocatalysed cyclopropanation where RP is radical precursor. | ||
[Ir(dF(CF3)ppy)2(bpy)]PF6, [Ru(bpy)3](PF6)2 and 4CzIPN all performed similarly in the cyclopropanation reaction involving styrene and a silicate radical precursor (Fig. 56a, 94%, 88% and 87%, respectively);2054CzIPN was chosen as the PC due to cost considerations. Interestingly, in the cyclopropanation with trifluoromethyl-substituted alkenes (Fig. 56c), 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 afforded the same high product yield (94% and 92%, respectively) while [Ru(bpy)3](PF6)2 and fac-Ir(ppy)3 provided lower yields (71% and 74%, respectively).206 The low oxidation potential of silicates (Eox = 0.4–0.7 V) should allow the SET to be thermodynamically feasible for most of PCs studied (Ered* = 1.32 V, 1.21 V and 1.35 V for [Ir(dF(CF3)ppy)2(bpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 4CzIPN, respectively) but this step may be more challenging for the less photooxidising PCs (Ered* = 0.77 V and 0.31 V for [Ru(bpy)3](PF6)2 and fac-Ir(ppy)3, respectively). For the cyclobutanation reaction (Fig. 56b), 4CzIPN significantly outperformed alternative PCs (70% yield for 4CzIPN, but only 46% for [Ir(dF(CF3)ppy)2(bpy)]PF6).207 This difference in yield is a result of the iridium PC producing a Giese-type side product in addition to the cyclised product (Fig. 58). The Giese-type side product is only observed with 4CzIPN if the reaction is conducted in the presence of water.
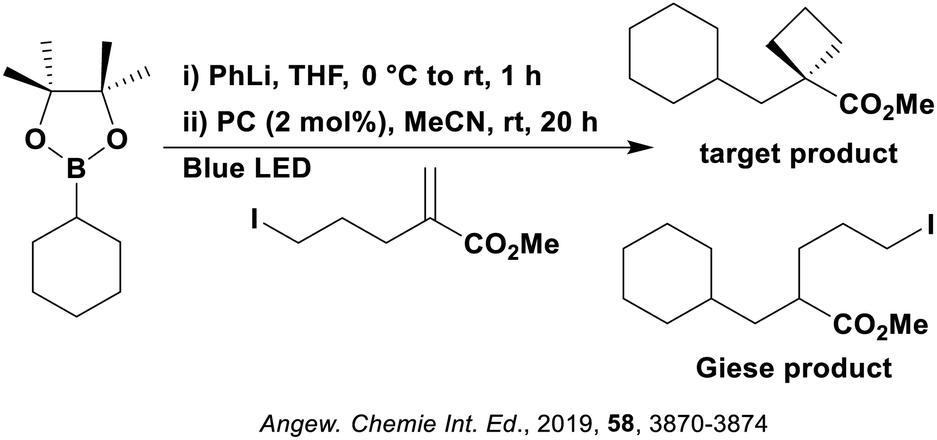 | ||
| Fig. 58 Reaction scheme for the cyclobutanation reaction showing the different products than can be obtained. | ||
Photocatalytic cyclisation reactions can be applied to the formation nitrogen heterocycles from imino-tethered DHPs (Fig. 59).208 This reaction proceeds via a proton coupled electron transfer (PCET) mechanism, as shown in Fig. 60, with the excited PC being reductively quenched by the imino-DHP, facilitated by the presence of the Brønsted-basic phosphate anion. The α-heteroatom-stabilised radical that eventually forms, undergoes a 6-endo-trig cyclisation to produce the cyclised product containing a nitrogen-centred radical. This radical intermediate can then be reduced by the reduced PC, closing the photocatalytic cycle, and generating the final product. Since the imino-DHPs have oxidation potentials ranging from Eox = 1.01 V to 1.23 V, the PC should have a moderate to strong photooxidising power. 4CzIPN was considered alongside iridium and ruthenium PCs. Only [Ir(dF(CF3)ppy)2(bpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 were shown to provide comparable yields as 4CzIPN (33%, 25% and 20% yields, respectively); the poor yield of 9% from [Ru(bpy)3](PF6)2 is likely related to its poor photooxidising ability (Ered* = 0.77 V compared to 1.35 V for 4CzIPN).
 | ||
| Fig. 59 Reaction scheme for the photocatalytic cyclisation of imino tethered DHPs to form N-containing heterocycles. | ||
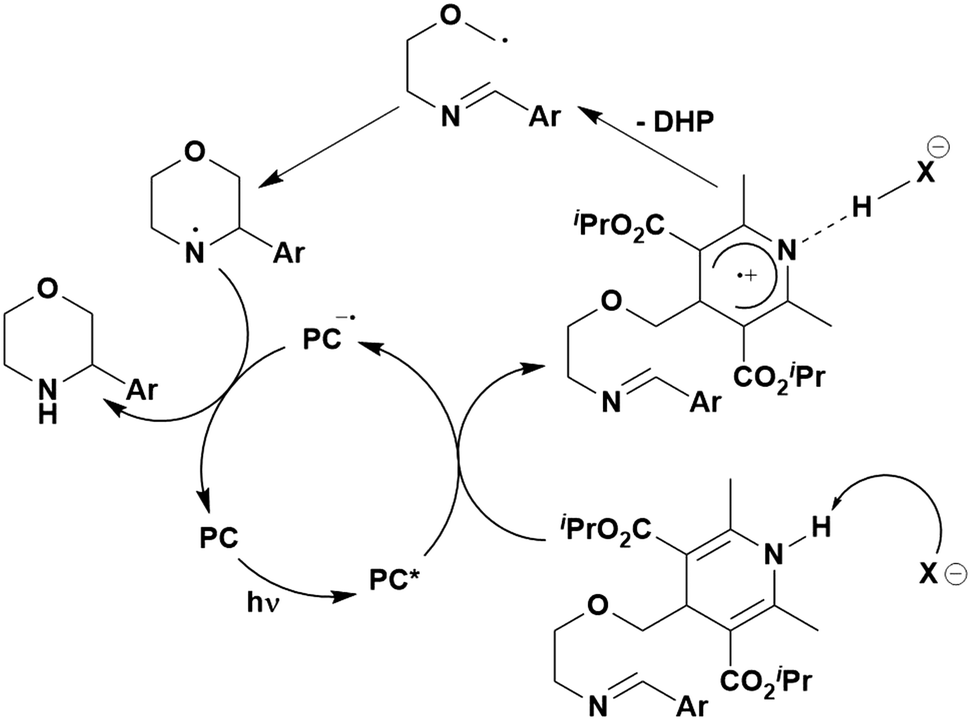 | ||
| Fig. 60 Suggested mechanism for the photocatalytic cyclisation of imino-tethered DHPs to form heterocycles. | ||
The preceding examples involve a C(sp3)–C(sp2) type coupling, but C(sp3)–C(sp) coupling reactions have also been reported using 4CzIPN as the photocatalyst. Ye et al. reported the coupling of alkynyl bromides with Hantzsch esters to produce internal alkynes (Fig. 61).209 In an analogous fashion to the work of Molander et al. with heteroaryl sulfones,177 a Hantzsch ester was used as the alkyl radical precursor under reductive quenching conditions. The nucleophilic alkyl radical then adds to the electrophilic alkynyl carbon, producing a vinylic radical intermediate (Fig. 62). This collapses to give a bromyl radical and alkylated alkyne. The photocatalytic cycle is closed by reduction of the bromyl radical. Similar yields were obtained for [Ru(bpy)3]Cl2, fac-Ir(ppy)3 and 4CzIPN (76%, 71% and 70%, respectively), with 4CzIPN being chosen as the optimal PC as it was considered to be more environmentally benign.
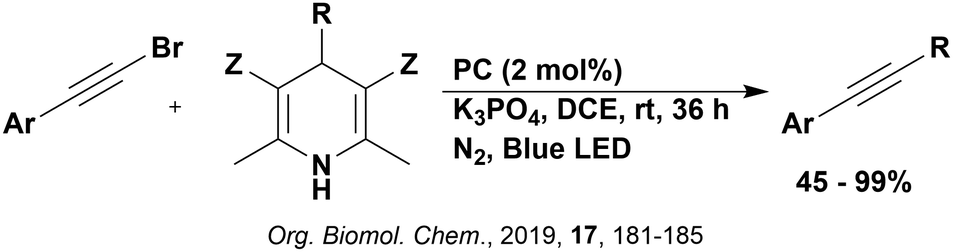 | ||
| Fig. 61 Reaction scheme for the coupling of alkynyl bromides and Hantzsch esters to synthesise internal alkynes where Z = CO2Et or CN. | ||
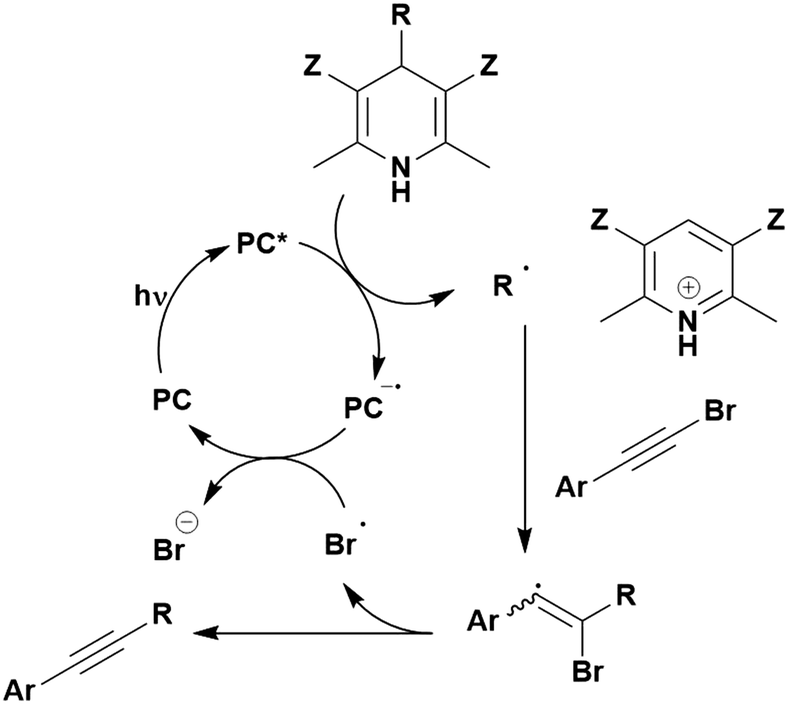 | ||
| Fig. 62 Potential mechanism for the coupling of alkynyl bromides with Hantzsch esters using visible light mediated photocatalysis, where Z = CO2Et or CN. | ||
Photocatalytic C–C and C–X bond formation can occur via an energy transfer mechanism, rather than via an electron transfer as has been the case for the aforementioned examples. A first example of PEnT involves the cross-dehydrogenative coupling of C(sp3)–H-containing compounds with N-heteroarenes (Fig. 63).210 Two energy transfer pathways are proposed to be operational (Fig. 64). The minor pathway involves PEnT from the excited PC to (NH4)2S2O8, resulting in homolysis and the formation of two sulfate radicals. Hydrogen atom transfer occurs between the sulfate radical and cyclohexane, the C(sp3)–H source in this example, to form a cyclohexyl radical. In the dominant PEnT pathway, the excited PC transfers energy to the heteroarene, lepidine, which can then react with the cyclohexyl radical. The sulfate radical then abstracts a hydrogen from this adduct to yield the final product.
 | ||
| Fig. 63 Reaction scheme for the dehydrogenative cross-coupling of C(sp3)–H with N-heteroarenes. | ||
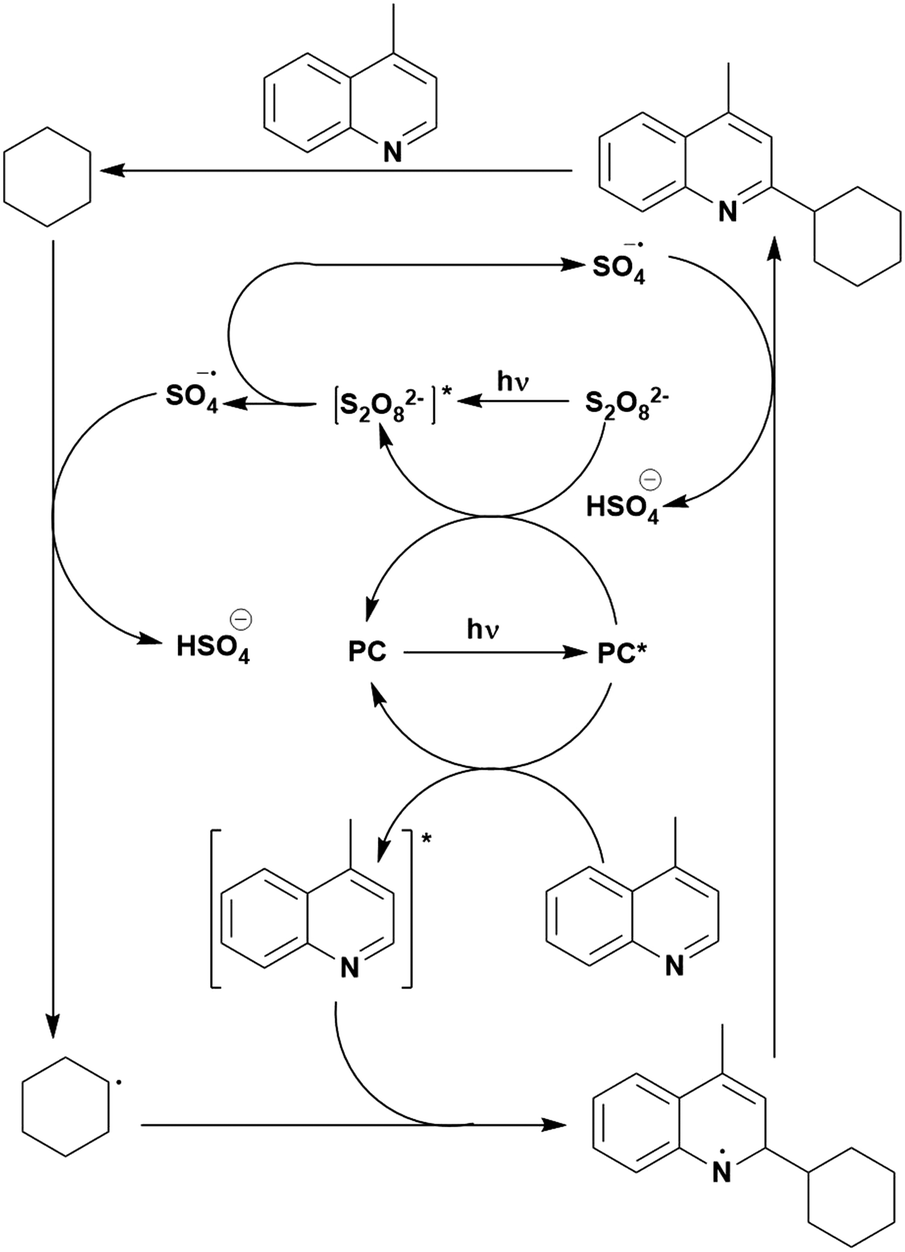 | ||
| Fig. 64 Viable mechanism for the dehydrogenative cross-coupling of cyclohexane with lepidine. | ||
Support for the dual PEnT mechanism is provided in Stern–Volmer quenching experiments, whereby both (NH4)2S2O8 and lepidine can quench the excited PC, although the latter does so at a significantly faster rate (KSV = 0.038 and 0.28, respectively). Kinetic isotope effect studies suggest that the C–H activation is the rate determining step. The process was confirmed to involve radicals as the addition of TEMPO inhibited the reaction and the presence of diphenylethene resulted in the formation of a cyclohexyl-containing adduct. EPR spectroscopy confirmed the presence of the sulfate radicals and that irradiation was necessary for their formation. No additional radical signals were observed when adding PC to the peroxydisulfate under irradiation, suggesting that the reaction proceeds via PEnT rather than PET.
Both 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 afforded the same yields (82% and 77%, respectively), which is likely due to their similar triplet state energies [ET = 2.58 eV (249 kJ mol−1) and 2.61 eV (251 kJ mol−1), respectively]. Other PCs tested provided much low yields (6–22%), which is likely due to their lower triplet state energies [1.77 eV (171 kJ mol−1) − 2.51 eV (242 kJ mol−1 for eosin Y and fac-Ir(ppy)3, respectively].
Photoinduced energy transfer is also proposed to be involved in the cross-coupling of DHPs with thiosulfonates or selenium sulfonates (Fig. 65); however, in this case, the PEnT proceeds in tandem with photoinduced electron transfer.211 In the putative mechanism, reductive quenching of the excited PC by the DHP radical precursor to release the alkyl radical first occurs (Fig. 66). This alkyl radical reacts with the thiosulfonate, forming the functionalised product and a sulfone radical intermediate. Reduction of this intermediate by the reduced PC followed by protonation yields a benzosulfinic acid. When conducted under nitrogen, the reaction stops here; however, in the presence of air, the authors suggest the excited PC can also undergo energy transfer to form singlet oxygen, which is then responsible for oxidizing the sulfide to the corresponding sulfoxide. To probe the reaction mechanism, TEMPO was added, which resulted in inhibition of product formation, confirming this is a radical process. When the thiosulfonate was replaced with PhSSPh no product was formed, implying that PhSSPh is not an intermediate in this process. However, no mechanistic support was provided to substantiate the energy transfer portion of this proposed mechanism. The same yields were obtained using fac-Ir(ppy)3, 4CzIPN and eosin Y as PCs under N2 (77%, 81% and 76%, respectively) while the use [Ir(ppy)2(dtbbpy)]PF6 proved to be slightly less successful (64%). There is no obvious correlation between the redox potentials and the yields obtained.
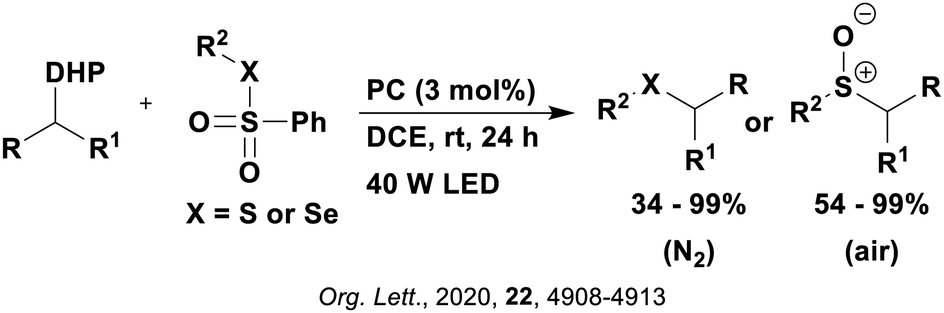 | ||
| Fig. 65 Reaction scheme for the cross-coupling of DHPs with thiosulfonates or selenium sulfonates. | ||
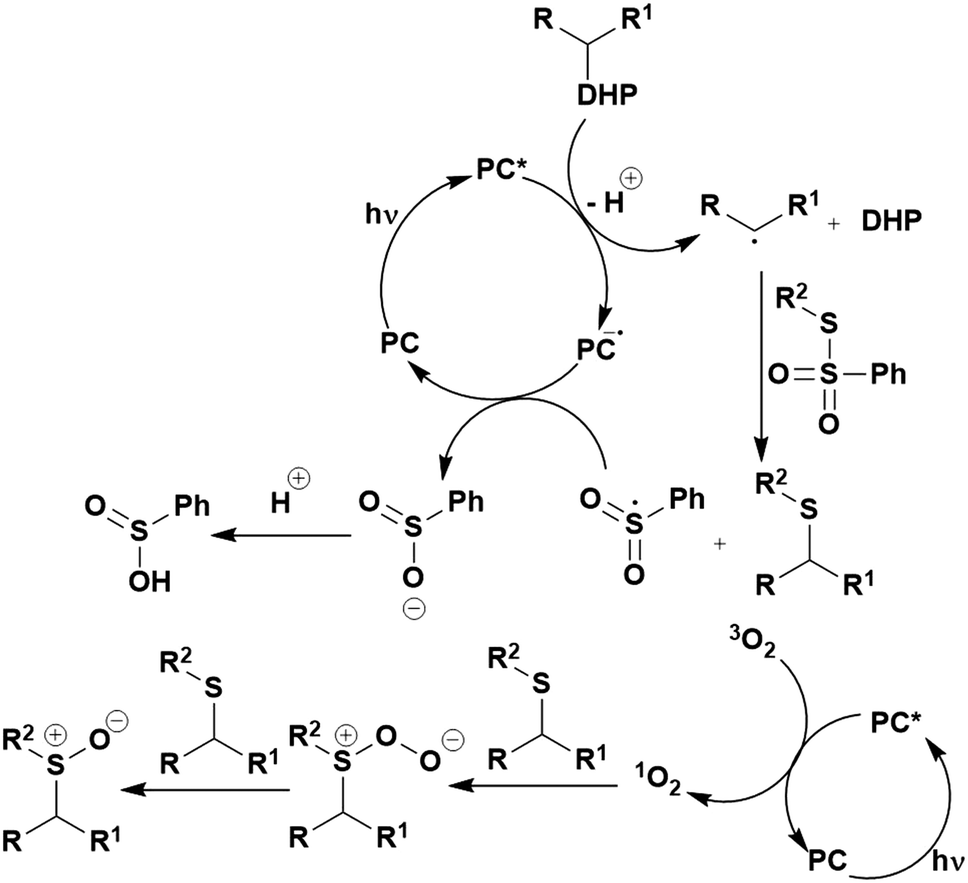 | ||
| Fig. 66 Putative mechanism for the cross-coupling of DHPs with thiosulfonates. | ||
Oxidation and reduction reactions
Photoredox catalysis also has broad applications in oxidation and reduction reactions. For instance, photocatalytic reduction of enamides to form α-amino carbanions can be reacted with CO2 in order to generate α,α-disubstituted α-amino acids (Fig. 67a).212 This reaction is hypothesized to proceed via a reductive quenching cycle of the photocatalyst, although a complete mechanism is not proposed. From Stern–Volmer quenching studies, it was surmised that the excited PC undergoes SET from DIPEA to generate the reduced photocatalyst, which then reduces the enamide to form an α-amino carbanion, which is then trapped by CO2 synthesising the conjugate base of the α,α-disubstituted α-amino acid. 4CzIPN gave the highest yield of all photocatalysts tested at 85% compared with 65% for [Ir(ppy)2(dtbbpy)]PF6, 62% for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 17% for fac-Ir(ppy)3. The higher yield obtained by 4CzIPN may be related to its photooxidising strength compared to the other PCs (Ered* = 1.35 V for 4CzIPN compared to 0.66 V for [Ir(ppy)2(dtbbpy)]PF6, 1.21 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 0.31 V for fac-Ir(ppy)3), especially given the oxidation potential of DIPEA is Eox = 0.81 V.35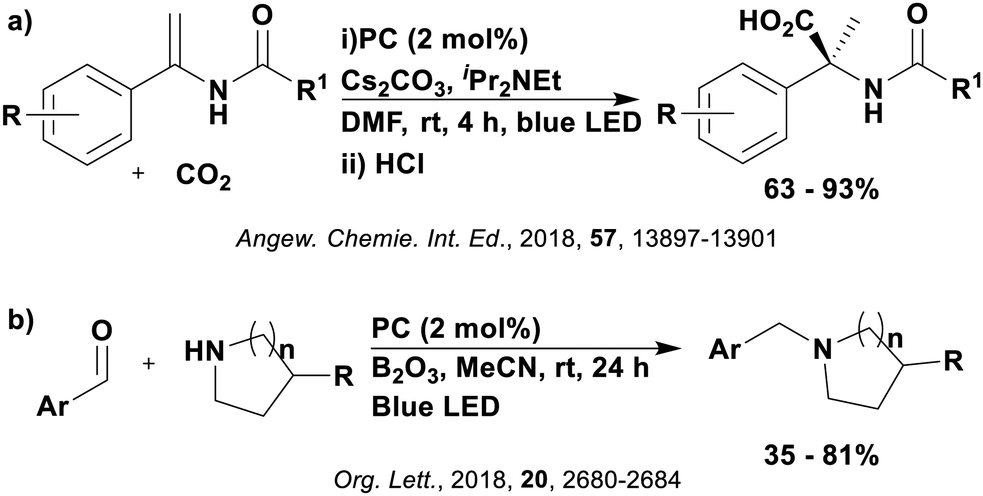 | ||
| Fig. 67 Reaction scheme for (a) reduction of enamides and (b) reductive amination. | ||
Direct reductive amination is also possible using photocatalysis whereby aromatic aldehydes may be converted to tertiary amines (Fig. 67b).213 This is possible though oxidation of an in situ formed aminal intermediate by the excited photocatalyst, forming the α-amino radical (Fig. 68), which then undergoes fragmentation, reduction by the photocatalyst and protonation, affording the desired product. For photocatalyst screening, 2 mol% of [Ir(dF(CF3)ppy)2(bpy)]PF6 produced superior yields of 78% while when 5 mol% of 4CzIPN was used, only 53% of product was isolated. This difference in PC loading makes a direct comparison of the photocatalysts challenging.
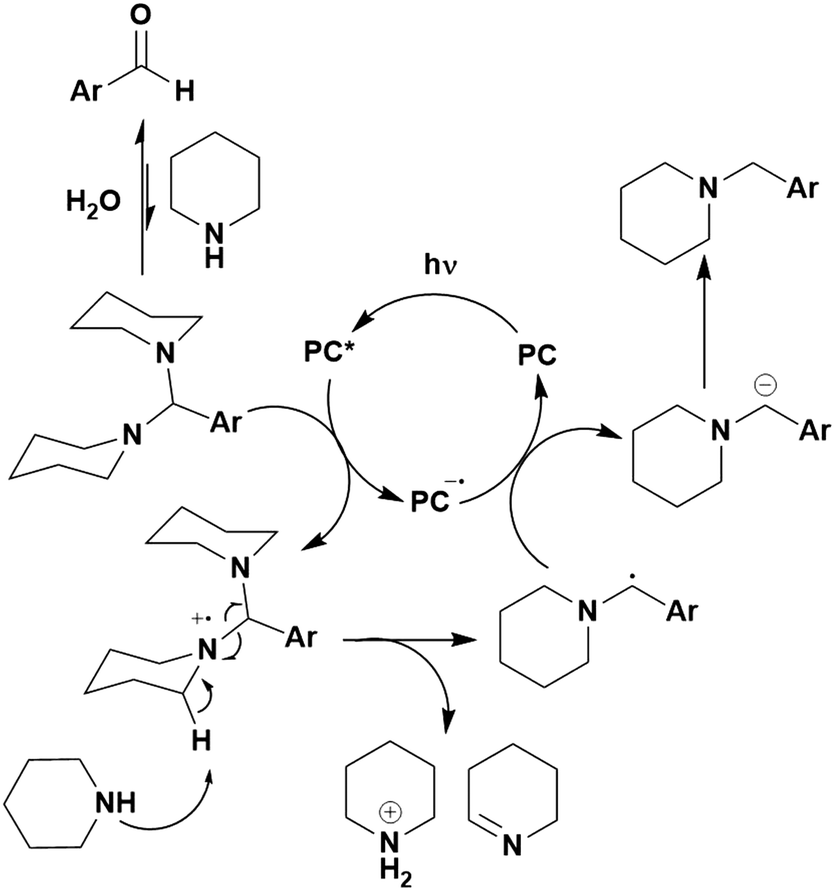 | ||
| Fig. 68 Plausible mechanism for photoredox catalysed reductive amination. | ||
An example of the use of 4CzIPN as a PC in a rather different context is directed towards the heterogeneous photocatalytic degradation of lignin. The degradation of lignin, which is typically treated as a waste product, to obtain small molecules or value-added energy fuels has received significant attention.214,215 The popular two-step approach to degrade lignin involves both oxidation and reduction steps; selective oxidation of the benzylic β-O-4 alcohol to benzylic β-O-4 ketone followed by a reduction to cleave the C–O bond (Fig. 69a).216 Porous organic frameworks (POFs) have been considered as photocatalysts for this process217 and the following is an example of their use. Zhang et al. synthesised copolymers of 1,4-bis(9H-carbazol-9-yl)benzene (DCB) (Fig. 69b) and 4CzIPN to form carbazolic copolymers (CzCPs).35 In these polymers, DCB acts as the electron donor component to allow for the reduction of the lignin models while 4CzIPN is the electron acceptor moiety responsible for the oxidation step. Varying the ratio of DCB and 4CzIPN results in CzCPs with tuneable redox properties. Nine of these copolymers were synthesised with D:A ratios varying from 100:0 (CzCP0) and 0:100 (CzCP100). Increasing the ratio of the 4CzIPN acceptor component results in a stabilisation of the LUMO energy level, making the photocatalyst increasingly oxidising in the ground state.
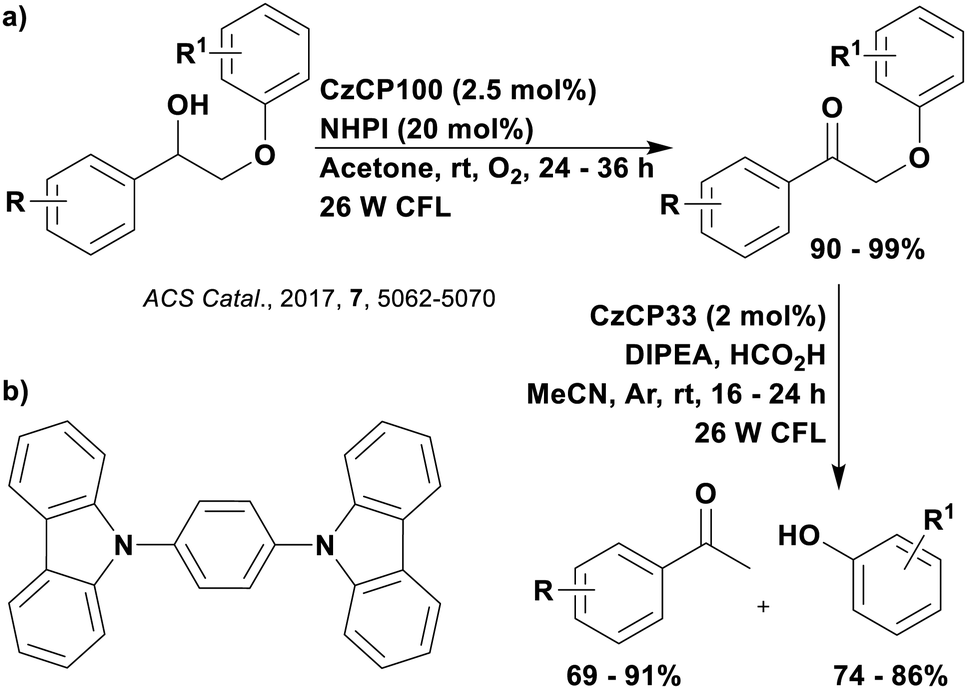 | ||
| Fig. 69 a) Reaction scheme for the degradation of lignin models and b) the structure of the donor molecule used in the POFs of CzCPs. | ||
To evaluate the effectiveness of the CzCPs towards lignin degradation, the initial oxidation step of β-O-4 lignin alcohols was first considered, for which an oxidative quenching mechanism of the photocatalyst is observed (Fig. 70). The photocatalyst CzCP100 (0 DCB: 100 4CzIPN) proved most successful, obtaining 100% conversion and 99% yield. As the ratio of 4CzIPN in the copolymer decreases, the conversion and yield also decreased accordingly; for example, CzCP0 (100 DCB: 0 4CzIPN) resulted in 55% conversion and 52% yield. These results are reflective of the stronger ground state oxidising capacity of CzCP100 (Eox = 1.83 V) compared to CzCP0 (Eox = 1.23 V), facilitating the required SET. For comparison, [Ru(bpz)3]2+ provides a comparable yield as CzCP100 (15% and 11% yield of product, respectively, as N-hydroxyphthalimide (NHPI) was not used), likely owing to its similar Eox = 1.86 V. Alternative catalytic systems used for the degradation of lignin, such as tert-butyl nitrate/DDQ218 and 1,4-benzoquinone219 were also tested with the former producing a yield of 99% and the latter a 59% yield in comparison to the 99% yield from the CzCP100/NHPI/O2 system.
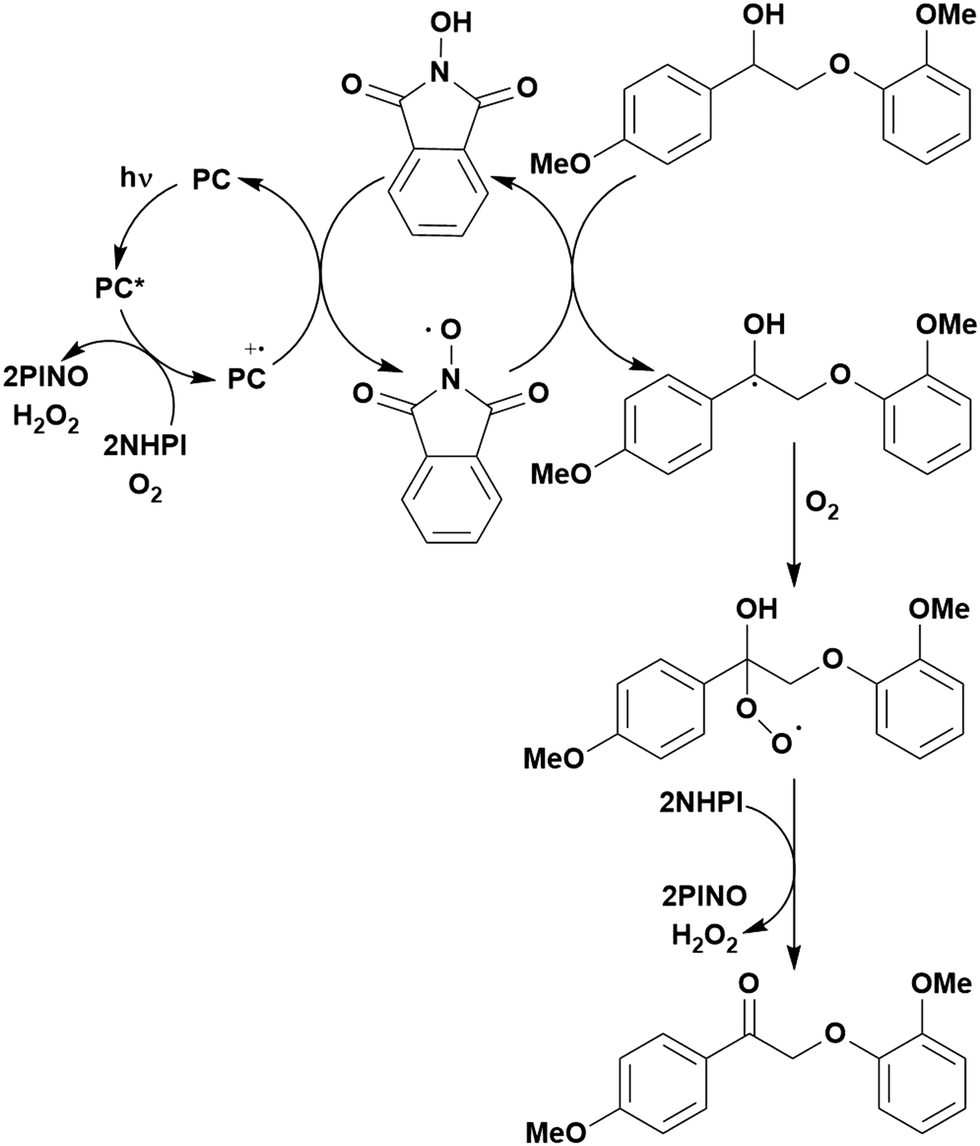 | ||
| Fig. 70 Photocatalytic oxidation of β-O-4 lignin alcohols. | ||
The reduction step involving C–O bond cleavage of β-O-4 ketones was next investigated, whereby the ground state reduction potential of the PC was identified as the key parameter (Fig. 71). Since a higher ratio of the donor component results in a more reducing ground state reduction potential (Ered = −2.09 V and −0.99 V for Cz:CP33 and CzCP100, respectively), it was predicted that the polymers with a greater amount of the donor would prove better in this reaction. In reality, the copolymer Cz:CP33 obtained the highest yield of 89%, with Cz:CP0 only yielding 30% of product. Ground and excited state reduction potentials have not been defined for the CzCP0 copolymer. Unfortunately, comparison with reference photocatalysts used for lignin degradation were not provided.
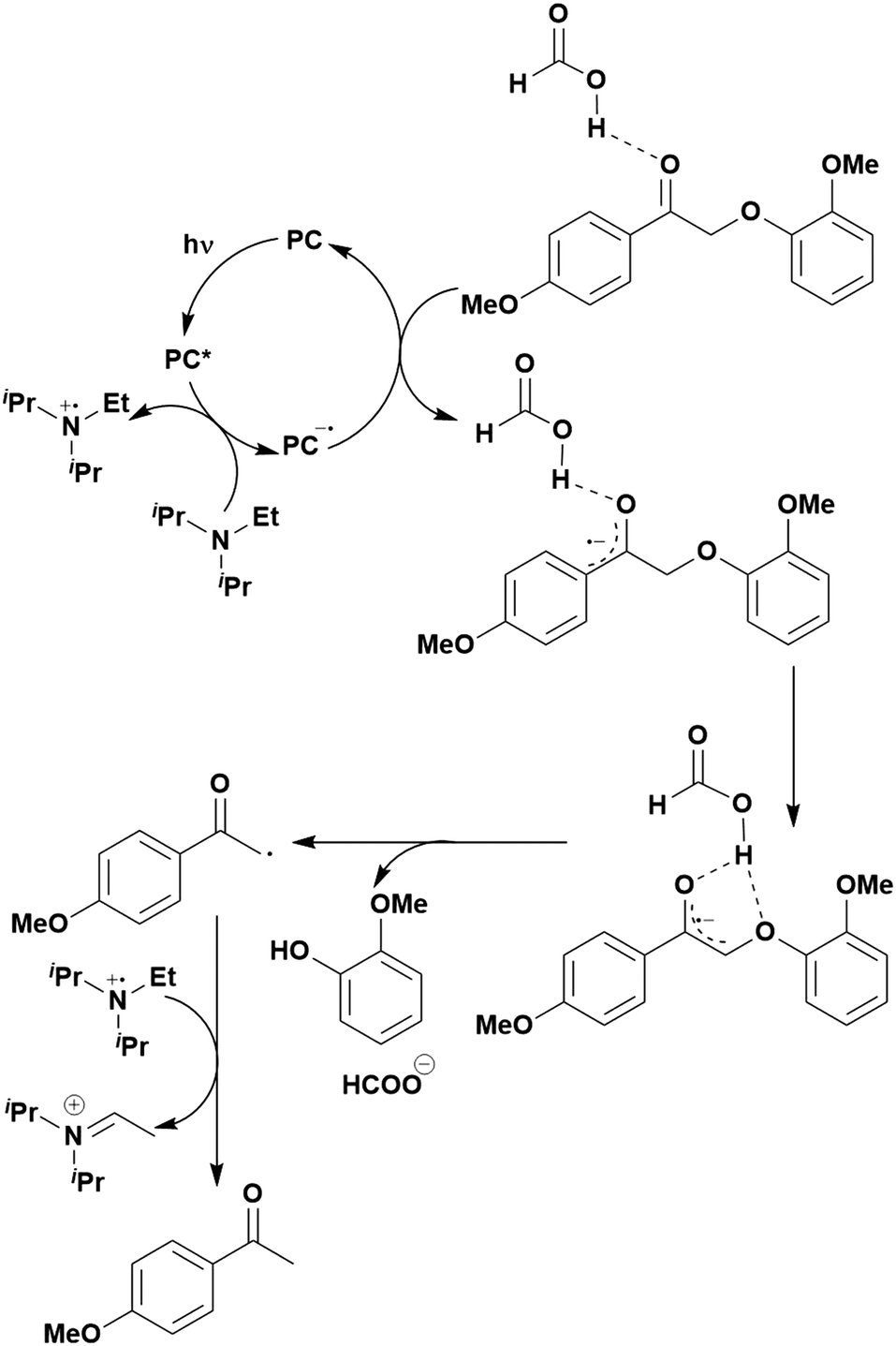 | ||
| Fig. 71 Photocatalytic reduction of lignin β -O-4 ketones. | ||
Cyclisation reactions
A phosphorus radical-mediated cascade cyclisation of 1,6-diynes (Fig. 72a) that proceeds via an oxidative quench of the photocatalyst (Fig. 73) has been reported.220 This mechanism was supported by Stern–Volmer quenching studies alongside radical trapping experiments. In this study, Xu et al. developed a family of highly photoreducing, substituted 5,12-dihydroquinoxalino[2,3-b]quinoxaline organic PCs (Fig. 72b) with Eox* = −1.79 to −2.31 V. These gave higher product yields (51–80%) than 4CzIPN (Eox* = −1.04 V, 40% product yield), although this can be rationalised by their stronger reducing capacity in the excited state, which is required to reduce the pyridinium oxidant.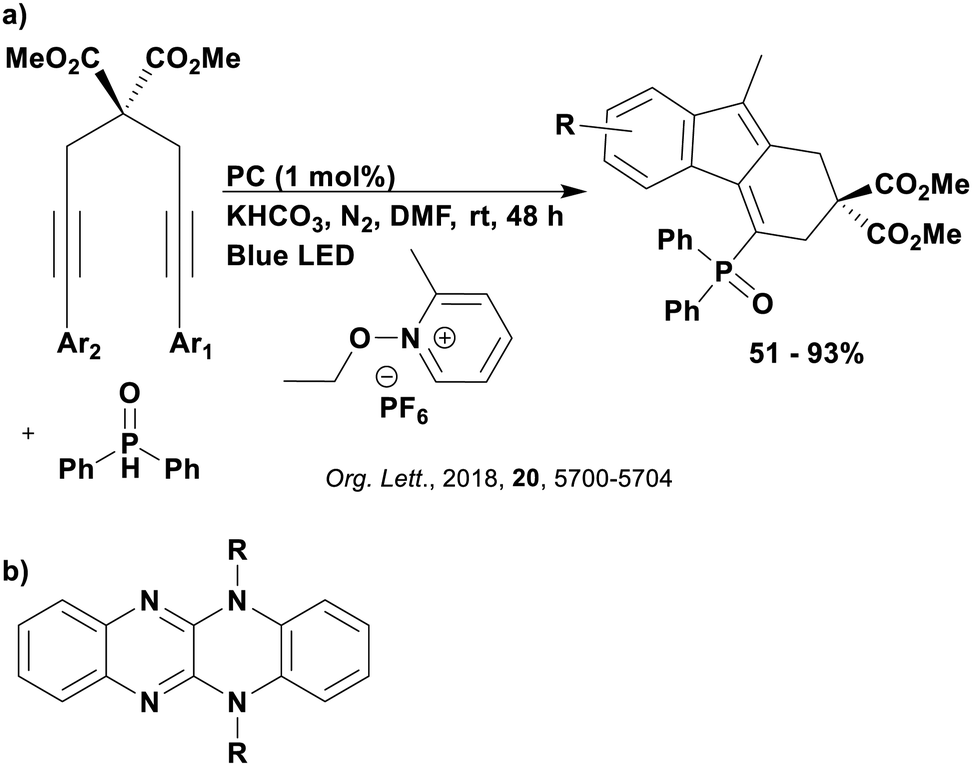 | ||
| Fig. 72 (a) Reaction scheme for the cyclisation of 1,6-diynes and (b) generic structure of the organic photocatalyst developed for the cyclisation of 1,6-diynes where R = H, Et, Ph, p-OMeC6H5 or p-CF3C6H5. | ||
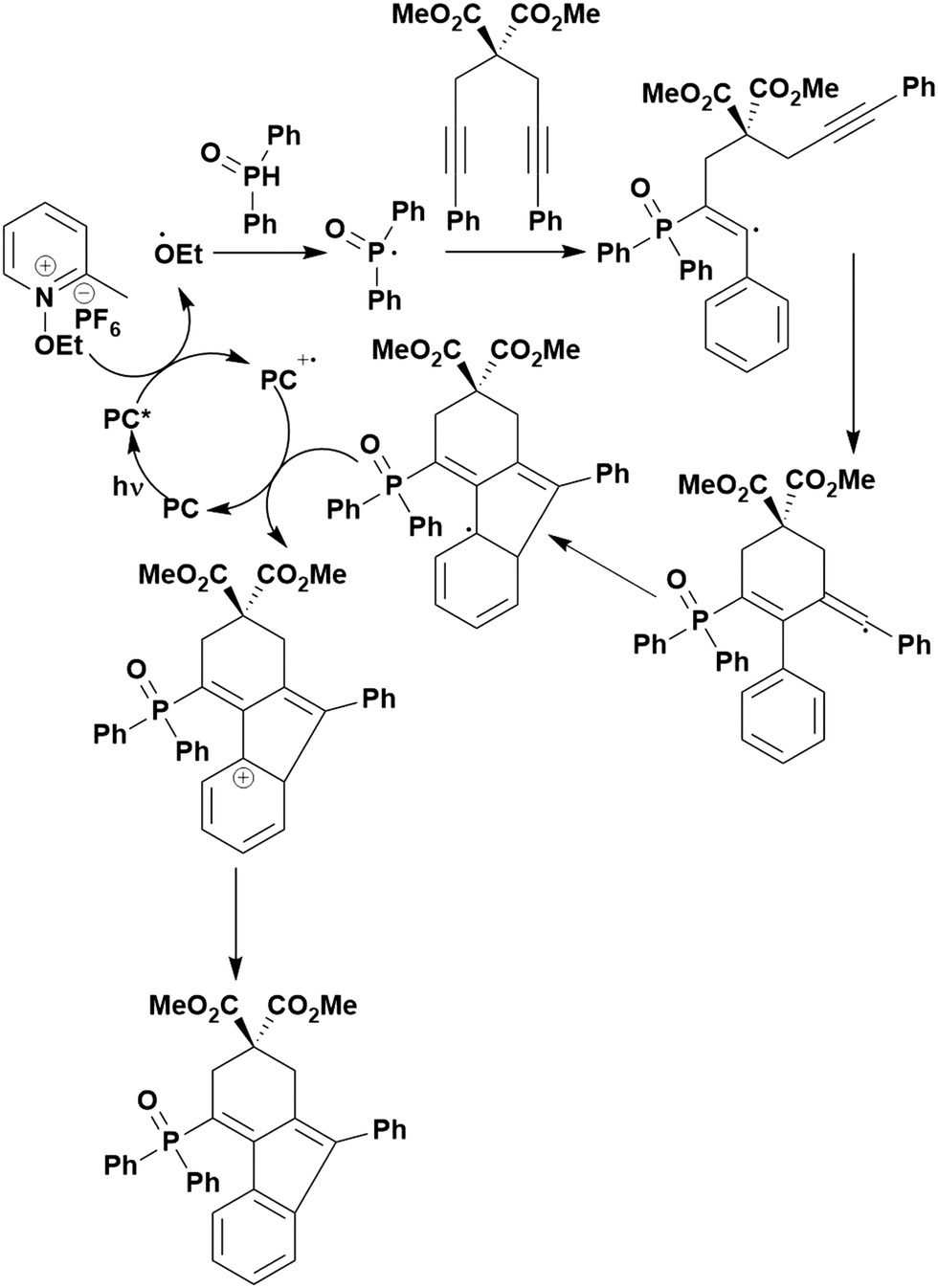 | ||
| Fig. 73 Proposed mechanism for the phosphorus radical-mediated cascade cyclisation of 1,6-diynes using photoredox catalysis. | ||
Cyclisation reactions are also possible through a reductive quenching mechanism, for example in the preparation of heterocycles via the selenation of olefins (Fig. 74a),221 the synthesis of 2-phosphorylated thioflavones (Fig. 74b),222 the synthesis of 3-phosphorylated benzothiphenes (Fig. 74c),223 the formation of polysubstituted γ-lactones (Fig. 74d)224 and the dearomative arylcarboxylation of indoles (Fig. 74e).225 The first involves aerobic dehydrogenative cyclisation of alkenes with diaryl diselenides.221 The diselenide undergoes SET to the excited PC, with the resultant radical cationic diselenide intermediate activated towards attack by pendant alkenes. This forms a reactive cationic phenylselenium species, which rearranges to the selenophane-decorated oxazoline (Fig. 75). Molecular oxygen is used as a sacrificial oxidant to close the photocatalytic cycle. Only organic PCs were screened for this reaction, of which 4CzIPN produced the highest product yield (94% obtained after 4 hours), which was matched by [Mes-Acr]+ (90% after 2 hours). Since the oxidation potential of diphenyl selenide is Eox = 1.35 V, photocatalysts with a more positive Ered* should be thermodynamically capable of completing the SET (Ered* = 2.06 V and 1.35 V for [Mes-Acr]+ and 4CzIPN, respectively), which contributes to the success of both PCs in this reaction.
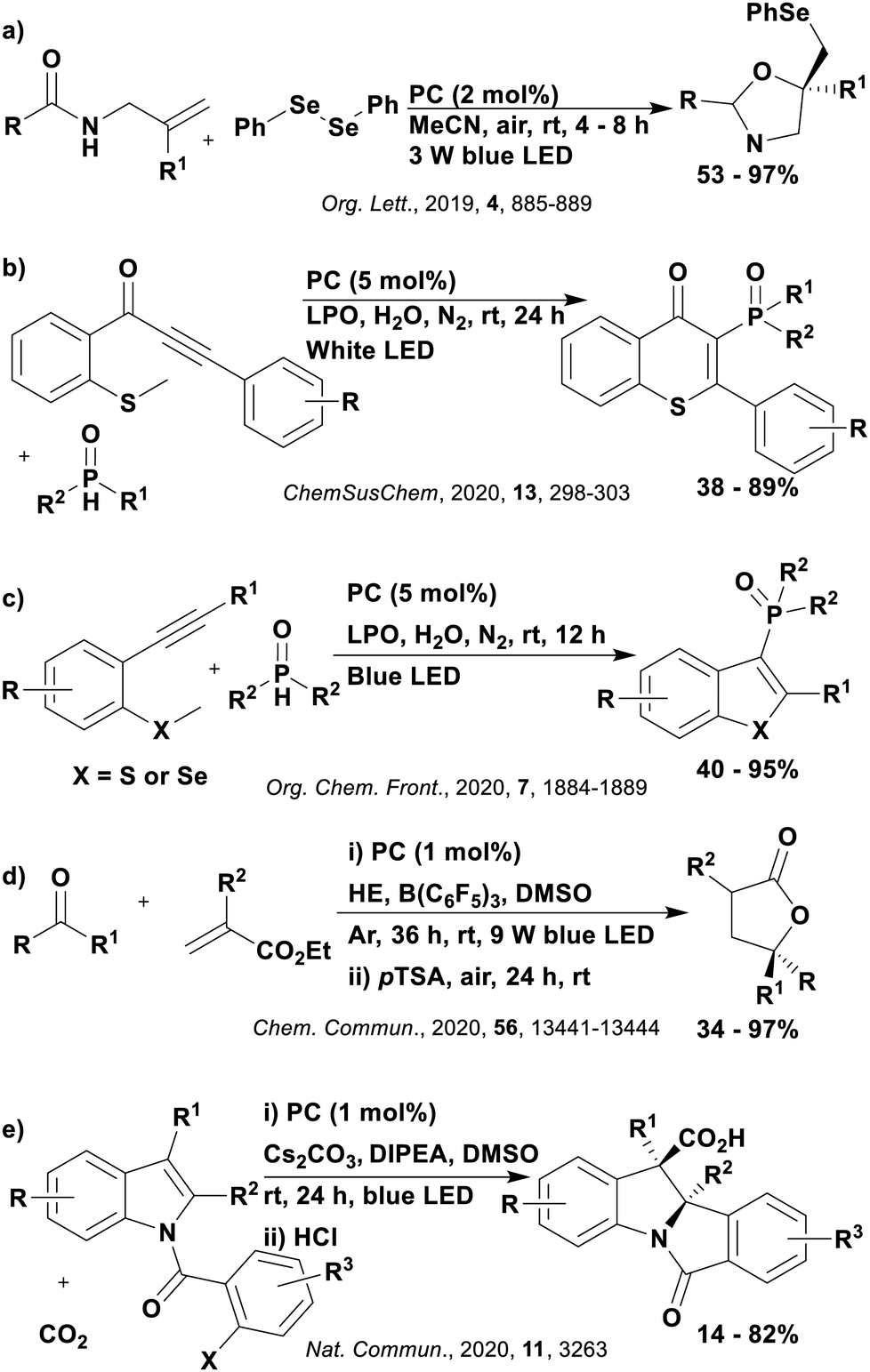 | ||
| Fig. 74 Reaction scheme for cyclisation reactions that proceed via a reductive quenching cycle: (a) preparation of heterocycles, (b) synthesis of 2-phosphorylated thioflavones, (c) synthesis of 3-phosphorylated benzothiophenes, (d) polysubstituted γ-lactone formation and (e) the dearomative arylcarboxylation of indoles. | ||
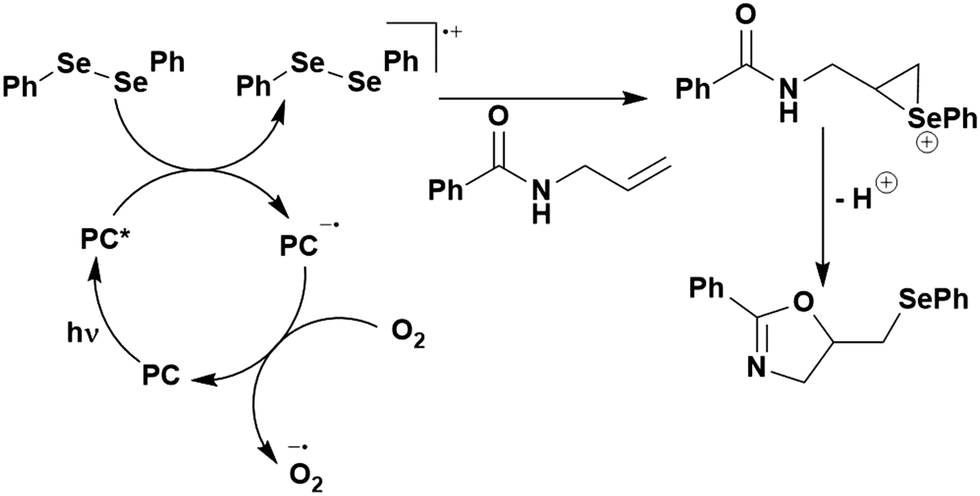 | ||
| Fig. 75 Possible mechanism for photocatalytic heterocycle formation. | ||
In the synthesis of 2-phosphorylated thioflavones (Fig. 74b), the excited PC is reductively quenched by the diphenylphosphine oxide, resulting in a phosphoryl radical, which adds to the alkyne of the methylthiolated phenylpropynone (Fig. 76).222 Intramolecular cyclisation affords the final product. The photocatalyst is regenerated by SET to dilauroyl peroxide (LPO). Evidence for this mechanism was obtained by experiments that showed the suppression of the reaction upon addition of TEMPO, suggesting a radical process, while a quantum yield of 0.24 indicates a radical chain mechanism is unlikely. By contrast, in the proposed mechanism for the synthesis of 3-phosphorylated benzothiophenes (Fig. 74c),223 the excited photocatalyst is quenched by the methylthiolated alkyne. The resulting radical couples with the phosphoryl radical generated from irradiation of diphenylphosphine oxide in the presence of LPO. Consistent with the proposed mechanism in Fig. 76, the reduced photocatalyst reduces LPO to close the photocatalytic cycle. Stern–Volmer quenching studies support the putative mechanism as the alkyne was observed to quench the luminescence of the PC. The addition of a radical scavenger inhibited the reaction, confirming that the reaction involves radical processes. These conflicting mechanistic proposals for two extraordinarily similar reactions highlight the uncertainty that typically surrounds photocatalysis mechanism elucidation.
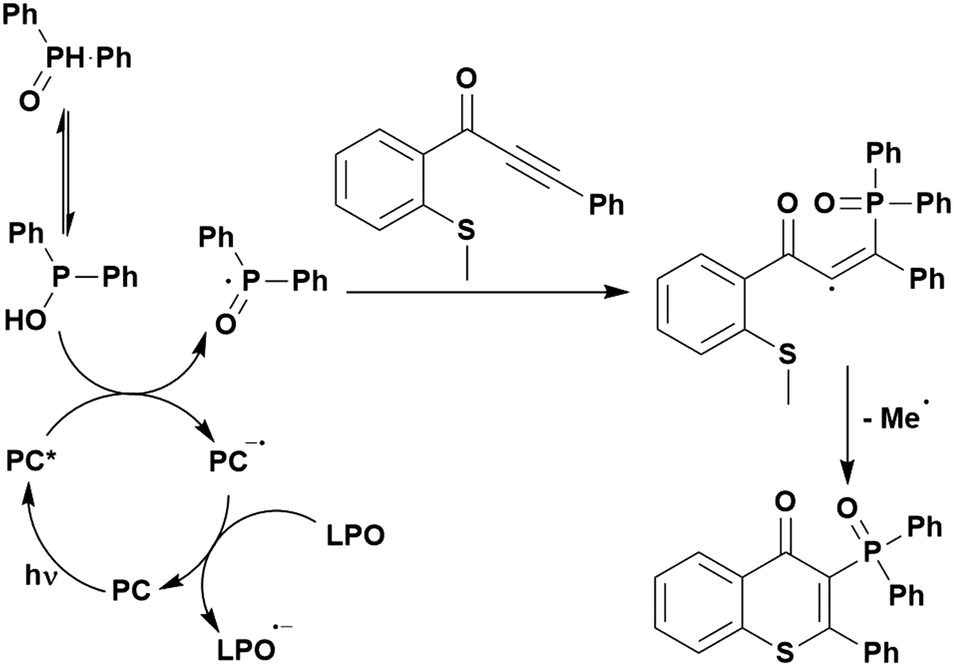 | ||
| Fig. 76 Proposed mechanism for the synthesis of 2-phosphorylated thioflavones. | ||
A range of organic photocatalysts were trialled in both phosphorylation reactions, with 4CzIPN proving the best in both instances. When forming 2-phosphorylated thioflavones, 4CzIPN yielded 45% while organic dyes such as eosin Y (24%) and rose bengal (19%) proved themselves to be poorer options. These results are most likely related to the photooxidising ability of the PC, which, according to the proposed mechanism, must be capable of oxidising the diphenylphosphine oxide (Eox ≈ 1.0 V).226 The relative yields may be correlated to 4CzIPN's more positive Ered* (Ered* = 1.35 V) compared to those of eosin Y or rose bengal (Ered* = 0.83 V and 0.81 V, respectively). For the synthesis of 3-phosphorylated benzothiophenes, only 4CzIPN provided any detectable yield (90%) while the other organic PCs, such as eosin Y or rose bengal, proved ineffective. Again, this is likely related to the greater photooxidising ability of 4CzIPN.
In the formation of polysubstituted γ-lactones (Fig. 74d), the putative mechanism involves reductive quenching of the excited PC by the Hantzsch ester followed by the reduction of the aldehyde by the reduced PC in a PCET step facilitated by one of the Lewis acid or the oxidised Hantzsch ester224 The ketyl radical adds to the α,β-unsaturated ester and this resultant radical is reduced by the Hantzsch ester radical. Protonation and acid-catalysed intramolecular transesterification yields the final product. Addition of TEMPO to the reaction inhibits product formation, implicating a radical process. Deuterium labelling of the Hantzsch ester and the absence of deuterium incorporation in the final product imply that a HAT process is unlikely to be in operation. Stern–Volmer quenching experiments indicated that the Hantzsch ester was the only species capable of significantly quenching the luminescence of the PC. In the PC screen for the reaction involving ketones as the carbonyl reagent, 4CzIPN fared slightly more poorly than [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (32% and 42%, respectively), while when aldehydes were used as substrates, the opposite trend in reactivity was observed (97% and 59% for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively). The authors proposed that since the aromatic ketones are more difficult to reduce, the stronger ground state reducing ability of the Ir PC provides the greater driving force for turning over the photocatalytic cycle (Ered= −1.37 V and −1.21 V, respectively), while for aldehydes, 4CzIPN is sufficient reducing and the reaction proceeds without the need for the Lewis acid additive.
For the dearomative arylcarboxylation of indoles with CO2 (Fig. 74e),225 the mechanism differs from the previous examples of photoinduced cyclisation in that two photocatalytic cycles are invoked, implying a two photon process. In both cycles, the excited PC is reductively quenched by DIPEA (Fig. 77). The reduced PC is then suggested to reduce the aryl halide, which after fragmentation to release the halide anion, produces an aryl radical. This species undergoes fast intramolecular radical addition to CC double bond of the indole to form a benzylic radical. Reduction of this species by the reduced PC, closing the second photocatalytic cycle, forms a carbanion, which undergoes nucleophilic addition to CO2, followed by protonation to yield the final product. The radical nature of this process was confirmed by the addition of TEMPO, which reduced the yield of the final product as an adduct with the benzylic radical was formed. Deuterium labelling studies eliminated the possibility of an HAT mechanism and provided support for the formation of the benzylic carbanion intermediate. Stern–Volmer experiments demonstrated that DIPEA was the predominant species responsible for the luminescence quenching of the PC. Only three PCs were considered for this reaction: 4CzIPN, [Ir(dF(Me)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2. These provided yields of 88%, 78% and 52%, respectively, which correlate with the photooxidisng ability of the PC (Ered* = 1.35 V, 0.92 V and 0.77 V, respectively).
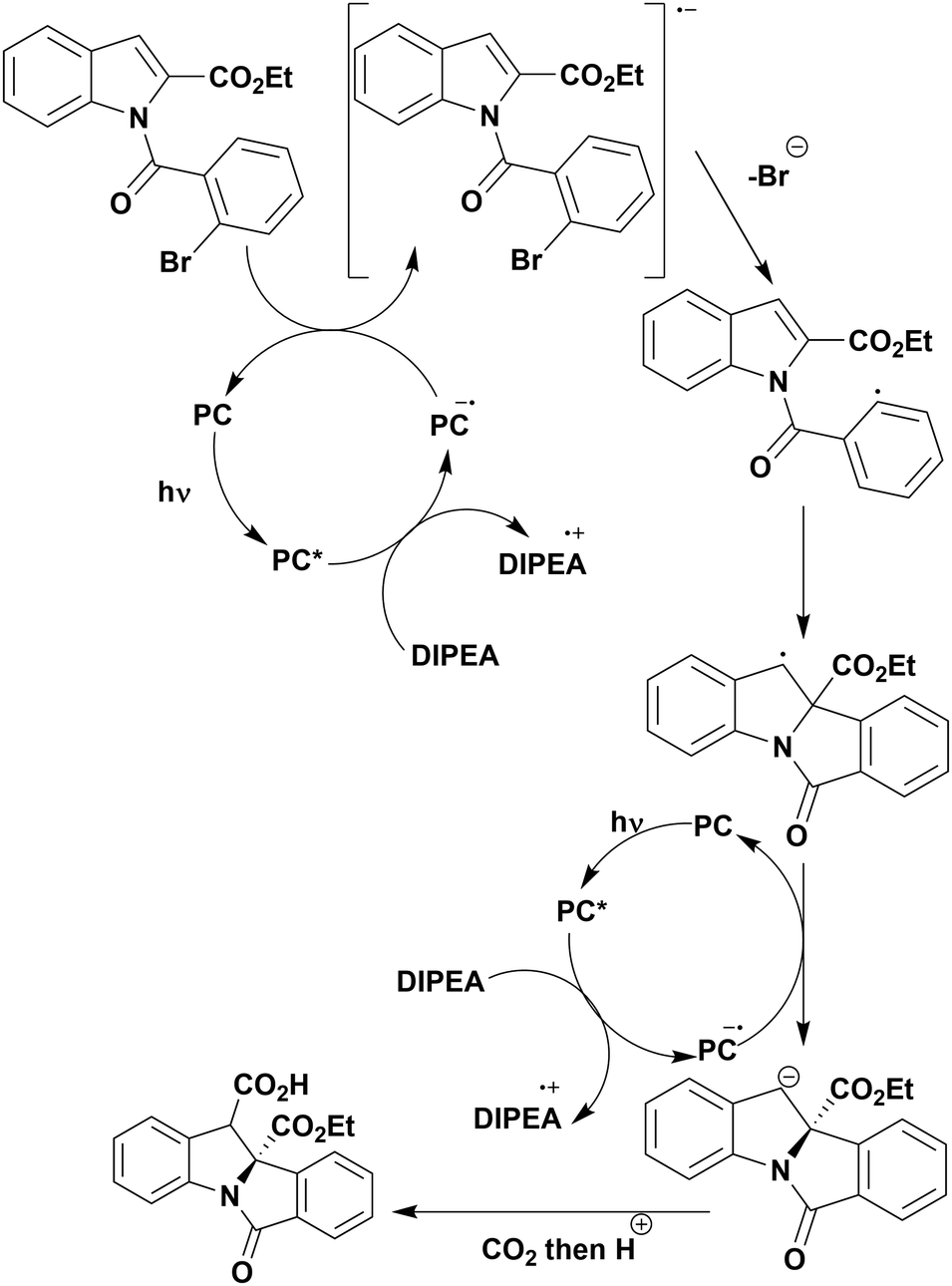 | ||
| Fig. 77 Viable mechanism for the dearomative arylcarboxylation of indoles. | ||
Distinct from the previous examples, Yu et al. report an example of cyclisation that is proposed to proceed through an energy transfer mechanism. In this example, cyclisation of N-arylpropiolamides to 3-phosphorylated, trifluoromethylated or thiocyanated azaspiro[4.5]trienones occurs (Fig. 78a and 78b).227 Energy transfer from the excited PC to the N-arylpropiolamide is suggested, with the excited N-arylpropiolamide undergoing SET to LPO (Fig. 79). Another SET event occurs from the diphenylphosphine to the nitrogen centred radical cation, regenerating the initial N-arylpropiolamide. The phosphoryl radical then adds to the alkyne, generating an alkenyl radical intermediate, which undergoes intramolecular cyclisation to give an azaspiro radical. Oxidation of this species by LPO, followed by a cascade sequence of steps, including addition of H2O, methanol elimination and deprotonation, yields the final product.
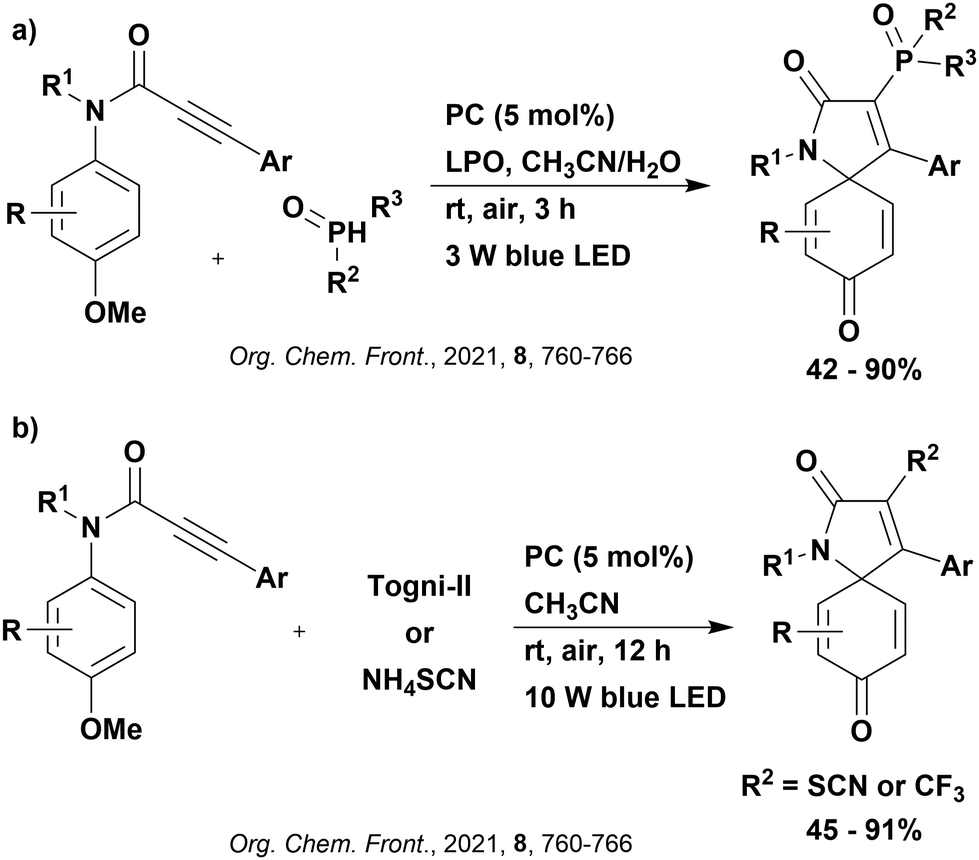 | ||
| Fig. 78 Reaction scheme for the cyclisation of N-arylpropiolamides. | ||
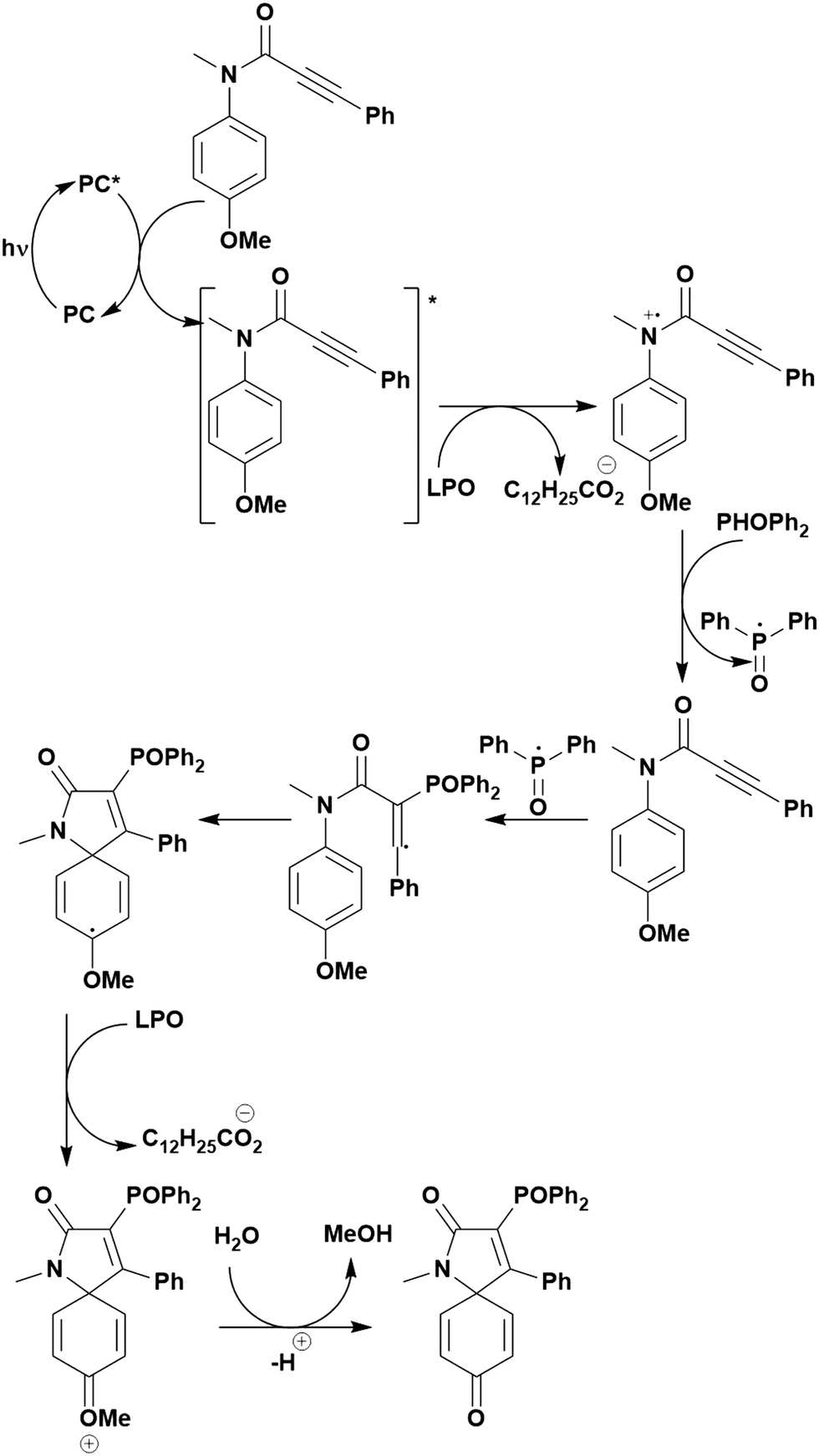 | ||
| Fig. 79 Possible mechanism for the cyclisation of N-arylpropiolamides. | ||
Stern–Volmer experiments revealed that N-arylpropiolamide quenches the luminescence of the PC. A PET mechanism was ruled out based on an analysis of the redox potentials; the N-arylpropiolamide (Eox = 1.58 V vs. SCE) is thermodynamically incapable of being oxidised by the excited PC (Ered* = 1.35 V for 4CzIPN). Control experiments with radical scavengers TEMPO and BHT suppressed the reaction, confirming that radical intermediates exist in the reaction. 4CzIPN far outcompeted the other organic PCs considered in this reaction, such as eosin Y and rose bengal, with respective yields of 83%, 15% and trace. Since an energy transfer process is proposed, there should be a correlation between the triplet energies of the PCs and the reaction yields; however, as the triplet energy of the N-arylpropiolamide was not provided, it is not possible to infer this link.
Polymerisation reactions
4CzIPN also functions as a suitable photocatalyst for polymerisation reactions, including atom transfer radical polymerisation (ATRP) and reversible addition–fragmentation transfer (RAFT) polymerisation reactions. Methyl methacrylate (MMA) can be polymerised using an ATRP mechanism using 4CzIPN at photocatalyst loading of parts per million (ppm) and ethyl α-bromophenyl-acetate (EBPA) as the initiator (Fig. 80a).228 The photocatalyst activates the initiator in an oxidative quenching cycle to generate the propagating radical species (Fig. 81). The PC is then regenerated by SET from the propagating chain to the oxidised photocatalyst, thus terminating the polymerisation. Comparison of yields with other photocatalysts under the same conditions was not conducted; however, polymers of similar dispersity have been reported with other organic dyes, although under very different reaction conditions.229 For example, Zhu et al. reported 90% conversion of the monomer to poly(methyl methacrylate) (PMMA) with Mw/Mn = 1.50 (where Mw = weight average molecular weight and Mn = number average molecular weight) after 3 hours using 0.0015 mol% of 4CzIPN as the photocatalyst.228 By comparison, Theriot et al., when using perylene as a photocatalyst (0.11 mol %) and α-bromophenyl-acetate (BPA) as the initiator, obtained PMMA with Mw/Mn = 1.49.229 This similarity confirms the success of 4CzIPN as a PC in polymerisation reactions, especially since it can be used at such low loadings.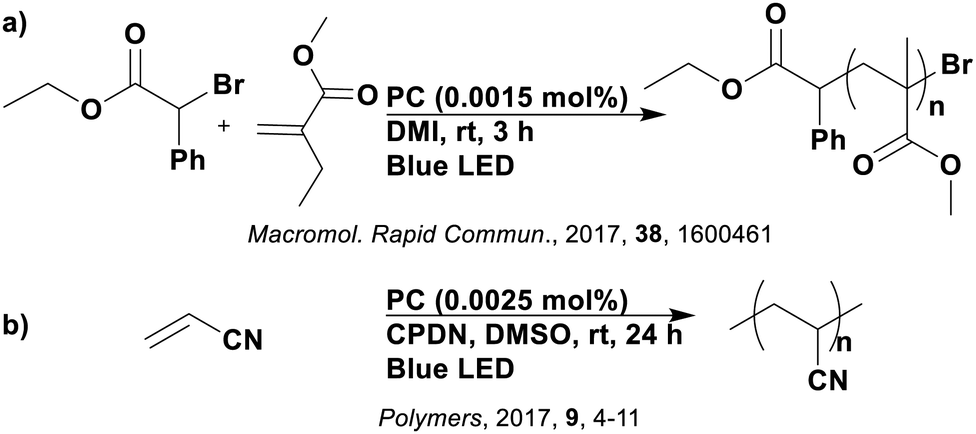 | ||
| Fig. 80 Reaction schemes for photocatalytic polymerisations of (a) methyl methacrylate and (b) acrylonitrile. | ||
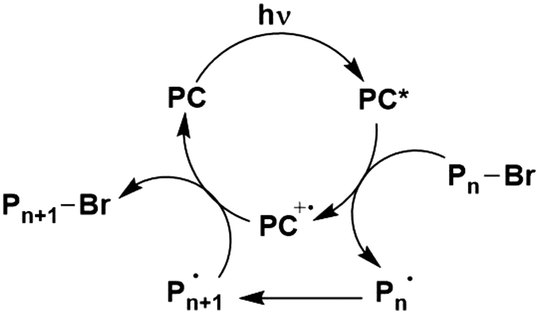 | ||
| Fig. 81 Photocatalyzed ATRP mechanism. | ||
4CzIPN has been shown to act as a radical initiator in the RAFT polymerisation of acrylonitrile when used in combination with the chain transfer agent 2-cyanoprop-2-yl-1-dithionaphthalate (CPDN) (Fig. 80b).230 The results indicated that 4CzIPN provided molecular weight control of the resulting polymer of a similar level to commonly used ultra-violet photoinitiators such as diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide (TPO) and 2,2′-azobis(2-methylpropionitrile) (AIBN).
Halogenation
Visible light promoted chlorination of electron-rich arenes has also been demonstrated using 4CzIPN as the photocatalyst (Fig. 82).231 An in situ bromination of the arene is first implicated before a halogen exchange occurs, generating the corresponding chlorinated product (Fig. 83). Here, the photoexcited catalyst is used to oxidise both bromide and chloride ions to their respective radical counterparts. Oxygen, acting as a sacrificial oxidant, is then used to regenerate the photocatalyst, closing the photocatalytic cycle. Other organic PCs were tested but all gave very poor yields in comparison with 4CzIPN. For example, using 2 mol% of 4CzIPN yielded 63% of the chloroanisole product while the second highest yield was obtained from eosin Y (5 mol%) at 16%. This large difference in yield is likely to be correlated with the much greater excited state oxidising capacity of 4CzIPN in comparison to eosin Y (Erex* = 1.35 V and 0.83 V for 4CzIPN and eosin Y, respectively). However, [Mes-Acr]+ (Ered* = 2.06 V) also only gave a 16% yield of product using 5 mol% catalyst loading. Since the oxidation potential of anisole is Eox = 1.75 V, it is possible that the excited [Mes-Acr]+ could be also oxidising anisole, potentially leading to a non-productive side product formation that may explain the low product yield in this case. | ||
| Fig. 82 Photochlorination reaction scheme. | ||
 | ||
| Fig. 83 Proposed mechanism for the photochlorination of arenes. | ||
3. 4CzIPN in dual catalysis
Diverse in its applications, 4CzIPN has also been used as a photocatalyst in dual catalytic reactions for example, with transition metals and hydrogen atom transfer (HAT) co-catalysts. In this section, we highlight examples of 4CzIPN in synergistic catalysis.Photoredox/nickel cross-coupling reactions
Visible light photocatalysis working in combination with transition metal catalysis, unsurprisingly termed metallaphotocatalysis, has become an alluring and fruitful strategy to access new bond-forming reactions.232,233 There are multiple examples where 4CzIPN is used in combination with a nickel co-catalyst to promote cross-coupling reactions. These reactions typically involve the cross coupling of C(sp3) nucleophiles with electrophiles under mild conditions, typically through a Ni(I)/Ni(III) catalytic cycle.234 Radical precursors, generated photocatalytically via a reductive quenching cycle, add to the Ni(0) catalyst to generate a transient Ni(I) complex. Oxidative addition with an aryl halide to generate an intermediate Ni(III) complex followed by reductive elimination generates the coupled product. Alternatively, oxidative addition of the aryl halide to Ni(0) occurs first, generating a Ni(II) species, followed by addition of the C(sp3) radical to form the Ni(III) complex, which can then undergo reductive elimination. Regardless, the resultant Ni(I) complex is reduced by the reduced PC, closing both the catalytic and photocatalytic cycles (Fig. 84). The photocatalyst must have a wide redox window to accomplish both the alkyl radical formation and the nickel complex reduction.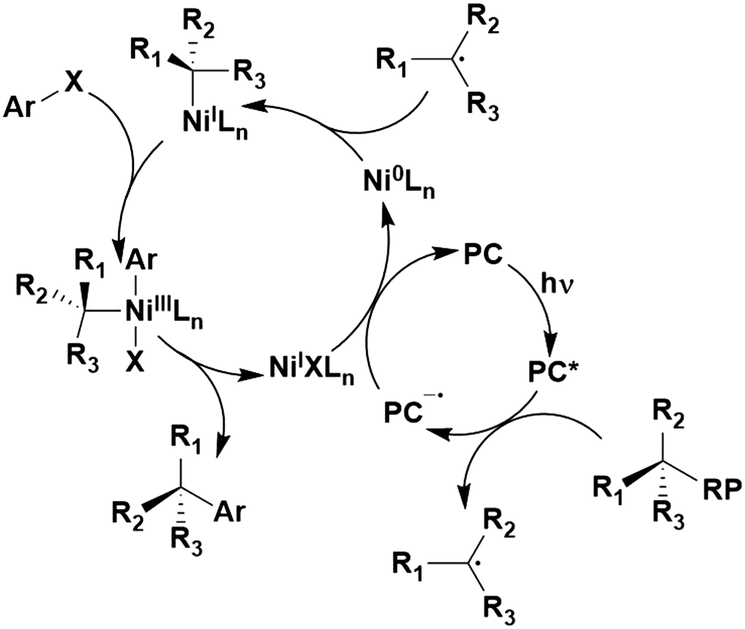 | ||
| Fig. 84 Generic mechanism applicable to photoredox-nickel catalysed cross coupling reactions for C(sp3)–C(sp2) bond formation going through a Ni(I)/Ni(III) catalytic cycle where RP signifies radical precursor. | ||
The most common photoredox/nickel cross-coupling reaction involves C(sp3)–C(sp2) coupling, using a nickel catalyst formed from one of NiX2.dme or Ni(COD)2 (COD = 1,5-cyclooctadiene) in the presence of a diine ligand that is typically 4,4′-di-tert-butyl-2,2′bipyridine (dtbbpy). A variety of radical precursors have been reported, including alkyl or aryl trifluoroborate salts,235–237 trifluoroboratochromanones,238 4-alkyl-1,4-dihydropyridine derivatives (DHPs),183,234,239 carboxylic acids,21,240–242 alkyl bis(catecholato)silicates,243 aliphatic amines244 or aziridines245 and cycloalkanols.246 Upon undergoing SET to the excited photocatalyst, these C(sp3) radicals can then be cross-coupled with alkyl, aryl or hetero aryl halides,21,183,234–236,238,240–246 aryl triflates,21 vinyl halides,21,243 carboxylic acids239 or anhydrides (Fig. 85 and 86).237
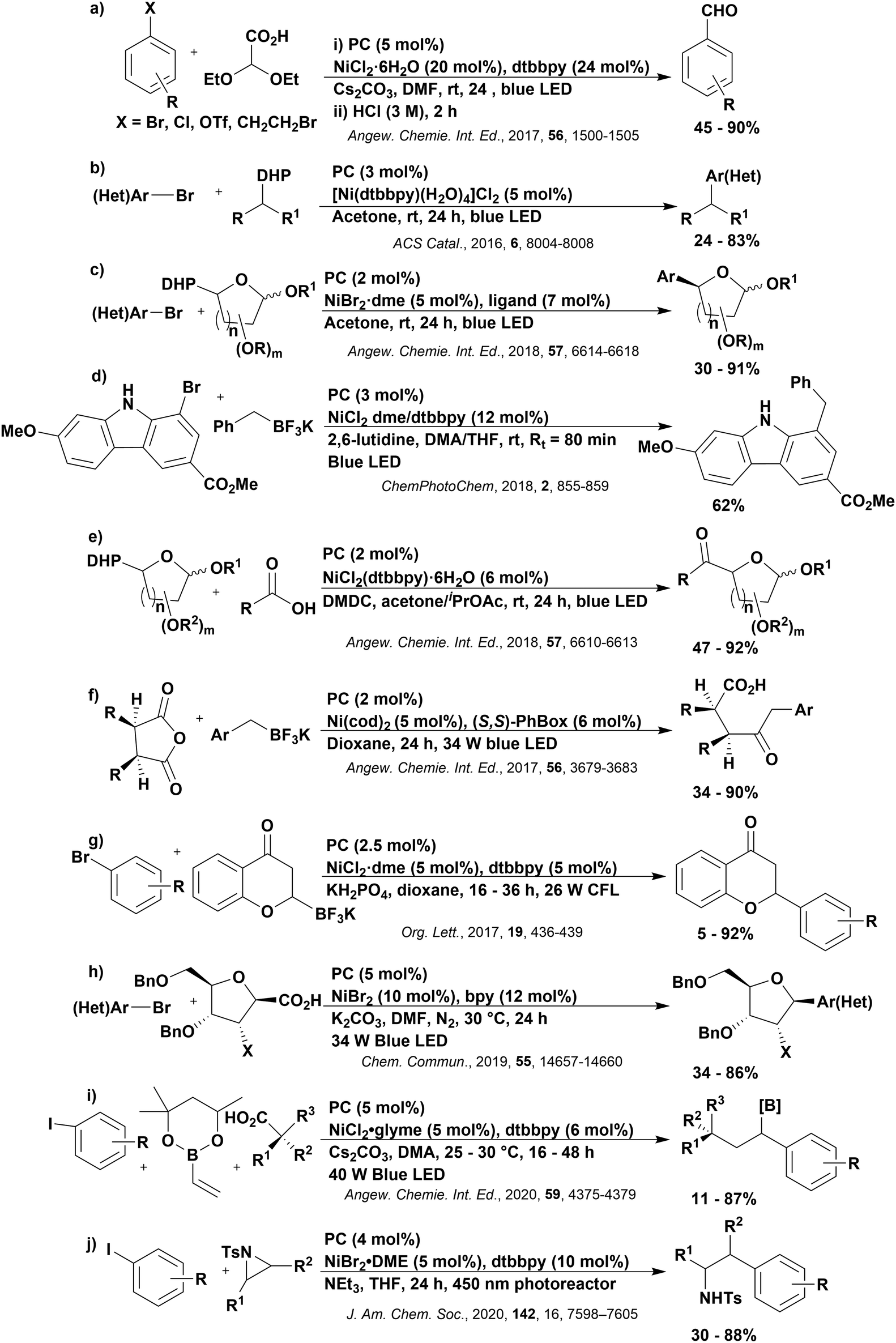 | ||
| Fig. 85 Reaction schemes for the dual catalytic C(sp3)–C(sp2) cross-coupling reactions with Ni where 4CzIPN has proven to be the best photocatalyst. | ||
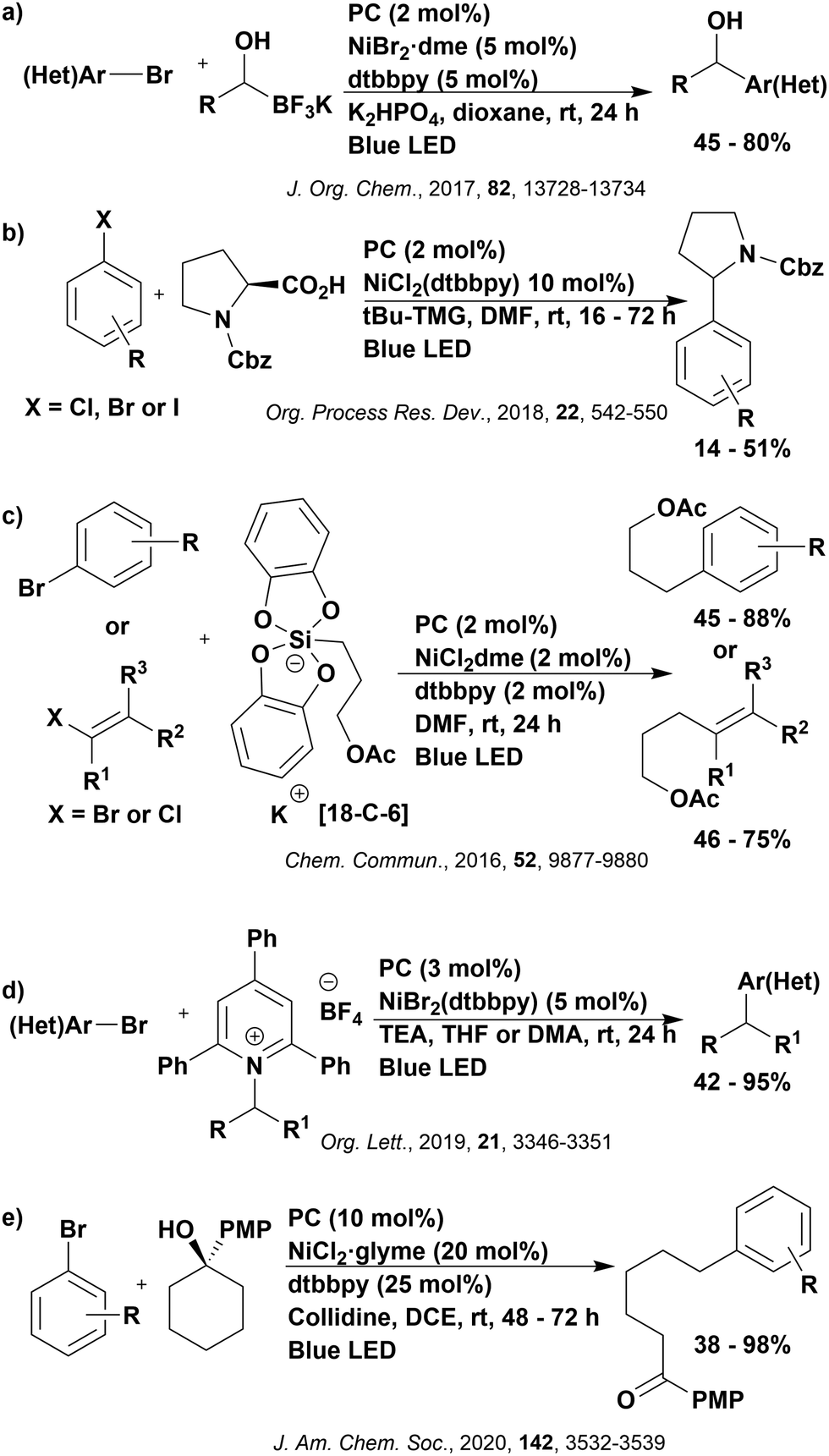 | ||
| Fig. 86 Reaction schemes for the metallaphotocatalysis involving Ni for C(sp3)–C(sp2) cross coupling where 4CzIPN does not give the highest final product yield. | ||
For this subset of reactions, 4CzIPN was shown to be the most efficient PC in terms of final product yield for the formylation of aryl halides and triflates (Fig. 85a),21 alkylation of aryl and hetero aryl halides (Fig. 85b),183 synthesis of arylated C-saccharides (Fig. 85c),234 cross coupling of aryl halides with trifluoroborate salts in flow (Fig. 85d),235 synthesis of non-anomeric C-acyl glycosides (Fig. 85e),239 enantioselective desymmetrization of cyclic meso-anhydrides (Fig. 85f),237 α-arylation/heteroarylation of 2-trifluoroboratochromanones (Fig. 85g),238 synthesis of aryl/heteroaryl-C-nucleosides (Fig. 85h),241 synthesis of complex alkyl boronic esters (Fig. 85i)242 and the synthesis of β-phenethylamines (Fig. 85j).245
Although 4CzIPN has been shown to be highly successful in most of these dual catalytic C(sp3)–C(sp2) cross coupling reactions, the two commonly used iridium complexes, [Ir(dF(CF3)ppy)2(bpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, can outperform 4CzIPN in terms of yield in examples including the synthesis of secondary benzylic alcohols (Fig. 86a), decarboxylative arylation reaction (Fig. 86b), alkylation of aryl halides or alkenes (Fig. 86c), deaminative reductive arylation (Fig. 86d) and site specific arylation of ketones (Fig. 86e).236,240,243,244,246 This could be as a result of their more reducing ground state reduction potentials (Ered = −1.37 V for both iridium photocatalysts vs. −1.21 V for 4CzIPN), since reduction of these Ni(I) species is typically around Ered ≈ −1.1 V.247 However, factors other than final product yield must be considered when evaluating which photocatalyst to employ in these types of reactions. For example, 4CzIPN produced the highest E/Z isomeric ratio despite a lower yield in the stereoselective alkenyl-alkyl cross-coupling reaction (Fig. 86c).243 For the decarboxylative arylation cross-coupling reaction (Fig. 86b),240 higher product yields were reached much faster with 4CzIPN than with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; although, conversion plateaued and a lower overall yield was observed than with the iridium photocatalyst. Finally, in many cases the differences in product yields in examples with 4CzIPN and iridium-based photocatalysts argue based on cost considerations for the choice of the former.244 For the remote site specific arylation of ketones using tertiary alcohols (Fig. 86e),246 only highly photooxidising PCs were capable of oxidation of the cycloalkanol (Eox = 1.57 V for 1-(4-methoxyphenyl)cyclohexan-1-ol), rendering 4CzIPN (Ered* = 1.35 V, 0% yield) incapable whereas [Mes-Acr]ClO4 flourished (Ered* = 2.06 V, 86% yield).
Although C(sp3)–C(sp2) dual catalytic cross-coupling tends to follow the mechanism shown in Fig. 84, the excited PC could instead be reductively quenched by a sacrificial electron donor and the reduced PC be used to reduce the radical precursor as well as the Ni(I) species. The nickel catalytic cycle generally follows the process shown in Fig. 84 but in this case, two photocatalytic cycles are proposed, suggesting this is a two-photon process. Examples where this alternative mechanism may be in operation are the coupling of α-chloro esters with aryl iodides (Fig. 87a)248 and the site-selective 1,2-dicarbofunctionalization of vinyl boronates (Fig. 87b).249 In the former reaction, however, it should be noted that the pathway for the formation of the α-carbonyl radical has not been conclusively proven, it may instead be formed by reduction from a Ni(I) or Ni(0) species rather than from the reduced PC. [Ir(ppy)2(dtbbpy)]PF6 and 4CzIPN were considered in the coupling of α-chloro esters with aryl iodides; however, direct comparison is difficult since neither were tested under the exact same reaction conditions. The former gave 25% yield using Cy2NMe as the sole reductant while the latter gave 95% when using a Hantzsch ester, HEH, as the reductant.
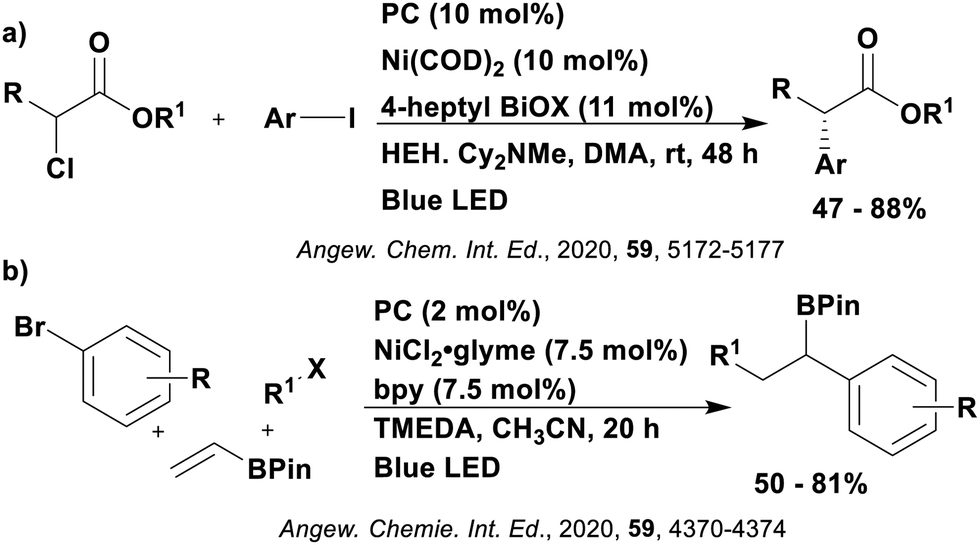 | ||
| Fig. 87 Reaction scheme for the C(sp3)–C(sp2) cross coupling involving (a) α-chloro esters with aryl iodides and (b) 1,2-dicarbofunctionalization of vinyl borates. | ||
In the 1,2-dicarbofunctionalization of vinyl borates (Fig. 87b)249 the excited PC is proposed to be reductively quenched by TMEDA in both photocatalytic cycles with the regeneration of the PC occurring through either SET to the Ni(I) species (as in Fig. 84) and SET to the tertiary alkyl halide to generate an alkyl radical. Addition of the alkyl radical to the vinyl borate forms another carbon-centred radical, which is captured by the Ni catalyst; at this point the nickel catalytic cycle proceeds as normal. Stern–Volmer quenching studies reveal that TMEDA is responsible for quenching the luminescence of the excited PC. The presence of the alkyl radical was confirmed by EPR spectroscopy and corroborated by radical trapping experiments with additives such as TEMPO. Only three PCs were considered in this reaction: 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 and [Ir(ppy)2(bpy)]PF6, providing yields of 86%, 53% and 62%, respectively. Notably, the organic PC was present in 5 mol% while the iridium PCs were used at 2 mol%. When the catalyst loading of 4CzIPN was decreased to 3 mol% the yield decreases to 55%, comparable with the iridium PCs.
A different mechanism is proposed for the C(sp3)–C(sp2) cross coupling of alkyl bromides with vinyl bromides (Fig. 88).250 Reductive quenching of the excited PC by a bromide ion yields a bromyl radical, which can abstract a hydrogen atom from (TMS)3SiH (as shown in Fig. 53). The silyl radical formed can then abstract a bromine from the alkyl bromide, generating the alkyl radical. The nickel catalytic cycle occurs as usual (shown in Fig. 84), with closure of the photocatalytic cycle occurring in tandem with the reduction of the Ni(I) species. However, no mechanistic evidence is provided. In the PC screen, 2 mol% of organic PCs were compared against 1 mol% of organometallic PCs, making direct comparison between these results difficult. For example, 4CzIPN obtained the highest yield of 81% while [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 performed the next best, giving 72% of product. However, it is clear that 4CzIPN could photocatalyse this reaction while eosin Y and rose bengal could not. This may be due to the stronger photooxidising ability of 4CzIPN (Ered* = 1.35 V, 0.83 V and 0.81 V for 4CzIPN, eosin Y and rose bengal, respectively).
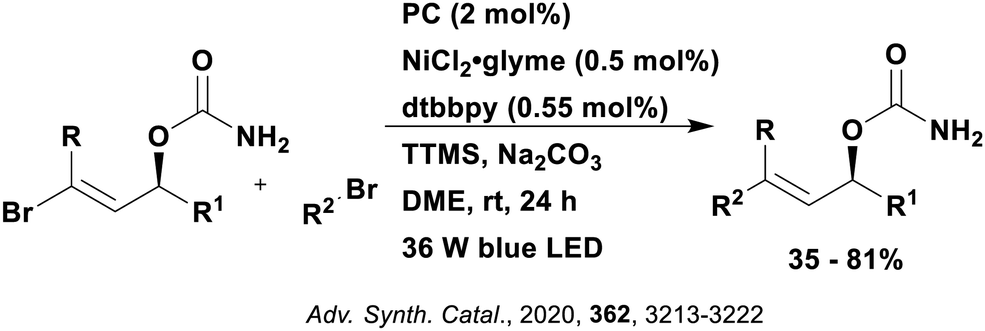 | ||
| Fig. 88 Reaction scheme for the coupling of alkyl bromides with vinyl bromides. TTMS = tris(trimethylsilyl)silane. | ||
The C(sp3)–C(sp2) dual catalytic cross coupling can also be observed in the remote arylation of nitriles using aryl halides (Fig. 89a).251 Utilisation of alkynes instead allows for vinylation of nitriles (Fig. 89b) while alkyl bromides can be used to alkylate the nitrile via a C(sp3)–C(sp3) cross coupling (Fig. 89c). The proposed mechanism involves reductive quenching of the excited PC by the oxime radical precursor, subsequently generating an iminyl radical (Fig. 90), which undergoes rapid ring opening, forming a distal nitrile radical. This radical is trapped by the Ni(II) species, forming a Ni(III) complex, which upon reductive elimination, allows for release of the arylated product. The proposed mechanism was based on similar work from Leonori et al.252 A quantum yield of 0.19 provides further support that this reaction is not a radical chain process. For the arylation reaction (Fig. 89a), 4CzIPN was identified as the second best photocatalyst, obtaining a yield of 24% in comparison to the superior 41% yield attained with [Ir(ppy)2(dtbbpy)]PF6. This is likely related to the superior ground state reducing capacity of the iridium photocatalyst (Ered = −1.51 V for [Ir(ppy)2(dtbbpy)]PF6 and Ered = −1.21 V for 4CzIPN), which is needed to reduce the Ni(I) species (Ered = −1.20 V in DMF).253
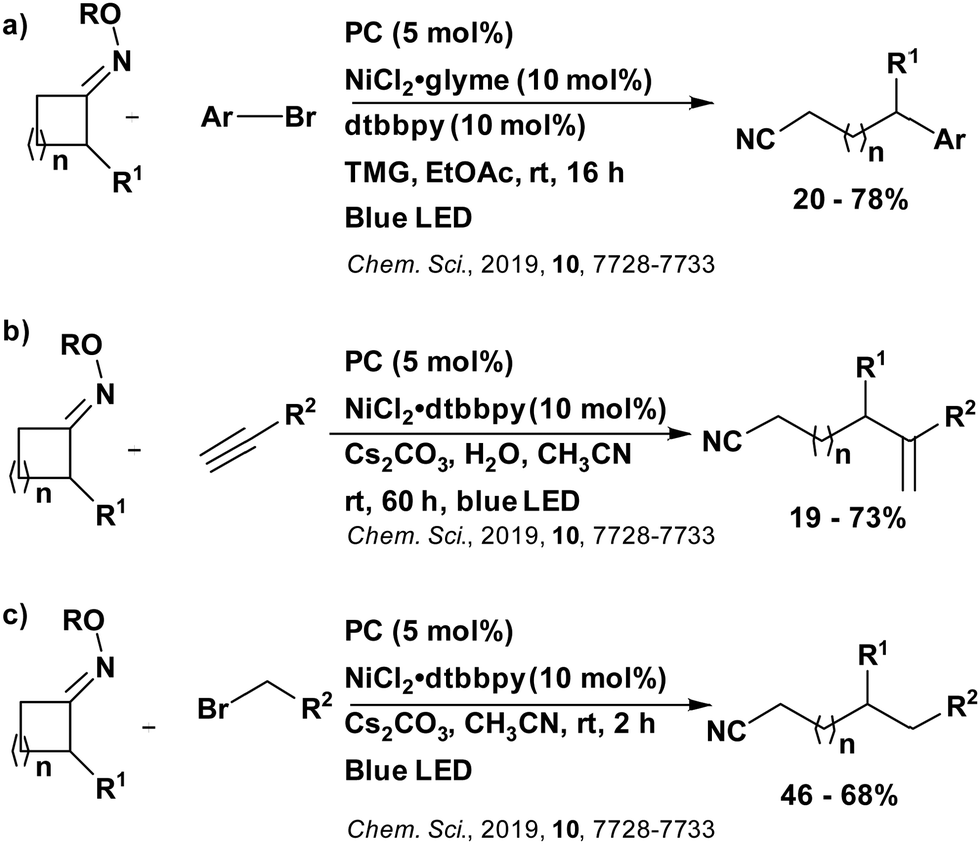 | ||
| Fig. 89 Reaction scheme for the (a) arylation, (b) vinylation and (c) alkylation of nitriles. | ||
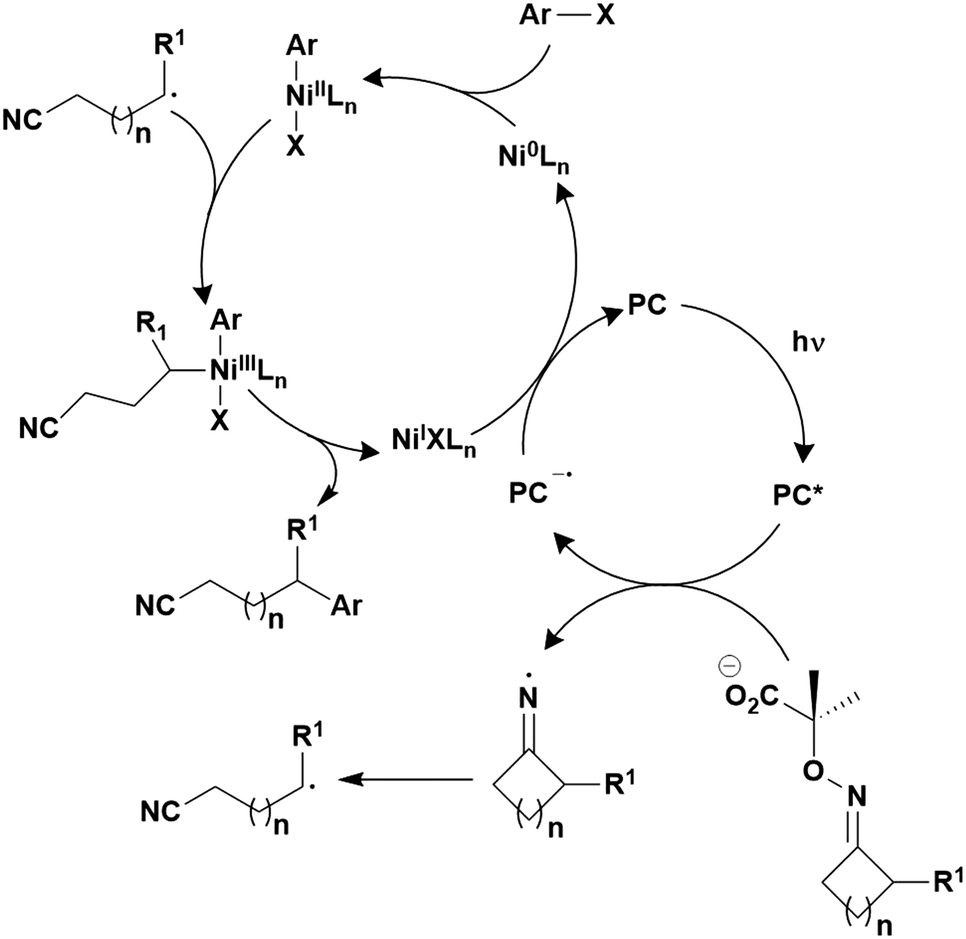 | ||
| Fig. 90 Suggested mechanism for the dual catalytic arylation of nitriles. | ||
Interestingly, in the reaction of the oxime with an alkyne (Fig. 89b), 4CzIPN was identified as the optimal photocatalyst, yielding 41% of product while [Ir(ppy)2(dtbbpy)]PF6 managed only 33%. When using alkyl bromides as the coupling partner (Fig. 89c), the most successful catalyst was shown to be [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which gave 5% yield of product. Both 4CzIPN and [Ir(ppy)2(dtbbpy)]PF6 yielded no product in this case.
Although less common, C(sp3)–C(sp3) cross-coupling has also been reported using this form of dual catalysis. Cross coupling of alkyl halides with silicate radical precursors (Fig. 91a)254 or with ethers (Fig. 91b),255 allylic alcohols with DHP radical precursors (Fig. 91c)256 or oxabenzonorbornadiene with alkylamines (Fig. 91d)257 have been investigated. In the former, Ni(COD)2 with bpy acting as the ancillary ligand, was utilised to generate the coupled poduct.254 Only three PCs were tested, with both 4CzIPN and [Ru(bpy)3](PF6)2 giving 22% yield of the desired product and [Ir(dF(CF3)ppy)2(bpy)2]PF6 performing slightly better, yielding 34% of the coupled product. Undesired homocoupling between the alkyl halides prevented higher yields from being achieved under these conditions; follow on mechanistic studies are currently underway by Fensterbank et al.
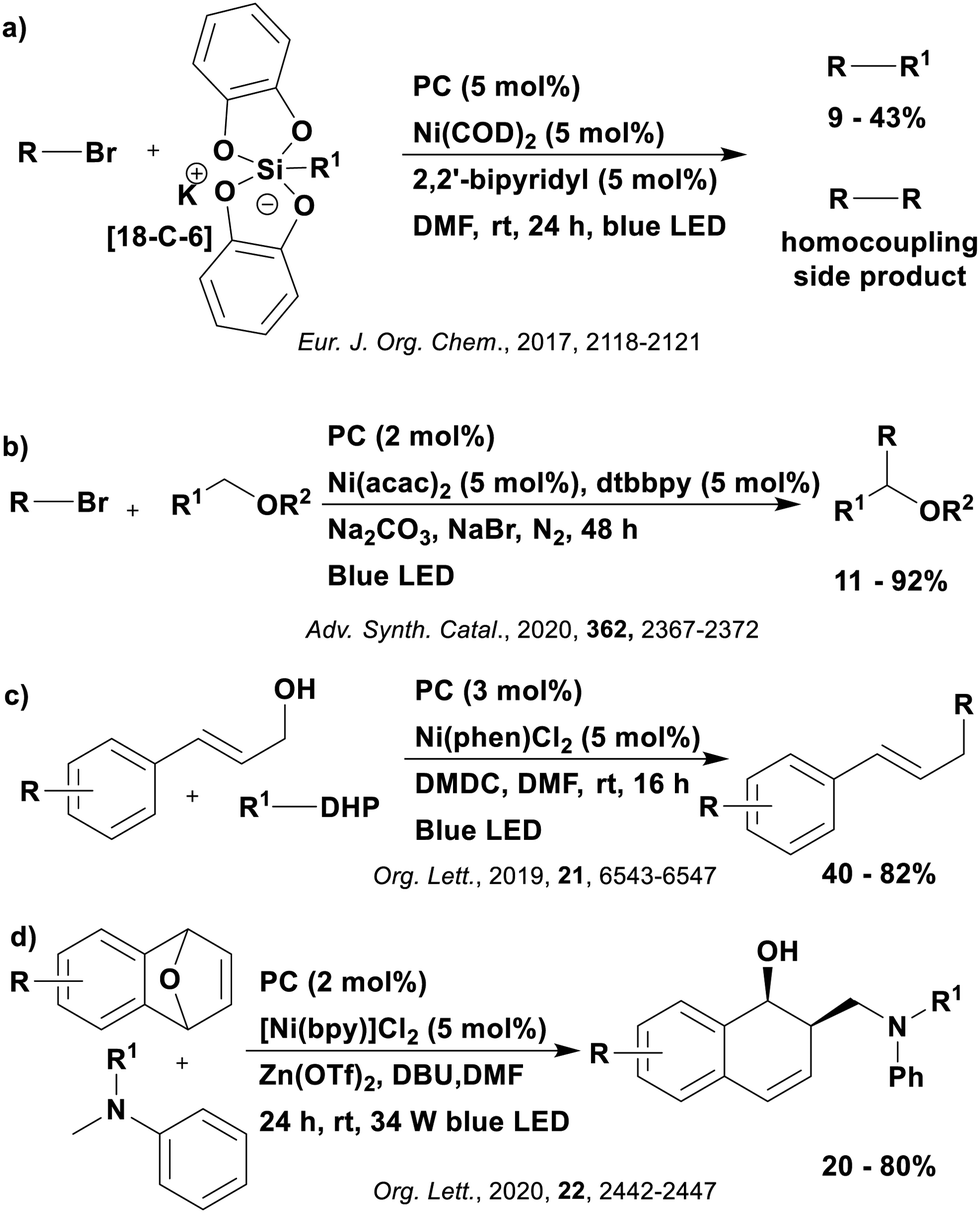 | ||
| Fig. 91 Reaction scheme for the C(sp3)–C(sp3) metallaphotoredox cross coupling involving a) alkyl halides and silicates, b) alkyl halides and ethers, c) allylic alcohols with DHP radical precursors and d) oxabenzonorbornadiene and alkylamines. DMDC = dimethyl carbonate. | ||
In the coupling of alkyl bromides with ethers (Fig. 91b), a proposed mechanism is shown in Fig. 92.255 Oxidative addition of the alkyl bromide to the Ni(0) species produces a Ni(II) alkyl halide complex, which reductively quenches the excited PC. Photoelimination of the Ni(III) species releases an electrophilic bromyl radical, which undergoes HAT with the ether. The as formed carbon-centred radical is then captured by the Ni(II) species. Reductive elimination from the resultant Ni(III) complex releases the product. Both catalytic cycles close following a SET from the reduced PC to the Ni(I) species. Alkyl iodides or triflates proved unsuccessful as substrates in this reaction because they cannot promote the HAT step. The reaction yield was dependent on the power of the LED source, supporting a proposed two-photon mechanism. In the PC screen, only [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 4CzIPN provided any product, 82% and 65%, at 2 mol% and 5 mol%, respectively. The higher yields with the iridium PC may be due to its stronger reduction potential in the ground state (Ered = −1.37 V and −1.21 V, respectively).
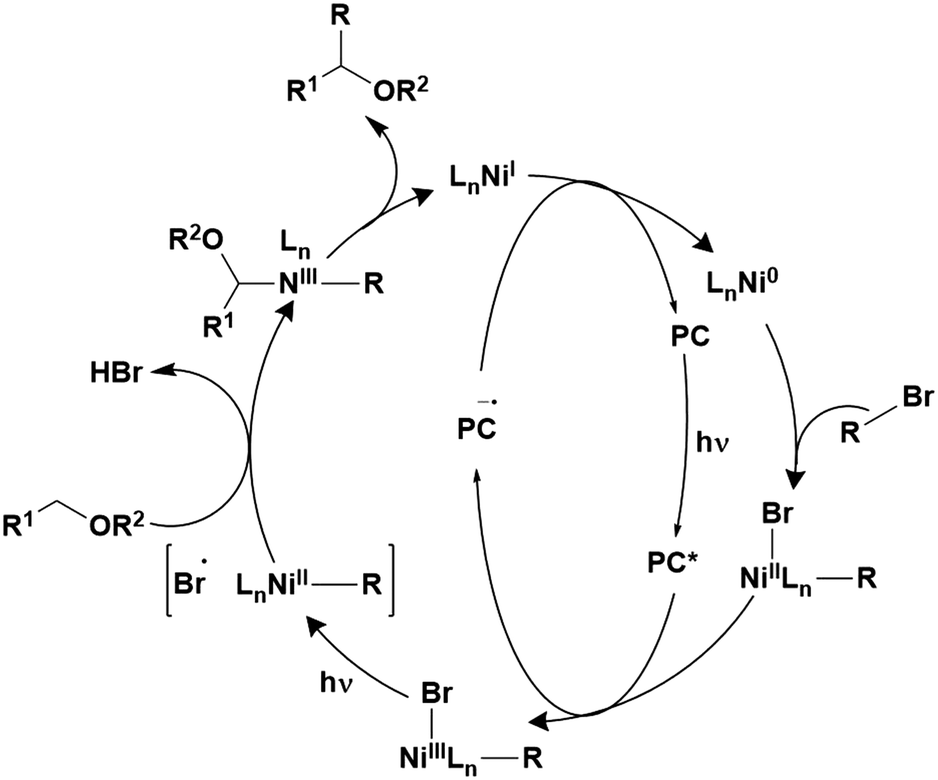 | ||
| Fig. 92 Viable mechanism for the C(sp3)–C(sp3) cross coupling of alkyl halides and ethers. | ||
Molander et al. demonstrated the photocatalyzed C(sp3)–C(sp3) Tsuji–Trost cross coupling reaction (Fig. 91c).256 The proposed mechanism (Fig. 93) involves formation of the alkyl radical from DHP radical precursors through SET to the excited PC. This radical is trapped by the Ni(0) complex, followed by oxidative addition of the in situ-generated allyl methyl carbonate. Reductive elimination releases the final product and the resultant Ni(I) is reduced by the reduced PC, simultaneously closing both catalytic cycles. Stern–Volmer quenching studies were conducted to support this proposed mechanism, which indicated that neither the nickel catalyst nor the allyl methyl carbonate could quench the excited PC. A secondary kinetic isotope effect (kH/kD = 1.15) was observed, supporting the hypothesis that oxidative addition of the allyl methyl carbonate onto the Ni(I) species is the rate-determining step. High throughput screening was conducted to identify the optimum conditions, with only 4CzIPN and [Ir(dF(CF3)ppy)2(bpy)]PF6 considered as PCs, and both providing comparable yields.
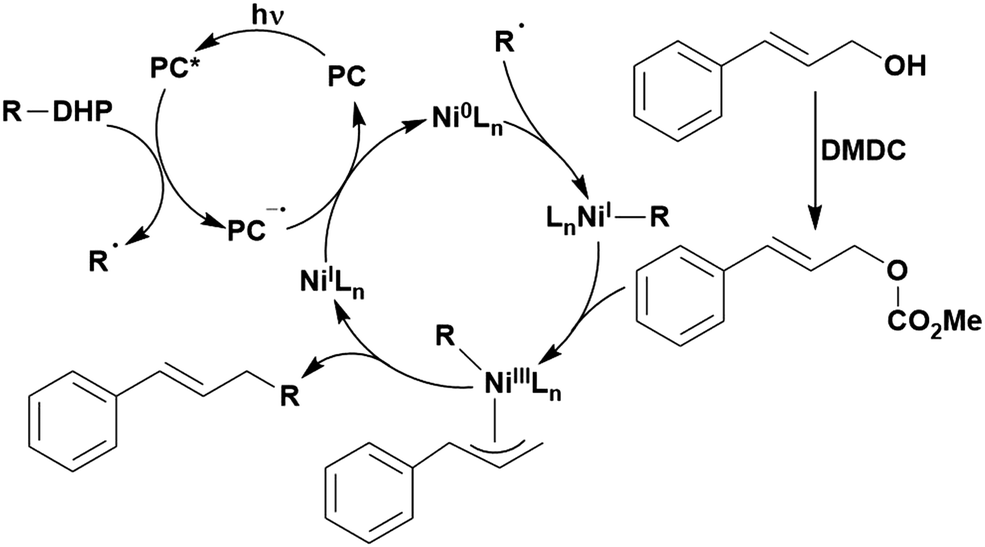 | ||
| Fig. 93 Proposed mechanism for the metallaphotoredox cross coupling of allylic alcohols with DHP radical precursors. | ||
Aminomethylation/ring opening of oxabenzonorbornadienes (Fig. 91d) also provides a route for the formation of C(sp3)–C(sp3) bonds via dual photoredox/Ni catalysis.257 Operating in a similar mechanism to that illustrated in Fig. 93, the excited PC is reductively quenched by the alkylamine, furnishing an α-aminoalkyl radical after deprotonation. Simultaneously, oxidative addition of Ni(0) into the C–O bond of the oxabenzonorbornadiene, facilitated by Zn(OTf)2, forms a σ-allyl intermediate, which then rearranges to a π-allyl Ni(II) species. Addition of the α-aminoalkyl reagent to the Ni(II) species forms a Ni(III) complex. Reductive elimination affords the final product. SET from the reduced PC to the Ni(I) species closes both catalytic cycles. The addition of TEMPO quenches the reaction, confirming this to be a radical process. The most efficient quenching of the excited PC, as confirmed by Stern–Volmer experiments, was by the alkylamine, while DBU only moderately quenched the luminescence. No photocatalyst screen was conducted in this study, only 4CzIPN was employed and provided yields of 20–80%. However, for five alkylamine substrates, 4CzIPN gave 0% yield and instead, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was used as the PC, providing yields between 45–80% for these substates. The authors propose no reasoning for this and acknowledge that both PCs should be capable, thermodynamically, of photooxidising all the alkylamines utilised in the substrate scope.
A further example of C(sp3)–C(sp3) cross-coupling, although proceeding slightly differently mechanistically, involves the coupling of aliphatic alcohol derivatives with alkyl halides (Fig. 94a). The same protocol can also be applied to C(sp2)–C(sp3) cross-coupling when used with aryl halides (Fig. 94b).258 The putative mechanism is shown in Fig. 95 and reflects the general mechanism shown in Fig. 84, except the formation of the alkyl radical differs. In this instance, β-scission of the aliphatic alcohol derivative generates an alkyl radical. The excited PC is reductively quenched instead by a Hantzsch ester. An association constant of 2.9 M−1 between the Hantzsch ester and the alcohol derivate implies an electron donor–acceptor (EDA) complex is formed between the two prior to homolytic cleavage. Both 4CzIPN and [Ir(ppy)2(dtbbpy)]PF6 afforded the same amount of product (84% and 87%, respectively), while [Mes-Acr]ClO4 and [Ru(bpy)3]Cl2 provided similar yields (54% and 51%, respectively). These results correlate generally to the ground state reduction potential of the PC (Ered = −1.51 V, −1.21 V and −0.57 V for 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 and [Mes-Acr]ClO4, respectively). For [Ru(bpy)3]Cl2, however, the low photooxidising ability of this PC may be responsible for its lower yield (Ered* = 0.77 V and 1.35 V for [Ru(bpy)3]Cl2 and 4CzIPN, respectively).
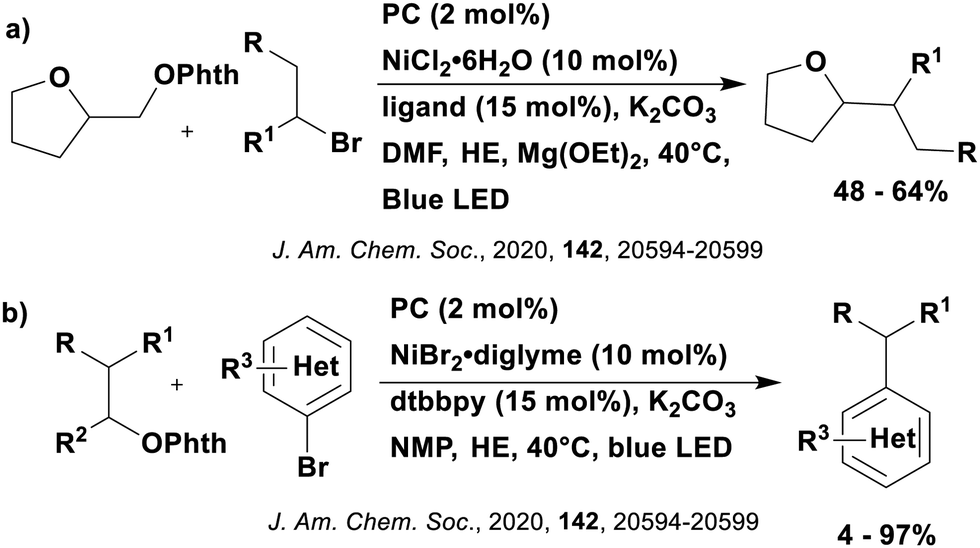 | ||
| Fig. 94 Reaction scheme for the C(sp3)–C(sp3) and (b) C(sp2)–C(sp3) cross coupling of aliphatic alcohol derivatives with a) alkyl and (b) aryl halides. HE = Hantzsch ester. | ||
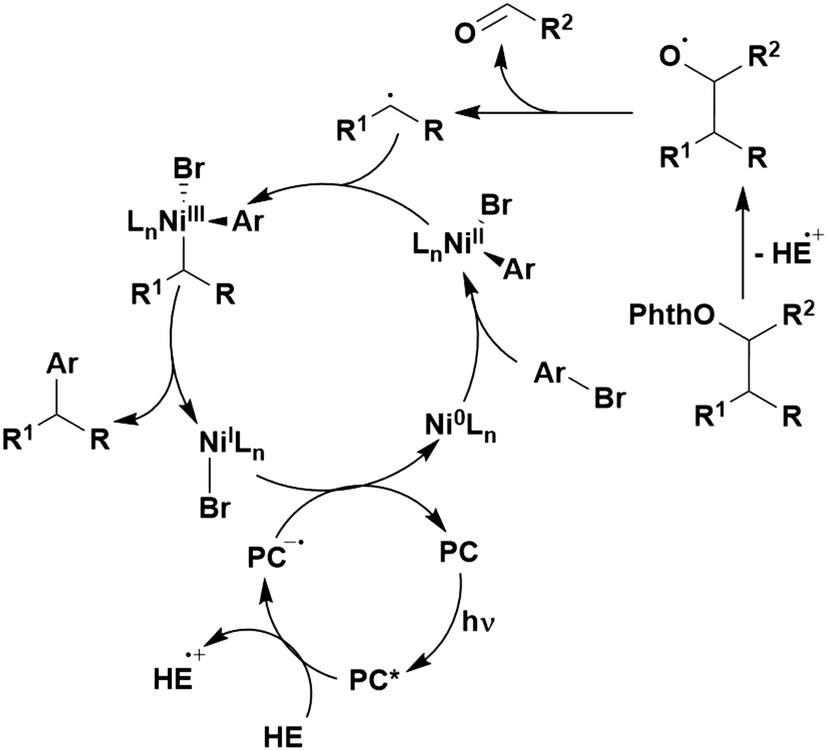 | ||
| Fig. 95 Putative mechanism for the C(sp2)–C(sp3) cross-coupling of aryl halides with aliphatic alcohol derivatives. | ||
Photoredox/nickel cross-coupling reactions have been developed to promote the carboxylation of aliphatic and aromatic bromides and triflates (Fig. 96)259 which may be classified as C(sp2)–C(sp) cross-coupling. In this case, the nickel catalyst is NiBr2.glyme in combination with neocuproine. The initial Ni(II) catalyst is first thought to be reduced to a Ni(0) species, before undergoing oxidative addition with the aryl halide. Reductive quenching of the photocatalyst by a Hantzsch ester is observed before the reduced photocatalyst is used to reduce the Ni(II) species. The putative mechanism involves carboxylation of the in situ formed Ni(I) complex, followed by product dissociation and regeneration of the Ni(II) catalyst (Fig. 97). In the carboxylation of bromobenzene, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN and [Ir(dF(CF3)ppy)2(bpy)]PF6 all yielded essentially the same product yield (46%, 45% and 40%, respectively). Other PCs considered were considerably worse, including [Ru(bpy)3](PF6)2 (3%) and fac-Ir(ppy)3 (5%). The excited photocatalyst must first be capable of oxidising the Hantzsch ester (Eox = 0.887 V),260 which explains why low yields were obtained for [Ru(bpy)3](PF6)2 (Ered* = 0.77 V) and fac-Ir(ppy)3 (Ered* = 0.31 V). Secondly, the ground state reduction potential of the photocatalyst must be sufficiently negative in order to reduce the Ni(II) species (Ered ≈ −1.2 V),261 which is easily achieved by [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(bpy)]PF6 and 4CzIPN. It should also be noted that the NiBr2.glyme (Ered = −1.70 V vs. Fc/Fc+) was shown by Stern–Volmer quenching studies to be capable of oxidatively quenching 4CzIPN, as does the in situ generated LNiBr2 (Ered = −1.27 V vs. Fc/Fc+), despite this not being a thermodynamically favourable process (Eox* = −1.04 V for 4CzIPN). Both of these processes occur at a slower rate than the quenching from the Hantzsch ester (KSV = 0.18 M−1, 0.20 M−1 and 0.61 M−1 for NiBr2, LNiBr2 and Hantzsch ester, respectively).
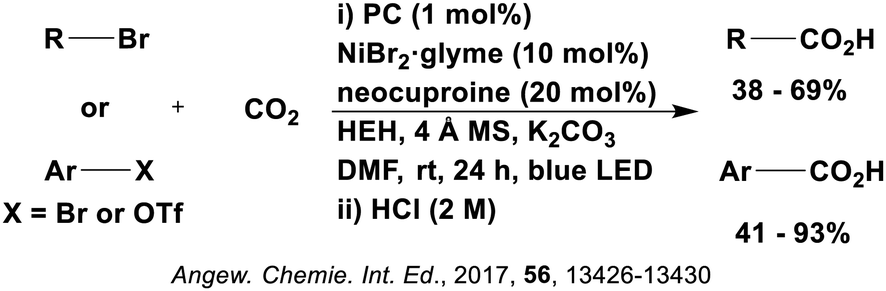 | ||
| Fig. 96 Reaction schemes for the metallaphotocatalysis mediated carboxylation of alkyl or aryl halides or triflates. | ||
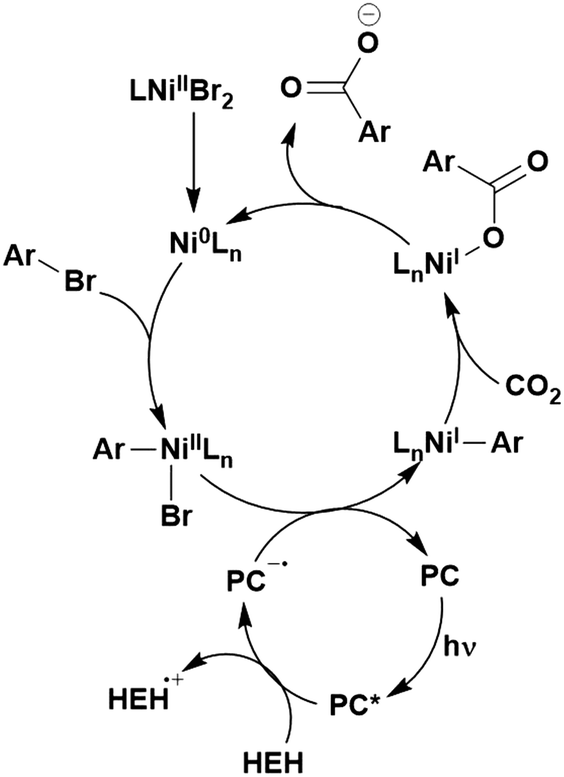 | ||
| Fig. 97 Viable mechanism for the carboxylation aryl bromides using combined photo and nickel catalysis (where L = neocuproine and HEH = Hantzsch ester). | ||
The combination of photocatalysis with nickel catalysis for cross-coupling reactions has also been extended to C(sp)-S bond formation to form alkynyl sulfides in flow (Fig. 98).262 The proposed mechanism invokes a two photon process, whereby the excited PC is first reductively quenched by the thiol, which after deprotonation, results in a thiyl radical. The reduced PC is then proposed to reduce the nickel catalyst, NiCl2.dme (in the presence of pyridine acting as the ancillary ligand), to generate the active Ni(I) species. This Ni(I) complex traps the thiyl radical, generating a Ni(II) complex, which is subsequently reduced by the reduced photocatalyst. Oxidative addition to the resultant Ni(I) complex followed by reductive elimination closes the nickel catalytic cycle as well as generating the required alkynyl sulfide (Fig. 99). Although two reduction steps involving the PC are proposed, no mechanistic support of this is provided. This reaction has been shown to work in both the presence and absence of the nickel catalyst (96% and 28%, respectively), although a much higher product yield is obtained when the dual catalysis is in operation. In choice of PC, 4CzIPN proved far superior in comparison to the other PCs, obtaining 96% yield of product while the second-best PC, [Ir(dF(CF3)ppy)2(bpy)]PF6, resulted in only 46% yield and the third best, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 formed 24%. There seems to be a correlation between the decreasing excited state oxidation potential and the decreasing yield (Ered* = 1.35 V, 1.32 V and 1.21 V, respectively).
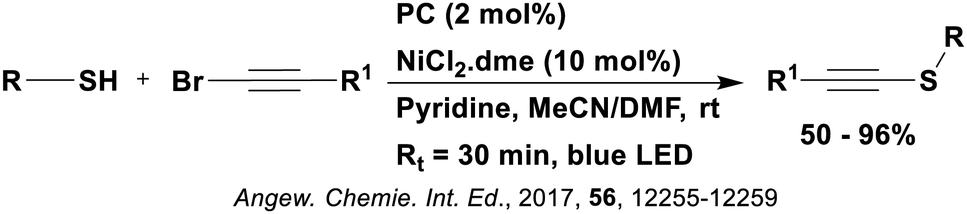 | ||
| Fig. 98 Reaction scheme for the formation ok alkynyl sulfides. | ||
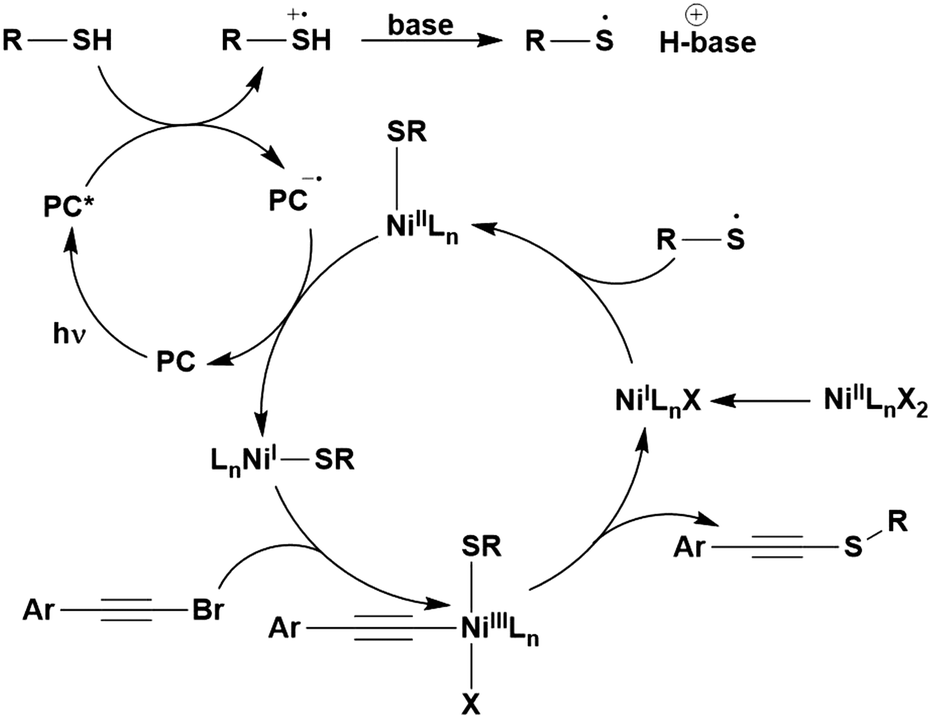 | ||
| Fig. 99 Plausible mechanism for the photo/nickel catalysed formation of alkynyl sulfides. | ||
Finally, photoredox/nickel cross coupling can be applied to a cascade coupling reaction, generating three new bonds in one synthetic step when using a combination of a diene, a substituted sulfinates and an aryl(hetero) halide (Fig. 100).263 The sulfinate reductively quenches the excited PC, generating the sulfonyl radical, which then undergoes a radical cascade cyclisation with the diene, yielding a C-centred radical (Fig. 101). This radical is then captured by the Ni(0) catalyst followed by oxidative addition of the aryl halide. The resultant Ni(III) complex undergoes reductive elimination to release the product, with the Ni(I) being reduced by the reduced photocatalyst, concomitantly closing both catalytic cycles. This proposed mechanism has been supported by Stern–Volmer quenching experiments, showing the sulfinate to be responsible for the PC quenching. Radical inhibition experiments with TEMPO also confirmed the presence of radicals in this process, while stoichiometric studies with a Ni(II)–ArCl complex resulted in none of the desired product, suggesting a Ni(II)–aryl species is not present in the catalytic cycle. Photocatalysts 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 all provided comparably high yields (89%, 91% and 88%, respectively) although 4CzIPN was used with 2.5 mol% loading while the iridium photocatalysts were used at 1 mol%. Ruthenium photocatalysts tested, including [Ru(bpy)3](PF6)2, proved far inferior, managing only 20% yield of product. Rueping et al. suggested this difference in yield is due to the lower photooxidising capacity of the ruthenium photocatalyst in comparison to the others (Ered* = 0.77 V for [Ru(bpy)3](PF6)2 in comparison to Ered* = 1.35 V and 1.21 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), making the oxidation of PhSO2Na (Eox = 0.5 V)264 more challenging. However, this explanation does not account for the success of [Ir(ppy)2(dtbbpy)]PF6, which is an even worse photooxidant (Ered* = 0.66 V), which suggests something other than these thermodynamic parameters is significant here.
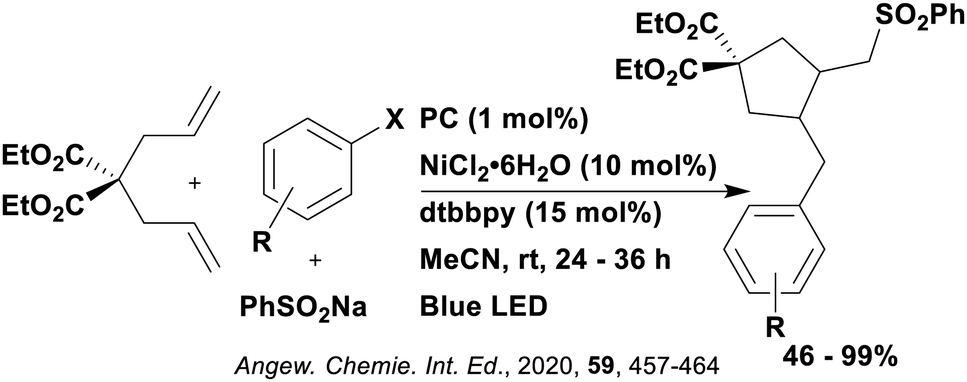 | ||
| Fig. 100 Reaction scheme for the dual catalytic cascade coupling reaction of dienes, sulfinates and aryl halides. | ||
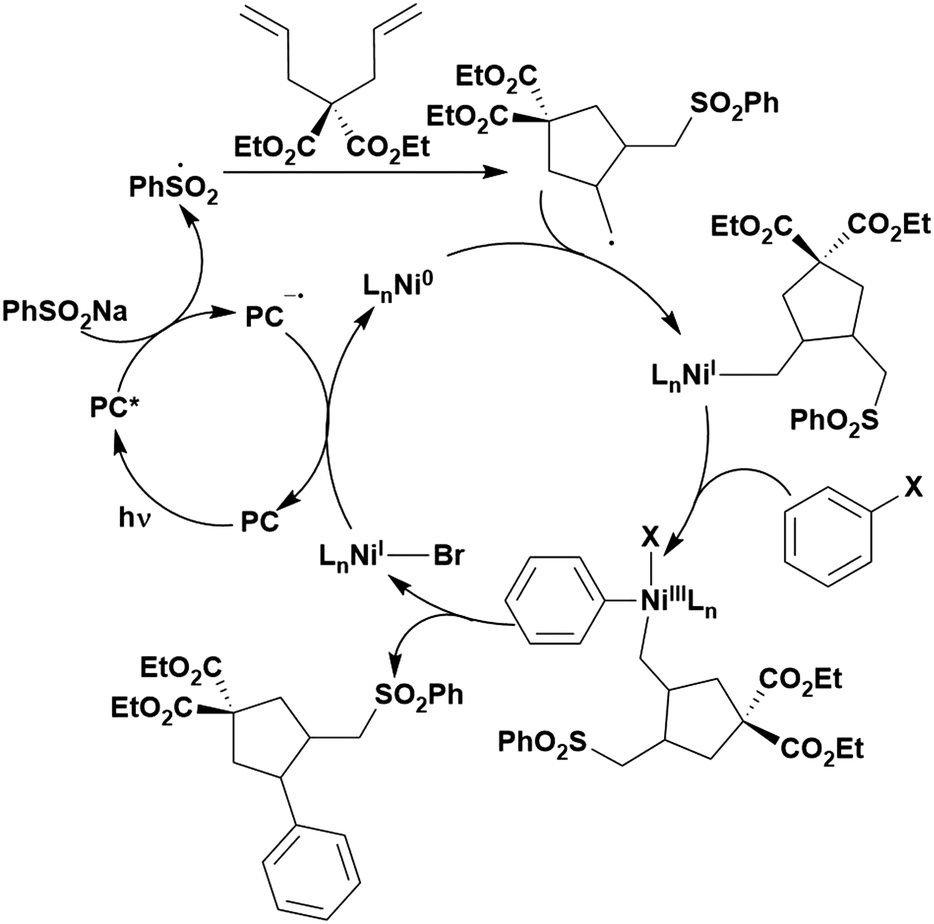 | ||
| Fig. 101 Proposed mechanism for the dual catalytic radical cascade coupling involving dienes, sulfinates and aryl halides. | ||
Photoredox/palladium cross-coupling reactions
Several reports exist merging photocatalysis with palladium-catalysed cross-coupling, for example decarboxylative formylation of aryl halides (Fig. 102a), acetoxylation of oximes (Fig. 102b), hydroxylation of arenes (Fig. 102c and d), and decarboxylative alkenylation of aliphatic carboxylic acids (Fig. 102e). In the former reaction, photoinduced decarboxylation of glyoxylic acid under a reductive quenching cycle, results in the formation of a formyl radical.265 This then coordinates to a palladium(II) species generating transiently a Pd(III) complex that is reduced to a Pd(II) complex by the reduced PC, closing the photocatalytic cycle. This Pd(II) complex then undergoes reductive elimination, ejecting the formylated product, regenerating the Pd(0) catalyst, which is then free to undergo another oxidative addition with the aryl halide (Fig. 103). 4CzIPN gave higher product yields in comparison to [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (78% and 51%, respectively), although both achieved 100% reaction conversion. This suggests that the iridium photocatalyst allowed for a greater formation of the hydrodeiodination by-product, which was observed in only ≈ 10% yield for 4CzIPN. This reaction can be considered a C(sp2)–C(sp2) coupling and proceeds in a very similar manner to the photoredox/Ni dual catalysis described previously. Although none of the Ni examples given involved C(sp2)–C(sp2) coupling, a study by Wang et al. also involved the formylation of aryl halides; however, in this particular case the carboxylic acid was used to generate the C(sp3) radicals.21 In the formylation of 4-bromobenzonitrile, Wang et al. obtained a 75% yield using Ni as the co-catalyst, while Fu et al. obtained 68% with a Pd co-catalyst, demonstrating that both procedures are similarly successful for the formylation of arenes.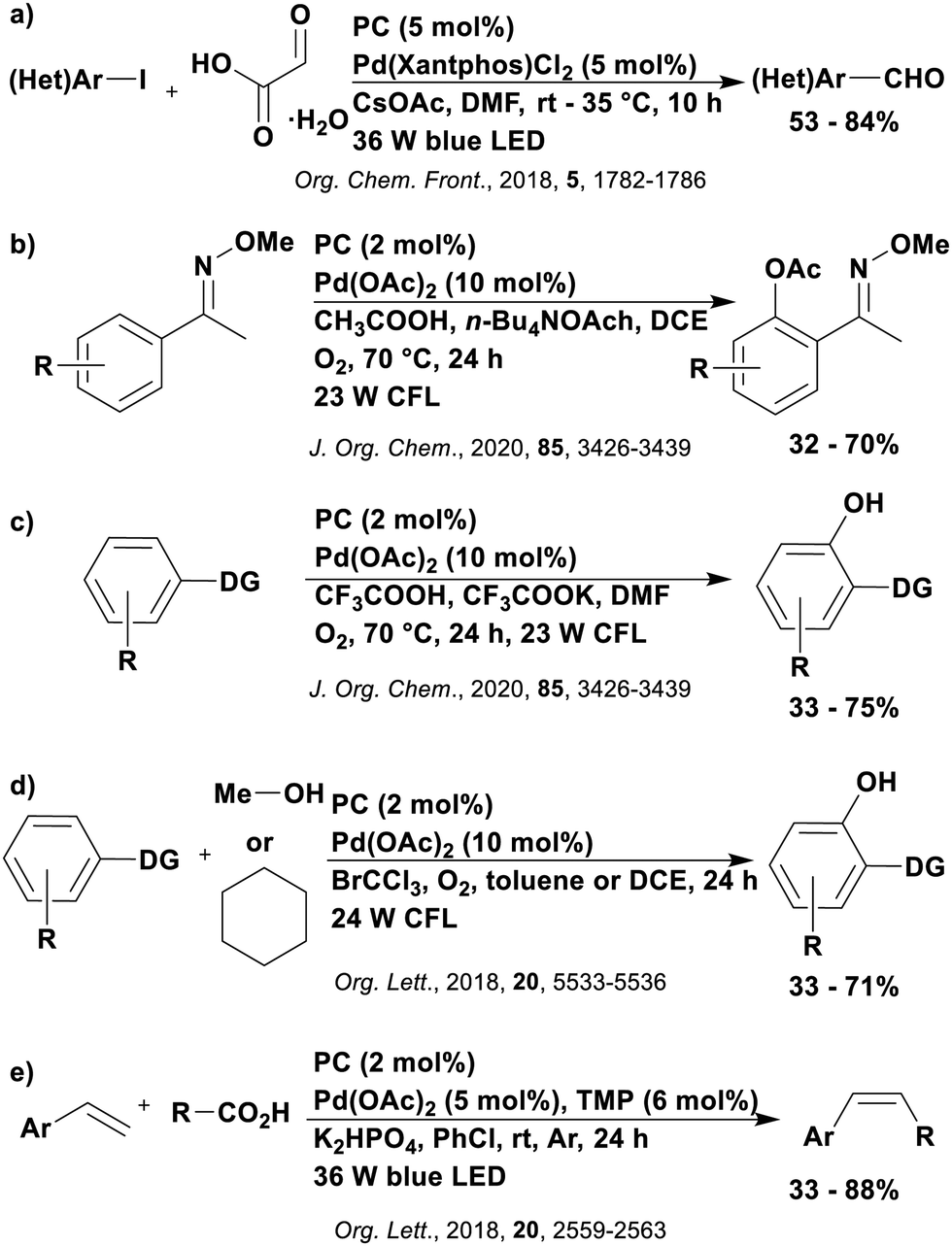 | ||
| Fig. 102 Reactions scheme for metallaphotocatalysis involving palladium where (a) decarboxylative formylation of aryl halides, (b) acetoxylation of oximes, (c and d) hydroxylation of arenes (DG = donor group) and (e) decarboxylative alkenylation of aliphatic carboxylic acids with vinyl arenes. | ||
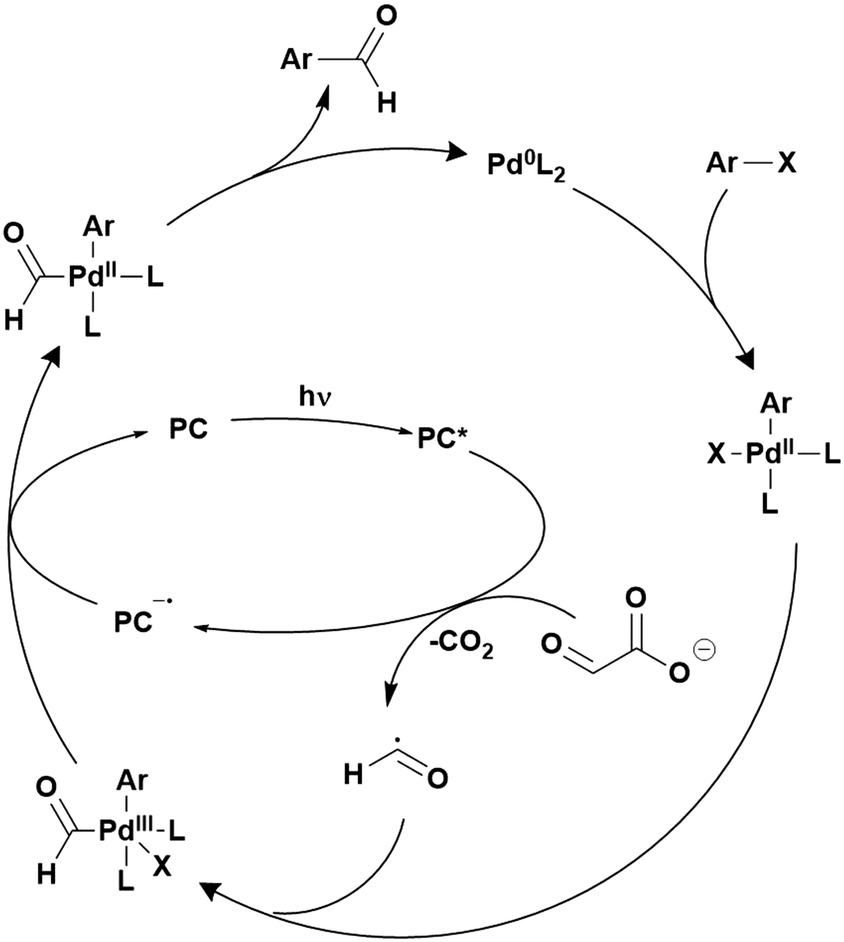 | ||
| Fig. 103 Working mechanism for the photoredox/Pd catalysed formylation of aryl halides. | ||
More commonly, 4CzIPN has been reported to work in tandem with the palladium catalyst Pd(OAc)2 under an oxidative quenching mechanism. Examples include the acetoxylation of oximes (Fig. 102b)266 or the hydroxylation of arenes (Fig. 102c and 102d).266,267 The proposed mechanism for these reactions is shown in Fig. 104.266 Oxidative quenching of the excited PC by oxygen generates the superoxide radical anion as well as the oxidised PC. Cyclopalladation of the oxime or arene generates the Pd(II) complex, which is oxidised by both the oxidised PC and the superoxide radical, resulting in a Pd(IV) species. Reductive elimination of the acetoxylated product closes the Pd-based catalytic cycle. A hydrolysis step then ensues for the hydroxylation reaction. The presence of the superoxide radicals was confirmed by EPR spectroscopy and an observed kinetic isotope effect suggested C–H bond cleavage may be the rate-determining step. Optimisation of the photocatalyst was considered for the acetoxylation reaction, with 4CzIPN proving superior to eosin Y, rose bengal and [Ru(bpy)3]Cl2 (69%, 21%, 17% and 57%). The photocatalyst is required to oxidise the Pd(II) complex, which has an oxidation potential of Eox = 0.83 V,268 which explains the lower yields of the other organic photocatalysts in comparison to 4CzIPN (Eox = 0.78 V, 0.84 V and 1.52 V for eosin Y, rose bengal and 4CzIPN, respectively). For [Ru(bpy)3]Cl2 the lower yield can be explained by the excited state oxidation potential, which must be sufficiently negative to reduce oxygen to the superoxide radical anion (Ered = −0.86 V);269 hence, this process is more thermodynamically challenging for the ruthenium photocatalyst in comparison to 4CzIPN (Eox* = −0.81 V and −1.04 V for [Ru(bpy)3]Cl2 and 4CzIPN, respectively).
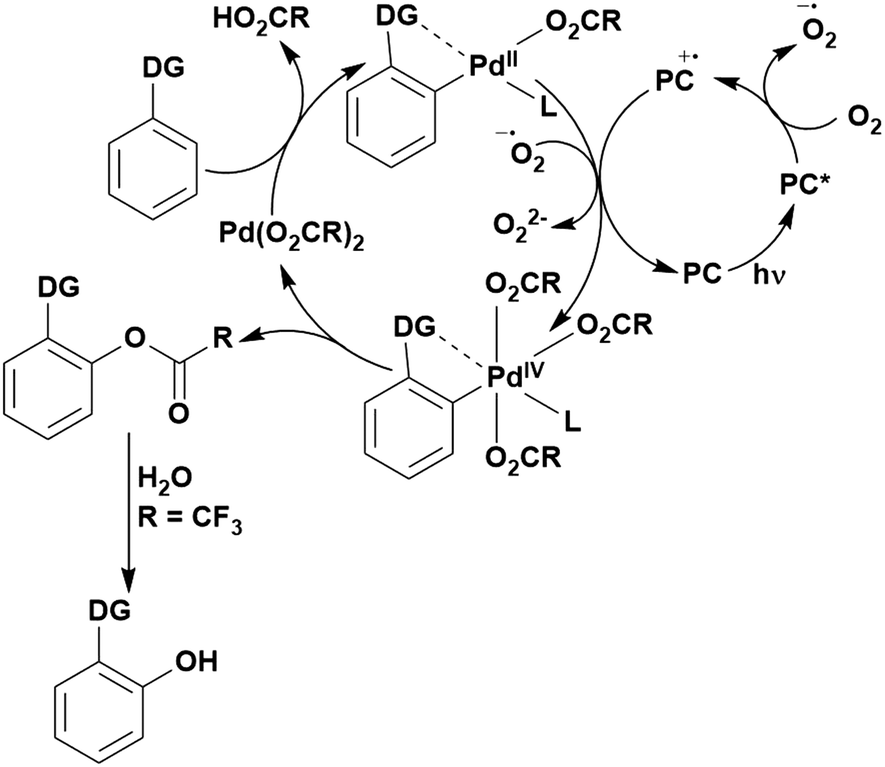 | ||
| Fig. 104 Proposed mechanism for the acetoxylation of oximes and hydroxylation of arenes. DG = oxime for acetoxylation reaction and donor group for hydroxlation reaction. | ||
In the case of hydroxylation of arenes (Fig. 102d), the excited PC is used to reduce the hydrogen abstractor BrCCl3 (Fig. 105) and then also to promote the oxidation of a transient Pd(III) species to Pd(IV), closing the photocatalytic cycle, prior to reductive elimination of product in the Pd-based catalytic cycle.267 Only organic photocatalysts were considered in this study of which 4CzIPN provided the highest yield of 67% in comparison to eosin Y at 45%, which was the next best PC. Analogously to the previous example, this is likely due to the greater ground state oxidising capacity of 4CzIPN (Eox = 1.52 V and 0.78 V for 4CzIPN and eosin Y).
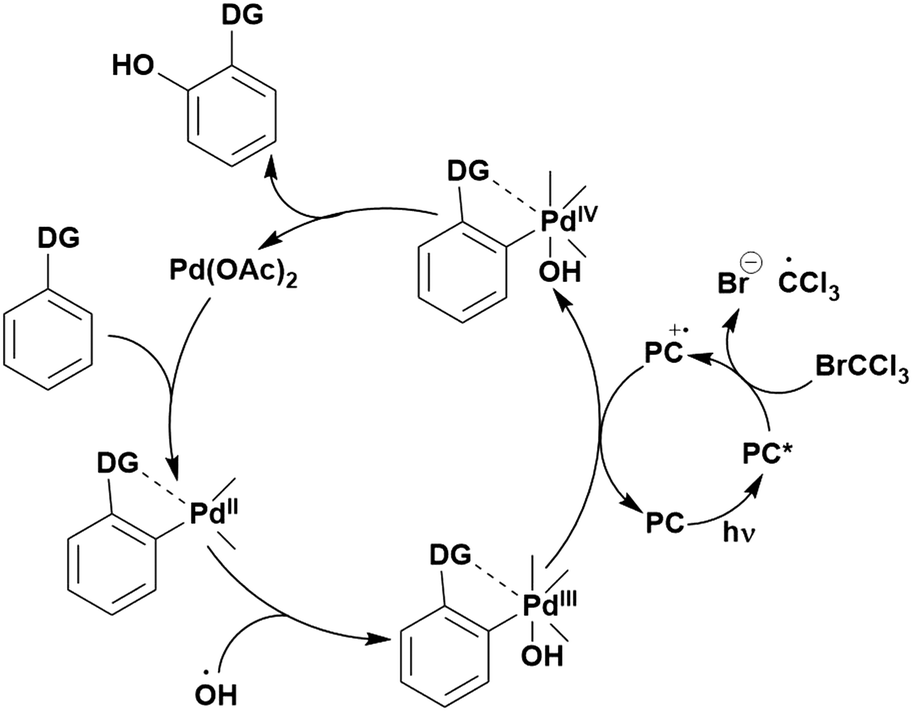 | ||
| Fig. 105 Proposed mechanism for the hydroxylation of arenes using photoredox and palladium dual catalysis where DG is pyridine or benzothiazole. | ||
Despite having compatible redox potentials, 4CzIPN was an unsuitable photocatalyst for the cis-selective decarboxylative alkenylation of aliphatic carboxylic acids (Fig. 102e), giving 0% yield.270 This is hypothesized to be due to its incompatibility with the palladium catalyst under the required reaction conditions, although no further investigation was undertaken. Instead, [Ir(dF(CF3)ppy)(dtbbpy)]PF6 was found to be the best photocatalyst, giving an 80% yield.
Photoredox/cobalt dual catalysis
A third example of metallaphotocatalysis employs a cobalt co-catalyst, examples of which are the cross-dimerization of two alkynes to form 1,3-enynes (Fig. 106a),271 the isomerisation of alkenes (Fig. 106b)272 the olefination of alkyl halides (Fig. 106c),186 the allylic alkylation (Fig. 106d),273 the desulfonylative allylic substitution (Fig. 106e)274 and the decarboxylation of α-ketoacids to allylic ketones (Fig. 106f).275 The optimized conditions identified for the former reaction used a combination of Co(BF4)2·6H2O with 1,3-bis(diphenylphosphino)propane (dppp) as an ancillary ligand and 4CzIPN as the photocatalyst. An oxidative quenching mechanism is proposed (Fig. 107) whereby two equivalents of the excited photocatalyst reduces the cobalt(II) catalyst to a Co(0) species, with DIPEA (N,N-Diisopropylethylamine) added as a putative sacrificial electron donor to regenerate the photocatalyst. A third equivalent of photocatalyst is then invoked by the authors to access a Co(I)/Co(III) catalytic cycle. Deuterium labelling experiments conducted indicated the likelihood of the oxidative addition and migratory insertion steps in the cobalt catalytic cycle. Only 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 were considered as photocatalysts, with respective yields of 71% and 28%. The difference in yields is possibly linked to the greater reducing capacity of the excited state for 4CzIPN (Eox* = −1.04 V and −0.89 V for 4CzIPN and the iridium PC, respectively), making the reduction of the Co(II) complex more facile, particularly since an analogous Co complex Co(dppp)Br2 exhibited a reduction potential of Ered ≈ −0.6 V.276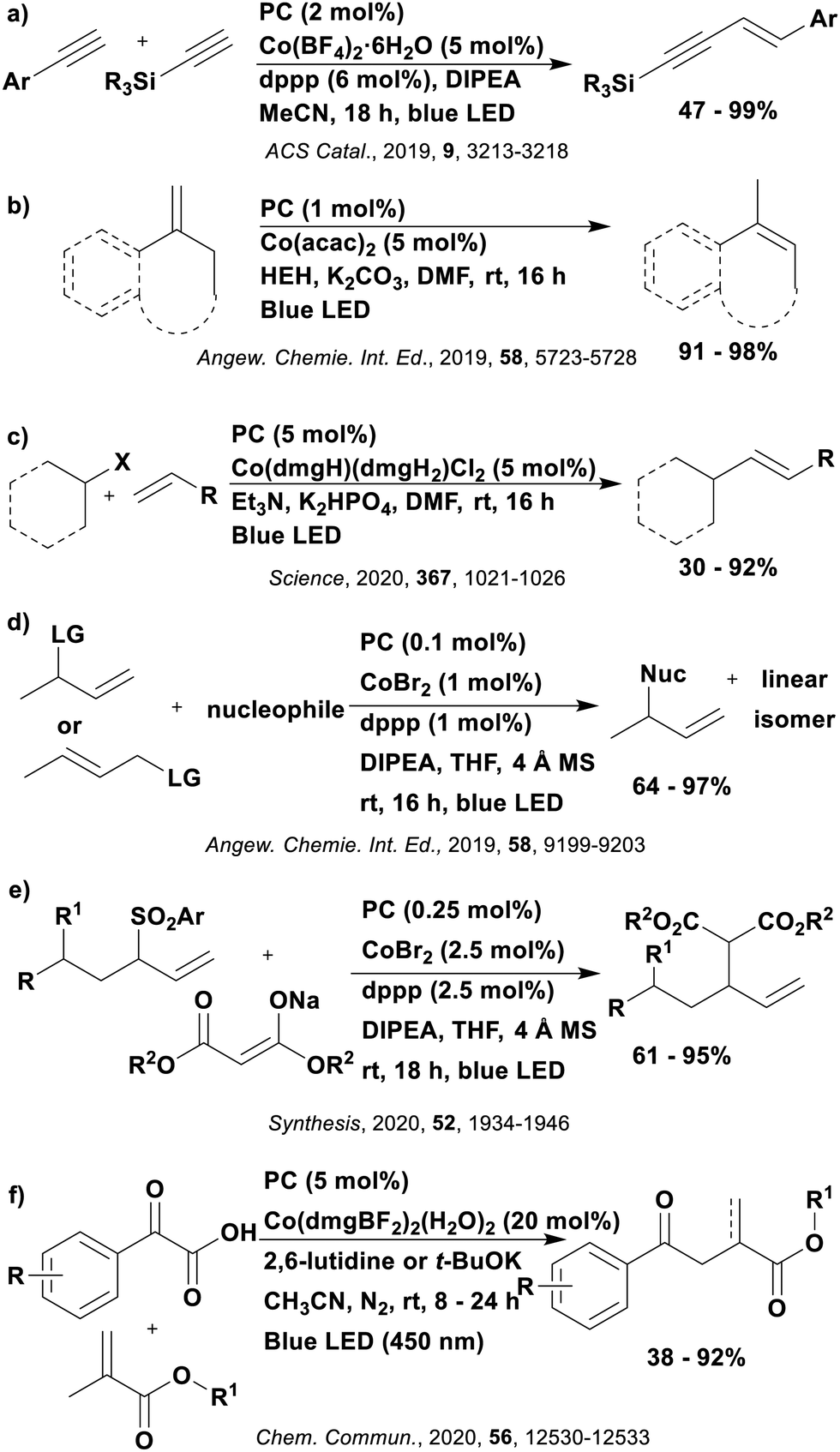 | ||
| Fig. 106 Reaction schemes for the Co/PC dual catalysis of (a) cross-dimerisation of two alkynes (b) isomerisation of alkenes, (c) olefination of alkyl halides, (d) allylic alkylation, (e) desulfonylative allylic substitution and (f) decarboxylation of α-ketoacids to allylic ketones. | ||
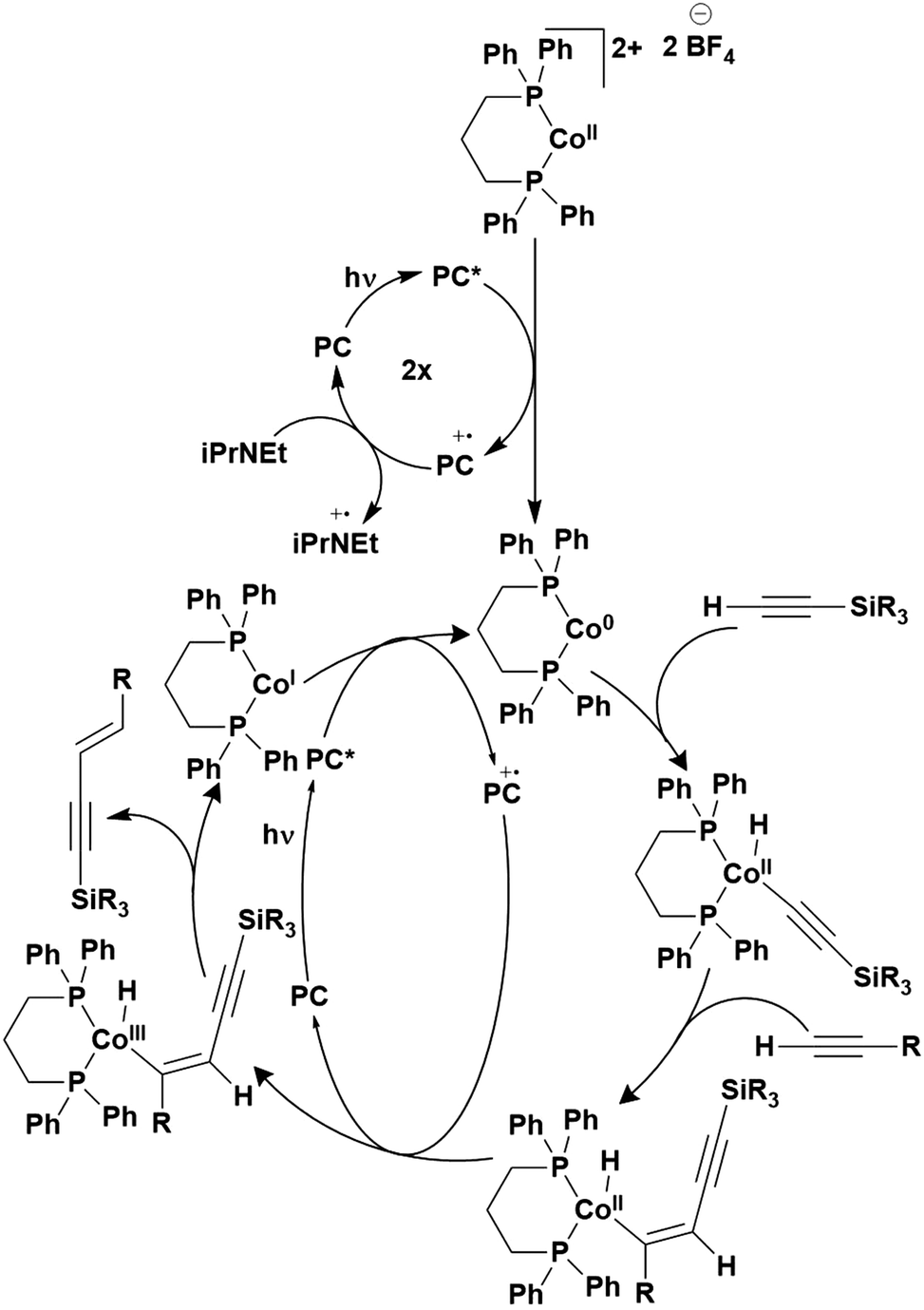 | ||
| Fig. 107 Proposed mechanism for the metallaphotocatalysis reaction involving a cobalt catalyst in the cross-dimerization of two alkynes. | ||
Dual catalysis involving 4CzIPN and Co(acac)2 has been reported for the isomerisation of alkenes (Fig. 106b). The presence of 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) or (oxydi-2,1-phenylene)bis(diphenylphosphine) (DPEphos) as a ligand is used to control the regio- and stereoselectivity of the reaction.272 A Hantzsch ester is employed as a sacrificial reductant of the excited PC, and the resulting reduced PC then catalyses the reduction of the Co(II) catalyst to a Co(I) species (Fig. 108). The reaction was shown to proceed in the absence of 4CzIPN, indicating direct reduction of Co(II) by the photoexcited Hantzsch ester is likely to occur as an alternative pathway. Photocatalysts other than 4CzIPN were not considered in this reaction; noteworthy is that 4CzIPN provided very high yields of 91–98% yield in the five substrates tested.
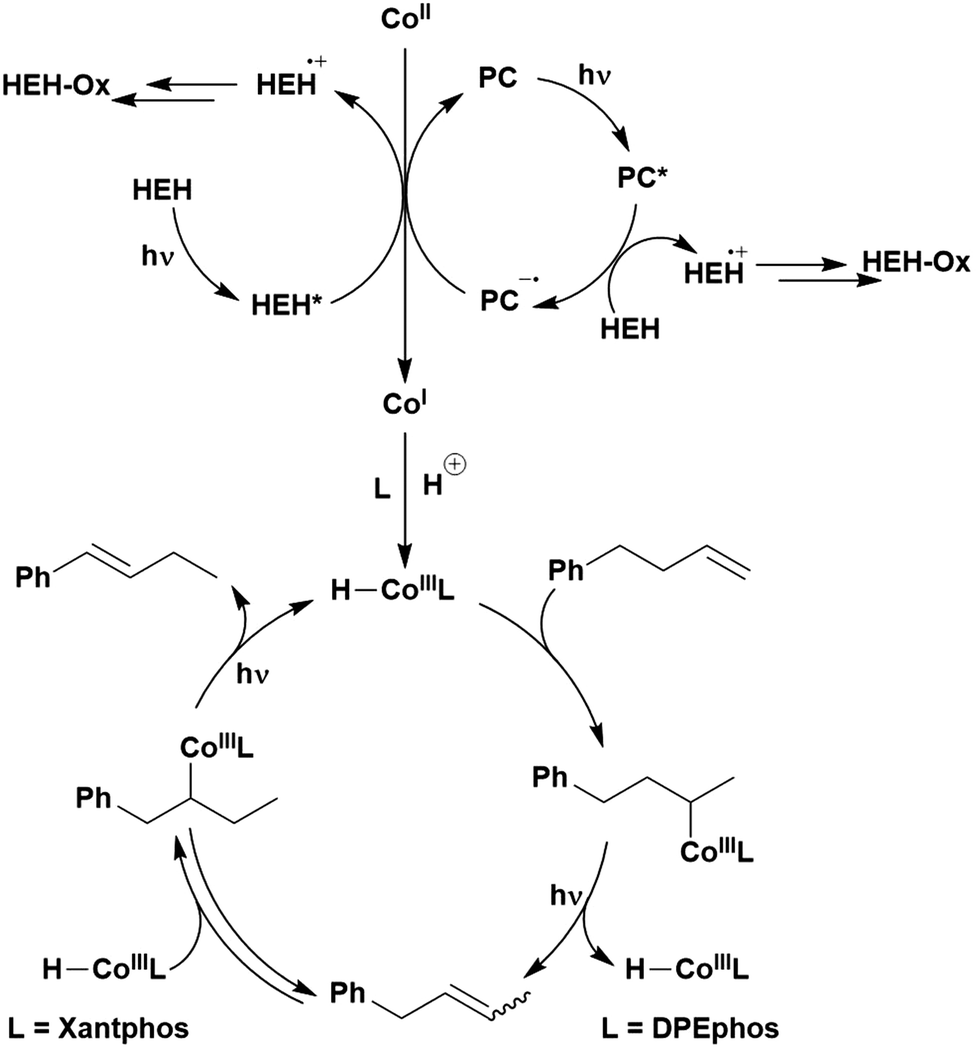 | ||
| Fig. 108 Plausible mechanism for the dual catalytic isomerisation of alkenes. | ||
The combination of a PC with Co(dmgH)(dmgH2)Cl2 (dmg = dimethylglyoximate) has been shown to promote the olefination of alkyl halides through the creative use of amino radicals as halogen atom transfer (XAT) agents. (Fig. 106c).186 The excited PC is reductively quenched by triethylamine (TEA), forming the amino radical cation, which abstracts a halogen from the alkyl halide (Fig. 109). Closure of the photocatalytic cycle involves the reduced photocatalyst reducing a Co(III) species. A quantum yield of 0.01 suggests this is not a radical chain process. Leonari et al. have utilised this technique of XAT agents in combination with hydrogen atom transfer (HAT) catalysts for deuteration, alkylation and allylation of aryl and alkyl halides; however, in the olefination of alkyl halides, a Co complex is also used as the second catalyst. Photocatalyst screening was conducted in the alkylation reaction and applied throughout the study; hence, only 4CzIPN was considered as the photocatalyst for this olefination reaction. For more information about the study, refer to section on dual catalysis with HAT catalysts.
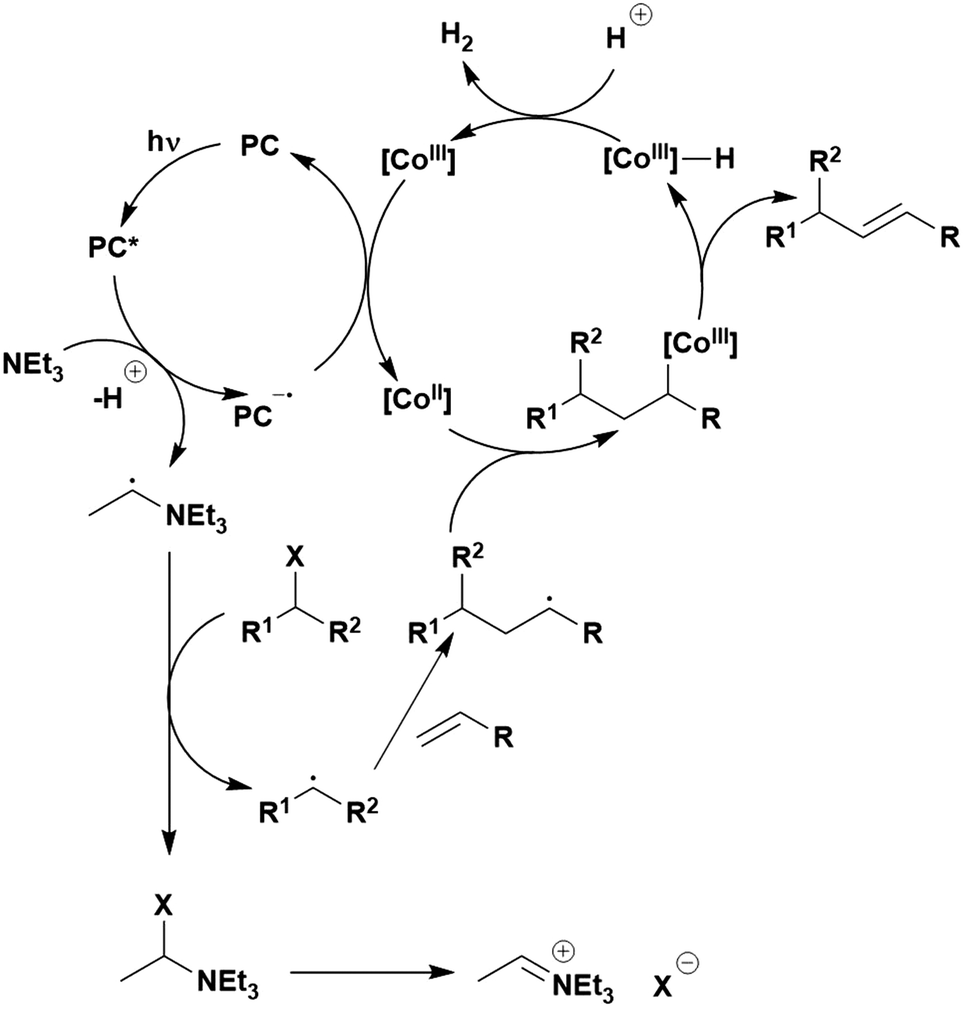 | ||
| Fig. 109 Suggested mechanism for the olefination of alky halides using dual PC and Co catalysis with amino radicals as XAT agents. | ||
In the allylic substitution reactions (Fig. 106d and e), CoBr2 in the presence of the ancillary ligand dppp is used in combination with 4CzIPN.273,274 As the desulfonylative substitution (Fig. 106e) is a subsequent study by the same group as the allylic alkylation reaction (Fig. 106d), the same optimized conditions and mechanistic suggestions proposed in the initial study were used. Thus, only in the allylic alkylation from Fig. 106d will be discussed. The basic mechanistic proposal involves reductive quenching of the excited PC by the additive, DIPEA, with the reduced PC then serving to reduce the Co(II) species to the catalytically active Co(I) complex. Oxidative addition of the allylic electrophile forms a π-allyl Co(III) intermediate, which upon attack from the nucleophile, releases the final product and regenerates the Co(I) species. A quantum yield of 0.13 was obtained, suggesting that this reaction does not proceed via a radical chain process.
[Ir(dF(CF3)ppy)2(dtbbpy)]PF6, eosin Y and 4CzIPN were tested in the PC screen, providing yields of 41%, 59% and 64%, respectively, hence 4CzIPN was selected for further optimization. As the most photooxidising PC, this may be why 4CzIPN performed the best (Ered* = 1.35 V, 0.83 V and 1.21 V for 4CzIPN, eosin Y and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively); however, the trends in Ered* do not explain the relative product yields. Eosin Y has the lowest Ered* but provided comparable yields to 4CzIPN and outperformed the Ir PC.
Finally, for the decarboxylation of α-ketoacids to allylic ketones (Fig. 106f),275 the proposed mechanism is similar to that shown in Fig. 109. Reductive quenching of the excited PC by the α-ketoacid carboxylate yields a benzoyl radical after decarboxylation. This radical then adds to the methacrylate derivative with the formed alkyl radical trapped by the Co(II) catalyst. Cleavage of Co-C bonds and β-H elimination allows release of the final product. Closure of both catalytic cycles occurs as in Fig. 109. Stern–Volmer quenching experiments indicated that the α-ketoacid could only quench the luminescence of the PC in the presence of base. Using TEMPO inhibited product formation and an adduct with the benzoyl radical was detected, providing evidence of the presence of this radical species. Of the PCs tested, only 4CzIPN yielded any product (85%) while the use of [Ru(bpy)3](PF6)2 and [Mes-Acr]ClO4 yielded no product. Oxidation of phenylglyoxylic carboxylate is moderately challenging (Eox = 0.98 V vs. SCE), which may explain why the ruthenium PC was unsuccessful in this reaction (Ered* = 0.77 V and 1.35 V for [Ru(bpy)3](PF6)2 and 4CzIPN, respectively). Despite [Mes-Acr]ClO4 being capable of this SET (Ered* = 2.06 V), the limited reducing ability in the ground state may act as a barrier to it being effective in this reaction (Ered = −0.57 V and −1.21 V for [Mes-Acr]ClO4 and 4CzIPN, respectively).
Photoredox/titanium dual catalysis
The fourth example of metallaphotocatalysis involves the use of a titanium complex as the additional catalyst. This can be applied to the spirocyclisation of epoxides (Fig. 110a),277 the Barbier allylation of aldehydes and ketones (Fig. 110b)278 and the allylation of carbonyls with 1,3-butadiene (Fig. 110c)279 all of which use Cp2TiCl2 as the titanium catalyst of choice. In all reactions, the plausible mechanism involves reduction of the Cp2TiCl2 catalyst to a Ti(III) species by the excited photocatalyst. Closing of the photocatalytic cycle is obtained by oxidation of a Hantzsch ester (HE) by the oxidised photocatalyst. For the spirocyclisation of epoxides, the proposed mechanism is depicted in Fig. 111. Reductive opening of the epoxide is stimulated by the Ti(III) complex, generating the Ti(IV) complex, whereby the carbon centred radical adds intramolecularly to the alkyne functionality, producing the spirocyclic structure. The resultant vinyl radical abstracts a proton from the oxidised HE and the product is released from the Ti(IV) complex, completing the titanium catalytic cycle.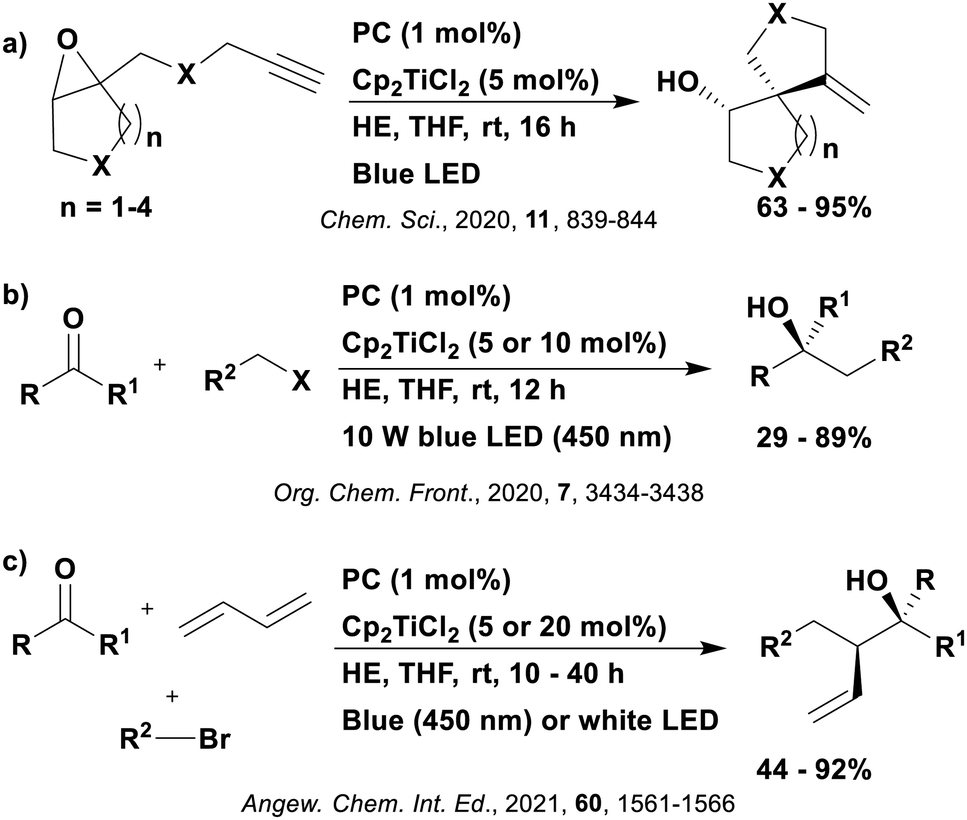 | ||
| Fig. 110 Reaction scheme for the (a) spirocyclisation of epoxides, (b) Barbier allylation of aldehydes and ketones and (c) the allylation of carbonyls with 1,3-butadiene. | ||
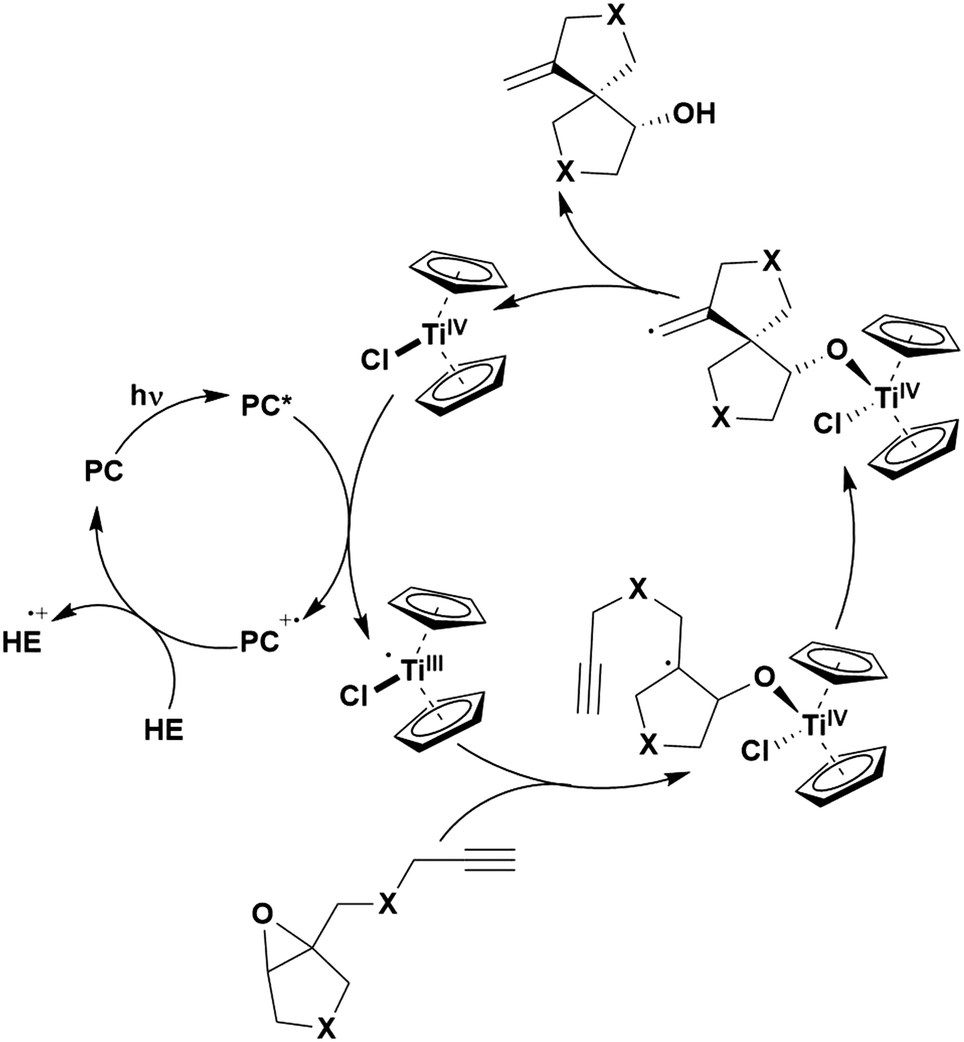 | ||
| Fig. 111 Possible mechanism for the spirocyclisation of epoxides using metallaphotocatalysis, where HE is the Hanztsch ester. | ||
Stern–Volmer experiments conducted in all cases indicated that both the titanium catalyst and the HE can quench the emission of the excited photocatalyst, suggesting the possibility of both an oxidative and reductive quenching mechanism, although Shi et al. proposed the oxidative quenching mechanism showing in Fig. 111 since quenching with the titanium catalyst occurs faster (kq = 3.9 × 109 L mol−1 s−1 and 4.5 × 107 L mol−1 s−1 for Cp2TiCl2 and HE, respectively, with [Ir(ppy)2(dtbbpy)]PF6). For the Barbier allylation reaction, allyl bromide could also quench the PC, although at a slower rate than Cp2TiCl2 (kq = 3.3 × 109 L mol−1 s−1 and 5.8 × 108 L mol−1 s−1 for Cp2TiCl2 and allyl bromide, respectively, with 4CzIPN).278 Again, in the allylation with 1,3-butadiene, BrCF2CO2Et could also act as a quencher but at a much slower rate (kq = 3.65 × 109 L mol−1 s−1 and 3.05 × 107 L mol−1 s−1 for Cp2TiCl2 and BrCF2CO2Et, respectively, with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6.
Quantum yields of 0.016 and 0.08 were obtained for the Barbier allylation reaction and the allylation reaction with 1,3-butadiene, respectively, which suggest that a radical-chain mechanism is unlikely in both reactions.
Photocatalysts including 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 all achieved the same high yields in the spirocyclisation of epoxides (94%, 95% and 94%, respectively) and in the allylation with 1,3-butadiene (85%, 86% and 89%, respectively) while [Ru(bpy)3](PF6)2 proved much less successful (24% and 0% yield, in the two reactions, respectively).277,279 Similarly, in the Barbier allylation, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 behaved comparably (92% and 86%, respectively), while [Ru(bpy)3](PF6)2 was unsuccessful (0% yield). Reduction of the Cp2TiCl species is fairly facile (Ered = −0.22 V) so in terms of thermodynamics, should be feasible for all photocatalysts (Eox* = −1.04 V, −0.96 V, −0.89 V and −0.81 V and for 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2 respectively). Hence, maybe the difference in yield is more related to regeneration of the photocatalyst (Eox = 1.52 V, 1.21 V, 1.69 V and 1.29 V for 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2), respectively). Alternatively, since the possibility of a reductive quenching mechanism has already been suggested, this could be a reason for low yields for some photocatalysts.
Photoredox/iron catalysis
Two examples exist depicting the use of 4CzIPN in combination with an iron co-catalyst: the acylarylation of unactivated alkenes towards the synthesis of 3-(α-acyl) indolines (Fig. 112a);280 and the reduction of CO2 (Fig. 112b).281 In the former, FeCl2 was used as the iron catalyst, which was proposed to reductively quench the excited PC (Fig. 113). The photocatalytic cycle is closed by SET from the reduced PC to di-tert-butyl peroxide (DTBP), forming a tert-butanolate anion and a tert-butoxyl radical. The latter abstracts a hydrogen from the aldehyde reagent, producing an acyl radical that can add to the alkene. From this adduct, rapid intramolecular radical trapping by the N-aryl moiety occurs, forming the dearomatized aryl radical, which is then oxidised by the Fe(III) species, closing the iron catalytic cycle. Finally, a deprotonation step occurs to yield the 3-(α-acyl) indoline. Addition of TEMPO prevented the formation of the final product and instead, and adduct with the acyl radical formed, demonstrating its presence as an active species in the reaction. A primary kinetic isotope effect value of 1.3 implies that C–H bond cleavage of the N-aryl moiety is unlikely to be involved in the rate determining step. A quantum yield of 0.24 was obtained; however, the authors suggested a radical chain process may exist.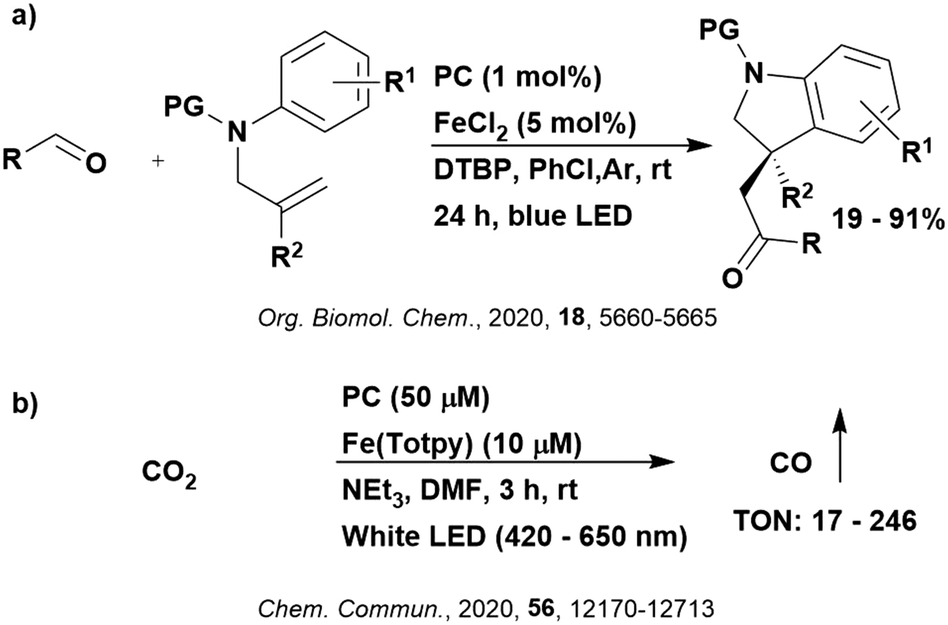 | ||
| Fig. 112 Reaction scheme for the dual photoredox/Fe catalysis for the acylarylation of unactivated alkenes. PG = protecting group. | ||
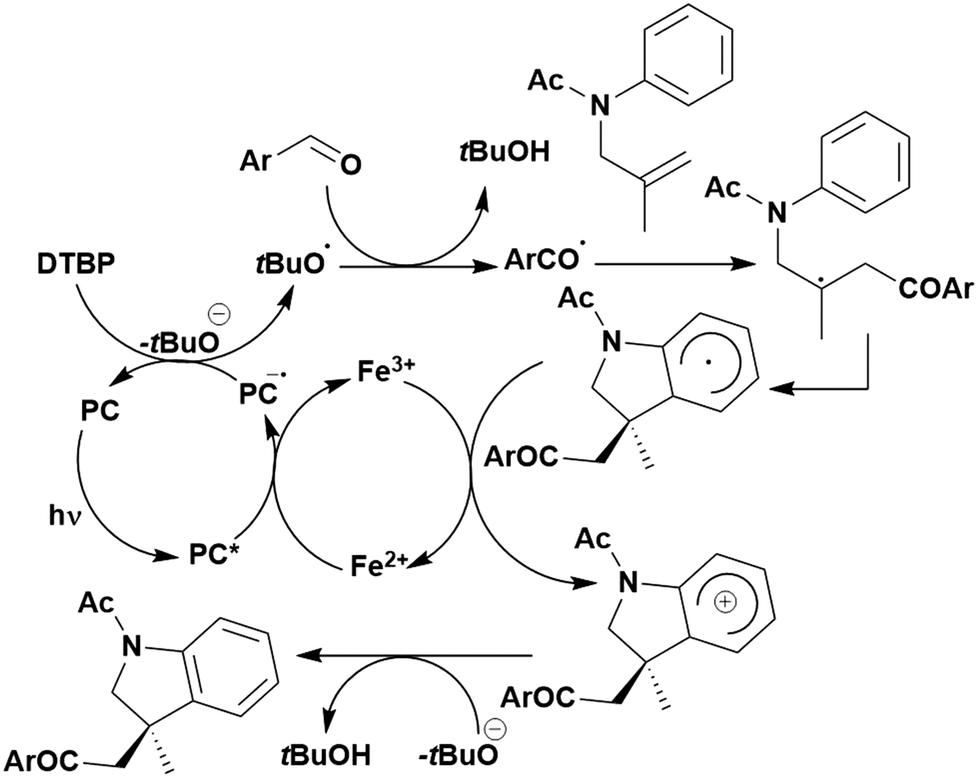 | ||
| Fig. 113 Viable mechanism for the dual photoredox/Fe catalysis for the acylarylation of unactivated alkenes. | ||
Of the six PCs screened, only 4CzIPN and fac-Ir(ppy)3 provided product (62% and 52%, respectively), with the likes of eosin Y and [Mes-Acr]ClO4 giving no/trace product. These results may be linked to the ground state reduction potential of the PC (Ered = −1.21 V, −2.19 V, −1.06 V and −0.57 V for 4CzIPN, fac-Ir(ppy)3, eosin Y and [Mes-Acr]ClO4, respectively). Additionally, the stronger photooxidising capacity of 4CzIPN in comparison to fac-Ir(ppy)3 may contribute to the difference in yields obtained for these two (Ered* = 1.35 V and 0.31 V, respectively).
A terpyridine–Fe(III) complex was used as the metal catalyst for the reduction of CO2 (Fig. 112b).281 Reductive quenching of the excited PC occurs by the sacrificial electron donor NEt3, as confirmed by Stern–Volmer quenching experiments. The reduced PC can then reduce the Fe(III) catalyst to Fe(II), with a second reduction step, proposed to occur from the reduced PC, invoked to generate a Fe(I) species (Fig. 114). The Fe(I) complex undergoes disproportionation to yield both an Fe(II) complex and the active Fe(0) species. Coordination of CO2 to this Fe(0) species is followed by protonation, the cleavage of a C–O bond and the release of water. The final CO is liberated by replacement with a chloride ion or solvent to close the iron catalytic cycle. The evolution of two absorption bands at 620 nm and 798 nm upon irradiation implied the formation of the Fe(I) species. 4CzIPN proved to be much more successful in this reaction than the other PCs investigated, providing a turnover number (TON) of 244 while the next best PC, [Ru(bpy)3]2+, could manage only 17. The higher TON obtained by 4CzIPN compared to [Ru(bpy)3]2+ (Ered* = 1.35 V and 0.77 V, respectively) may be related to its greater driving force for the photooxidation of NEt3 (Eox = 0.77 V vs. SCE).
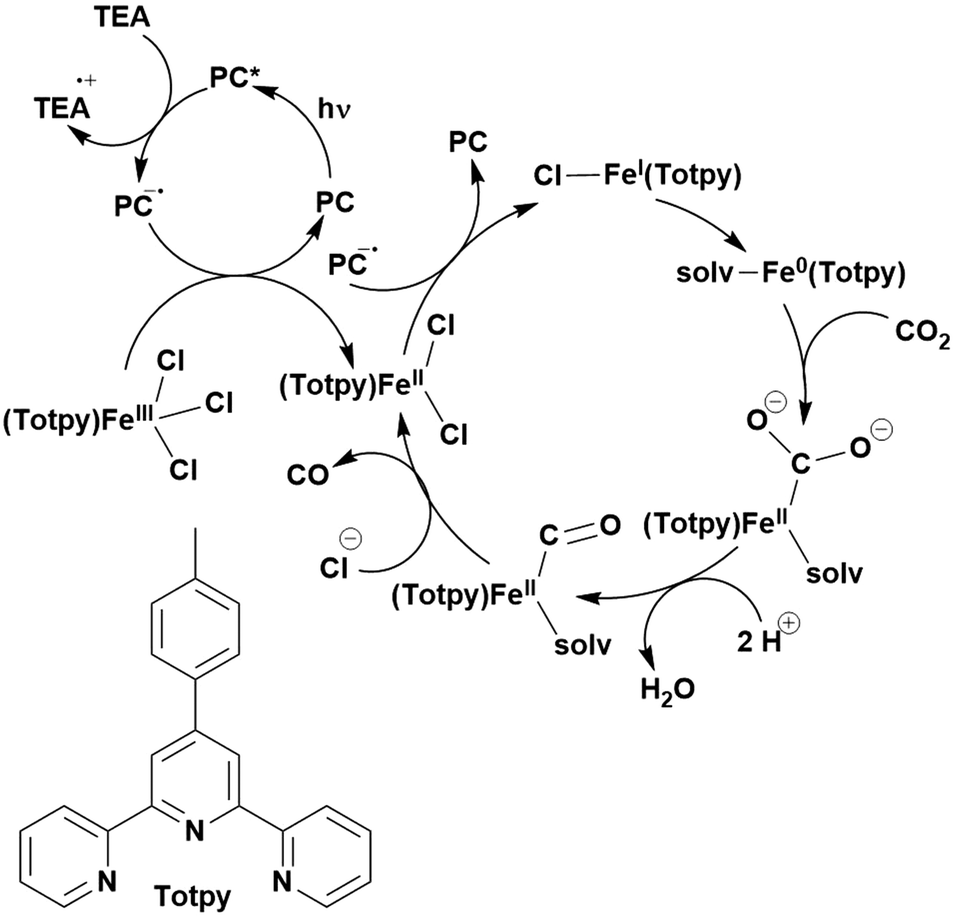 | ||
| Fig. 114 Putative mechanism for CO2 reduction using dual photoredox/Fe catalysis. | ||
Photoredox/chromium catalysis
The penultimate example of metallaphotocatalysis involves the use of chromium chloride as the metal co-catalyst in the dialkylation of 1,3-dienes (Fig. 115).282 Reductive quenching of the excited photocatalyst by a Hantzsch ester is proposed to occur, forming an alkyl radical and pyridinium (py) cation pyH+ (Fig. 116). The alkyl radical can then add to the diene, with the resultant radical being trapped by the Cr(II) catalyst. A chromium alkoxide is then proposed to form via a six-membered Zimmerman–Traxler transition state upon addition of the aldehyde.283,284 Release of the product is realised upon hydrolysis by the pyridinium cation, whereby closure of both catalytic cycles occurs through SET from the reduced photocatalyst to the Cr(III) complex. Support of this putative mechanism is obtained from Stern–Volmer quenching experiments, which reveal that only the Hantzsch ester can quench the excited PC. Addition of TEMPO inhibits the reaction, with an alkyl TEMPO adduct being isolated, indicating the formation of an alkyl radical in the process. The quantum yield of the reaction was determined to be 0.09 suggesting this is not a radical chain process. Only four photocatalysts were considered, with 4CzIPN greatly outperforming its competitors (76% isolated yield comparison to 35%, 25% and 5% GC-FID yields obtained with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2, respectively, under the same conditions). These yields correlate with the photooxidising ability of the PC (Ered* = 1.35 V, 1.21 V, 0.66 V and 0.77 V for 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ir(ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2, respectively). This is reasonable since the oxidation potential of the Hantzsch ester is Eox = 1.10 V.285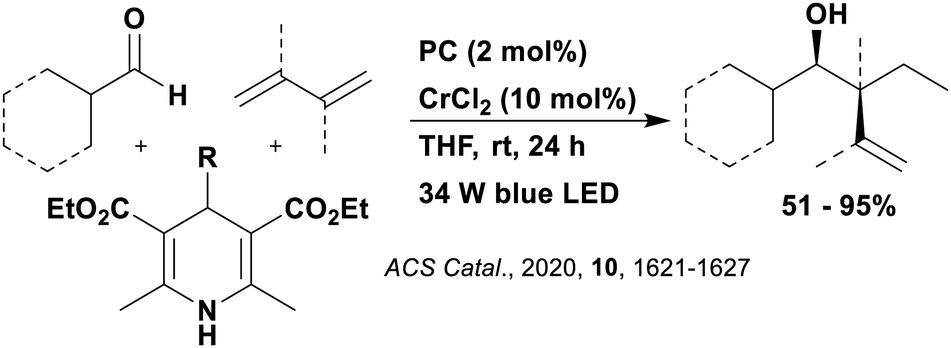 | ||
| Fig. 115 Reaction scheme for the dialkylation of 1,3-dienes using metallaphotocatalysis with chromium chloride. | ||
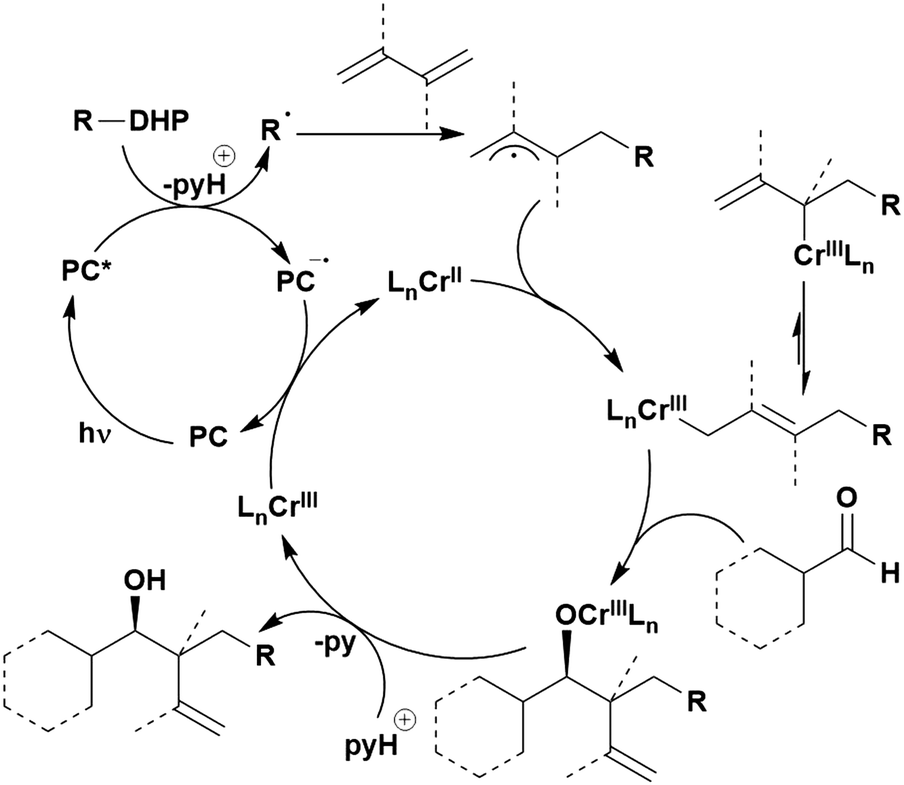 | ||
| Fig. 116 Putative mechanism for the dialkylation of 1,3-dienes using metallaphotocatalysis with chromium chloride. | ||
Photoredox/copper catalysis
The final example of metallaphotocatalysis utilises a copper complex co-catalyst, for example in the decarboxylative radical sulfonylation reaction to form C(sp3)-S bonds (Fig. 117a)286 or the decarboxylative hydroalkylation of alkynes (Fig. 117b).287 In the former reaction, the excited PC is proposed to reduce the N-hydroxyphthalimide, which readily forms an alkyl radical (Fig. 118). Simultaneously, the sulfinate anion is coordinated to the Cu(II) catalyst to produce the Cu(II)-SO2R intermediate, which is intercepted by the alkyl radical to yield the sulfone product and a Cu(I) species. Oxidation of the Cu(I) complex by the oxidised PC closes both catalytic cycles. Evidence for this mechanism is provided in the form of Stern–Volmer quenching experiments, which showed N-hydroxyphthtalimide to be the most efficient quencher of the excited PC. Radical trapping experiments, with 1,1-diphenylethylene for example, provided proof of the presence of alkyl radicals while reaction of Cu(II) p-toluenesulfinate with an ethyl radical precursor yielded an ethylsulfone as expected, suggesting the Cu(II) species does act to assist RSO2 group transfer. When optimising reaction conditions with primary alkyl acids, [Cu(NCMe)4]BF4 was first used as the copper source, which provided 59% yield with 4CzIPN as the PC. All other PCs considered were transition metal complexes and under these conditions these managed between 8–39% yield, with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 providing the highest yield. The greater yield provided by 4CzIPN seems linked to its photoreducing ability (Eox* = −1.04 V and −0.89 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively).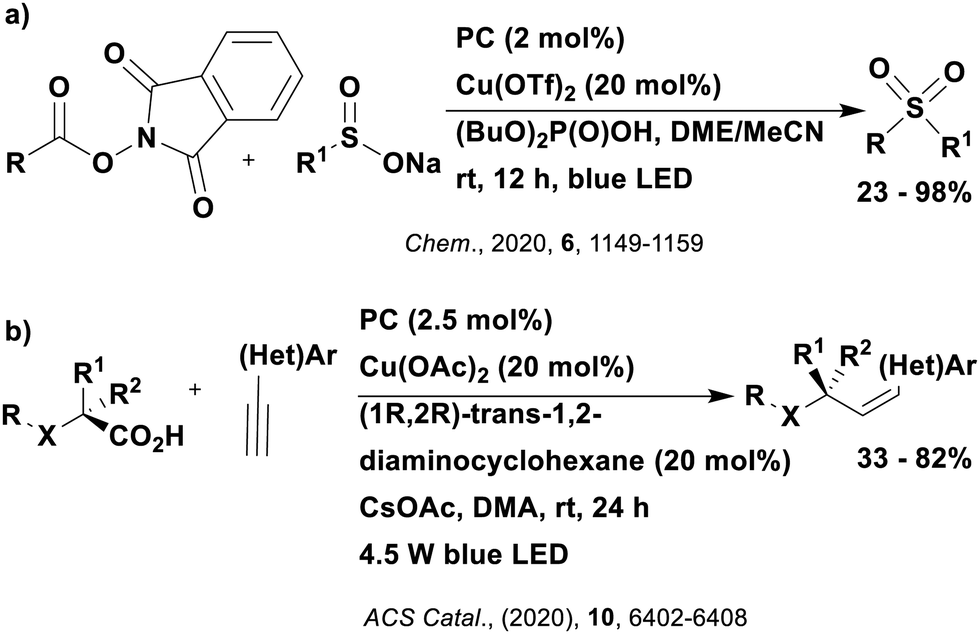 | ||
| Fig. 117 Reaction scheme for photoredox/Cu dual catalysis where (a) is the decarboxylative radical sulfonylation reaction and (b) is the decarboxylative hydroalkylation of alkynes. | ||
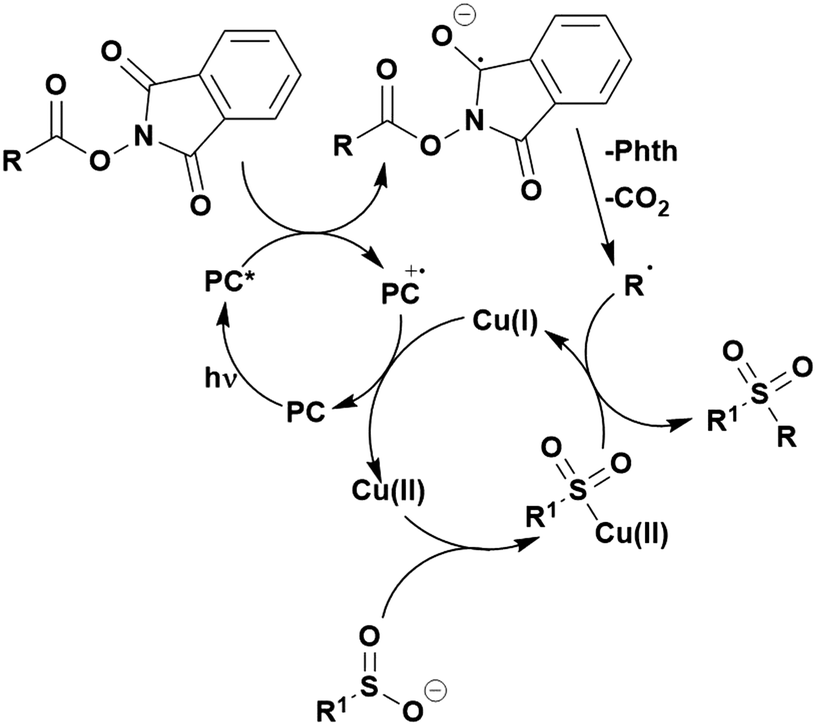 | ||
| Fig. 118 Putative mechanism for the photoredox/Cu catalysed decarboxylative radical sulfonylation reaction. | ||
The proposed mechanism for the dual photoredox/Cu catalysed decarboxylative hydroalkylation of alkynes (Fig. 117b) is shown in Fig. 119. In this case, photocatalytic cycles involving both electron and energy transfer are proposed. Firstly, the excited PC is proposed to be reductively quenched by both α-amino and α-oxy carboxylates, as confirmed by Stern–Volmer quenching experiments. The Cu(II) catalyst generated in situ can also reductively quench the excited PC, but with a slower quenching rate, thus it is undetermined whether the active Cu(I) species is generated from SET (either from the PC* or from PC˙−) or from a disproportionation reaction. Once the active Cu(I) species is formed, in the presence of base the alkyne can coordinate to the metal. The resultant adduct is photoexcited, which is thought to accelerate attack from the alkyl radical, itself generated from the α-amino carboxylate. The reduced photocatalyst reduces this intermediate to generate a vinyl anion. Closure of the copper catalytic cycle ensues by protonation and proto-demetalation. The authors then propose E/Z isomerisation of the alkene, mediated by energy transfer from the excited PC.
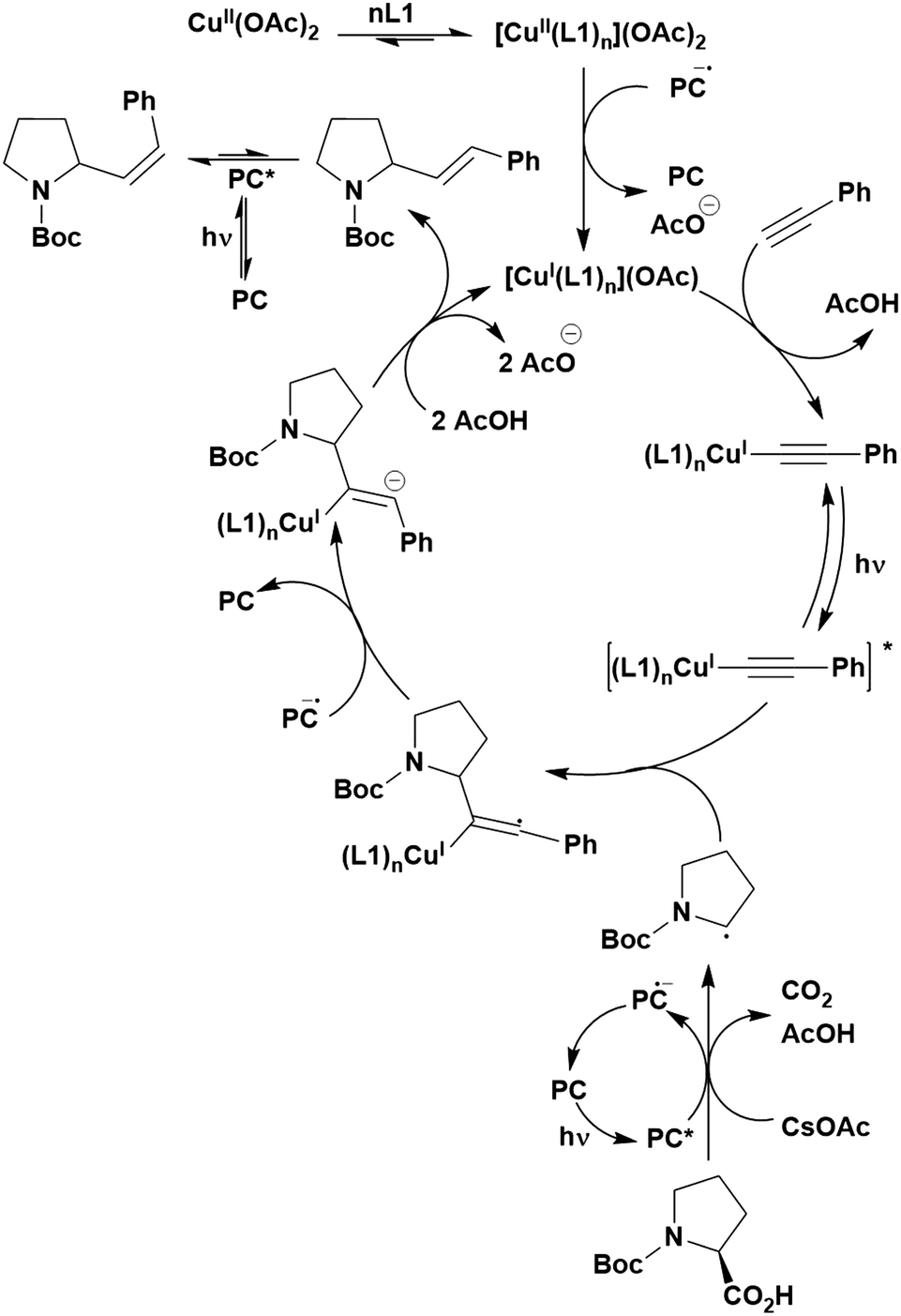 | ||
| Fig. 119 Suggested mechanism for the decarboxylative hydroalkylation of alkynes using dual photoredox/Cu catalysis where L1 is (1R,2R)-trans-1,2-diaminocyclohexane. | ||
When trying to understand the mechanism, Cu(OAc)2 was exchanged with Cu(I) phenylacetylide, and the reaction still proceeded with similar yield and Z-selectivity, suggesting this Cu(I) species is an active intermediate in the reaction. The addition of TEMPO quenches the reaction and instead an adduct is formed between tempo and the alkyl radical generated from the α-amino carboxylate, confirming the presence of the latter. A simple photocatalytic E/Z isomerization reaction of the alkene was undertaken using the PC, which yielded the same Z:E ratio, implying the PC is responsible for the isomerization. High throughput experimentation was used to identify the best conditions, with 4CzIPN being selected as the PC. Comparison of the performance with other PCs is difficult as their relative success is given only in terms of conversion of reactants, whereas the success of 4CzIPN is provided in terms of yield and selectivity of the final product.
Photoredox and HAT catalysis
There is now a wide body of literature that combines photoredox catalysis and a hydrogen atom transfer (HAT) catalyst. 4CzIPN has been demonstrated to be compatible with this dual catalysis mode with a number of HAT catalysts, which are typically thiols or amines (Fig. 120). In the examples reported of this type of dual catalysis, the PC always participates in a reductive quenching cycle, where the excited PC is generally reductively quenched by the substrate or a sacrificial electron donor. Following this step, the substrate abstracts a H atom from the thiol HAT catalyst, generating a thiyl radical, which is reduced to a thiyl anion by the reduced PC (Fig. 121a). Subsequent protonation regenerates the HAT catalyst to close both catalytic cycles. Alternatively, if the HAT catalyst is an amine, then SET from the amine to the excited photocatalyst occurs instead (Fig. 121b).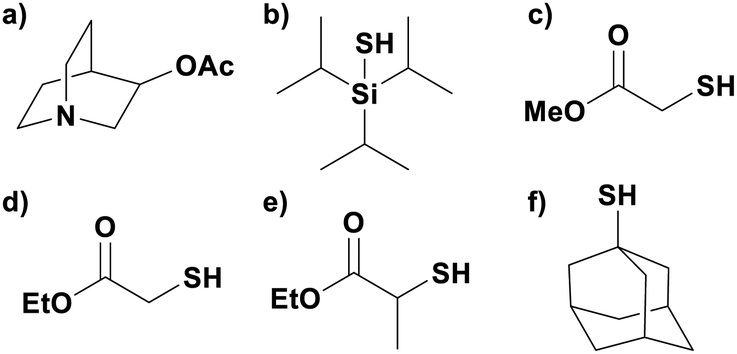 | ||
| Fig. 120 Common hydrogen atom transfer catalysts; (a) quinuclidin-3-yl acetate, (b) triisopropylsilanethiol, (c) methyl thioglycolate, (d) ethyl thioglycolate, (e) ethyl 2-sulfanylpropanoate and (f) adamantane thiol. | ||
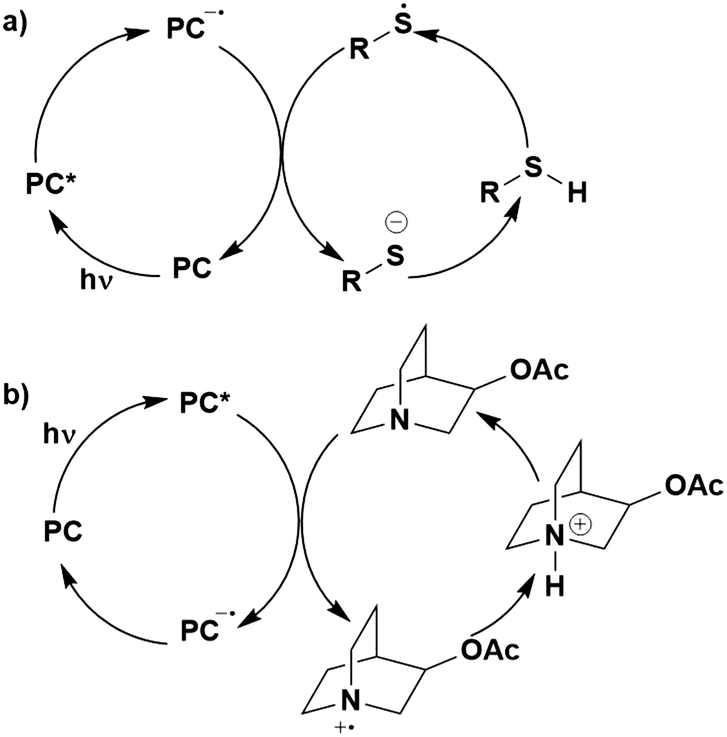 | ||
| Fig. 121 Two generic mechanistic options for the combination of photocatalysis with a HAT catalyst where the HAT catalyst is (a) a thiol and (b) an amine. | ||
Examples of this dual catalysis with 4CzIPN as the PC include the hydrosilylation288 and difunctionalisation289 of alkenes (Fig. 122a and b, respectively) and silylation of quinoxalinones and heteroarenes (Fig. 122c).290 In the former, the HAT catalyst chosen is dependent on the nature of alkene substituent, with quinuclidine-3-yl acetate being used for alkenes with electron-withdrawing groups and triisopropylsilanethiol for alkenes with electron-donating groups. A follow up study by Wu et al. instead focused on the difunctionalisation of alkenes using quinuclidine-3-yl acetate as the HAT catalyst. In both cases, 4CzIPN was shown to be the best photocatalyst (87% yield in the hydrosilylation of electron-deficient alkenes, and 75% in the difunctionalisation of alkenes), with only [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 providing somewhat comparable yields (80% yield in the hydrosilylation of electron deficient alkenes, and 19% in the difunctionalisation of alkenes). This may be related to the photooxidising ability of the photocatalyst (Ered* = 1.35 V and 1.21 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), particularly since the oxidation of quinuclidine-3-yl acetate is quite challenging (Eox = 1.22 V).291
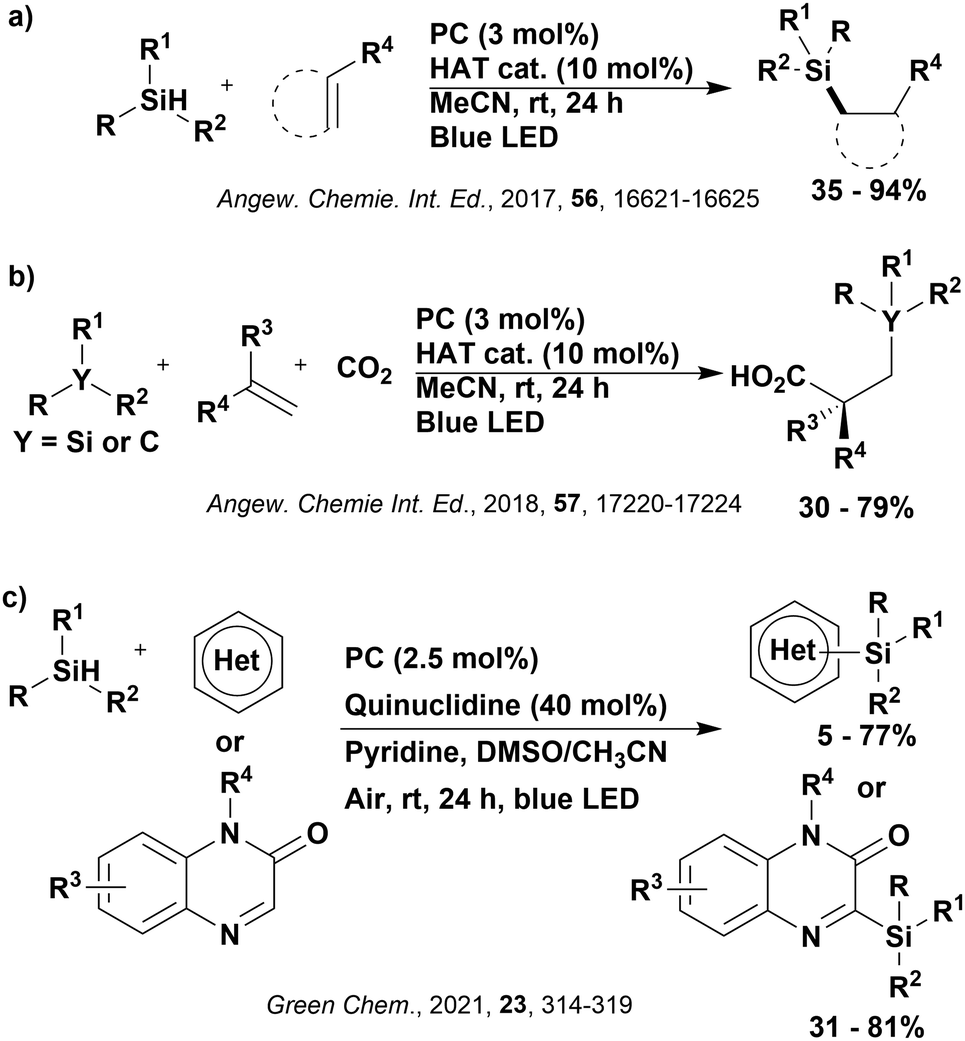 | ||
| Fig. 122 Reaction schemes for the synergistic photoredox and HAT catalysis for (a) hydrosilylation and (b) difunctionalisation of alkenes and (c) silylation of quinoxalinones and heteroarenes. | ||
Both electron and energy transfer of the PC are proposed as steps in the mechanism of the silylation of quinoxalinones and electron-deficient heteroarenes (Fig. 122c and 123).290 Interaction of the PC and the HAT catalyst, quinuclidine, occurs as in the general mechanism shown in Fig. 121b, whereby the HAT catalyst reductively quenches the excited PC. Simultaneously, the excited PC undergoes PEnT to oxygen, generating singlet oxygen, which in turn is reduced by the reduced PC, to yield a superoxyl radical. The silyl radical, generated through HAT with the quinuclidinium radical cation, adds to the quinoxalinone, with the resultant compound undergoing direct HAT to the superoxyl radical, giving the final product. When TEMPO was added to the mixture, the reaction was inhibited and instead an adduct with the silyl radical was formed, confirming the presence of silyl radicals in this reaction. Moreover, upon addition of the singlet oxygen quencher, 1,4-diazabicyclo[2.2.2]octane (DABCO), the reaction was arrested, suggesting 1O2 does indeed play a crucial role in the reaction. A primary kinetic isotope effect value of kH/kD = 1.27 implied cleavage of the Si–H bond was unlikely to be involved in the rate determining step. Finally, Stern–Volmer quenching studies showed that quinuclidine does indeed quench the luminescence of the PC. Only 3 PCs were investigated in this reaction, 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ir(ppy)2(dtbbpy)]PF6 giving yields of 47%, 31% and 0%, respectively. These results correlate with the photooxidising ability of the PC (Ered* = 1.35 V, 1.21 V and 0.66 V, respectively). For the remainder of the study, 4CzIPN was selected as the PC of choice.
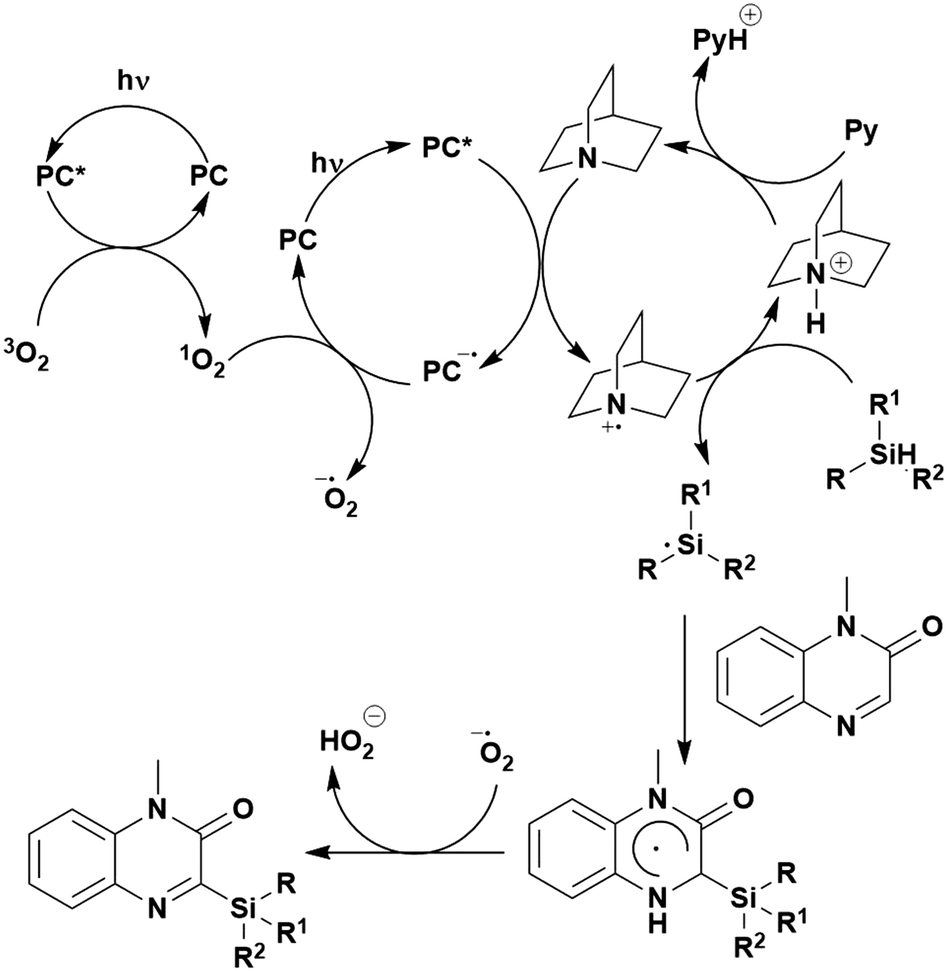 | ||
| Fig. 123 Putative mechanism for the silylation of quinoxalinones. | ||
Alternatively, alkenes can be used as the coupling partner in the α-tertiary amine synthesis of primary amines, using [Bu4N]N3 as the HAT catalyst (Fig. 124).292 The excited PC is proposed to oxidise the azide anion, forming an azidyl radical Fig. 125). This radical then abstracts a proton from the α-C–H position of the primary amine, forming an α-amino radical, which undergoes rapid addition to the Michael acceptor. This in situ-generated α-carboxy-stabilised radical is then reduced by the reduced PC, closing the photocatalytic cycle, with subsequent protonation required to form the final product. This final protonation step is thought to occur from HN3. The low quantum yield of 0.04 is consistent with this reaction not involving a radical chain process. In mechanistic investigations, Stern–Volmer quenching experiments indicated that both the azide anion and cyclohexylamine quenched the emission of the PC; however, the former is a much more efficient quencher (KSV = 2.605 × 103 M−1 and 12.5 M−1 for the azide anion and cyclohexylamine, respectively), supporting the proposed mechanism shown in Fig. 125. Only three PCs were considered in this reaction: 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ir(ppy)2(dtbbpy)]PF6, with all performing similarly well (85%, 83% and 79%, respectively). This difference in yield does correlate with photooxidising ability of the PC (Ered* = 1.35 V, 1.21 V and 0.66 V, for 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ir(ppy)2(dtbbpy)]PF6, respectively), which is needed to oxidise the azide anion (Eox = 0.87 V). However, since the yields obtained are very similar, despite the rather significant differences in Ered*, it is more likely that the photooxidising capacity of the PC is not the yield-determining factor in this reaction. Changes to the HAT catalyst have a much more substantial impact on the yield. Using 4CzIPN as the PC, well-established HAT catalysts such as tri(isopropyl)silanethiolate and quinuclidine provided lower yields in comparison to [Bu4N]N3 (70%, 42% and 85%, respectively).
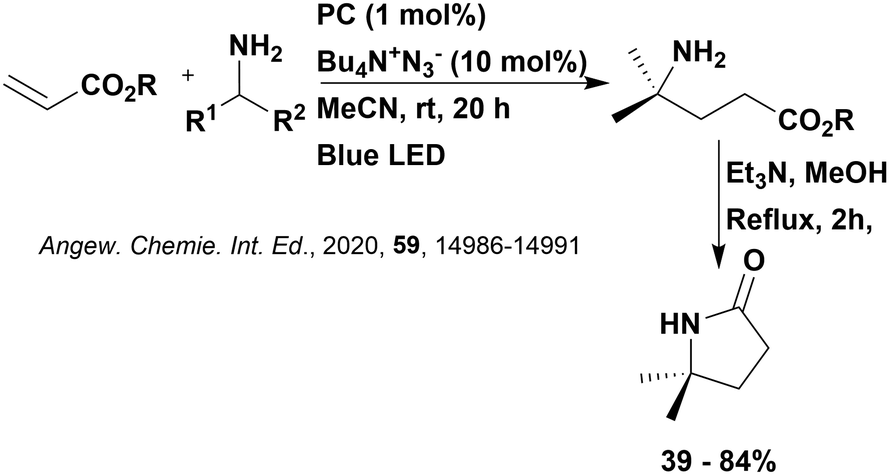 | ||
| Fig. 124 Reaction scheme for the α-C–H alkylation of primary amines. | ||
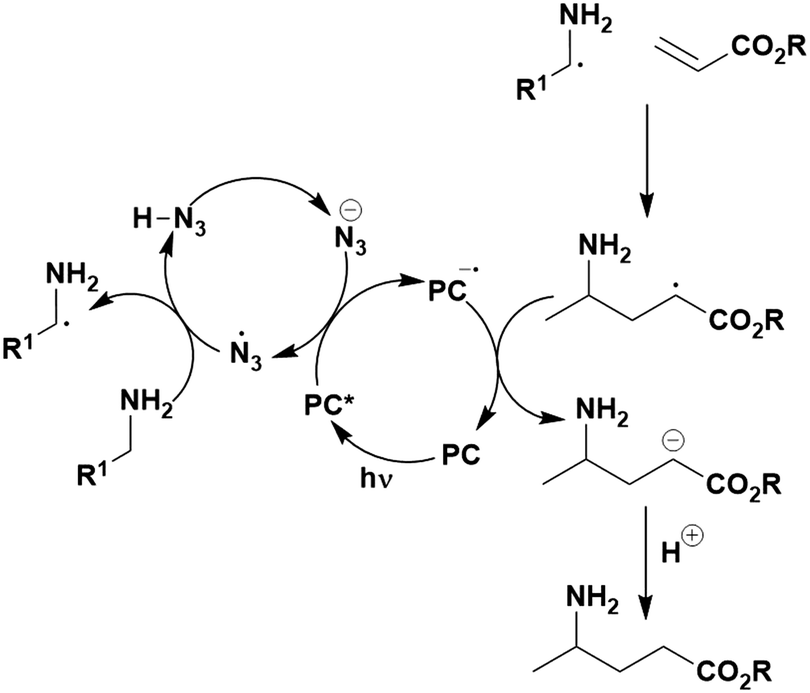 | ||
| Fig. 125 Proposed mechanism for the α-C–H alkylation of primary amines. | ||
Dialkylation of the primary amine is much less efficient, with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 performing comparably to 4CzIPN (7% and 2%). Cresswell et al. suggests the Ir PC exhibits enhanced stability to photobleaching, although there is no corroborating evidence provided to support this contention.
An additional example of functionalisation of alkenes, but with less mechanistic clarity, can be seen in the defluoroborylation of polyfluoroarenes, gem-difluoroalkenes and trifluoromethylalkenes (Fig. 126a–c, respectively).293 A radical-based pathway was confirmed by the use of TEMPO, and Stern–Volmer quenching experiments provided evidence that the thiol HAT catalyst was the species quenching the excited state of the PC, not the fluorinated reagents. Boryl radical intermediates are thought to be present in the mechanism, based on similar work from Taniguchi et al.294 while a study from Weaver et al.295 suggests the possibility of SET to the fluoroarene, generating the fluoroaryl radical anion, may occur. A quantum yield of 0.43 suggests this is not a radical chain process and the kinetic isotope effect observed indicates the HAT of the NHC borane may not contribute to the rate-determining step. Taking all of this into consideration, multiple reaction mechanisms are proposed, although all incorporate reductive quenching of the excited photocatalyst by the HAT catalyst, with the reduced PC being used to reduce one of the fluorinated intermediates. Optimisation involving the polyfluoroarenes indicated both 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as excellent PCs for this reaction. The iridium PC provided the greatest yield of 93% when used in concert with ethyl thioglycolate as the HAT catalyst, while 4CzIPN managed 76% yield under these conditions. Instead, 4CzIPN worked better when used with ethyl 2-sulfanylpropanoate as the HAT catalyst (90% yield) but use of this HAT catalyst with the iridium PC was not considered. For the gem-difluoroalkenes, [Ir(dF(CF3)ppy)2(5,5′-dFbpy)]PF6 was found as the optimal photocatalyst (80% yield in comparison to 50% yield for 4CzIPN). This is likely to be due to the ground state reduction potential (Ered = −1.16 V vs. SCE for [Ir(dF(CF3)ppy)2(5,5′-dFbpy)]PF6).296 Finally, for trifluoromethylalkenes, only 4CzIPN was employed as the photocatalyst, yielding 86% of product during the solvent optimisation studies.
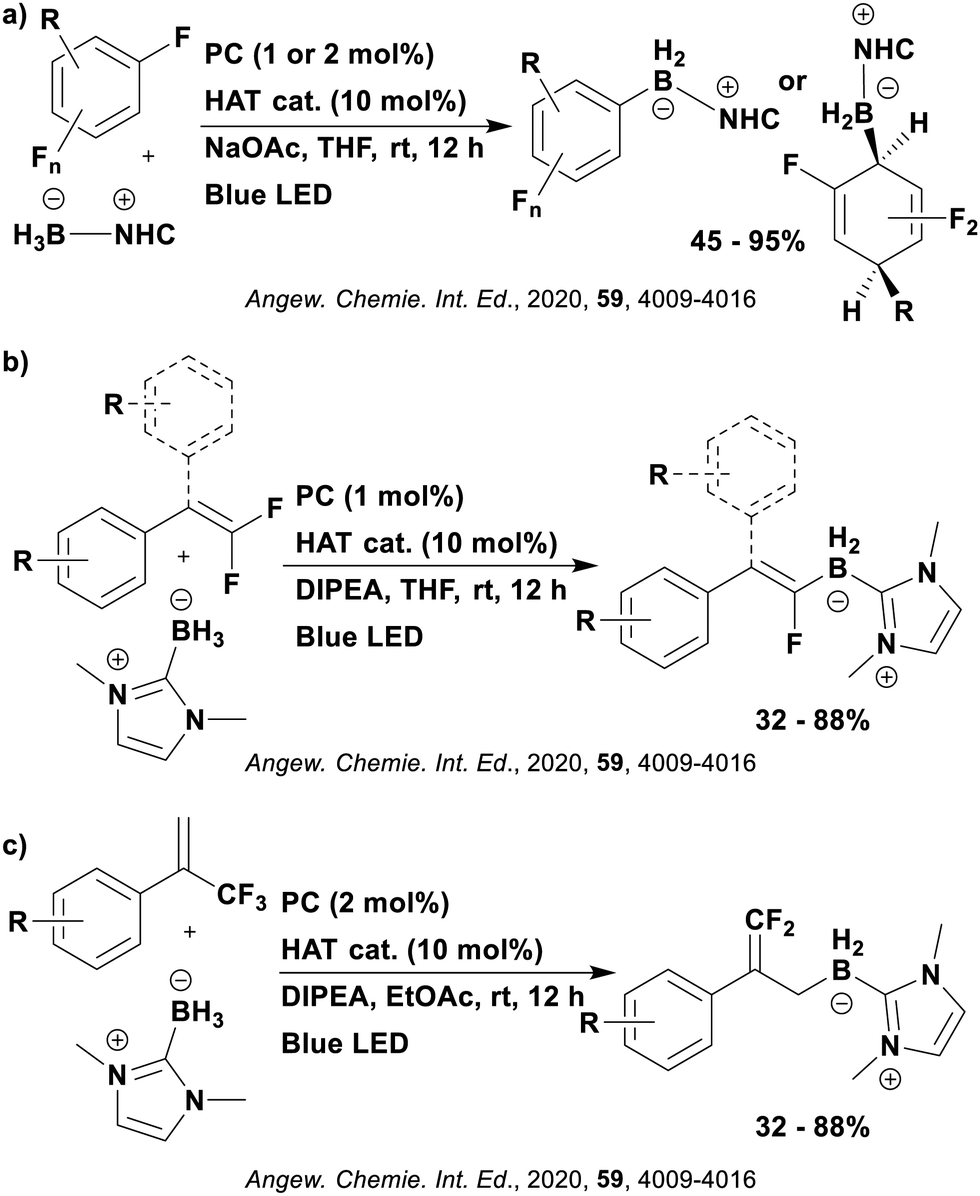 | ||
| Fig. 126 Reaction schemes for the defluoroborylation of (a) polyfluoroarenes, (b) gem-difluoroalkenes and (c) trifluoromethylalkenes. | ||
This form of dual catalysis can also be utilised in the redox neutral fragmentation of 1,2-diol derivatives, a reaction that can be applied to the fragmentation of lignin model compounds (Fig. 127).297 The excited PC is proposed to be reductively quenched by the HAT catalyst, producing a thiyl radical. This thiyl radical then abstracts the α H-atom of the 1,2-diol derivative to generate a ketyl radical, regenerating the HAT catalyst (Fig. 128). Oxidation of this ketyl radical by oxygen produces a ketone, which is reduced to the radical anion by the reduced PC, completing the photocatalytic cycle. Subsequent C–O bond cleavage and protonation generates the final fragmentation products. This mechanism was supported by Stern–Volmer quenching experiments that showed efficient quenching of the PC upon addition of HAT catalyst and base. Iridium PC [Ir(ppy)2(dtbbpy)]PF6 was identified as the best photocatalyst with 1 mol% loading, forming 91% of the ketone and 81% of the alcohol. Using 5 mol% of photocatalyst, eosin Y and perylene yielded no product while 4CzIPN managed 26% of both fragmentation products. Further optimisation of the conditions using 4CzIPN resulted in yields of 80% and 57% for the ketone and alcohol, respectively. König et al. decided to use 4CzIPN throughout the remainder of the study with these optimised conditions as it exhibited similar reaction efficiency but with less sensitivity to the reaction conditions (for example, doing the reaction under air rather than N2 increases the yield for 4CzIPN but decreases the yield dramatically for the iridium photocatalyst). In general, the difference in yield between the photocatalysts is likely linked with their ground state reduction potential (Ered = −1.51 V, −1.21 V and −1.06 V for [Ir(ppy)2(dtbbpy)]PF6, 4CzIPN and eosin Y respectively), which must be sufficiently negative in order to reduce the in situ-formed ketone (Ered = −1.72 V for 2-phenoxy-1phenylethan-1-one).
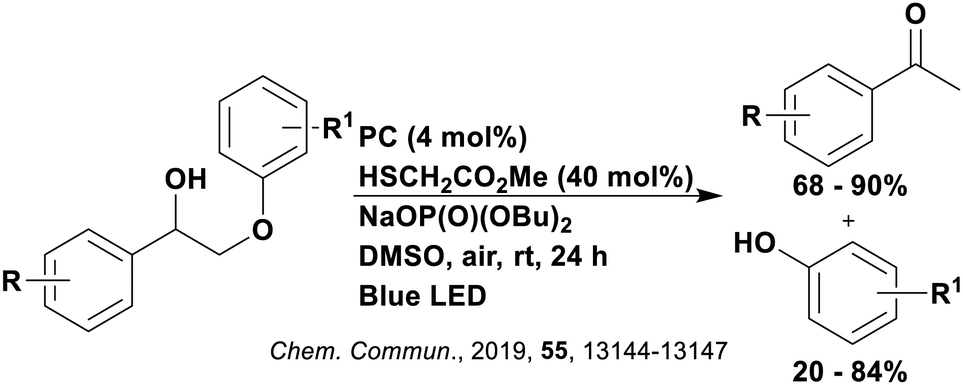 | ||
| Fig. 127 Reaction scheme for the fragmentation of 1,2-diol derivatives. | ||
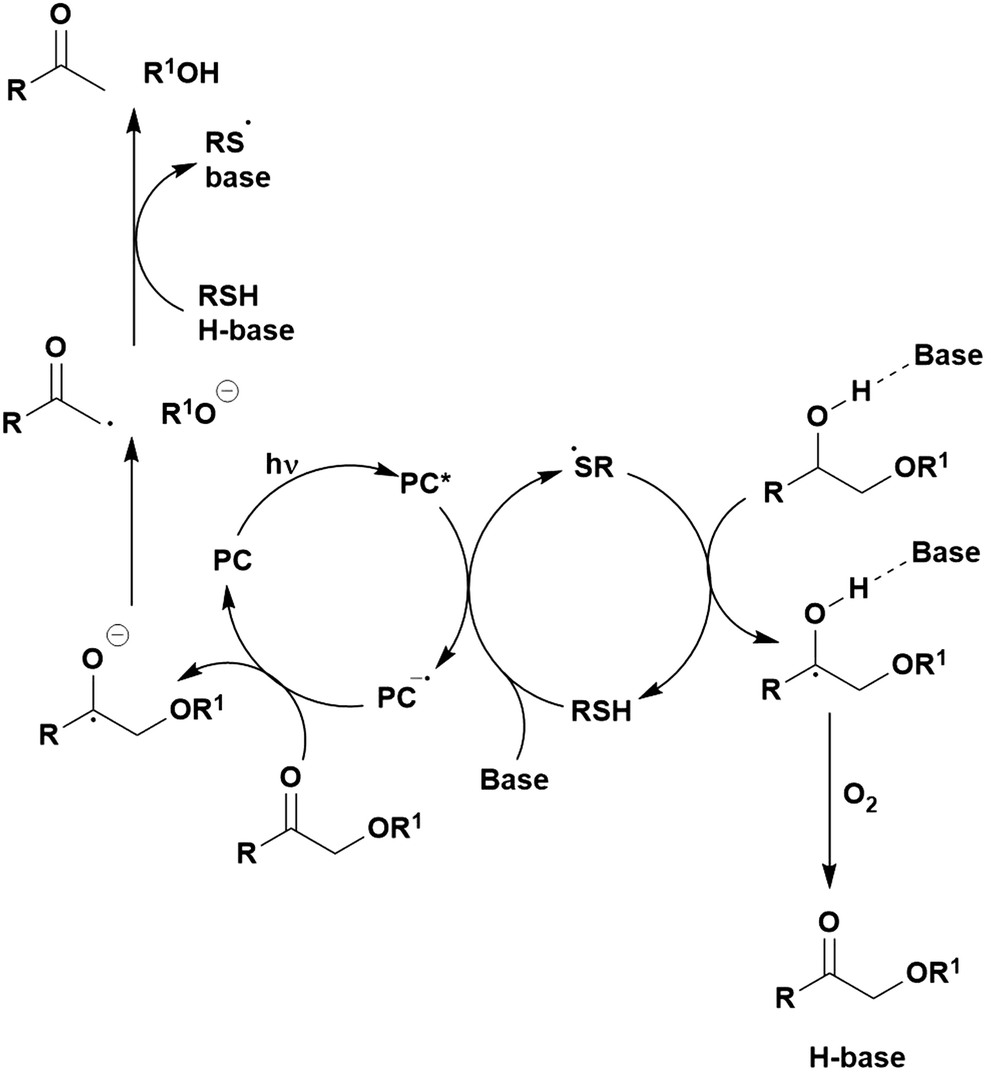 | ||
| Fig. 128 Plausible mechanism for the dual catalytic fragmentation of 1,2-diol derivatives.297 | ||
Additionally, this synergistic catalysis can be used to make more subtle changes, such as the incorporation of deuterium or tritium into a substrate. This can be observed in terms of halogen atom transfer (XAT) of alkyl or aryl halides (Fig. 129a)186 as well as hydrogen isotope exchange (HIE) at α-amino C(sp3)–H bonds (Fig. 129b),298 at the C1 position of aldehydes (Fig. 129c)299 or at α-amino C(sp3)–H bonds of amino acids and peptides (Fig. 129d).300 In the former reaction, amino radicals are used a XAT reagents in order to functionalise aryl and alkyl halides, as previously mentioned.186 In the proposed mechanism, the excited PC is used to oxidise TEA, forming the α-aminoalkyl radical. This radical then undergo XAT with 4-iodo-N-boc-piperidine and the resultant alkyl radical on the piperidine substrate can abstract a deuterium from the HAT catalyst, methyl thioglycolate (Fig. 130). The thiyl radical produced is reduced by the reduced PC before being deuterated from D2O closing both catalytic cycles. A wide variety of photocatalysts were considered for the standard dehalogenation of 4-iodo-N-boc-piperidine, with 4CzIPN proving the best (98% yield), followed by [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (83%), fac-Ir(ppy)3 (78%) and [Ir(ppy)2(dtbbpy)]PF6 (74%). This difference in yield seems to be reflective of the photooxidising capacity of the photocatalyst (Ered* = 1.35 V, 1.21 V, 0.31 V and 0.66 V for 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, fac-Ir(ppy)3 and [Ir(ppy)2(dtbbpy)]PF6, respectively), which must be capable of oxidising TEA (Eox = 0.77 V). Stern–Volmer quenching experiments confirm the amine XAT substrate is responsible for quenching of the PC, with negligible quenching observed with 4-iodo-N-boc-piperidine. The quantum yield of 0.065 for this reaction is suggestive that this is not a radical chain process.
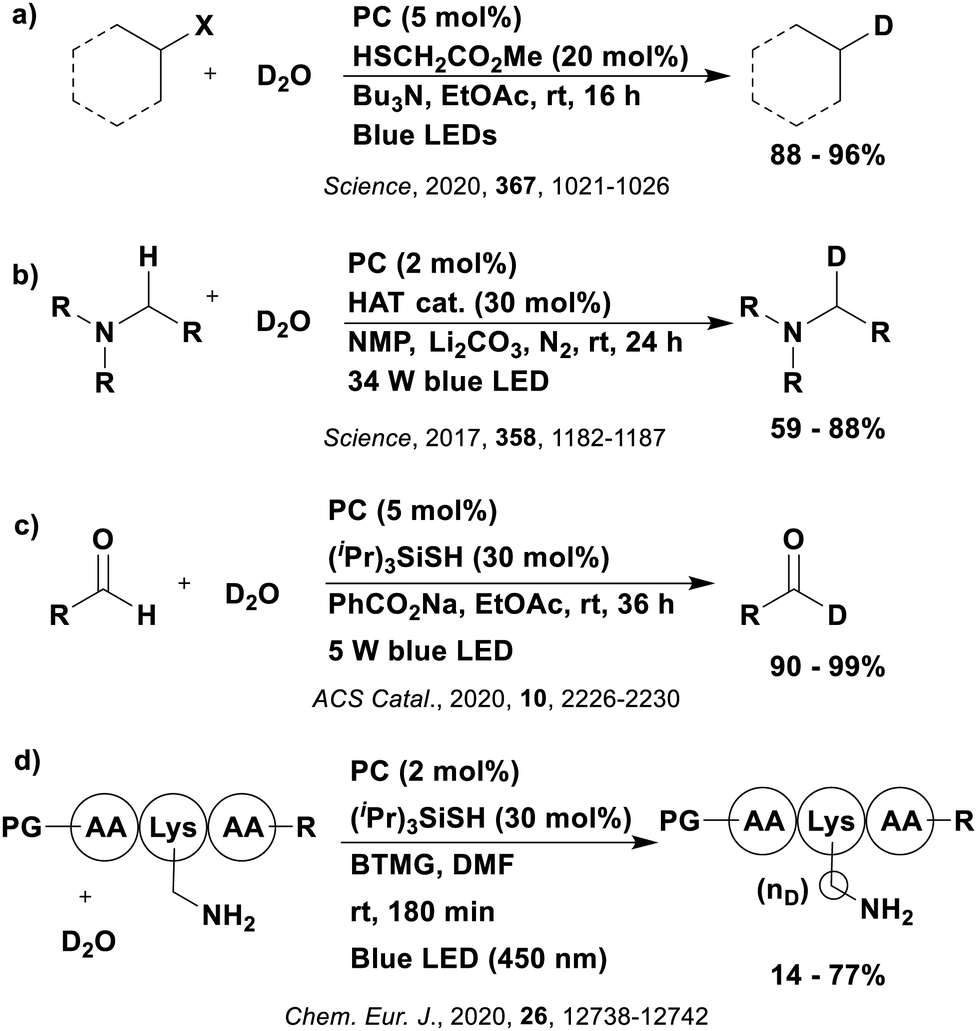 | ||
| Fig. 129 Reaction schemes for the deuteration of (a) alkyl or aryl halides, (b) α-amino C(sp3)–H bonds, (c) aldehydes and (d) amino acids and peptides. BTMG = 2-tert-butyl-1,1,3,3-tetramethylguanidine. | ||
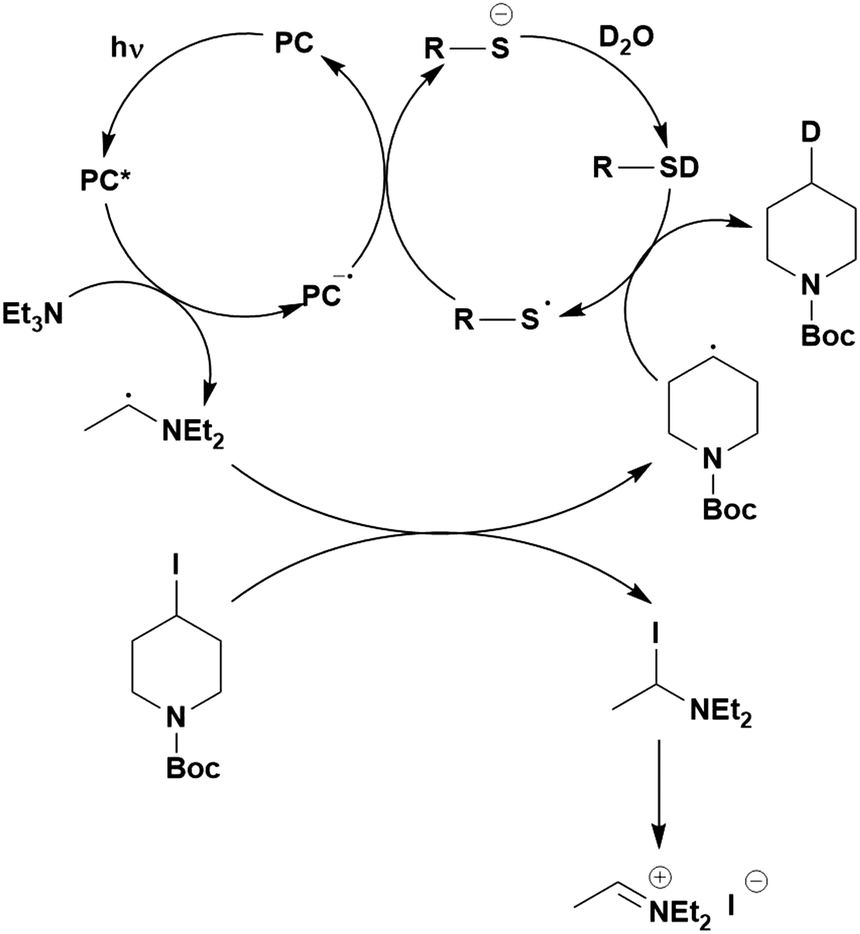 | ||
| Fig. 130 Proposed mechanism for the dehalogenation of an alkyl iodide. | ||
For HIE at α-amino C(sp3)–H bonds (Fig. 129b and d),298,300 a very similar mechanism was proposed whereby the α-amino radical produced by reductive quenching of the excited PC abstracts a deuterium or tritium atom from the HAT catalyst, which in this case is either methyl thioglycolate or triisopropylsilanethiol. In the deuteration of simple amines (Fig. 129b), the best photocatalyst identified was substrate-dependent, with 4CzIPN, [Ir(ppy)2(dtbbpy)]PF6 or [Ir(F(Me)ppy)2(dtbbpy)]PF6 being the photocatalysts that provided the highest product yields. For example, for substrates containing an aliphatic carboxylic acid group, less photooxidising photocatalysts were necessary in order to prevent photoredox-induced decarboxylation; hence, the iridium photocatalysts were more useful in this case (Ered* = 0.66 V, 0.94 V and 1.35 V for [Ir(ppy)2(dtbbpy)]PF6, [Ir(F(Me)ppy)2(dtbbpy)]PF6 and 4CzIPN, respectively). An example of such a substrate can be seen in Fig. 131. When undergoing HIE with amino acids and peptides (Fig. 129d), both 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6 provided the same deuterium incorporation (1.6 D molecule−1), while other PCs performed much worse (fac-Ir(ppy)3 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 afforded 0.4 and 0.1 D molecule−1, respectively).
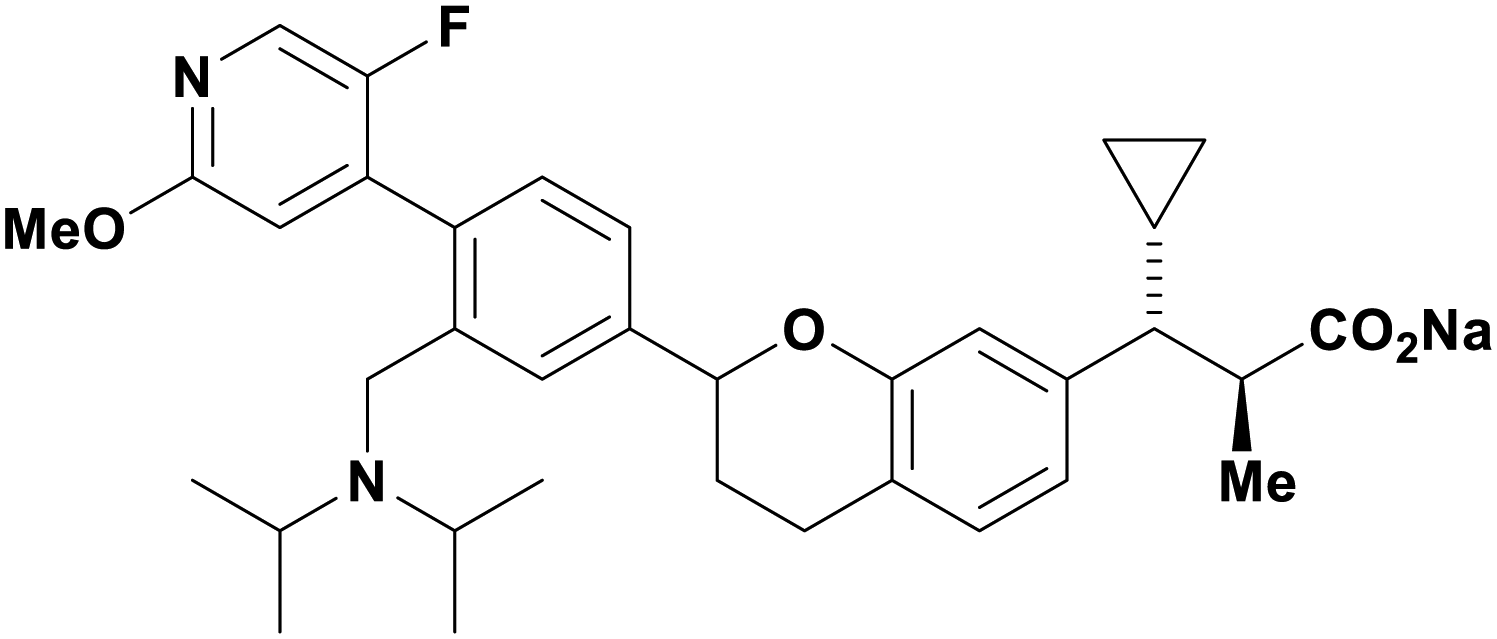 | ||
| Fig. 131 Example of substrate that can be oxidised by 4CzIPN in the deuteration reaction with a HAT catalyst. | ||
In the deuteration of the C1 position of aldehydes (Fig. 129c),299 the proposed mechanism is again similar to that shown in Fig. 130, except the excited PC is reductively quenched by sodium benzoate to give a benzoyloxy radical. The crucial acyl radical is formed by HAT to the benzoyloxy radical. A second HAT step between the deuterated HAT catalyst and the acyl radical affords the deuterated aldehyde. Only organic PCs were considered for this reaction, with 4CzIPN generating the highest percentage of D incorporation (40%) while PCs like eosin Y managing <5%. Further optimisation with 4CzIPN resulted in 93% D incorporation. The superior photooxidising ability of 4CzIPN may be linked to its higher success in this reaction (Ered* = 1.35 V and 0.83 V for 4CzIPN and eosin Y, respectively).
Finally, a report by Wendlandt et al. demonstrated the combination of photoredox catalysis with two distinct HAT catalytic cycles in the synthesis of rare sugar isomers through site-selective epimerisation (Fig. 132).125 The excited PC is reductively quenched by quinuclidine, generating the quinuclidine radical cation, which abstracts a proton from the sugar (Fig. 133). The protonated quinuclidine is then deprotonated by an exogenous base while the sugar radical abstracts a proton from adamantane thiol to form the isomerised product. The subsequently formed thiyl radical is reduced by the reduced photocatalyst, closing the photocatalytic cycle. Stern–Volmer quenching experiments support this mechanism, revealing that the PC is quenched by quinuclidine but not by the thiol. Using 2 mol% of 4CzIPN resulted in 92% yield of product while the only other photocatalyst considered, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, managed 88% yield at 1 mol% loading.
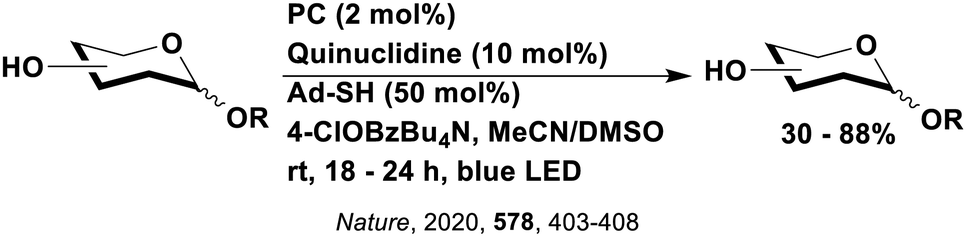 | ||
| Fig. 132 Reaction scheme for the synthesis of rare sugar isomers through site selective epimerisation. | ||
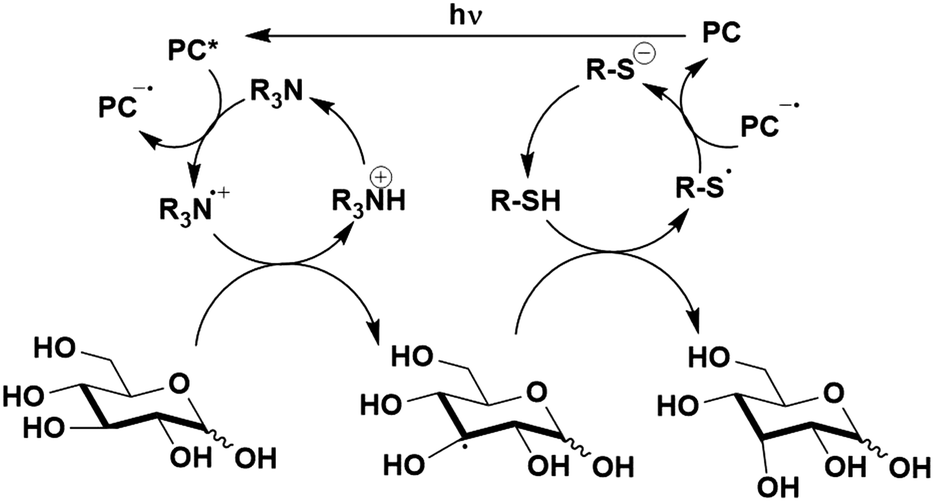 | ||
| Fig. 133 Viable mechanism for the site selective epimerisation of sugars. | ||
Photocatalysis/NHC catalysis
N-Heterocyclic carbene (NHC) catalysis can be used in conjunction with 4CzIPN in photoredox catalysis as a method of synthesising amides from aldehydes and imines (Fig. 134a)301 and making ketones from carboxylic acids (Fig. 134b).302 The proposed mechanism for the former first involves condensation of the aldehyde with the NHC to yield a Breslow intermediate, which reductively quenches the excited PC (Fig. 135). Concurrently, the reduced PC reduces the imine to form a N-centre radical that couples with the Breslow intermediate radical. The NHC catalyst is then released to give the final amide product. The presence of radicals in this mechanism was confirmed by the addition of TEMPO (effectively inhibiting the reaction) as well as EPR spectroscopy, which detected a nitrogen centred radical. The aldehyde, the base or the NHC catalyst, individually, showed no significant quenching of the excited PC; however, a combination of the three reagents together produced a strong quenching of the luminescence.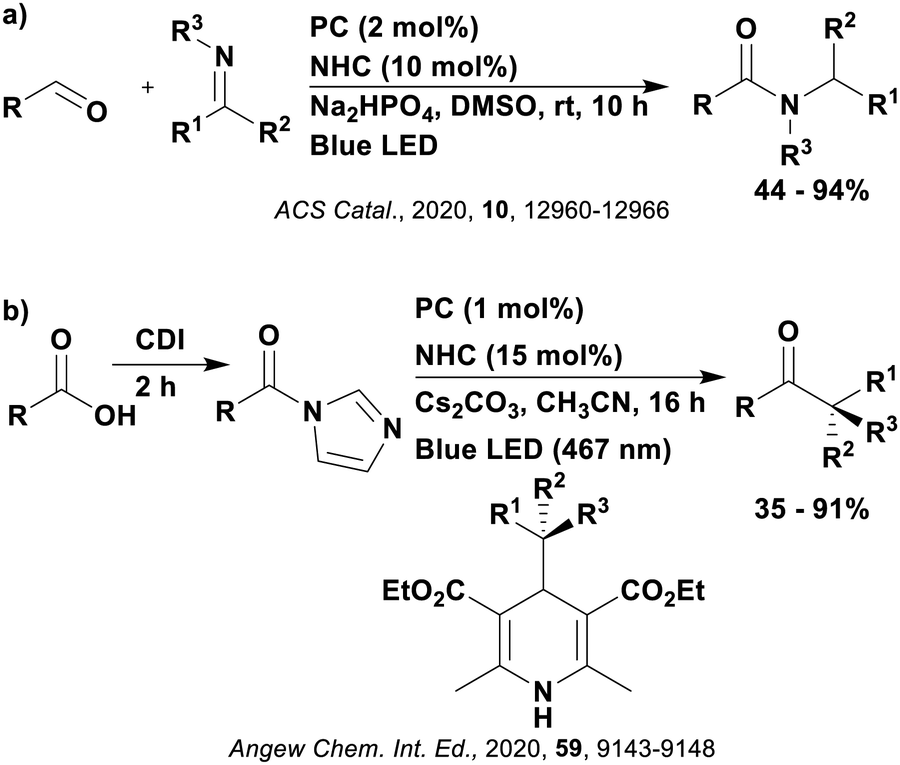 | ||
| Fig. 134 Reaction scheme for the dual photoredox/NHC catalysis to form (a) amides and (b) ketones. CDI = carbonyldiimidazole. | ||
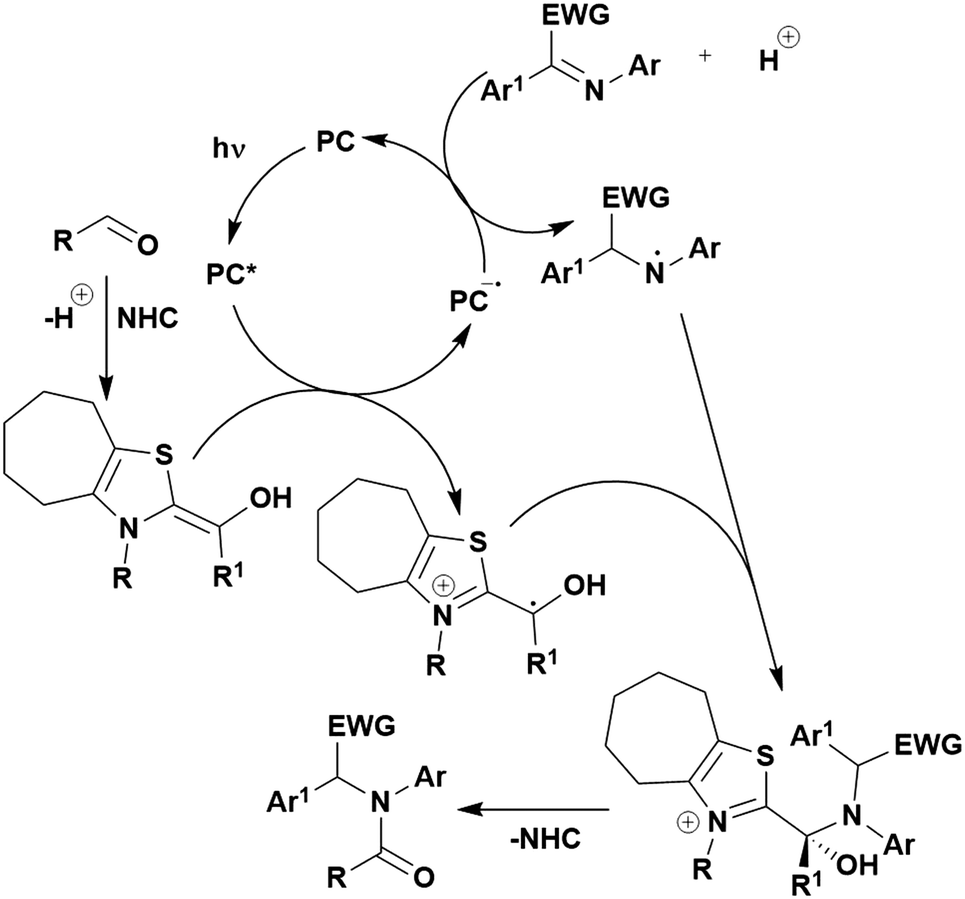 | ||
| Fig. 135 Putative mechanism for the dual photoredox/NHC catalysis to form amides. | ||
For this reaction, three Ru(II) and eleven Ir(III) complexes (10 cationic and one neutral) were evaluated as PCs, alongside six organic dyes. 4CzIPN outperformed the other PCs, yielding 43% of product. Yields obtained from the other PCs varied from 1–13%, except for [Ir(ppy)2(dtbbpy)]PF6, which managed 20%. The reduction of the imine is the challenging step (Ered = −1.16 V vs. SCE for methyl 2-((4-fluorophenyl)imino)-2-(phenyl)acetate in the presence of NaH2PO4), which may explain why some PCs struggled (e.g., eosin Y has Ered = −1.06 V and obtained 2% yield), especially since low conversions of the imine were typically obtained (58% conversion for 4CzIPN and between 8–42% for other PCs).
In the dual catalytic synthesis of ketones from carboxylic acids (Fig. 134b),302 the excited PC is reductively quenched by the DHP radical precursor, which after fragmentation, releases the alkyl radical. Meanwhile, the acyl triazolium species, generated by the addition of the NHC catalyst to the intermediate imidazole, is reduced by the reduced PC, affording the azolium radical. Loss of the NHC and radical coupling of the azolium and alkyl radicals, generates the final product. Addition of TEMPO inhibits the reaction, instead forming an adduct with the alkyl radical generated from the DHP precursor. Four PCs were considered, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN, fac-Ir(ppy)3 and [Mes-Acr]BF4, providing yields of 63%, 42%, 11% and 0%, respectively. These results generally correlate with the ground state reduction potential of the PC (Ered = −1.37 V, −1.21 V and −0.57 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN and [Mes-Acr]BF4, respectively), as the reduced PC is needed to reduce the acyl triazolium species (phenyl acyl azolium has Ered = −1.29 V vs. SCE). For fac-Ir(ppy)3, however, since Ered = −2.19 V, the poor yield is likely instead due to its poor photooxidising ability (Ered* = 0.31 V), which struggles to oxidise the DHP (benzyl Hantzsch ester has Eox = 1.00 V vs. SCE).303 The PC [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was chosen for the remainder of the study on account of it providing the highest yields.
Photocatalysis/bromine catalysis
The final example of dual catalysis involving 4CzIPN is the [3+2] cycloaddition reaction, facilitated by bromine radical catalysis (Fig. 136).304 The proposed mechanism involves energy transfer from the excited PC to the cinnamyl bromide pre-catalyst, generating the crucial bromyl radical (Fig. 137). Addition of this radical to the vinyl bond of the substituted cyclopropane generates a transient radical species that undergoes fast ring opening. The homoallyl radical intermediate then adds to the alkene reagent, followed by ring closure and elimination of the bromyl radical, closing the catalytic cycle. Stern–Volmer quenching experiments revealed that cinnamyl bromide acted as a quencher of the excited PC. Since cinnamyl bromide has a high oxidation potential of Eox = 2.01 V, SET between the precatalyst and the photocatalyst is thermodynamically unfavourable (for 4CzIPN, Ered* = 1.35 V and Eox = 1.52 V), which eliminated this mechanism from consideration, leaving PEnT as the alternative plausible mechanism. A correlation between the triplet energy ET of the PC and the yield of product was found, namely the higher the ET, the greater yield. For example, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 gave the highest yield (95%, ET = 2.65 eV, 256 kJ mol−1), closely followed by 4CzIPN (93%, ET = 2.52 eV, 243 kJ mol−1), whereas [Ir(ppy)2(dtbbpy)]PF6 was inadequate (11%, ET = 2.12 eV, 205 kJ mol−1). Since this is an energy transfer mechanism, emphasis on the spectral overlap of the emission of the PC and absorption of cinnamyl bromide must be highlighted. The aforementioned PCs emit at 470 nm, 535 nm and 581 nm, for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN and [Ir(ppy)2(dtbbpy)]PF6, respectively, in MeCN. From this, the more red-shifted the emission of the PC, the lower the yield of product indicating a poorer spectral overlap with the absorption of the precatalyst. 4CzIPN was selected as the photocatalyst of choice for the study, although no further mechanistic studies were undertaken to determine which energy transfer mechanism is in operation. A quantum yield of 0.183 suggests this is not a radical chain process. | ||
| Fig. 136 Reaction scheme for the [3+2] cycloaddition of substituted vinyl and ethynylcyclopropanes with alkenes. | ||
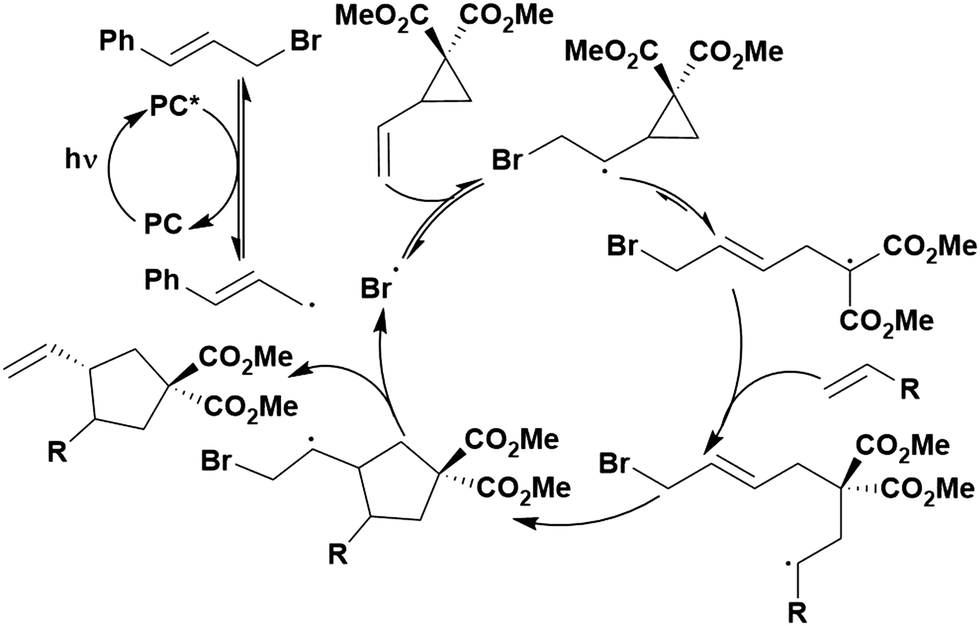 | ||
| Fig. 137 Suggested mechanism for the dual catalytic [3+2] cycloaddition of vinyl cyclopropanes with alkenes. | ||
4. Other CDCB-based TADF organic photocatalysts
As a photocatalyst, 4CzIPN has been shown to be effective in a wide range of transformations.305 Though it is the most widely used TADF-based photocatalyst in recent years, it is not the only TADF molecule that has been used, with 4DPAIPN in particular quickly becoming a popular choice.306 In this section, we compare and contrast organic TADF photocatalysts, confining at first to compounds within the carbazolyl dicyanobenzene (CDCB) family of compounds. While in most cases the TADF nature of the photocatalyst is not explicitly explored, the results obtained can nonetheless be used to gain insight into to the link between the photophysical parameters of the photocatalyst and the observed photocatalytic ability.Modification of the structure of the carbazole donor and dicyanobenzene acceptor moieties of 4CzIPN results in donor–acceptor compounds with modulated redox potentials and photophysical properties. A comprehensive listing of the properties of these compounds is provided in Table 2. Since the spatially separated HOMO is localised on the donor moieties and the LUMO is localised on the acceptor core, changes to these components can be done almost independently to alter the oxidative and reductive capabilities of the photocatalyst in both the ground and excited state.
| PC | λ abs/nm | λ PL/nm | E 0,0/eV (kJ mol−1) | ΔEST/eV | E ox/V | E red/V | E ox*/V | E red */V | τ pf/ns | τ df/μs | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a All potentials are given in volts versus SCE. Eox* = Eox − E0,0 and Ered* = Ered + E0,0. 1 eV = = (1.602 × 10−22 k J) × NA where NA = Avogradro's constant. λabs refers to the absorption maximum of the CT band. λPL refers to the photoluminescence maximum. Data reported in MeCN at room temperature unless otherwise noted. E0,0 determined from the intersection point of the normalised absorption and emission spectra unless otherwise noted.
b Determined in dichloromethane.
c Originally referenced versus Ag/AgNO3, converted to be referenced versus SCE using E (vs. SCE) = E(vs. Ag/AgNO3) + 0.30 V.326
d Determined in tetrahydrofuran.
e Determined in toluene.
f Determined in CH3CN/CH2Cl2 (5:1).
g Determined in dimethylformamide.
h Estimated by time-dependent (TD)DFT at the M06-2X/6-31+G(d) level.
i Measured at 77 K.
j Determined in 3 wt%-CzTPN:PzCz film measured at room temperature under N2 (PzCz = hexakis(9H-carbazol-9-yl) cyclotriphosphazene).
k Determined in DMA.
l
E
0,0 estimated using the medium wavelengths between the lowest fluorescence excitation peak (excitation λmax) and the fluorescence peak (emission λmax).120
m
E
0,0 obtained from the absorption spectrum.307
n
E
0,0 estimated from E0,0 = hc/λem.310
|
|||||||||||
| 3,5-2CzBN | 396e | 3.34e,m (322) | 0.40e | 3.9e | 307 | ||||||
| 2,3,6-3CzBN | 0.31 | 9.6 | 102.7 | 308 and 309 | |||||||
| 2,4,6-3CzBN | 431e | 3.10e,m (299) | 0.29 | 10.1 | 220.3 | 307–309 | |||||
| 3,4,5-3CzBN | 340 | 565 | 2.19n (211) | 2.07 | −1.58 | −0.51 | 1.00 | 310 | |||
| 4CzBN | 333b | 442e | 2.9b (280) | 0.25 | 1.61 | −1.63 | −1.29 | 1.27 | 5.4 | 36.9 | 309, 311 and 312 |
| 4CzBN-Br | 340b | 2.8b (270) | 0.22b | 1.62 | −1.50 | −1.18 | 1.30 | 7.8b | 311 | ||
| 5CzBN | 512 | 2.83 (273) | 0.07 | 1.41 | −1.52 | −1.42 | 1.31 | 16.2 | 7.8 | 114, 308 and 309 | |
| 5CzBN-OMe | 462 | 2.81 (271) | 1.02b | −1.66b | −1.79b | 1.15b | 114 | ||||
| 2CzPN | 365 | 530 | 2.77l (267) | 0.09e | 1.47 | −1.45 | −1.3 | 1.32 | 12.9 | 15.4 | 60, 120, 121 and 308 |
| 2CzIPN | 362 | 501 | 2.87l (277) | 0.05d | 1.46 | −1.51 | −1.41 | 1.36 | 120 and 313 | ||
| 2CzTPN | 430b | 511b | 2.64b,l (255) | 0.08j | 1.4b | −1.3b | −1.24b | 1.34b | 34j | 11.3j | 120 and 314 |
| 4CzPN | 435 | 535 | 2.56l (247) | 0.25e,i | 1.5 | −1.16 | −1.06 | 1.4 | 6.7 | 0.5 | 120, 121 and 315 |
| 4CzPN-Ph | 1.24c,d | −1.29c,d | 316 | ||||||||
| 4CzPN-tBu | 1.20c,d | −1.23c,d | 316 | ||||||||
| 4CzIPN | 435 | 535 | 2.67l (258) | 0.08e | 1.52 | −1.21 | −1.04 | 1.35 | 18.7 | 1.39 | 60, 114, 120 and 121 |
| 4CzIPN-Me | 456 | 603 | 0.307h | −1.23 | 1.31 | 317 and 318 | |||||
| 4CzIPN-Ph | 390 | 611 | 0.214h | 1.41 | −1.15 | 4 | 0.79 | 316 and 317 | |||
| 4CzIPN-tBu | 380 | 588 | 2.53 (244) | 0.308h | 1.22 | −1.32 | −1.31 | 1.21 | 10 | 1.4 | 316, 317, 319 and 320 |
| 4CzIPN-OMe | 482 | 2.61f (252) | 1.11 f | −1.34 f | −1.5 f | 1.27 f | 114 | ||||
| 4CzIPN-F | 2.60b (251) | 1.45b | −1.08b | −1.15b | 1.52b | 321 | |||||
| 4CzIPN-Cl | 2.68 (259) | 2.05 | −0.97 | −0.63 | 1.71 | 321 | |||||
| 4CzIPN-Br | 2.58b (249) | 1.76b | −1.06b | −0.82b | 1.52b | 321 | |||||
| 4CzTPNb | 463 | 556 | 2.43l (234) | 0.38 | 1.44 | −1.02 | −0.99 | 1.41 | 6.5 | 1.46 | 120 and 121 |
| 4CzTPN-Me | 561e | 9.2e | 1.5e | 60 | |||||||
| 4CzTPN-Ph | 577e | 0.25c,d | −1.29c,d | 9.0e | 1.1e | 60 and 316 | |||||
| 4CzTPN-tBu | 0.01e,i | 0.21c,d | −1.24c,d | 315 and 316 | |||||||
| 4CzTPN-Br | 349b | 546e | 2.72b (262) | 0.3e | 4.8b | 311 and 322 | |||||
| 3CzClIPN | 545 | 2.72 (262) | 1.79 | −1.16 | −0.93 | 1.56 | 6.9 | 114 | |||
| 4CzBnBN | 477k | 1.48g | −1.72g | −1.45 | 1.21 | 323 | |||||
| 4CzPEBN | 2.88g (278) | −1.69g | 1.19g | 324 | |||||||
| 4DPAPN | 448g | 559g | 2.46g (237) | −1.53g | 0.93g | 325 | |||||
| 4DPAIPN | 425 | 523 | 2.62l (253) | 1.34 | −1.52 | −1.28 | 1.1 | 120 | |||
| 4DPAIPN-Cl | 2.53 (244) | 1.23 | −1.44 | −1.30 | 1.09 | 320 | |||||
| 4DPAIPN-Br | 2.53 (244) | 1.12 | −1.55 | −1.41 | 0.98 | 320 | |||||
| 3DPAClIPN | 537 | 2.65 (256) | 1.31 | −1.41 | −1.34 | 1.24 | 11.5 | 114 | |||
| 3DPAFIPN | 525 | 2.68 (259) | 1.3 | −1.59 | −1.38 | 1.09 | 4.2 | 114 | |||
| 3DPA2FBN | 491 | 2.84 (274) | 1.24b | −1.92b | −1.6b | 0.92b | 4.2 | 114 | |||
For example, consider 4CzIPN and 4MeOCzIPN. Both contain the same isophthalonitrile acceptor component, hence should have a similar LUMO energy level and reduction potential (Ered = −1.21 V and −1.34 V for 4CzIPN and 4CzIPN-OMe, respectively, in MeCN and MeCN:DCM(5:1)). The difference between these two emitters arises in the donor groups; 4CzIPN has 4 carbazole (Cz) donor groups while 4CzIPN-OMe has 4 methoxy substituted carbazole (MeOCz) donor groups. The addition of the methoxy groups results in MeOCz being a stronger donor than Cz. Therefore, the HOMO energy level of 4CzIPN-OMe should be destabilised in comparison to 4CzIPN. This is indeed the case, as is reflected by the ground state oxidation potential (Eox = 1.52 V and 1.11 V for 4CzIPN and 4CzIPN-OMe, respectively, in MeCN and MeCN:DCM(5:1)). The smaller Eox value of 4CzIPN-OMe indicates it is a stronger ground state reductant than 4CzIPN.
When instead understanding the influence of structure on the ground state oxidising capability, consider 3DPAFIPN and 3DPA2FBN. In this case, both have three similarly disposed DPA donor groups about the central benzene ring and so the HOMO energy level and consequently the oxidation potential should be similar (Eox = 1.30 V and 1.24 V, for 3DPAFIPN and 3DPA2FBN, respectively, in MeCN and DCM). The main difference in these structures arises in the nature of the acceptor, where 3DPAFIPN has two cyano groups and one fluoro substituent on the phenyl ring while 3DPA2FBN has one cyano group and two fluoro substituents. Since cyano groups are more strongly electron-withdrawing than halogens, there is consequently a more stabilised LUMO in 3DPAFIPN. As a result, 3DPAFIPN is a stronger ground state oxidant, as reflected by the less negative ground state reduction potential (Ered = −1.59 V and −1.92 V for 3DPAFIPN and 3DPA2FBN, respectively, in MeCN and DCM).
As demonstrated, modification of the donor and acceptor groups clearly impacts upon the ground state redox potentials, which of course are linked to the excited state redox potentials, through Eox* = Eox − E0,0 and Ered* = Ered + E0,0. From these equations, it becomes clear that the changes in the optical gap must also be taken into consideration when discussing the impact on the excited state redox potentials. Destabilisation of the HOMO, as caused by the presence of a weaker donor, results in a larger E0,0. Again consider 4CzIPN and 4CzIPN-OMe. When acting as a photooxidant, the hole left in the HOMO of the PC after excitation of an electron to the LUMO, can readily accept an electron from a donor. Since both 4CzIPN and 4CzIPN-OMe have the same acceptor unit, Ered should be similar. However, the weaker carbazole donor means 4CzIPN has a larger optical gap (E0,0 = 2.67 eV (258 kJ mol−1) and 2.61 eV (252 kJ mol−1) for 4CzIPN and 4CzIPN-OMe, respectively, in MeCN and MeCN:DCM(5:1)). When considering Ered*, this means a more positive value for 4CzIPN in comparison to 4CzIPN-OMe (Ered* = 1.35 V and 1.27 V for 4CzIPN and 4CzIPN-OMe, respectively, in MeCN and MeCN:DCM(5:1)) owing to the larger optical gap in the former. This translates to 4CzIPN being a stronger photooxidant.
The same analysis can be used when considering the TADF compounds as photoreductants. In this case, the PC's electron that has been promoted to the LUMO can be donated to an acceptor substrate. Again, consider 3DPAFIPN and 3DPA2FBN. The more stabilized LUMO in 3DPAFIPN coupled with comparable HOMO levels results in a decreased optical gap for this compound (E0,0 = 2.68 eV (259 kJ mol−1) and 2.84 eV (274 kJ mol−1) for 3DPAFIPN and 3DPA2FBN, respectively, in MeCN). The larger optical gap observed for 3DPA2FBN will result in a more negative Eox* value (Eox* = −1.38 V and −1.60 V for 3DPAFIPN and 3DPA2FBN, respectively, in MeCN and DCM), meaning 3DPA2FBN in a stronger photoreductant than 3DPAFIPN.
These examples highlight that modifications can be made in terms of both the nature and number of donor and acceptor moieties in order to modulate the electrochemical properties of the PCs. A range of these donor–acceptor molecules, including many that have sufficiently small ΔEST to be considered TADF, from the CDCB family (Fig. 138 and 139) have been reported to act as photocatalysts.
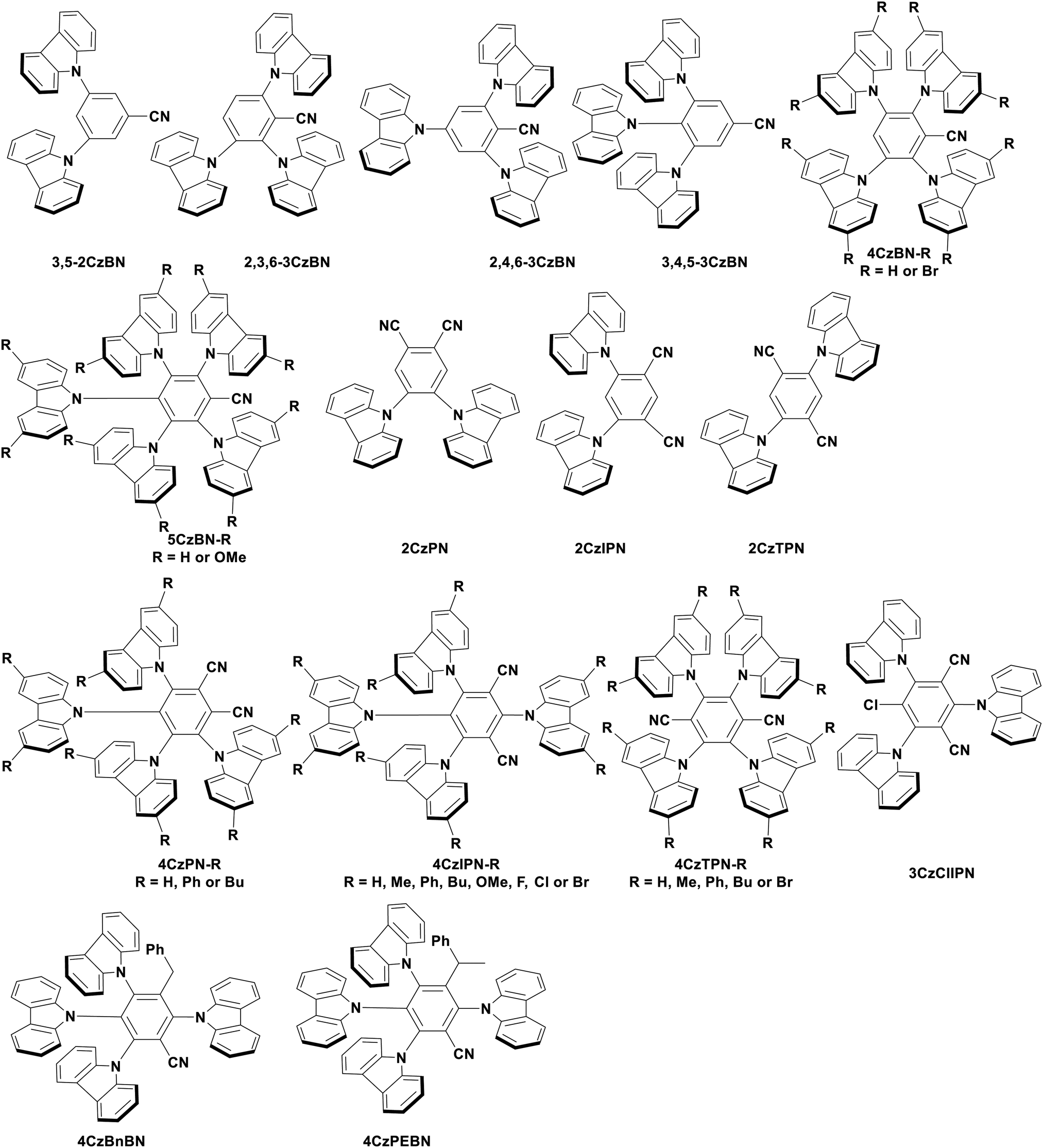 | ||
| Fig. 138 Structures of TADF compounds with benzonitrile and CDCB-based structure used as photocatalysts. | ||
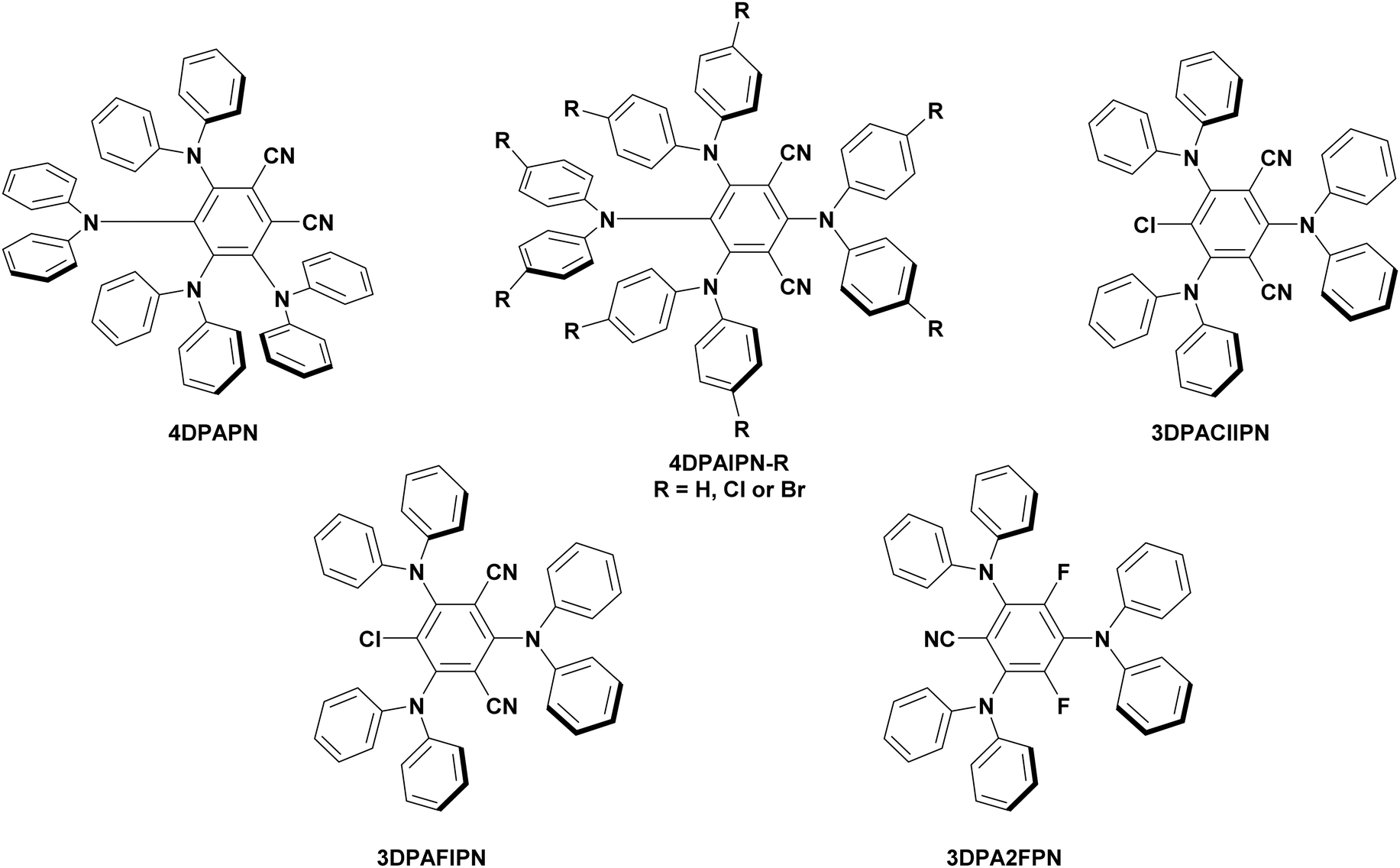 | ||
| Fig. 139 Structure of CDCB based TADF compounds which involve diphenylamine (DPA) as donor groups which have been considered as photocatalysts. | ||
5. CDCB based molecules acting as independent photocatalysts
Other TADF molecules within the CDCB family of compounds have also been reported as photocatalysts. This section highlights their use and contrasts their performance with 4CzIPN.Photoinduced decarboxylation of carboxylic acids in the formation of C–C and C–X bonds
Both the γ,γ-difluoroallylation of cycloketone oxime ethers (Fig. 140a) and the fragmentation–alkynylation of oxime ethers (Fig. 140b) proceed through a photoinduced oxidative decarboxylation mechanism as shown in Fig. 141, whereby the excited PC can be used to decarboxylate the cycloketone oxime ethers in a reductive quenching cycle.327 In the former reaction, the nucleophilic alkyl nitrile radical eventually is coupled to an electron-poor trifluoromethyl-substituted alkene and the resultant product undergoes single electron reduction by the reduced PC. Both 4CzIPN and 4DPAIPN were investigated as photocatalysts for this reaction, with 4CzIPN providing the higher product yields (64% and 34%, respectively, at 5 mol% of catalyst loading); however, both fell short of the high yields obtained by [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (76% yield). Oxidation of the oxime ether requires a strong photooxidant (Eox = 1.48 V for an analogous compound),321 which likely explains why 4DPAIN provides inferior yields (Ered* = 1.21 V, 1.35 V and 1.1 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN and 4DPAIPN, respectively). The other SET implicates the PC as a ground state reductant, for which the iridium photocatalyst has a superior redox potential (Ered = −1.37 V, and −1.21 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 4CzIPN), which may help to explain the differing yields obtained. Although 4DPAIPN is an even greater ground state reductant (Ered = −1.52 V), its poor photooxidising ability acts as a barrier to its success in this reaction.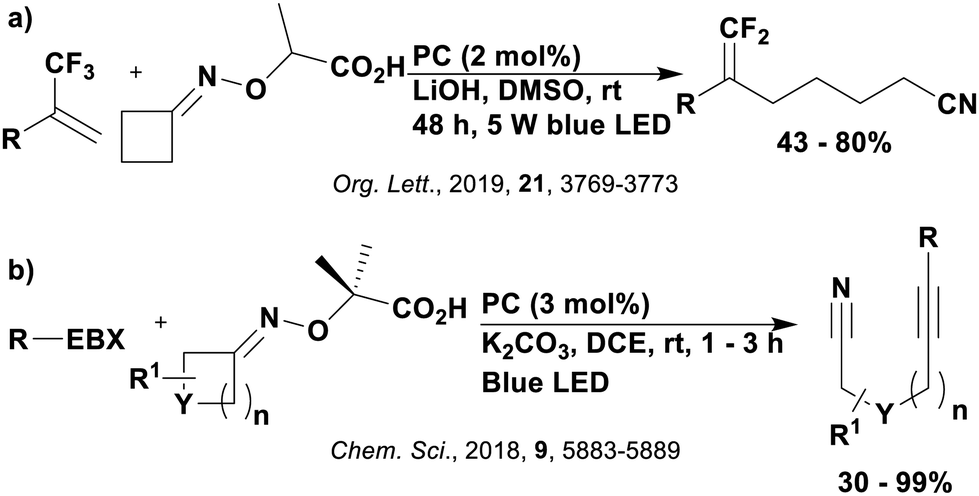 | ||
| Fig. 140 Reaction scheme for (a) the neutral γ,γ-difluoroallylation of cycloketone oxime ethers with trifluoromethyl alkenes and (b) fragmentation–alkynylation of oxime ethers where EBX = ethynyl benziodoxolone. | ||
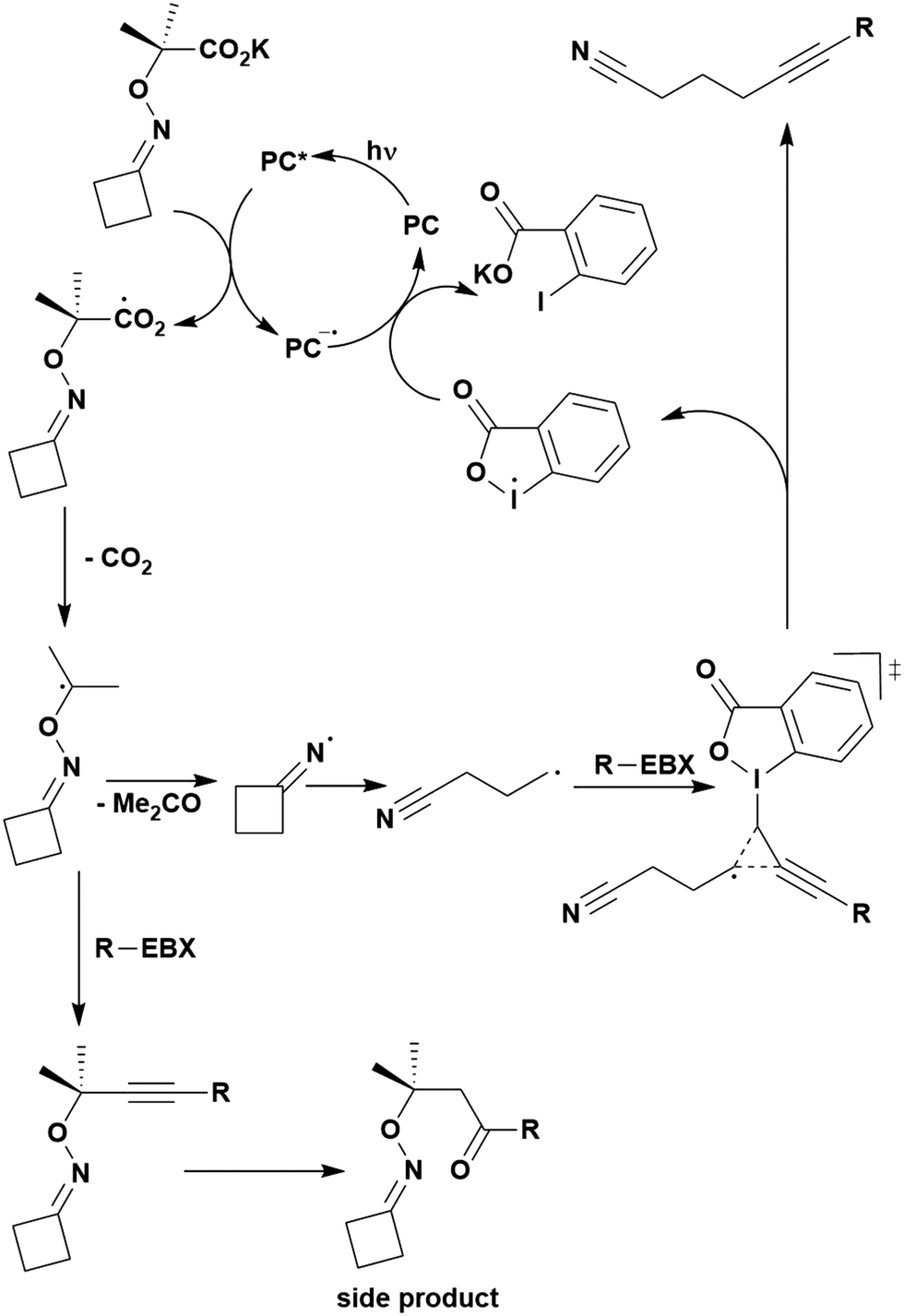 | ||
| Fig. 141 Proposed mechanism for the alkynylation of oxime ethers. | ||
For the reaction involving the alkynylation of oxime ethers (Fig. 140b), a similar mechanism is in operation.321 Again, the oxime ether is oxidatively decarboxylated by the excited photocatalyst, before the resultant alkyl nitrile radical is trapped by the ethynyl benziodoxolone (EBX) reagent (Fig. 141). Upon release of the product, the subsequent iodine radical is reduced by the reduced photocatalyst, closing the photocatalytic cycle. The TADF compounds surveyed were 4CzIPN, 4CzIPN-F, 4CzIPN-Cl and 4CzIPN-Br, which gave yields of 50%, 75%, 80% and 75%, respectively; all managed >95% conversion of oxime ether except for 4CzIPN, which showed only 60% conversion. The superiority of the halogenated compounds, in particular the chlorine-substituted compound, in comparison to 4CzIPN is reflected in their much greater photooxidising capacity (Ered* = 1.35 V, 1.52 V, 1.71 V, 1.52 V for 4CzIPN, 4CzIPN-F, 4CzIPN-Cl and 4CzIPN-Br, respectively), especially since 2-((cyclobutylideneamino)oxy)-2-methylpropanoate, the oxime ether carboxylate in Fig. 141, has an oxidation potential of Eox = 1.48 V. Notably, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was used as a reference PC and managed >95% conversion and 55% yield at 1 mol% PC loading, compared to the 5 mol% utilised for the TADF compounds. In further optimisation studies, it was found that decreasing the catalyst loading from 5 mol% to 3 mol% for the TADF compounds could be achieved without having an impact on the yield, therefore 3 mol% of PC was used in the substrate scope. The low yield of product obtained using the iridium photocatalyst is due to significant formation of the side product 2-oxo-2-phenylethyl 2-iodobenzoate. Waser et al. postulate this is formed through a decomposition of Ph-EBX when water or oxygen is present and may be catalysed by photocatalysis, although the mechanism is not yet known. An additional side product shown in Fig. 141 is also proven to exist by 1H NMR for the TADF compounds, which explains why their almost complete conversions are not reflected in the yields, however, high yields of the anticipated product are still obtained therefore, this side product is not thoroughly discussed.
Ethynyl benziodoxolone (EBX) reagents have also been used in conjunction with photocatalysis in the decarboxylative alkynylation of dipeptides (Fig. 142a) and peptide tetramers (Fig. 142b).320 Although no mechanistic investigations were undertaken or catalytic cycles shown, the mechanism proposed by Waser et al. is referenced to be likely to be in operation,328 which is reflective of the suggested mechanism shown in Fig. 141. A number of TADF compounds were considered as PCs in alkynylation of Cbz-Gly-Pro using Ph-EBX: 4CzIPN, 4CzIPN-Cl, 4CzIPN-tBu, 4CzIPN-OMe, 4DPAIPN, 4DPAIPN-Cl and 4DPAIPN-Br, which gave yields of 99%, 89%, 44%, <5%, 43%, 65% and 50%, respectively. The iridium PC [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was also considered, which provided 99% yield. The yields generally reflect the photoxidising capacity of the PC (Ered* = 1.35 V, 1.58 V, 1.21 V, 1.23 V, 0.90 V, 1.09 V, 0.98 V and 1.21 V for the TADF PCs and Ir PC, respectively). However, there are some exceptions to this, for example 4CzIPN-OMe (Ered* = 1.23 V, <5%) performs poorly despite being more strongly photooxidising than the Ir PC. This is suggested to be linked to fast BET from the excited CT state of the PC, as proposed by Zeitler et al.1144CzIPN-tBu also performed worse than expected (Ered* = 1.21 V, 44%), with Waser et al. conjecturing that issues of stability of the PC may be responsible for the lower yield. For the subsequent substrate scope, 4CzIPN was selected as the PC of choice.
 | ||
| Fig. 142 Reaction scheme for the decarboxylative alkylnylation of (a) dipeptides and (b) peptide tetramers. | ||
An additional example involves functionalisation of an alkene, whereby decarboxylation of Cbz-proline by single electron oxidation from the excited photocatalyst produces an electron-rich radical that then can be added to the electron-poor diethyl maleate (Fig. 143).114 The reduced photocatalyst is then used to reduce the resultant C-centred radical (Fig. 144). A variety of TADF compounds including 3CzClIPN, 4CzIPN, 5CzBN, 3DPAClIPN, 3DPAFIBN, 3DPA2FBN, 4CzIPN-OMe and 5CzBN-OMe were tested as photocatalysts in this model reaction. All photocatalysts produced the final product but with varying yield, depending highly on their respective ground and excited state redox potentials. For example, 4CzIPN and 3CzClIPN, possessing the highest Ered* values of the series (Ered* = 1.35 V and 1.56 V, respectively), and hence the best photooxidants, produced the highest yields (80% and 77%, respectively). The yields were also higher, but comparable, than those previously reported using Mes-4MeOAcr-Ph (73%, Ered* = 1.65 V), the best known photocatalyst for this reaction.23 The slight difference in yield between these photocatalysts can be correlated to the ground state reduction potentials (Ered = −1.21 V, −1.16 V and −0.82 V for 4CzIPN, 3CzClIPN and Mes-4MeOAc-Ph, respectively). Product yields varied from 1–37% for the other TADF photocatalysts and aligned with their reduced oxidising power in the excited state. Both methoxy-substituted photocatalysts, 4CzIPN-OMe and 5CzBN-OMe produced 3% and 1% yield, respectively, of the product despite having appropriate redox potentials. This is hypothesized by the authors to be as a result of fast, intramolecular BET from their excited charge transfer state, which is corroborated by their weak radiative emission intensity in comparison to the other compounds in the CDCB family. Zeitler et al. hypothesized that the stronger methoxy donor substituent triggers a competitive nonradiative relaxation process. It should be noted that these methoxy-substituted CDCB compounds have been shown as successful PCs in other reactions; for example, 5CzBN-OMe was shown to be highly efficient in the photocatalytic formation of amides (see Section 6: HAT catalysis).329 It is thus not evident what is the origin of their poor performance in the functionalisation of diethyl maleate.
 | ||
| Fig. 143 Reaction scheme for the photocatalytic decarboxylative conjugate addition of Cbz-proline to diethyl maleate. | ||
 | ||
| Fig. 144 Photoinduced decarboxylative conjugate addition of Cbz-protected proline to diethyl maleate. | ||
The photoinduced decarboxylation of benzylic carboxylic acids can be used towards the synthesis of secondary alcohols (Fig. 145a).323 As with the previous examples, the excited photocatalyst is used to oxidatively decarboxylate the carboxylic acid, generating a carbon-centred radical species, which is then reduced by the reduced photocatalyst to form the carbanion, closing the photocatalytic cycle (Fig. 146). The carbanion can then attack the electrophilic carbonyl carbon in the aliphatic aldehyde, producing the secondary alcohol product. Stern–Volmer quenching experiments confirmed that the excited PC is quenched by the carboxylate rather than the aldehyde. Three photocatalysts, 4CzIPN, 4CzPN and 3DPAFIPN, were considered, producing respective yields of 75%, 55% and 6% in the reaction of phenylacetic acid and n-pentanal. Other photocatalysts investigated, including [Ir(dF(CF)3ppy)2(dtbbpy)]PF6 provided very poor yields of 6% or less. Firstly, the excited PC must oxidise phenylacetate (PA) with NBu4PA having an oxidation potential of Eox = 1.27 V.173 The photooxidising capacity of the PCs is as follows: Ered* = 1.21 V, 1.35 V, 1.40 V and 1.09 V for [Ir(dF(CF)3ppy)2(dtbbpy)]PF6, 4CzIPN, 4CzPN and 3DPAFIPN, respectively. Hence from this, it is clear to why both the iridium PC and 3DPAFIPN perform poorly in this reaction, while 4CzIPN and 4CzPN are more than capable. Since only 4CzIPN and 4CzPN are photooxiding enough to complete the first step, only these two PCs will be discussed when considering the second SET, which is reduction of the benzylic radical (Ered = −1.43 V).330,331 Both 4CzIPN and 4CzPN have similar ground state reducing ability (Ered = −1.21 V and −1.16 V, respectively), so their great difference in yield is unexpected. To understand this variation in product yields, mechanistic investigations were undertaken. Photodecomposition of 4CzIPN was observed during the reaction, isolated as 4CzBnBN (Fig. 145b). Reaction of phenylacetic acid and n-pentanal using 4CzBnBN yields 39% of the desired product, leading König et al. to suggest it is the main active PC for carbanion generation, especially since it is very reducing in the ground state (Ered = −1.72 V). Photocatalytic carbanion generation has been discussed in detail in a recent review.332 From this, the authors propose the mechanism shown in Fig. 146, whereby they invoke 4CzBnBN as the PC. Since 4CzIPN photo decomposes to form this strong ground state reductant 4CzBnBN, this is likely why 4CzIPN outperforms 4CzPN.
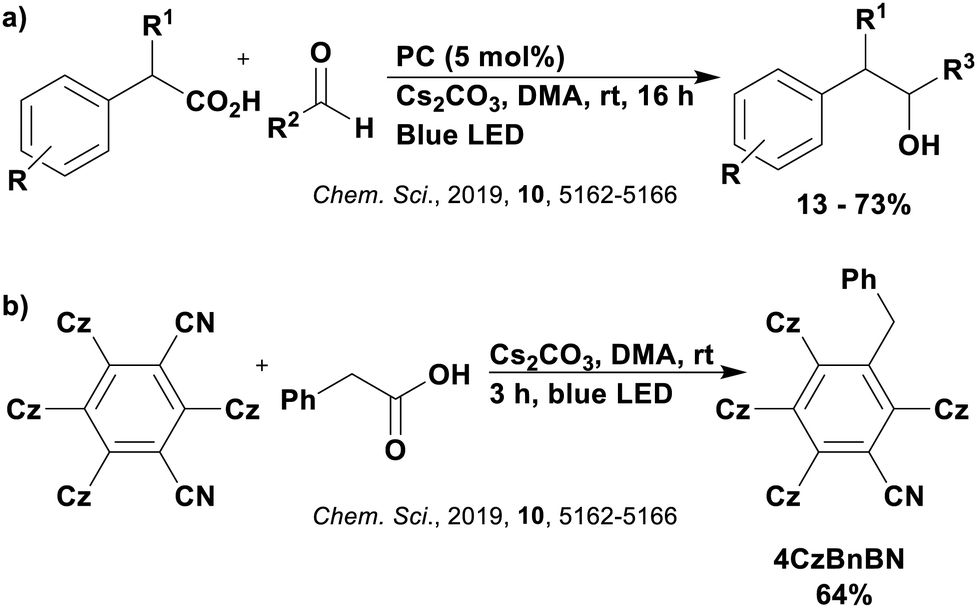 | ||
| Fig. 145 Reaction scheme for the (a) benzylation of aliphatic aldehydes to secondary alcohols and (b) formation 4CzBnBN. | ||
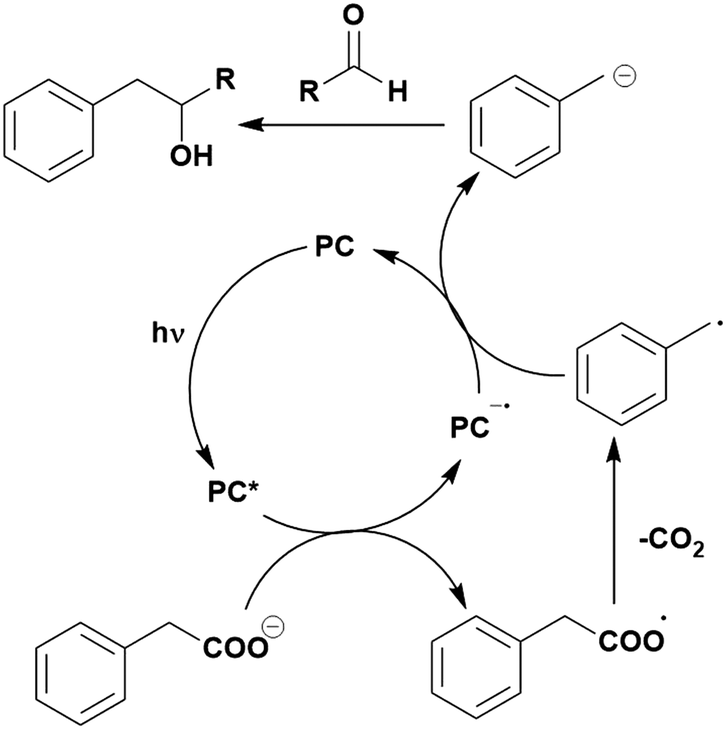 | ||
| Fig. 146 Plausible mechanism for the benzylation of aliphatic aldehydes to secondary alcohols. | ||
Moreover, an oxidative quenching mechanism may be in operation, which is exemplified by the decarboxylation of N-(acyloxy)phthalimides (Fig. 147a)333 or N-hydroxyphthalimide esters (Fig. 147b).334 The former was used as the starting reagent for the radical–radical coupling to access alkylated N-heterocycles (Fig. 148). Previously, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 had been identified as a superior photocatalyst, producing the alkylated isoquinoline in 90% yield.335 Both 4CzIPN and 2CzPN were evaluated as potential replacement PCs, producing slightly lower product yields of 84% and 80%, respectively.333 However, the CDCBs were present in a 1 mol% loading whilst the iridium photocatalyst was employed at 2 mol% loading, which may explain the slightly different yields obtained.
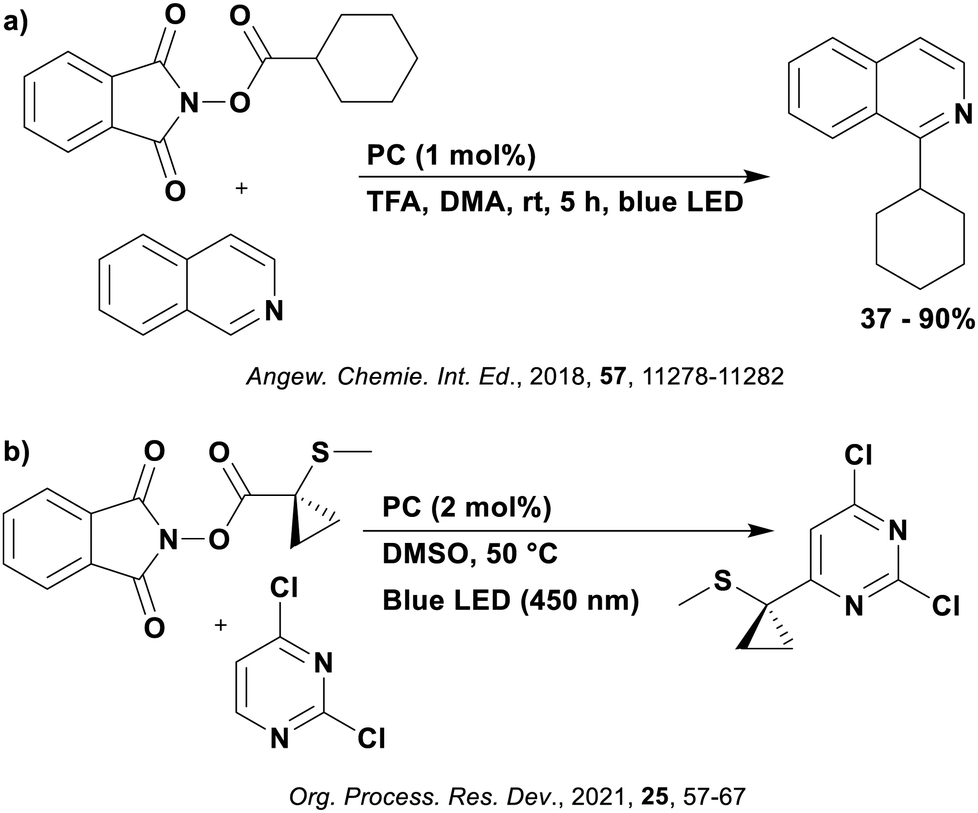 | ||
| Fig. 147 Reaction scheme for the (a) decarboxylative alkylation of heteroarenes using N-(acyloxy)phthalimides and (b) an additive free photoredox Minisci reaction. | ||
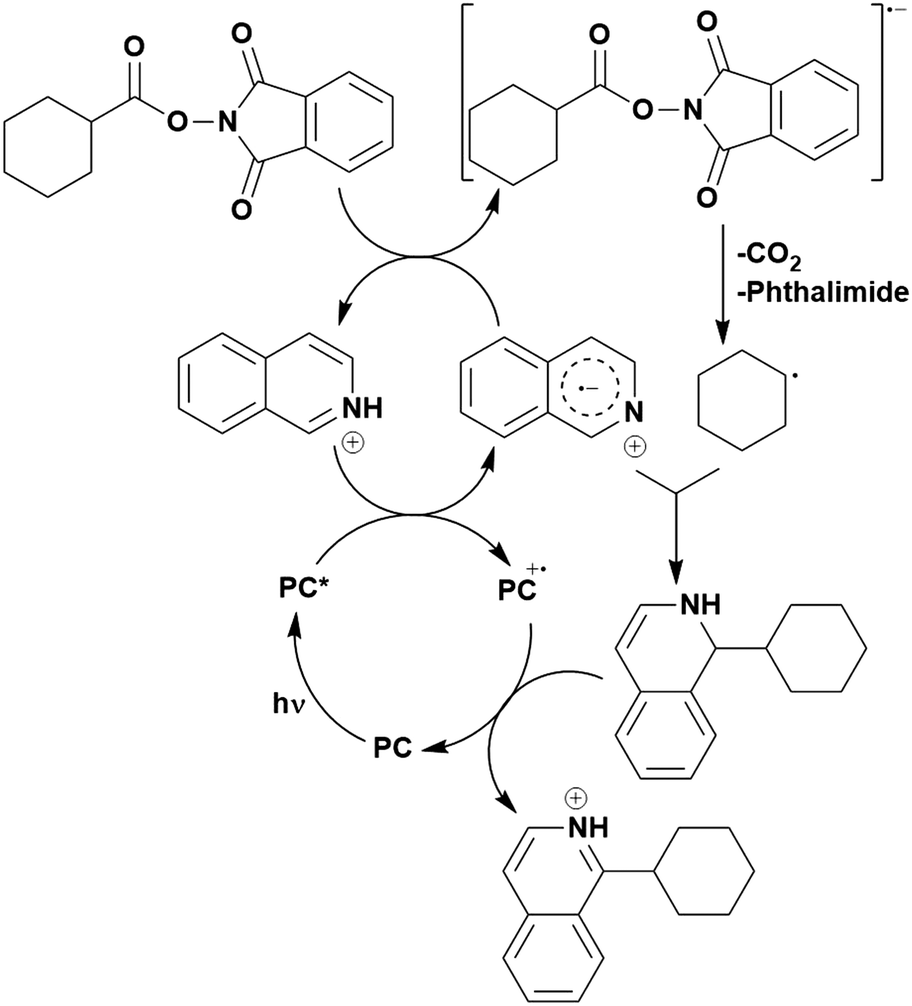 | ||
| Fig. 148 Plausible photocatalytic cycle for the decarboxylative alkylation of N-heteroarenes with N-(acyloxy)phthalimides. | ||
The latter reaction (Fig. 147b) is an example of an additive-free Minisci reaction.334 The excited PC is oxidative quenched by the N-hydroxyphthalimide ester to release a carbon-centred radical, which adds to the 2,4-dichloropyrimidine. The resulting adduct is oxidised by the oxidised PC to yield the final product. Organic TADF compounds 3CzClIPN, 5CzBN and 3DPA2FBN were investigated as PCs, providing respective product yields of 7%, 29% and 43% under the same additive-free reaction conditions. 4CzIPN also was considered and provided 26% yield, although this included the additive DIPEA. These yields generally align with the photoreducing ability of the PC (Eox* = −0.93 V, −1.42 V and −1.60 V for 3CzClIPN, 5CzBN and 3DPA2FBN, respectively), which must be capable of the reducing the N-hydroxyphthalimide ester (Ered = −1.22 V vs. SCE). The authors proposed that the addition of DIPEA results in a change to a reductive quenching mechanism, hence a discussion of the Eox* value of 4CzIPN is not relevant.
Formation of a sulfoxide by-product was observed for the 4CzIPN-DIPEA system, which occurs through oxidation of the final sulfide product (Eox = 1.90 V vs. SCE). The authors suggested that since the radical cation of 3DPA2FBN is less oxidising than that of 4CzIPN (Eox = 1.24 V and 1.52 V, respectively), this by-product cannot be formed by 3DPA2FBN, although thermodynamically, neither should be capable of initiating this SET.
Quantum yield determination of the 4CzIPN-DIPEA system and the 3DPA2FBN system gave values of 0.21 and 0.61, respectively. A radical chain process may be in operation in both cases, whereby the authors suggest that the final oxidation step to form the product can occur through either SET to the PC or to another molecule of the redox-active ester. No further investigations into the possible radical chain nature of this mechanism were conducted.
Stern–Volmer quenching experiments were conducted for 4CzIPN, 3DPA2FBN and 5CzBN; however, the authors noted that since the systems were not degassed, the measurements applied only to prompt fluorescence quenching. It should be noted that the presence of O2 does not guarantee full quenching of triplet excitons nor should it be assumed that O2 behaves benignly to singlet excitons. The Stern–Volmer quenching constant KSV with the hydroxyphthalimide ester was determined to be 2.32, 9.52 and 12.64, for 4CzIPN, 3DPA2FBN and 5CzBN, respectively, indicating that the kinetics of electron transfer do vary considerably between PCs. When correcting these KSV values with the lifetimes of the PC provided in Table 2, the quenching rate constant can be determined as 1.2 × 108 M−1 s−1, 2.3 × 109 M−1 s−1 and 7.9 × 108 M−1 s−1 for 4CzIPN, 3DPA2FBN and 5CzBN, respectively. The values align with the reported isolated yields implying both thermodynamic and kinetic factors play a role in influencing the product yield.
A similar example, although with less mechanistic clarity, involves the investigation of the impact of solvent and additives on the regioselectivity of the Minisci-type addition of amino acid-derived redox-active esters to quinolines (Fig. 149).336 More polar solvents, such as DMA or DMSO, favour the C4 addition product while less polar solvents, such as toluene or dioxane, result in the C2 addition product. No discussion of the mechanism is presented, although similar reactions proceed through an oxidative quenching cycle.337,338 Organic TADF compounds 4CzIPN, 5CzBN and 3DPAFIPN were investigated in the PC screen alongside [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 providing yields of 48%, 30%, 45% and 39%, respectively, of the C2 addition product; all PCs achieved 8–10% of the C4 addition product as the PC screen was conducted in toluene, which favours formation of the C2 addition product. No further PC screen was conducted in the more polar DMA or DMSO solvents. 4CzIPN was selected as the PC when synthesising the C4 addition product, providing yields of 32–65% and 3DPAFIPN was chosen for making the C2 addition product, forming product in 32–45% yield, although there is no justification provided for this choice given they both provided similar yields of product in the PC screen. There is no obvious correlation between the redox potentials required for an oxidative quenching cycle and the yields obtained (for example, Eox = 1.52 V, 1.41 V, 1.30 V, 1.69 V and Eox* = −1.04 V, −1.42 V, −1.38 V, −0.89 V for 4CzIPN, 5CzBN, 3DPAFIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively).
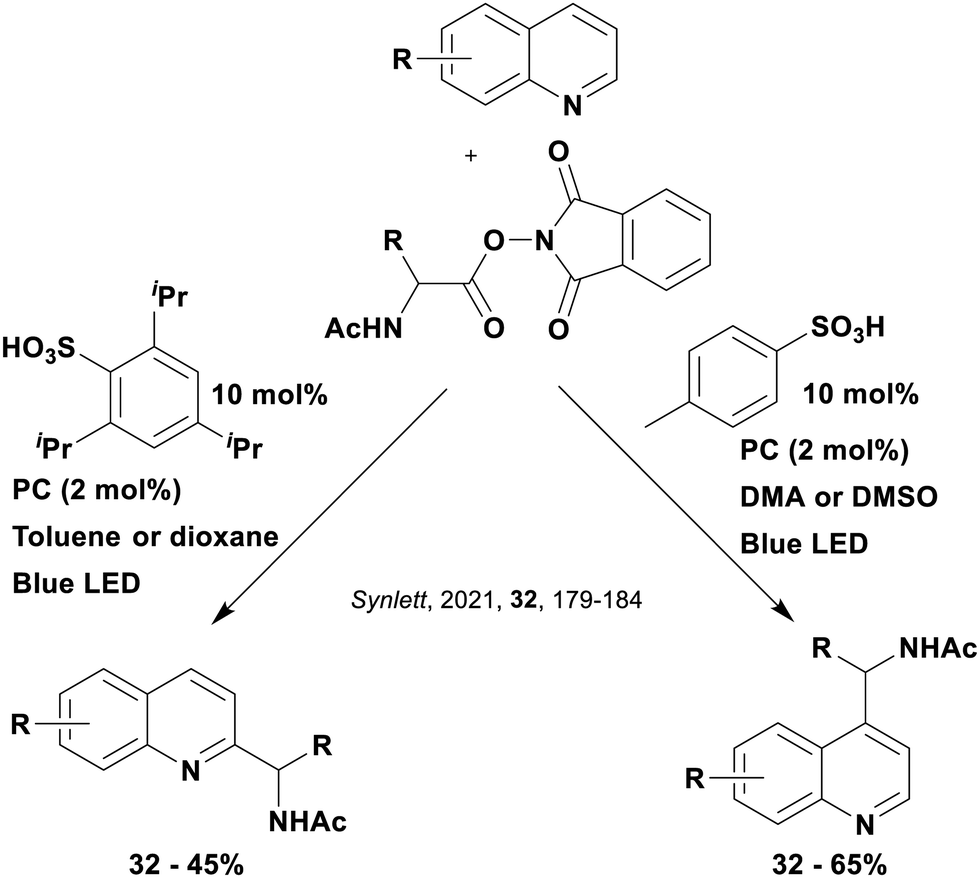 | ||
| Fig. 149 Reaction scheme for the investigation into the regioselectivity of a Minisci type addition reaction when altering solvent and additives. | ||
Alternative radical precursors in the formation of C–C and C–X bonds
Utilisation of alternative radical precursors in the formation of C–C and C–X bonds is also possible using TADF compounds as photocatalysts, for example, in the arylation of arylidene malonates using cyanoarenes (Fig. 150a)339 or the addition of malonates to alkenes (Fig. 150b).340 For the former, the proposed mechanism involves two photocatalytic cycles (Fig. 151), with the excited PC being quenched in both cases by a sacrificial electron donor, TEA, as confirmed by Stern–Volmer quenching experiments. The radical cation generated on TEA exists in equilibrium with the α-amino radical, which itself can be deprotonated by neutral TEA. Both TEA or protonated TEA can engage in the PCET mechanism with the arylidene malonate. The reduced photocatalyst is then used to reduce both the arylidene malonate and the protonated cyanoarene, with these resultant radicals coupling together, and following elimination of HCN, generating the final arylated product. From the proposed mechanism, two photons are required (one for each catalytic cycle), to generate one molecule of product hence a quantum yield <0.5 would act in support of this closed photocatalytic mechanism proposed; the experimentally determined quantum yield of 0.28 provides some corroboration. An inverse secondary kinetic isotope effect is also observed for the formation of the arylidene malonate radical as expected due to the change in hybridisation from sp2 to sp3 upon reduction, again acting as corroboration of the proposed mechanism.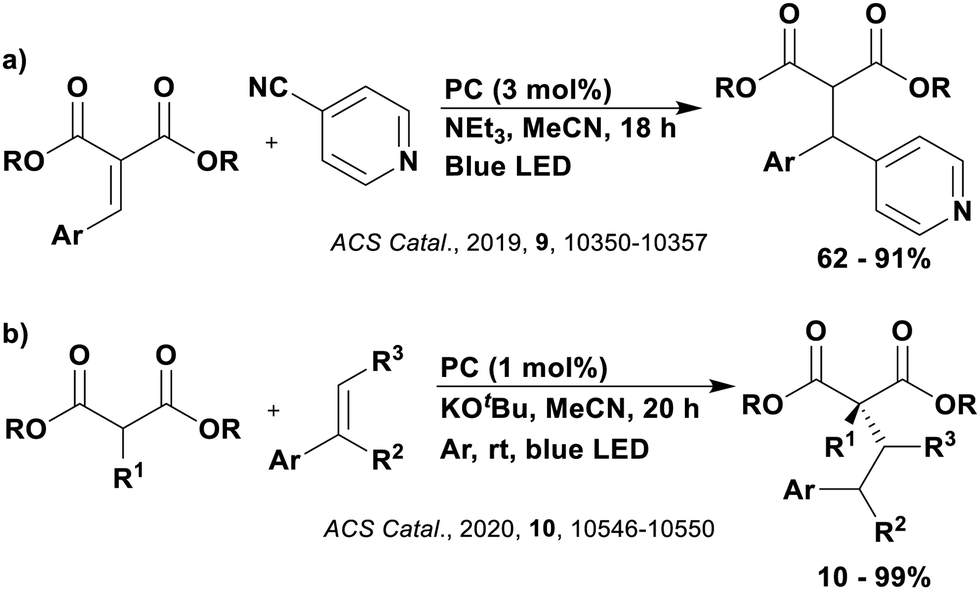 | ||
| Fig. 150 Reaction schemes for the (a) arylation of arylidene malonates and (b) reaction of malonates with alkenes. | ||
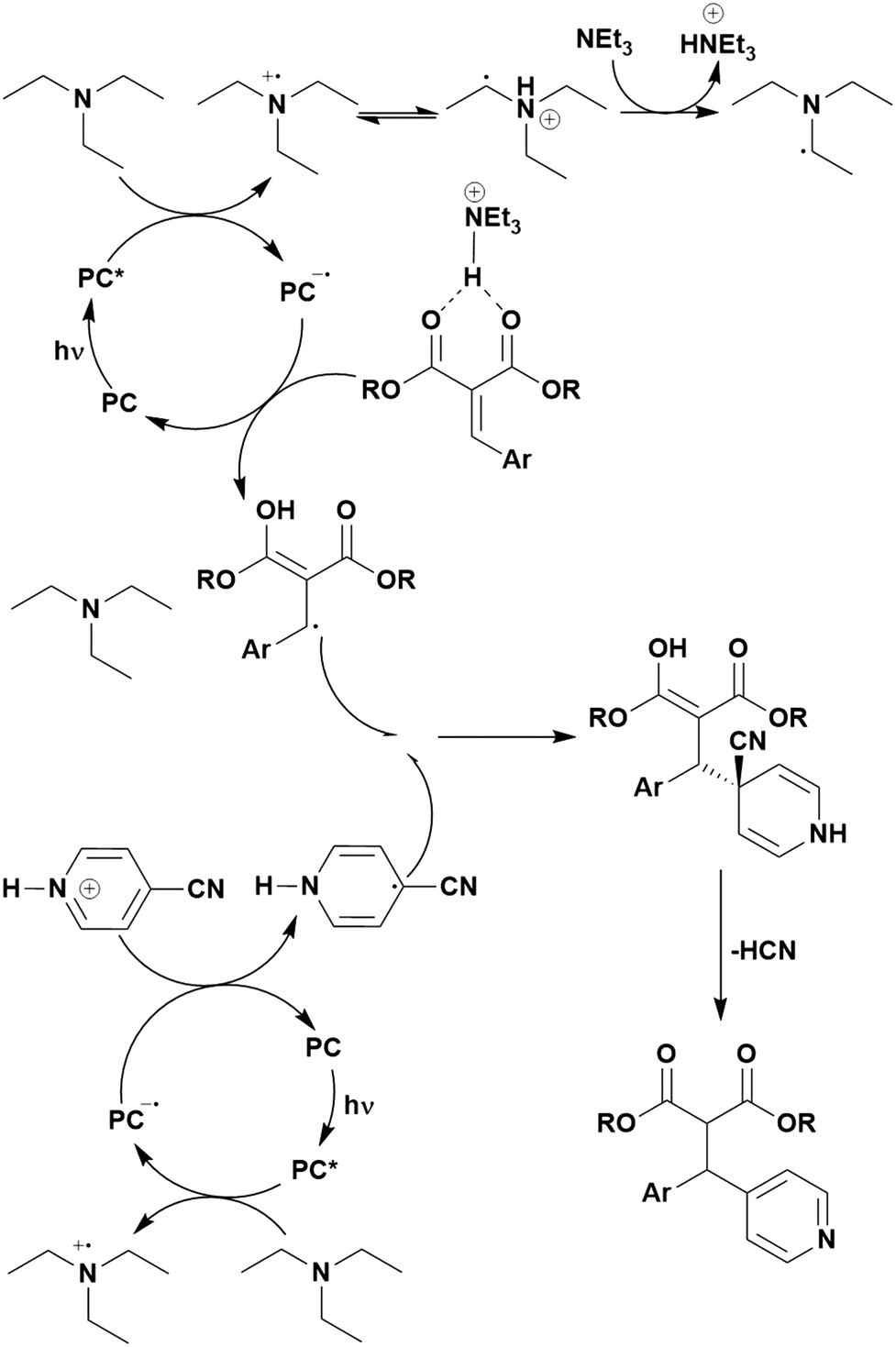 | ||
| Fig. 151 Proposed mechanism for the arylation of arylidene malonates. | ||
High throughput experimentation (HTE) was used to quickly identify optimal conditions, with 4DPAIPN being the only TADF compound considered. A high yield of 85% was obtained with 4DPAIPN and TEA as the sacrificial electron donor in MeCN, with the next highest yield of 70% being obtained with [Ir(ppy)2(dtbbpy)]PF6 under the same conditions. Other photocatalysts under these conditions provided little (33% with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6) to no yield (0% with fac-Ir(ppy)3 and [Ru(bpy)3](PF6)2. These results can be rationalized by considering the ground state reducing capacity of the PCs, which correlates with the final product yield (Ered = −1.52 V, −1.51 V, −1.37 V −1.33 V for 4DPAIPN and [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2). Although fac-Ir(ppy)3 does have an appropriate ground state reduction potential (Ered = −2.19 V), it has very weak excited state oxidising capacity (Eox* = 0.31 V), which may explain why it generates no product, especially since TEA has an oxidation potential of Eox = 0.83 V.160
Upon identification of the optimal conditions in terms of photocatalyst, additives and solvent, additional conditions were investigated, for example the impact of the light irradiation power on the reaction yield. The irradiation intensity is based on the intensity control allowed on a Kessil PR160 456 nm light, with low intensity (irradiation: 25) giving an average yield of 79%, medium intensity (irradiation 75, standard conditions) providing an average yield of 87% and high intensity (irradiation: 100) resulting in 88% average yield. As expected, a higher yield is observed when increasing the intensity from low to medium; however, the yield seems to plateau upon further increase.
In the reaction of malonates with alkenes (Fig. 150b), the proposed mechanism is simpler in that only one photocatalytic cycle is suggested.340 The excited PC is reductively quenched by the malonate enolate tautomer, with the resultant radical adding to the alkene to form a benzyl radical. The reduced PC is then invoked to reduce this benzyl radical, which after protonation, yields the final product. Radical trapping experiments with TEMPO provided evidence for the radical nature of the proposed mechanism. Only organic PCs were considered in this reaction and in the initial PC screen, 4CzIPN was the only CDCB-based structure. 4CzIPN vastly outcompeted the other PCs, yielding 87% of product while the next best PC, eosin y, could achieve only 5%. The enolates of malonates typically have low oxidation potentials (e.g., the enolate of diethyl malonate has Eox = 0.59 V vs. SCE), thus it is likely that the step involving regeneration of the PC is the more thermodynamically challenging step, and the one best correlated to the reaction yield (Ered = −1.21 V and −1.06 V for 4CzIPN and eosin Y, respectively). This hypotehesis is supported by the lower yields obtained when the benzyl radical contained electron-donating groups rather than electron-withdrawing groups, as the electron-donating groups cause the benzyl radical to be more difficult to reduce. For example, when using p-MeOC6H4 as the arene, the benzyl radical has a reduction potential of Ered = −1.82 V, and a yield of 0% was obtained with 4CzIPN. By contrast, the benzyl radical of p-NCC6H4 has a reduction potential of Ered = −0.77 V and consequently a yield of 56% was obtained. For the arenes bearing electron-donating groups, 3DPA2FBN was instead employed as the PC, and higher yields could be obtained (e.g., 28% yield of product when using p-MeOC6H4) on account of its greater reducing capacity in the ground state (Ered = −1.92 V).
Cyanoarenes have also been used in conjunction with oximes and iminium chlorides for primary amine synthesis (Fig. 152a and b, respectively),341 as well as in reaction with primary or secondary amines to form heteroarylamines (Fig. 152c).342 High throughput experimentation was used to determine the optimal conditions when using oximes as the coupling partner, with 4CzIPN and 4DPAIPN being the TADF PCs under investigation. The best photocatalyst was identified as [Ir(dF(Me)ppy)2(dtbbpy)]PF6 giving a 71% yield with DIPA, while 4CzIPN and 4DPAIPN managed only 2% and 4%, respectively. For oxime reactants, the mechanism is proposed to involve both photocatalytic electron and energy transfer, with triplet–triplet sensitisation of the oxime being suggested. Photocatalysts with a triplet energy higher than that of the oxime (ET ≈ 2.34 eV, 226 kJ mol−1 for o-benzoyl oxime under investigation) are capable of undergoing the triplet sensitisation process, for example [Ir(dF(Me)ppy)2(dtbbpy)]PF6 has a triplet energy of ET = 2.42 eV (233 kJ mol−1). However, this doesn’t provide an explanation for the lack of success with 4CzIPN, which has a triplet energy ET = 2.52 eV (243 kJ mol−1).304
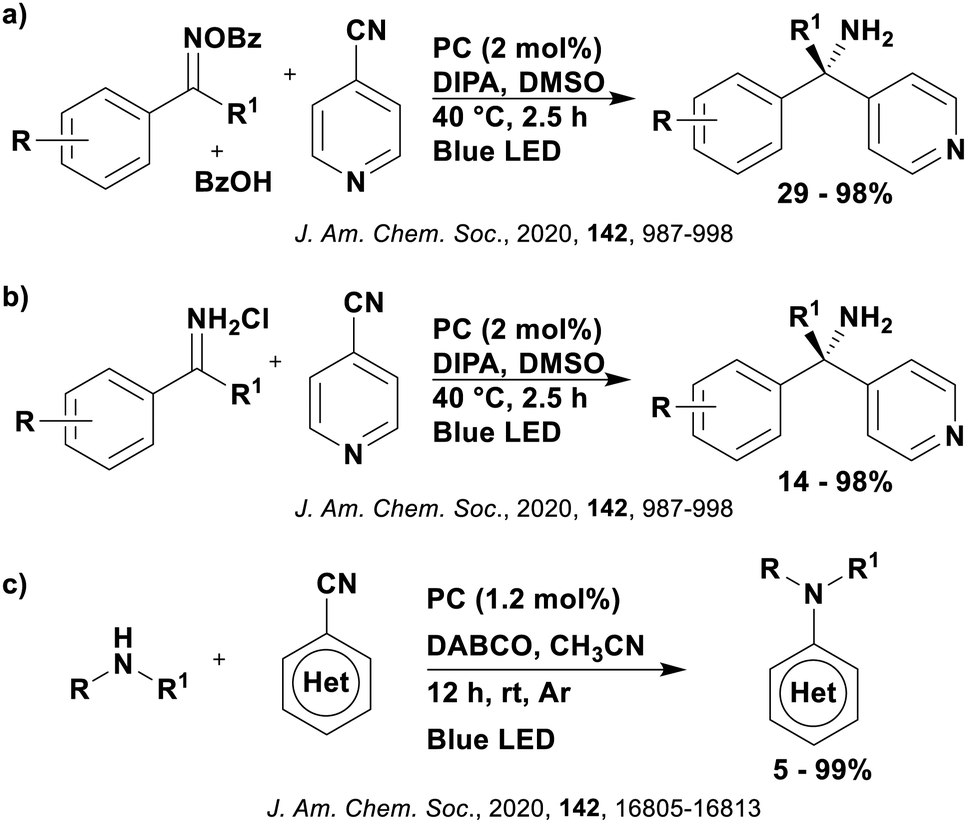 | ||
| Fig. 152 Reaction scheme for the reaction of cyanoarenes with (a) oximes, (b) iminium chlorides and (c) amines. | ||
The proposed mechanism for the coupling of cyanoarenes with amines (Fig. 152c) is somewhat similar to that shown in Fig. 151 in that the PC is reductively quenched by the amine reagent and the reduced PC is used to reduce the cyanoarene.342 These two radicals then couple together, releasing HCN, to form the final product. A range of spectroscopic techniques were used to support these mechanistic studies, including flash vacuum photolysis and transient absorption spectroscopy. 3DPAFIPN provided the highest yield in the PC screen and thus was subject to this spectroscopic analysis. Upon excitation of the PC, a characteristic absorption at 590 nm was observed and assigned to the triplet excited state of the photocatalyst (3PC*). After addition of the amine reagent, this band decreased in favour of two new absorption bands at 308 nm and 466 nm, which correspond to the amine radical cation and PC˙−, suggesting the triplet state is responsible for the SET. Addition of the cyanoarene consequently resulted in quenching of the 466 nm band. The decrease in the lifetimes of 3PC* when in the presence of the amine (from 10.0 μs to 4.1 μs in MeCN) and of PC˙− when in the presence of the cyanoarene (from 109.1 μs to 12.6 μs in MeCN), were used to determine the rate of electron transfer as 5.1 × 108 M−1 s−1 for SET between the amine and PC*, and 7.0 × 107 M−1 s−1 for the SET between PC˙− and the cyanoarene.
3DPAFIPN, 4DPAIPN and 4CzIPN were tested as the PCs, providing yields of 99%, 99% and 0%, respectively. Transient absorption spectroscopy indicated that 4CzIPN could be reductively quenched by the amine reagent, as indicated by the new absorption band at 465 nm; however, upon addition of the cyanoarene, no change in this absorption band was observed, implying that 4CzIPN˙− could not reduce the cyanoarene, despite having suitable redox potentials (Ered = −1.21 V for 4CzIPN and −0.87 V for p-cyanopyridine) in the presence of the amine radical cation.
fac-Ir(ppy)3 was also investigated as a PC in this reaction and provided a yield of 88%, although this PC was shown to proceed through an oxidative quenching pathway.
Alkyl radicals generated from silicate or trifluoroborate radical precursors can add to olefins that are embedded within bicyclic scaffolds. Radical polar crossover can ensue to generate polycarbocyclic and polyheterocyclic cyclopropanes (Fig. 153).343 The mechanism is thought to proceed as shown in Fig. 43, although no mechanistic studies were undertaken. Organic PCs 4CzIPN and 4CzIPN-Cl were considered in this reaction alongside iridium and ruthenium complexes. The ratio of product to internal standard (IS) as determined by HPLC was 1.31 and 1.40 for 4CzIPN and 4CzIPN-Cl, respectively, which was lower than that obtained for [Ru(bpy)](PF6)2 (1.63) and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1.52). However, the ratio of reactant to the IS was also found to be lower for the organic PCs (0.27, 0.77, 1.11 and 1.14 for 4CzIPN, 4CzIPN-Cl, [Ru(bpy)](PF6)2 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), suggesting a higher conversion was possible for the organic PCs. Since the photooxidising ability of the organic PCs is higher (Ered* = 1.35 V, 1.71 V, 0.77 V and 1.21 V for 4CzIPN, 4CzIPN-Cl, [Ru(bpy)](PF6)2 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), perhaps this may be why they achieve higher conversion of the reagents, although this does not explain why they achieve a lower product ratio.
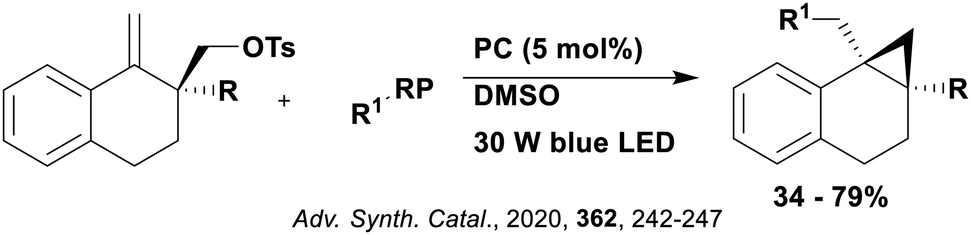 | ||
| Fig. 153 Reaction scheme for the generation of polycarbocyclic and polyheterocyclic cyclopropanes. RP = radical precursor. | ||
Radical precursors involving DHPs have typically been used to form alkyl radicals; however, they can also be used to form carbamoyl radicals that can be added to olefins (Fig. 154).344 Reductive quenching of the PC by the 4-carboxamido-Hantzsch esters releases the nucleophilic carbamoyl radical after aromatization-driven fragmentation. This radical adds to the electron-deficient olefin, the product of which is reduced by the reduced PC. Protonation of the resultant carbanion yields the final product. This proposed mechanism was supported by Stern–Volmer quenching studies, which showed that the luminescence quenching by the 4-carboxamido-Hantzsch ester was much faster than by the olefin (kq = 9.22 × 1010 M−1 s−1 and 2.96 × 109 M−1 s−1, respectively). Addition of TEMPO inhibited the reaction, instead forming an adduct with the carbamoyl radical. Only three PCs were investigated in the PC screen: 3DPAFIPN (80%), 3DPA2FBN (78%) and [Mes-Acr]ClO4 (65%) with 3DPAFIPN being selected for the remainder of the study. The origin behind the TADF compounds ability to outcompete [Mes-Acr]ClO4 may be linked to the relative ground state reduction potentials of the PC (Ered = −1.59 V, −1.92 V and −0.57 V for 3DPAFIPN, 3DPA2FBN and [Mes-Acr]ClO4, respectively).
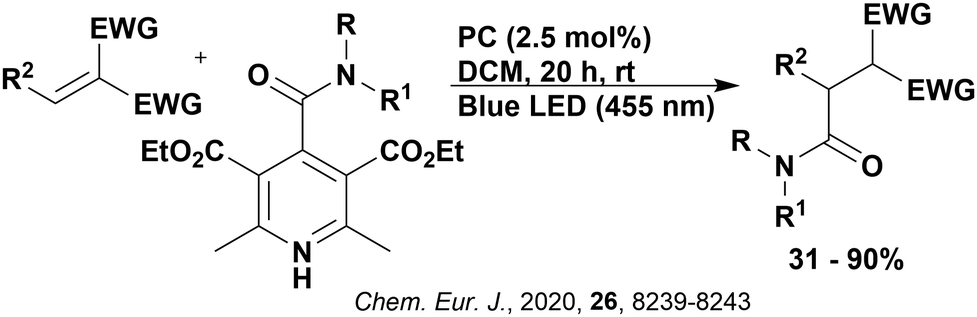 | ||
| Fig. 154 Reaction scheme for the addition of carbamoyl radicals to olefins. | ||
Additionally, sodium trifluoromethylsulfinate may be used as radical precursor, which was used in the difunctionalisation of vinyl ureas (Fig. 155).345 The excited photocatalyst is proposed to oxidise the trifluoromethylsulfinate anion, yielding the electrophilic trifluoromethyl radical (Fig. 156). This radical adds to the double bond on the urea, forming a tertiary radical which is reduced by the reduced PC. The carbanion formed then undergoes intramolecular nucleophilic aromatic substitution to produce the final difunctionalised product. Radical inhibition experiments with TEMPO confirm this is a radical process and radical trapping with 1,1-diphenylethylene demonstrated the presence of the trifluoromethyl radical. Stern–Volmer quenching experiments confirmed that the excited PC is quenched more efficiently by the trifluromethylsulfinate salt rather than the urea, while a quantum yield of 0.18 suggests that the reaction is not a radical chain mechanism. Both 4CzIPN and 3DPAFIPN were considered as TADF photocatalysts, with the former greatly outperforming the latter (89% yield and trace yield, respectively). Only [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 provided some competition for 4CzIPN, obtaining 70% yield. This difference in yield is reflective of the photooxidising ability of the photocatalyst (Ered* = 1.35 V, 1.21 V and 1.09 V for 4CzIPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 3DPAFIPN, respectively), which needs to be sufficiently high to oxidise CF3SO2Na (Eox = 1.05 V).
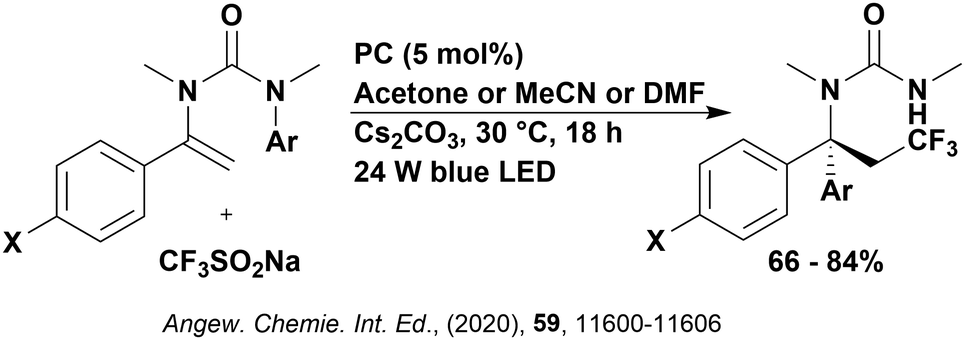 | ||
| Fig. 155 Reaction scheme for the difunctionalisation of vinyl ureas. | ||
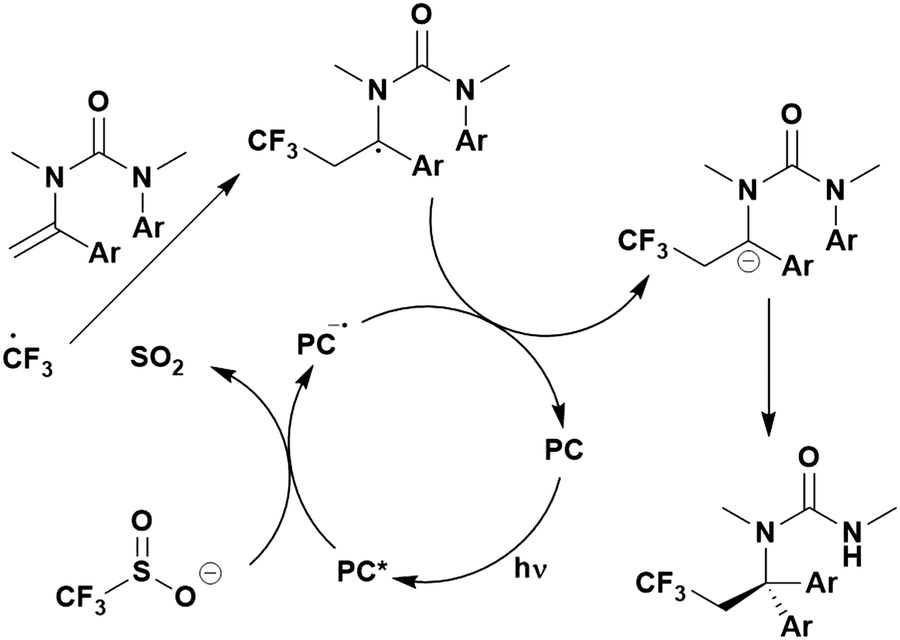 | ||
| Fig. 156 Suggested mechanism for the difunctionalisation of vinyl ureas. | ||
Furthermore, cyanation of aryl or heteroaryl bromides is possible using photocatalysis (Fig. 157).346 The excited PC is proposed to oxidise tris(trimethylsilyl) silanol (supersilanol), which forms a silicon-centred radical that can abstract a bromine from an aryl bromide (Fig. 158). The resultant weakly nucleophilic aryl radical adds to tosyl cyanide (TsCN) and, through a Barton nitrile transfer, yields the final product,347,348 as well as the tosyl radical, which is reduced by the reduced photocatalyst. The mechanism was corroborated by addition of TEMPO, which suppressed formation of the expected product, instead yielding an aryl-TEMPO adduct, confirming the formation on an aryl radical. Only organic TADF compounds were considered as photocatalysts, providing yields of 71%, 55% and 0% for 4CzIPN, 4CzTPN and 4DPAIPN, respectively. This difference in yield is correlated with the differing photooxidising ability of the PCs. Oxidation of supersilanol is quite challenging (Eox = 1.54 V vs. SCE in MeCN)349 and only strongly photooxidising PCs can complete the required SET (Ered* = 1.35 V, 1.41 V and 1.1 V for 4CzIPN, 4CzTPN and 4DPAIPN, respectively), which explains the absence of yield for 4DPAIPN. The lower yield obtained by 4CzTPN may be related to the ground state reduction potential (Ered = −1.21 V and −1.02 V for 4CzIPN and 4CzTPN, respectively), which must be sufficiently negative to reduce the tosyl radical (Ered = −0.50 V vs. SCE in MeCN);264,350 hence, this process is more thermodynamically favourable for 4CzIPN.
 | ||
| Fig. 157 Reaction scheme for the photocatalytic cyanation of arenes. | ||
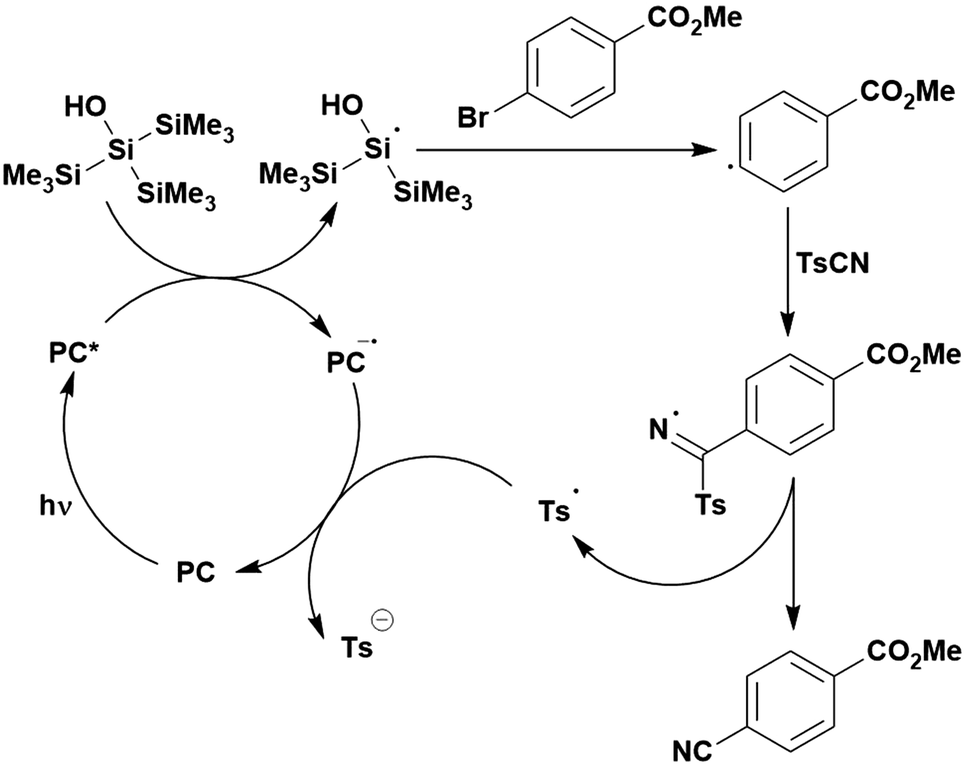 | ||
| Fig. 158 Proposed mechanism for the photoredox mediated cyanation of aryl bromides via silyl radical mediated bromine abstraction. | ||
Organic halides can be used as radical precursors for the formation of C–P bonds to produce asymmetrical phosphines and phosphonium salts (Fig. 159). The mechanism involves reductive quenching of the PC by sacrificial electron donors like NEt3 or DIPEA, followed by reduction of an aryl or alkyl iodide to close the photocatalytic cycle. The resultant radical abstracts a hydrogen atom from the phosphine to yield phosphorus-centred radicals, which homocouples to form a diphosphine compound. Additional aryl or alkyl radicals, formed from the excess of organohalide, attack the P–P bond to yield the tertiary phosphine product. Upon addition of TEMPO, product formation is inhibited, providing evidence that this is a radical process. Further confirmation of the presence of organic radicals was provided by EPR spectroscopy. Monitoring of the reaction using 31P{1H} NMR spectroscopy indicated the consumption of the original phosphine and the formation of the self-coupled diphosphine species. Finally, Stern–Volmer experiments indicated that DIPEA (or NEt3) was responsible for the quenching of the excited PC.
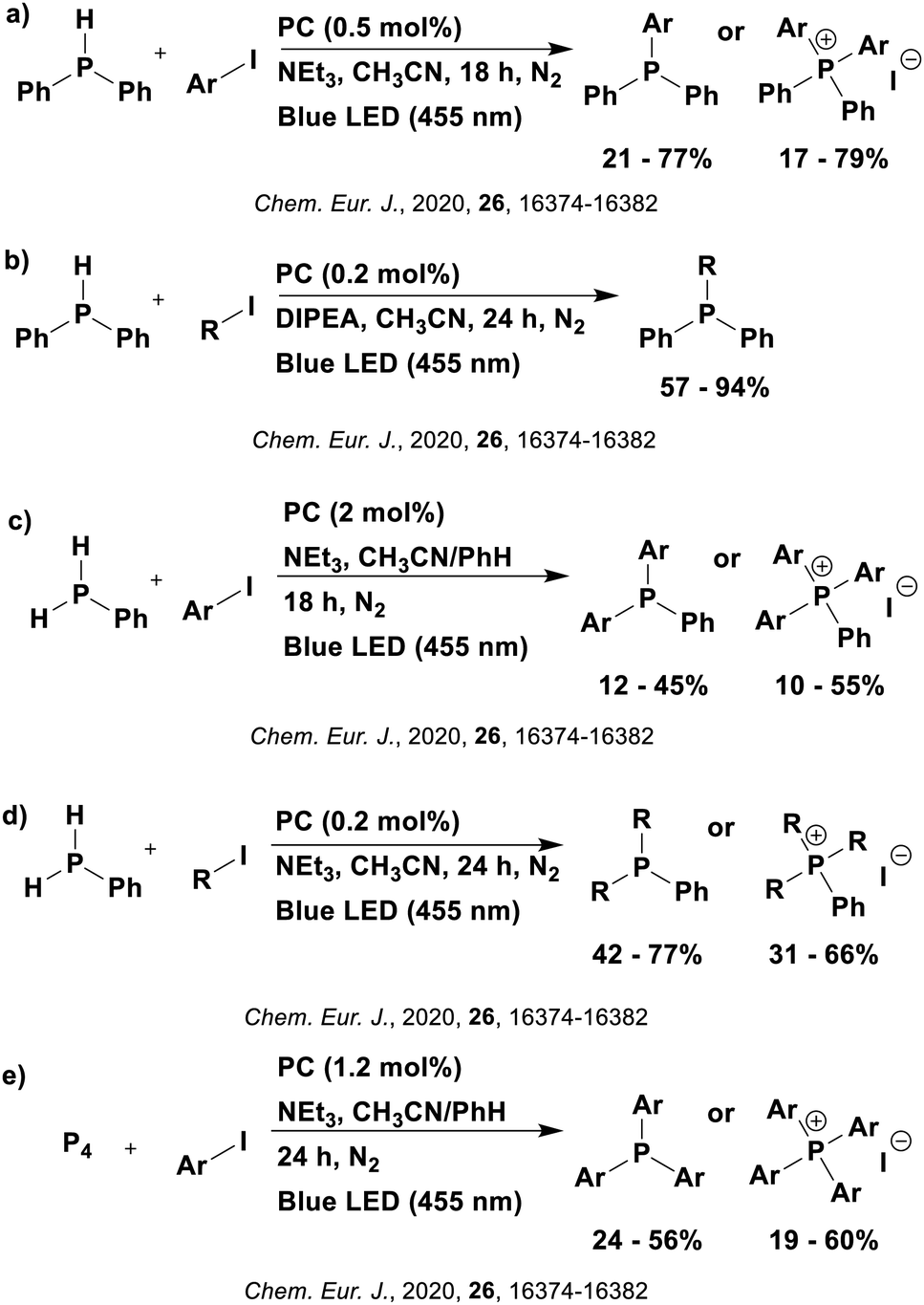 | ||
| Fig. 159 Reaction scheme for the synthesis of phosphines and phosphonium salts using photocatalysis. | ||
A photocatalyst screen was conducted for the functionalisation of P4 with aryl iodides to yield the respective phosphonium salt (Fig. 159e). 3DPAFIPN was identified as the optimal PC as it outperformed the other organic TADF PCs in the study, yielding 60% of product while 4CzIPN, 5CzBN, 4DPAPN, 4DPAIPN and 3DPA2FBN all yielded between 11–22%; 3DPAClIPN, 5CzBN-OMe and 4CzPN did not lead to product formation. The PCs 4CzIPN, 3DPAClIPN, 5CzBN, 5CzBN-OMe and 3DPA2FBN instead yielded between 8–21% of the tertiary arylated phosphine. No comment is provided by the authors as to the variance in product formation or yield.
Additionally, with the aid of benziodoxolones (EBXs), the 1,2-oxyalkynylation of ene-carbamates and enol ethers is possible to synthesise 1,2-amino alcohols and diols (Fig. 160).351 A reductive quenching cycle is proposed to be in operation (Fig. 161), in which SET from the ene-carbamate to the excited PC occurs, supported by Stern–Volmer quenching experiments. The resultant radical is trapped by the carboxylate radical from EBX, with the product adding to a second molecule of EBX to yield the final functionalised product, alongside the iodanyl radical. This iodanyl radical oxidises the reduced PC to close the photocatalytic cycle and form the previously mentioned carboxylate radical. The authors additionally propose that 1-acetoxy-1,2-benziodoxol-3-(1H)-one (BlOAc) acts to oxidatively quench the excited PC, releasing more of the iodanyl radical species. The oxidised PC can then also be used to oxidise the ene-carbamate. This hypothesis was supported by the observation that in the absence of BlOAc, the yield decreases (65% after 5 days, compared to 89% after 18 h in the presence of BlOAc).
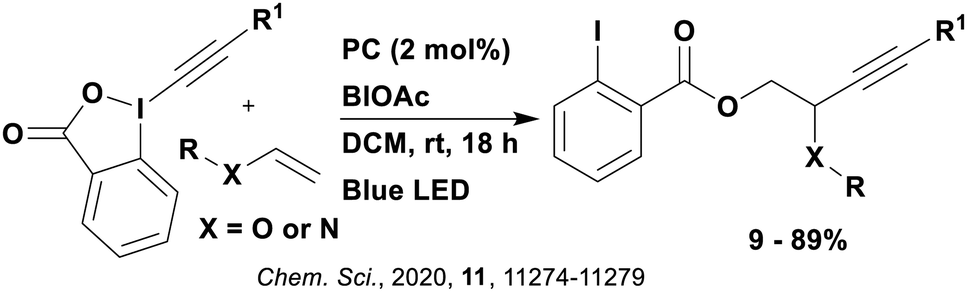 | ||
| Fig. 160 Reaction scheme for the 1,2-oxyalkynylation of ene-carbamates and enol ethers. | ||
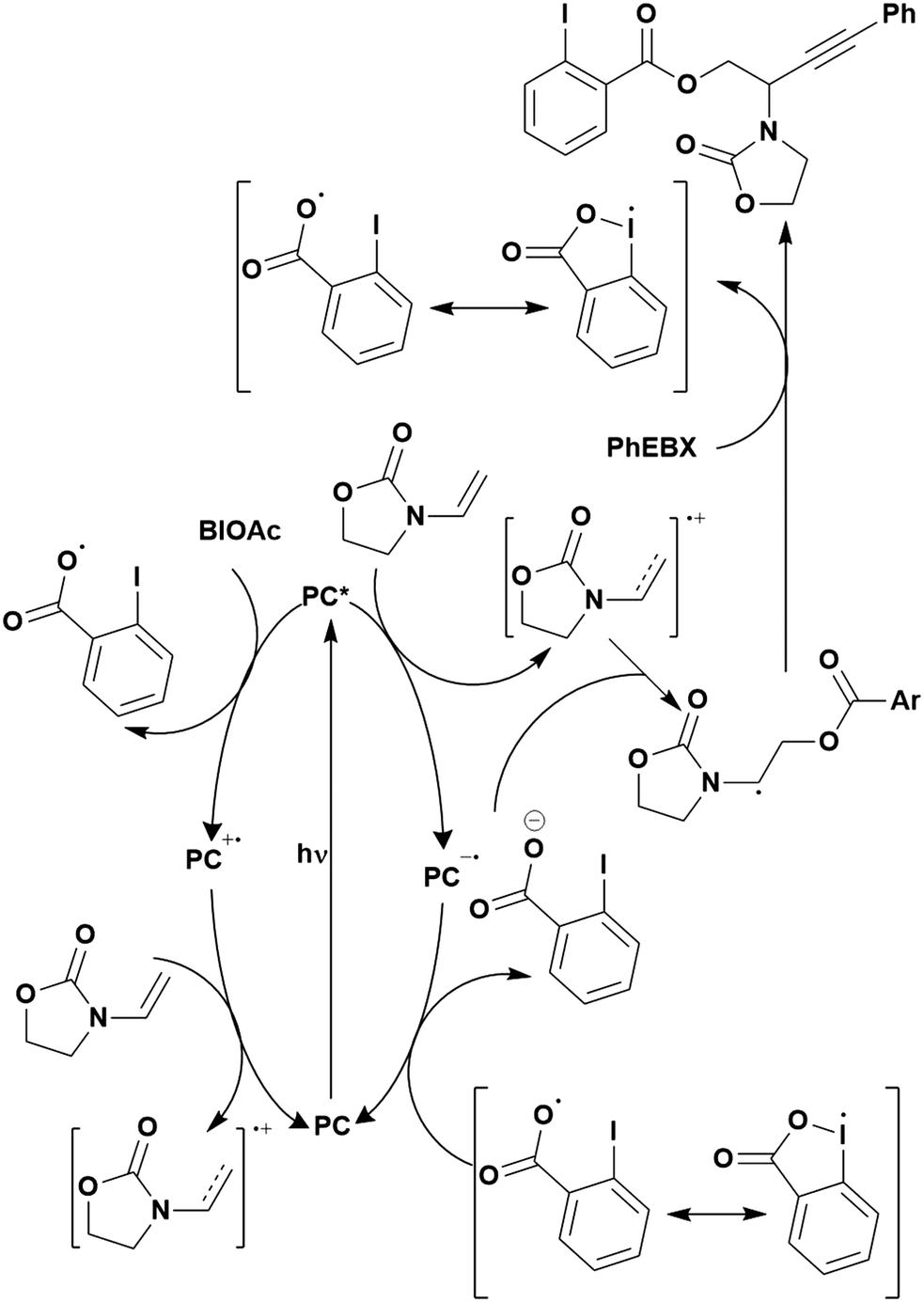 | ||
| Fig. 161 Viable mechanism for the 1,2-oxyalkynylation of ene-carbamates. | ||
4CzIPN, 4CzIPN-Cl and [Mes-Acr]ClO4 were considered as PCs and in the absence of additives, yielded 30%, 42% and 5%, respectively. Thus, additional reaction optimisation was initiated with 4CzIPN-Cl and a yield of 80% was obtained in the presence of BlOAc. This outcome was then compared with the use of organometallic PCs, with [Ru(bpz)3](PF6)2 (21%) and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (24%) affording significantly lower product yield in the presence of BlOAc. Since the ene-carbamate, N-vinyloxazolidinone, has an oxidation potential of Eox = 1.30 V vs. SCE, it is unsurprising that some PCs may struggle with this transformation (Ered* = 1.35 V and 1.21 V for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively, in comparison with Ered* = 1.71 V for 4CzIPN-Cl). Although this SET should be facile for both [Mes-Acr]ClO4 and [Ru(bpz)3](PF6)2 (Ered* = 2.06 V and 1.45 V, respectively), their lower yields may be linked to their more anodically shifted ground state reduction potentials (Ered = −0.57 V and −0.80 V, respectively) in comparison to 4CzIPN-Cl (Ered = −0.97 V).
Finally, C–H bonds can be constructed using this form of photocatalysis, as in the hydrodefluorination of trifluoromethylarenes (Fig. 162).352 The proposed mechanism, shown in Fig. 163, suggests the thiolate anion generated by deprotonation of 4-hydroxythiophenol (4-HTP) under basic conditions, is capable of reductively quenching the excited PC. The reduced PC then reduces the trifluoromethylarene to form a radical anion, which undergoes mesolytic cleavage of fluoride. The resultant C-centred difluorobenzylic radical is trapped by 4-HTP to form the desired product. Stern–Volmer quenching experiments indicated that a 1:1 combination of TMP:4-HTP was responsible for emission quenching of the PC. Addition of TEMPO resulted in the formation of an adduct between the C-centred difluorobenzylic radical and TEMPO, providing evidence for the formation of the radical species. The presence of this radical was also confirmed by the product identified upon addition of styrene to the reaction mixture. Moreover, utilisation of d2-4-HTP resulted in incorporation of deuterium into the product, suggesting 4-HTP as the hydrogen atom donor. High throughput screening of the PC was conducted, with 28 PC considered. Of these, 13 were from the TADF CDCB family: 2CzPN, 2CzIPN, 2CzTPN, 4CzPN, 4CzIPN, 4CzIPN-OMe, 4CzTPN, 5CzBN, 5CzBN-OMe, 3CzClIPN, 4DPAIPN, 3DPAClIPN and 3DPAFIPN. Specific individual yields for each PC were not provided, but from the graphical results of the PC screen, 4DPAIPN was clearly identified as the best PC, with fac-Ir(ppy)3 being the next best. The only other TADF PCs to provide any significant product yield were 5CzBN-OMe, 3DPAClIPN and 3DPAFIPN. For further optimisation studies, the respective yields of 4DPAIPN and fac-Ir(ppy)3 were provided as 62% and 53%, respectively, which is likely linked with the photooxidising ability of the PC (Ered* = 1.10 V and 0.31 V for 4DPAIPN and fac-Ir(ppy)3, respectively). The failure of the other PCs to promote the reaction is probably linked to their ground state reduction potentials, which must be significantly reducing to reduce the trifluormethylarene, where, for example, 4-(trifluoromethyl)benzonitrile has Ered = −1.79 V.
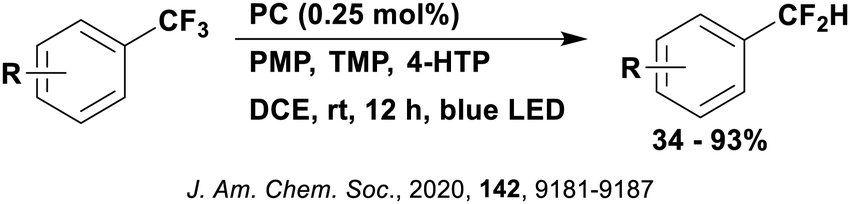 | ||
| Fig. 162 Reaction scheme for the hydrodefluorination of trifluromethylarenes where PMP = 1,2,2,6,6- pentamethylpiperidine, TMP = 2,2,6,6-tetramethylpiperidine and 4-HTP = 4-hydroxythiophenol. | ||
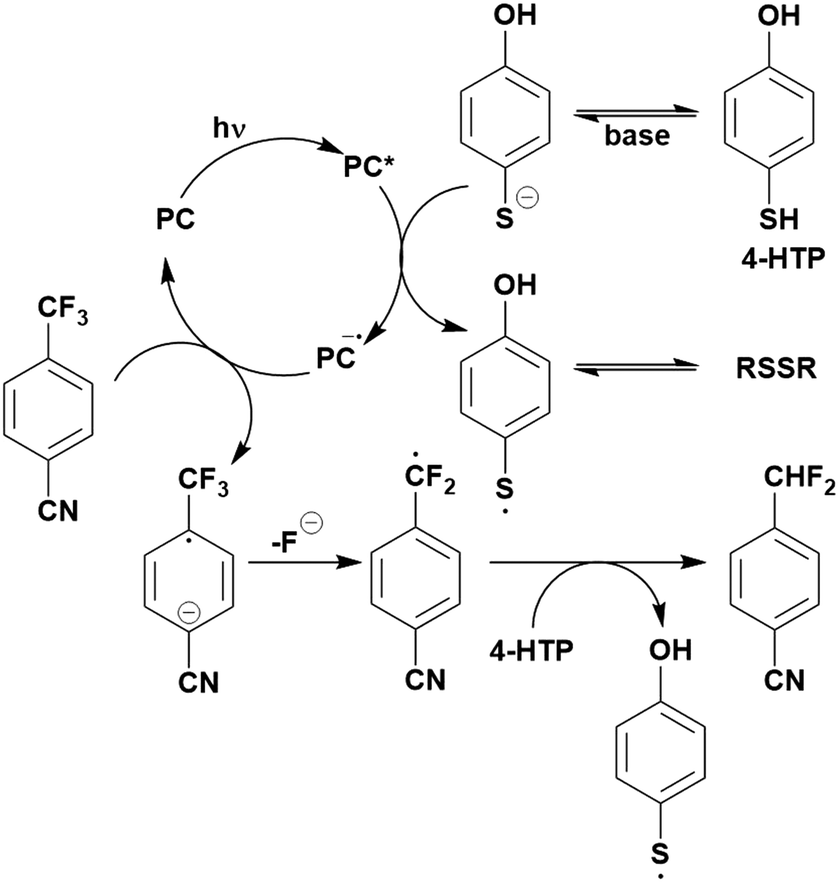 | ||
| Fig. 163 Plausible mechanism for the hydrodefluorination of trifluoromethylarenes. | ||
Bromination
The photocatalytic bromination of anisole has also been investigated using a TADF photocatalyst (Fig. 164), whereby the crucial oxidation step (Eox (MeOAr/MeOAr˙+) = 1.79 V) is particularly challenging,114 hence requiring a strongly oxidising photocatalyst. Impressively, 3CzClIPN was demonstrated to perform comparably to Fukuzumi's catalyst Mes-Acr-Me+ (89% and 87%, respectively).114 There are two potential mechanisms (Fig. 165) for this transformation, although the oxidative quenching mechanism is hypothesized to be in operation as a result of the redox potentials of 3CzClIPN. The excited state reduction potential of 3CzClIPN (Ered* = 1.56 V vs. SCE) is slightly too low to initiate the required SET while the ground state oxidation potential (Eox = 1.79 V vs. SCE) is sufficient.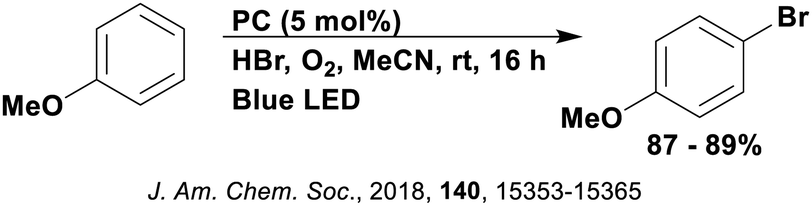 | ||
| Fig. 164 Reaction scheme for the bromination of anisole. | ||
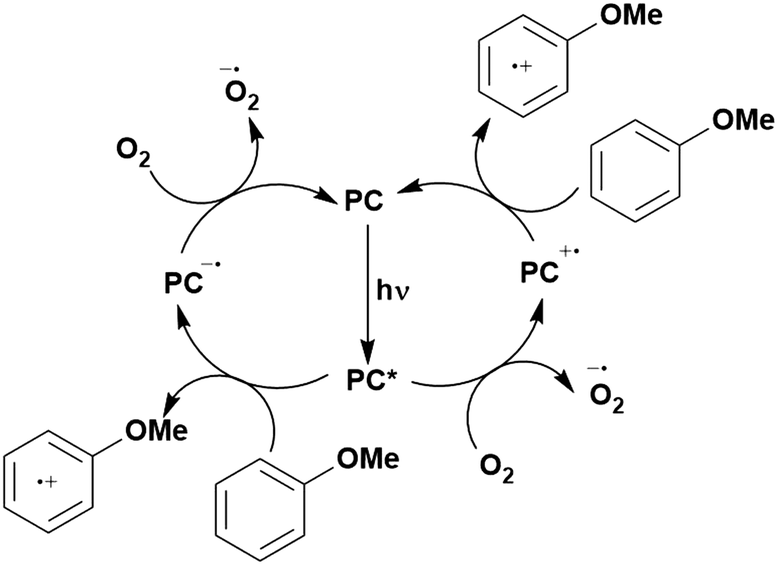 | ||
| Fig. 165 Oxidative and reductive quenching cycles possible for the photocatalytic bromination of anisole. | ||
Cyanation
Conversion of benzyl alcohols and methylated arenes to benzonitriles using sodium azide (Fig. 166)353 is also possible using photoredox catalysis. A reductive quenching mechanism is invoked in which the azide is proposed to undergo SET to the excited PC, forming an azidyl radical (Fig. 167). Closure of the photocatalytic cycle proceeds by oxidation of the reduced PC by O2. Meanwhile, the azidyl radical abstracts a hydrogen atom from the Lewis acid-coordinated benzyl alcohol, generating an α-alkoxy radical, which in the presence of oxygen, forms an aldehyde. This benzaldehyde reacts in a Schmidt reaction with hydrazoic acid, followed by loss of water and N2 to yield the final benzonitrile. The reactions proceed in similar way with toluene derivatives where the azidyl radical abstracts a proton from the weak benzylic C–H bond to form a benzyl radical. The benzyl radical is intercepted by O2 to yield a benzyl peroxo radical, which is converted to the benzaldehyde. The conversion to nitrile proceeds as before. EPR spectroscopy reveals the presence of the azidyl radical, while confirmation of the radical photocatalytic mechanism was corroborated by the use of radical scavengers and radical trapping agents. Organic TADF compounds 4CzIPN, 4CzIPN-Br and 4DPAIPN were each tested, providing yields of 89%, 92% and 0%. These differences in yield seem to correlate with the photooxidising ability of the PC (Ered* = 1.35 V, 1.52 V and 1.1 V for 4CzIPN, 4CzIPN-Br, and 4DPAIPN, respectively), as the PC must be capable of oxidising the azide anion (Eox = 1.08 V vs. SCE). 4CzIPN was, however, used as the PC for the remainder of the study.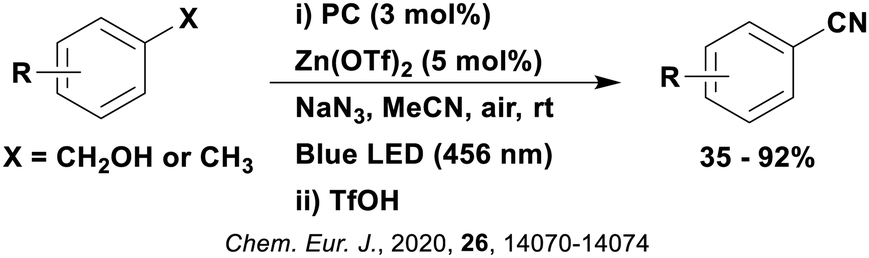 | ||
| Fig. 166 Reaction scheme for the photocatalytic cyanation of benzyl alcohols/methyl arenes. | ||
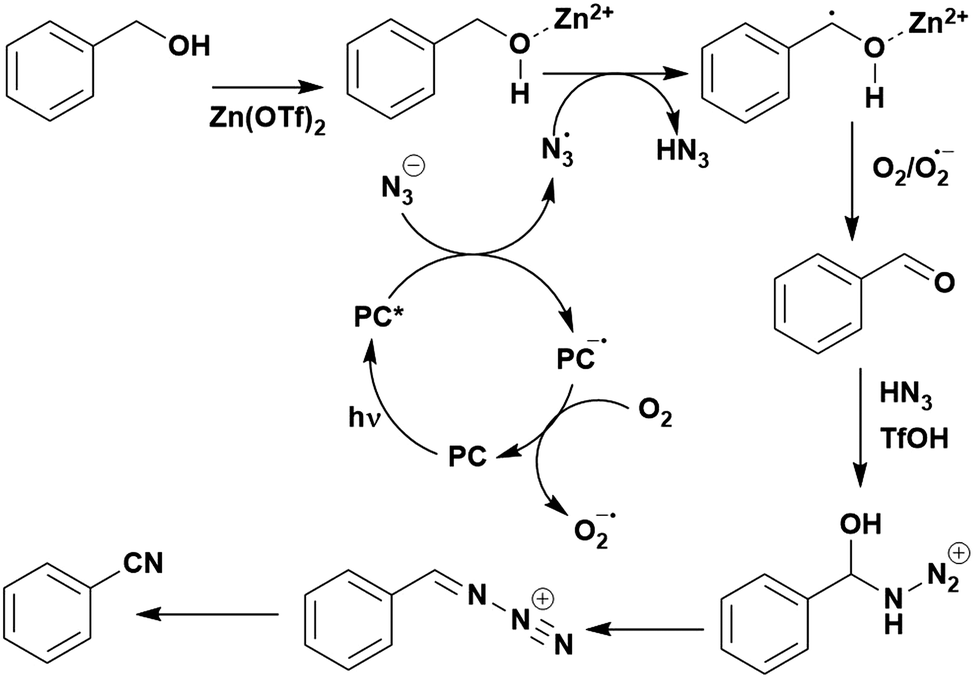 | ||
| Fig. 167 Proposed mechanism for the photocatalytic cyanation of benzyl alcohols/methyl arenes. | ||
Reduction
The reductive capacity of the photocatalysts 3CzClIPN, 4CzIPN, 5CzBN, 3DPAClIPN, 3DPAFIBN, 3DPA2FBN, 4CzIPN-OMe and 5CzBN-OMe, was considered in the reductive C–O bond cleavage of lignin derivatives (Fig. 168).114 Both steps, reduction to p-methoxy acetophenone (cf., e.g., Ered = −1.74 V vs. SCE for 2-oxo-2-phenylethyl acetate) and reductive dimerization of acetophenone derivatives to their corresponding pinacols (cf., e.g., acetophenone, Ered = −2.64 V vs. SCE),354 require a strongly reducing photocatalyst in the ground state (Fig. 169). The yields observed using the aforementioned TADF molecules track with their corresponding redox potentials. For instance, 3DPA2FBN with the most negative ground state reduction potential (Ered = −1.92 V vs. SCE) delivered the highest yield of 77% after 18 hours, outperforming [Ir(ppy)2(dtbbpy)]PF6 as the typical organometallic catalyst for this reaction (56% yield after 12 hours), although this is unsurprising since the iridium photocatalyst is less reducing (Ered = −1.51 V). 3CzClIPN, with the least negative reduction potential in the series (Ered = −1.16 V vs. SCE), produced no pinacol. Similarly, the methoxy-substituted photocatalysts did not have enough reducing power to deliver the final pinacol product but were capable of mediating the first reduction step, producing the p-methoxy acetophenone.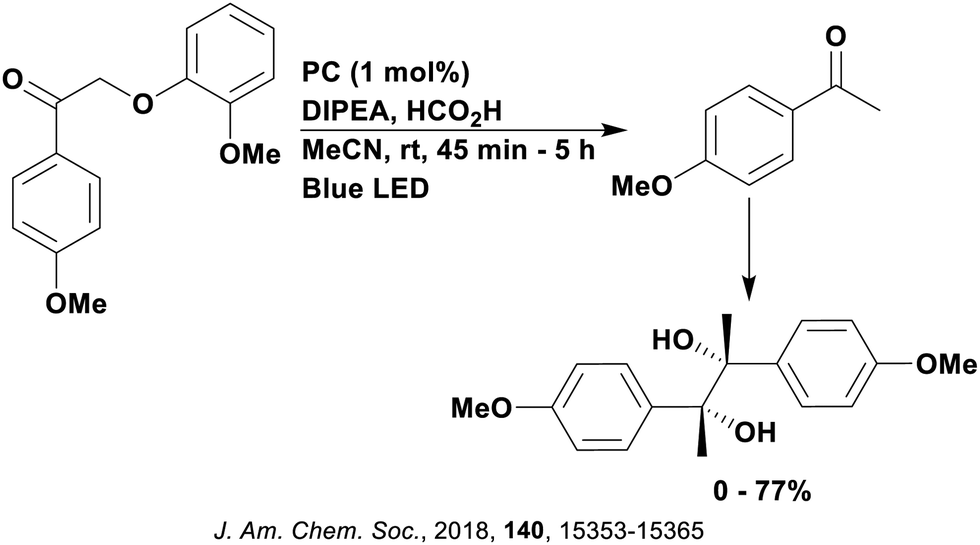 | ||
| Fig. 168 Reaction scheme for the reduction of lignin model derivative. | ||
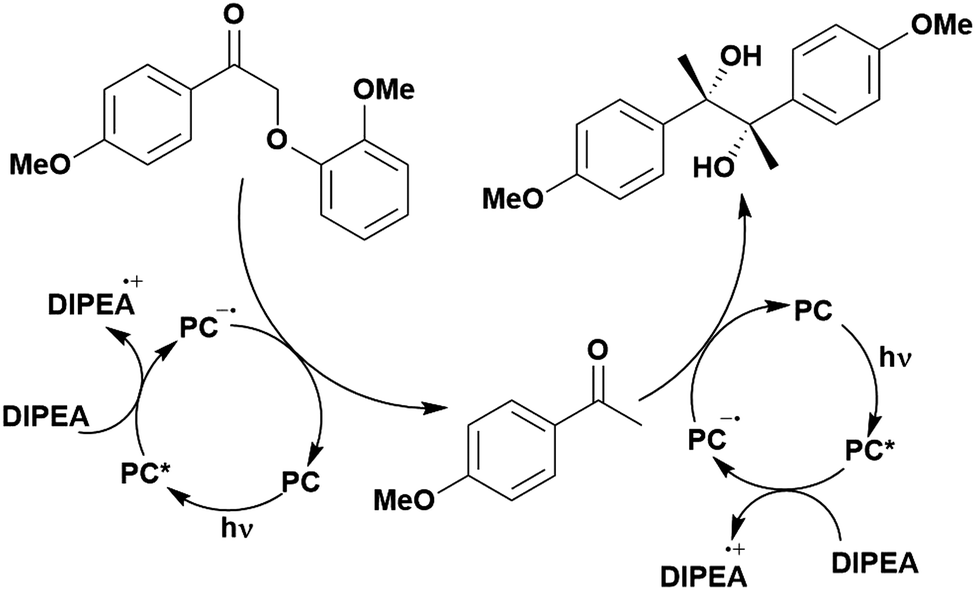 | ||
| Fig. 169 Proposed mechanism for the double photocatalytic reduction of lignin derivatives to pinacols via a p-methoxy acetophenone intermediate. | ||
Additionally, reduction of aryl halides has been shown to be feasible using CDCB-based organic compounds as PCs (Fig. 170), with the reduced aryl halide capable of engaging in a coupling reaction with an additional aryl group in C(sp2)–C(sp2) type coupling (Fig. 170b).310 The proposed mechanism involves reductive quenching of the excited PC by NEt3, with the reduced PC being used to reduce the aryl halide. After loss of the halide, the aryl radical can either abstract a proton from NEt3˙+ to yield the formally reduction product or add to an unsaturated bond such as to a heterocycle to form the C(sp2)–C(sp2) coupled product. Stern–Volmer quenching studies confirmed that NEt3 can effectively quench the luminescence of the PC while product inhibition was observed upon addition of TEMPO.
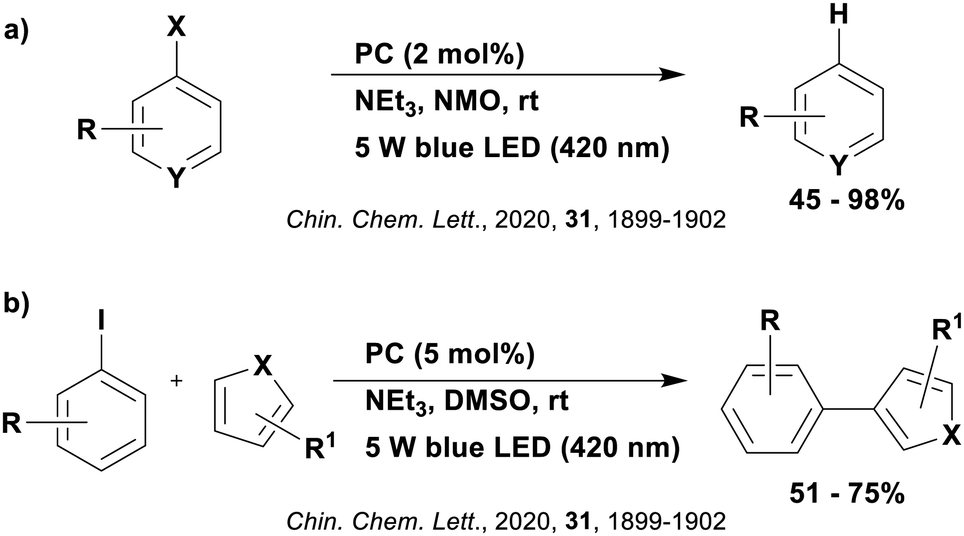 | ||
| Fig. 170 Reaction schemes for the reduction of aryl halides. | ||
Six CDCB compounds were investigated as PCs: 4CzPN, 4CzIPN, 5CzBN, 4DPAPN, 3,4,5-3CzBN and 2CzPN, providing yields of 51%, 54%, 70%, 35%, trace and trace, respectively. For comparison, [Ru(bpy)3](PF6)2 was also tested and this produced only trace amounts of product. Reduction of aryl halides is thermodynamically challenging and so the ground state reducing ability of the PCs is likely to govern this process (Ered = −1.16 V, −1.21 V, −1.52 V, −1.53 V, −1.58 V, −1.45 V, −1.33 V, for 4CzPN, 4CzIPN, 5CzBN, 4DPAPN, 3,4,5-3CzBN, 2CzPN and [Ru(bpy)3](PF6)2 respectively). The authors suggested that the superior performance of 5CzBN is attributed to its strong reduction ability; however, other PCs with equally strong Ered values struggled in this reaction, while 4CzPN, with the least reducing Ered value, performed adequately. This suggests that another factor is responsible for the relative success of the PCs in this reaction.
Detriflation
The reductive capabilities of 3DPA2FBN have been exploited in the photocatalytic detriflation of nonactivated aryl triflates (Fig. 171).114 This type of reaction had only previously been reported for aryl triflates with electron-withdrawing substituents on the aryl group (e.g. p-CN-benzene derivatives).355 The defunctionalisation of naphthyl triflate (Ered = −2.01 V vs. SCE)356 was considered, with both 3DPA2FBN and fac-Ir(ppy)3 under investigation owing to their very negative ground state reduction potentials (Ered = −1.92 V and −2.19 V, respectively). Both photocatalysts proved successful in this reaction with comparable yields of 86% and 90%; the slightly higher yield obtained by the iridium photocatalyst correlates with its more negative reduction potential.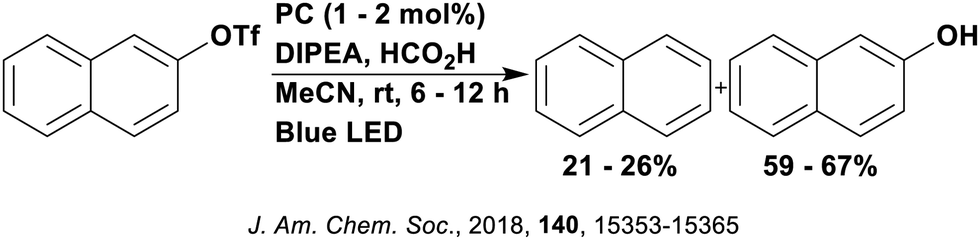 | ||
| Fig. 171 Reaction scheme for the defunctionalisation of naphthyl triflate. | ||
Polymerisation
Free radical and cationic polymerisations can be initiated using CDCB-based photocatalysts, which through reaction with an additive, either bis(4-tert-butylphenyl)iodonium hexafluorophosphate (Iod) or ethyl 4-(dimethylamino)benzoate (EDB) or both, produce the required species necessary to initiate the respective polymerisation reaction.311 This can be achieved through both reductive and oxidative quenching mechanisms (Fig. 172). The molecules considered as photocatalysts were 4CzTPN, 4CzTPN-Br, 4CzBN and 4CzBN-Br and all were shown to work in both two and three component systems in the polymerisation reactions. The success of the polymerisation in this case is defined by the rate of polymerisation and functional conversion (FC) of the monomer to the polymer (e.g., conversion of a diepoxide functionality in the monomer to the polymer without this epoxy functional group). In the polymerisation reactions, 4CzBN was the most efficient, followed by 4CzBN-Br, 4CzTPN-Br and finally 4CzTPN, for example, in the polymerisation of EPOX in a two-component system, FC = 54%, 46%, 34% and 27%, respectively under 455 nm excitation. All four TADF molecules outperformed the traditional photoinitiator bis(2,4,6-trimethylbenzoyl)phenylphosphine oxide (BAPO), which could not initiate the polymerisation in the aforementioned reaction conditions; no other photocatalysts were considered. The efficiency rate of the CDCBs seems to follow the electron transfer quantum yield in the photooxidation process generating PC+˙ and Ar˙ to initiate cationic and free radical polymerisations, respectively. These quantum yields were calculated by using Stern–Volmer quenching constants from the equation Φet(S1) = KSV[Iod]/(1 + KSV[Iod]). These high electron transfer quantum yields also follow the energy of the S1 state (S1 = 2.90 eV (280 kJ mol−1), 2.80 eV (270 kJ mol−1), 2.72 eV (262 kJ mol−1) and 2.50 eV (241 kJ mol−1) for 4CzBN, 4CzBN-Br, 4CzTPN-Br and 4CzTPN, respectively), suggesting the singlet excited state is important in this process.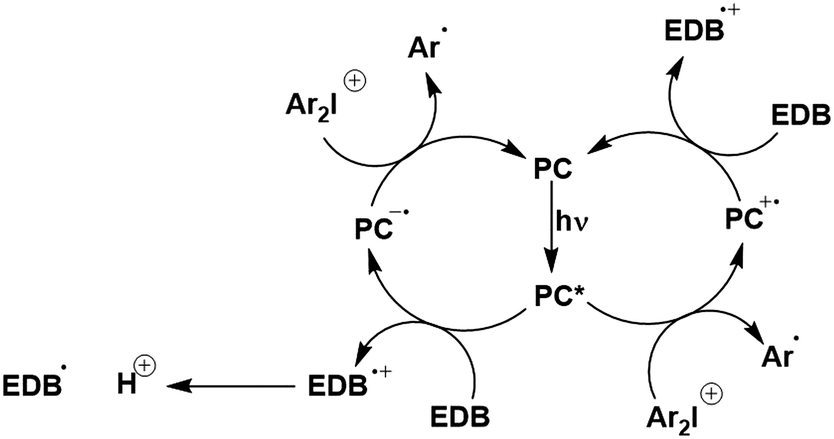 | ||
| Fig. 172 Photocatalytic polymerisation initiations involving both oxidative and reductive quenching cycles where EDB is ethyl 4-(dimethylamino)benzoate and Ar2I+ is bis(4-tert-butylphenyl)iodonium. | ||
Cyclisation
Borylcyclopropanation reactions can be photocatalysed by CDCB-based TADF molecules (Fig. 173).357 Diiodoborylmethane is used as the radical precursor, which after reduction by the exited photocatalyst and cleavage of a carbon–iodine bond, generates a borylmethyl radical (Fig. 174). This radical then adds to the double bond of α-MIDA-boryl styrene, forming a benzylic radical, which can then undergo cyclopropyl ring closure. Sodium thiosulfate was used a sacrificial reductant to regenerate the photocatalyst and close the catalytic cycle. Three TADF molecules were considered as the photocatalyst, 4CzIPN, 4CzIPN-tBu and 4CzIPN-Br, providing respective yields of 67%, 58% and 26%. These yields are reflective of their photoreducing capability (Eox* = −1.04 V, −1.31 V and −0.82 V for 4CzIPN, 4CzIPN-tBu, and 4CzIPN-Br, respectively), especially given that diiodoborylmethane has a reduction potential of Ered = −1.01 V. However, this correlation fails to explain the yield obtained using 4CzIPN-tBu, which obtained a lower yield than when using 4CzIPN despite being a stronger photooxidant. The reason for the poorer performance of 4CzIPN-tBu may be linked to its reduced oxidising capacity in the ground state (Eox = 1.22 V and 1.52 V for 4CzIPN-tBu and 4CzIPN, respectively). Alternative photocatalysts were not considered by Ooi et al. in this study; however, a study by Charette et al. showed the borylcyclopropanation of styrene using diiodoborylmethane was possible in 78% yield using xanthone as a photocatalyst (Eox* = −1.42 V)358 although this result was obtained using a flow set-up.359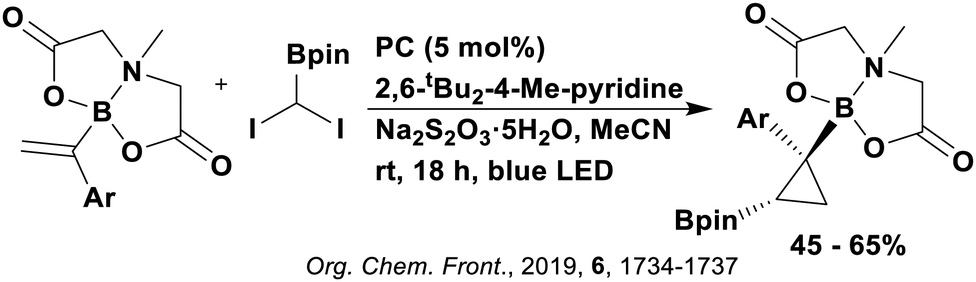 | ||
| Fig. 173 Reaction scheme for photocatalysed borylcyclopropanation. | ||
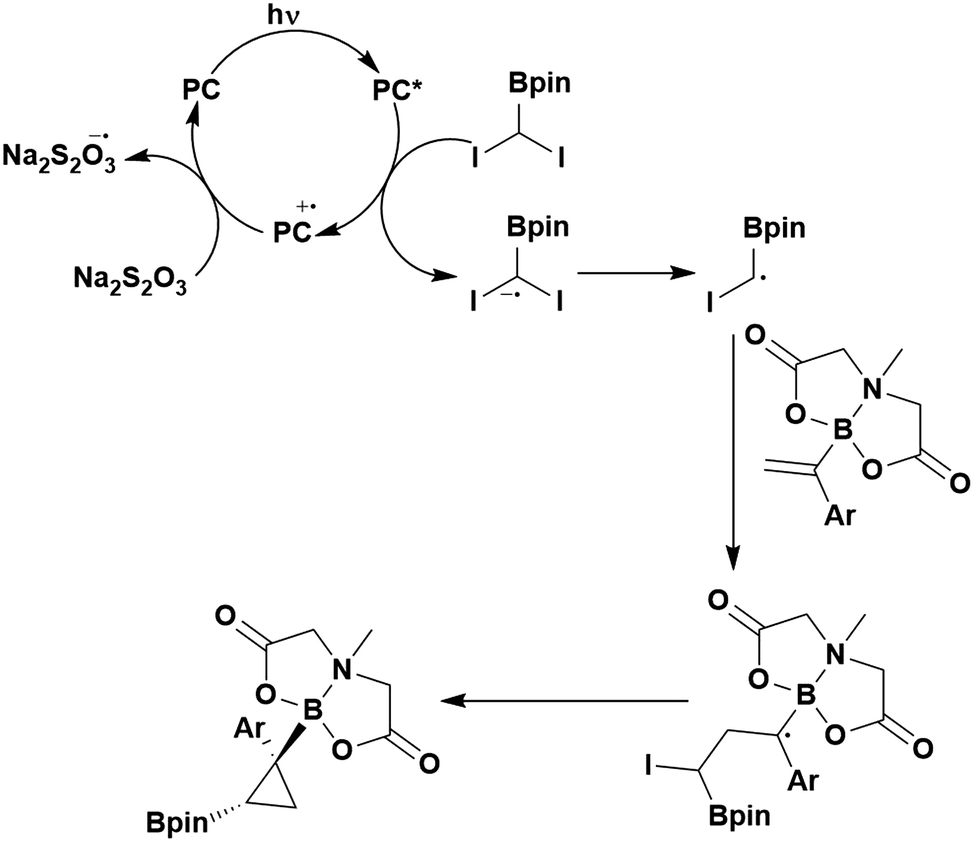 | ||
| Fig. 174 Proposed mechanism for the photocatalytic borylcyclopropanation. | ||
Photocatalysed cyclisation reactions have also been reported in the context of the hydroarylation of arenes (Fig. 175).360 The proposed mechanism involves reductive quenching of the PC by DIPEA. The resultant reduced PC then reduces the aryl halide to form an aryl radical, which undergoes 5-exo-trig cyclisation to form a cyclohexadienyl radical. The reduced PC is invoked again to reduce this radical to the anion, which is then protonated to yield the final product. Stern–Volmer quenching experiments reveal DIPEA to be an efficient quencher of the excited PC, while executing the reaction in D2O resulted in deuterium incorporation at the C(sp3) on the dearomatized ring, providing further confirmation of the radical-polar crossover mechanism. For this reaction, 3DPAFIPN proved to be the superior PC (86% product yield), in comparison to 4CzIPN (49%), 5CzBN (46%), 3DPA2FBN (66%), [Ir(ppy)2(dtbbpy)]PF6 (66%) and fac-Ir(ppy)3 (31%).
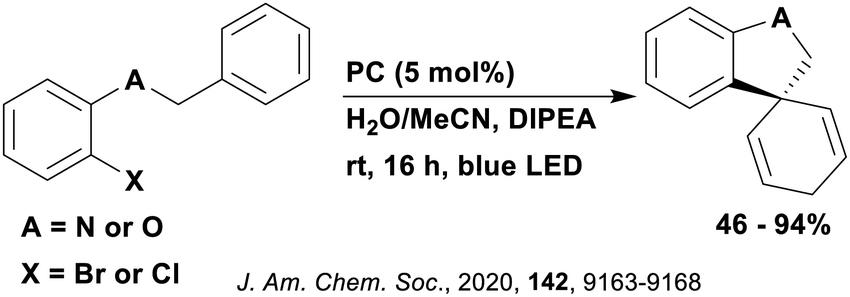 | ||
| Fig. 175 Reaction scheme for the hydroarylation of arenes. | ||
Reduction of the cyclohexadienyl radical (Ered = −1.34 V) is possible for all PCs (Ered = −1.59 V, −1.52 V, −1.92 V, −1.51 V and −2.19 V for 3DPAFIPN, 5CzBN, 3DPA2FBN, [Ir(ppy)2(dtbbpy)]PF6 and fac-Ir(ppy)3, respectively) except for 4CzIPN (Ered = −1.21 V), which explains why it performed poorly. The excited PC must also be capable of oxidation of DIPEA (Eox = 0.81 V),35 which is facile for 3DPAFIPN, 5CzBN and 3DPA2FBN (Ered* = 1.09 V, 1.31 V and 0.92 V, respectively), but explains the reduced yields obtained with [Ir(ppy)2(dtbbpy)]PF6 and fac-Ir(ppy)3 (Ered* = 0.66 V and 0.31 V, respectively). From these redox potentials, it is clear to understand how 3DPAFIPN performed so effectively in comparison to the other PCs; however, this analysis does not explain why the use of 5CzBN and 3DPAF2BN gave such low yields, suggesting something other than the thermodynamics of the PC is responsible for the poor yields in these two reactions.
An additional cyclisation example is exemplified in the photosynthesis of phosphorylated heteroaromatics (Fig. 176).318 The proposed mechanism is reminiscent of that shown in Fig. 76. The excited photocatalyst is reductively quenched by the tautomer of diphenylphosphine oxide, hydroxydiphenylphosphine, in the presence of base, in a PCET step. The resultant phosphoryl radical undergoes radical addition to the isocyano group of the aryl reagent, with the adduct proceeding to undergo an intramolecular cyclisation, oxidation, and deprotonation to afford the final product. Simultaneously, the photocatalytic cycle is closed by SET to TBHP (tert-butyl hydroperoxide). Radical scavengers were employed to verify that this reaction includes radical processes, with an adduct being formed with the phosphoryl radical. Further confirmation of the presence of phosphoroyl radicals was provided by EPR spectroscopy. A quantum yield of 0.3 suggests that this reaction does not proceed via a radical chain process while a primary kinetic isotope effect kH/kD of 1.2 implied that cleavage of the C(sp2)–H bond is unlikely to be the rate determining step.
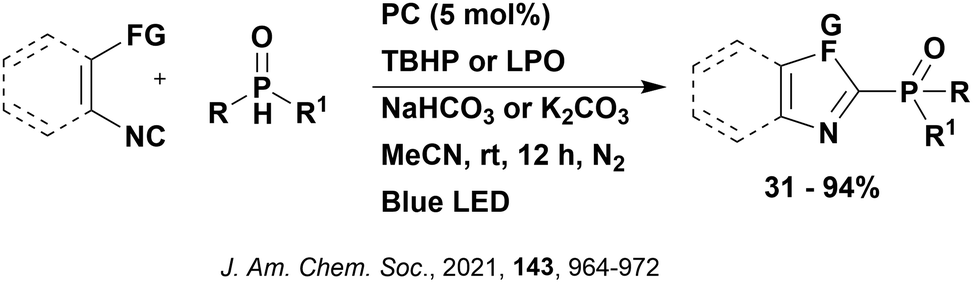 | ||
| Fig. 176 Reaction scheme for the synthesis of phosphorylated heteroaromatics. | ||
A wide variety of CDCB-based organic compounds were considered in this reaction: 4CzIPN-Br (0%), 4CzTPN (0%), 4CzIPN-Cl (19%), 4CzPN (34%), 4DPAIPN (40%), 4CzIPN-Ph (44%), 4CzIPN-Me (45%), 4CzIPN-OMe (66%), 4CzIPN (78%) and 4CzIPN-tBu (80%), with 4CzIPN-tBu being selected for the remainder of the study. The authors noted that for the substituted 4CzIPN analogues, the yield increases in line with a decrease in the photooxidising ability (from Ered* = 1.73 V for 4CzIPN-Br to 1.21 V for 4CzIPN-tBu), although they provide no mechanistic reasoning for this. The low yield of the halo-substituted compounds was attributed to poor solubility of the PCs in acetonitrile.
Isomerisation
A large number of organic TADF molecules have been studied as photocatalysts for the photosensitised E/Z isomerisation of stilbene (Fig. 177).325 These include 3,5-2CzBN, 2,3,6-3CzBN, 2,4,6-3CzBN, 4CzBN, 5CzBN, 2CzTPN, 2CzPN, 2CzIPN, 4CzTPN, 4CzPN and 4CzIPN. This reaction proceeds through a Dexter energy transfer mechanism via a triplet biradical intermediate (Fig. 178), implicating the triplet state of the PC. As discussed in Section 1, DET requires spectral overlap (eqn (10)), and typically the triplet energies of the PC and substrate are used as a surrogate to determine whether the reaction is thermodynamically feasible. In E/Z isomerization, the thermodynamics of the photochemoselective isomerization of the E-isomer to the Z-isomer require ET (E-isomer) < ET (PC) < ET (Z-isomer).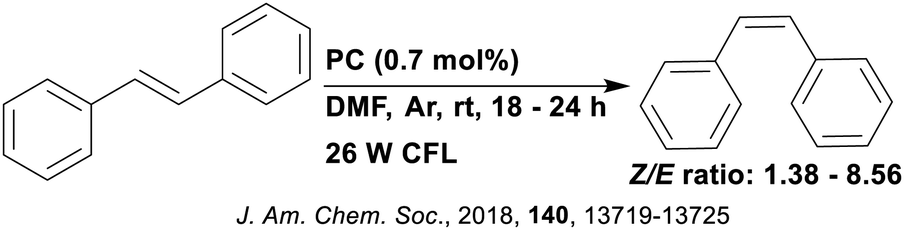 | ||
| Fig. 177 Reaction scheme for the E/Z isomerisation of alkenes. | ||
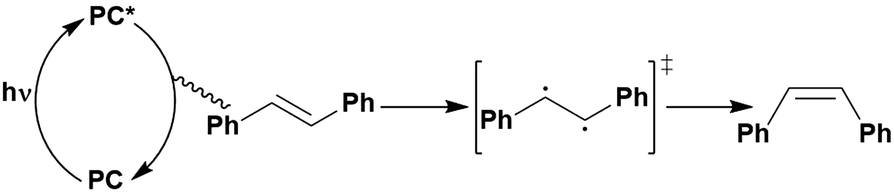 | ||
| Fig. 178 Proposed mechanism for the E/Z isomerisation of alkenes. | ||
The E/Z isomers of stilbene have triplet energies of 2.2 eV (212 kJ mol−1) and 2.5 eV (241 kJ mol−1), respectively).361 Zhang et al. found the greatest Z/E ratio is generated by 4CzTPN (Z/E = 8.56, 90% yield of Z-isomer, ET = 2.34 eV (226 kJ mol−1)), followed by 4CzPN (Z/E = 8.10, 89% yield of Z-isomer, ET = 2.45 eV (236 kJ mol−1)) while the lowest is obtained by the compounds whose triplet energy is too high for example 2,4,6-3CzBN (Z/E = 1.38, 58% yield of Z-isomer, ET = 2.87 eV, 277 kJ mol−1) and 2,3,6-3CzBN (Z/E = 1.50, 60% yield of Z-isomer, ET = 2.77 eV, 267 kJ mol−1), whereby the ET is sufficiently large that the PC can sensitise both E and Z isomers.325 Reference PC [Ru(bpy)3](PF6)2 (ET = 2.03 eV, 196 kJ mol−1)88 yields 87% of the Z-isomer (Z/E = 6.69).
When rationalising the obtained results, Zhang et al. invoke the 3CT state of the TADF compounds as the dominant state from which this energy transfer can occur. This is supported by a transient absorption spectroscopy study of 2CzPN and 4CzIPN where both were shown to exhibit 3LE and 3CT states using femtosecond and nanosecond transient absorption spectroscopy. The kinetic traces of the nanosecond transient absorption spectra are then studied with increasing amounts of E-stilbene, causing both the 3LE and 3CT spectral regions to decrease, implying both are involved in the energy transfer process. Global fitting of this data indicates that for 2CzPN, energy transfer from the 3LE state is quicker than from the 3CT while for 4CzIPN, the opposite is observed. Since using 4CzIPN as a PC results in a greater proportion of the Z-isomer (Z/E = 6.7 and 1.7 for 4CzIPN and 2CzPN, respectively), it seems more significant to have the energy transfer operating from the 3CT state.
Cycloadditions
The final class of reactions to be discussed is cycloaddition reactions, starting with the [2+2] cycloaddition of enone substrates (Fig. 179).362 This reaction is known to proceed via a Dexter energy transfer mechanism with fac-Ir(ppy)3 as the photosensitizer.363 Ten TADF compounds were considered as PCs in this reaction: 4CzIPN, 4CzIPN-tBu, 4CzIPN-Ph, 4CzIPN-OMe, 4CzPN, 4CzPN-tBu, 4CzPN-Ph, 5CzBN, 5CzBN-OMe and 3DPAFIPN, all of which giving yields ranging from 60–71%, except 4CzIPN-OMe, which managed only 23%. These yields are comparable to that obtained with fac-Ir(ppy)3 under the same conditions (64%); notably, there is a background reaction where the product forms in 21% in the absence of any PC. Since 4CzIPN-tBu gave the highest yield of product, this compound was selected as the PC for the substrate scope. Mechanistically, since similar yields were reported with most of the TADF compounds despite their very different reduction potentials, this suggests that SET to chalcone (Ered = −1.48 V)363 is not occurring as it would be expected that more strongly reducing PCs would yield more product whereas this is not the case. Addition of TEMPO inhibits the reaction, confirming the presence of the biradical intermediate. Luo et al. postulated that a FRET pathway does not occur due to the lack of spectral overlap between the absorption of the chalcone and the emission of the PC, suggesting instead that energy transfer must result from the triplet state through a DET mechanism. Stern–Volmer quenching experiments were conducted using both 4CzIPN-tBu and 4CzIPN-OMe, indicating that chalcone quenches the former much more efficiently than the latter (KSV = 0.0624 and 0.0304, respectively). Luo et al. contented that the low triplet energy of 4CzIPN-OMe (2.27 eV) is responsible for its poor performance. | ||
| Fig. 179 Reaction scheme for the [2+2] dimerization of substituted olefins. | ||
The effect of the excitation wavelength upon the reaction was also considered using 4CzIPN as the PC. Upon 24 h of irradiation, the same yields were obtained using 425 nm and 455 nm LEDs (68%), while 395 nm yielded only 52%. At a reduced reaction time of 15 h, the 425 nm LED was identified as the superior light choice (63% and 58% yields from 425 nm and 455 nm LEDs, respectively). The reduced yield obtained using 395 nm was suggested to be linked to the irreversible side reactions that may ensue under high energy irradiation,364 although no evidence of this was provided.
A second cycloaddition reaction that has been shown to be effectively photocatalysed by TADF compounds is a dearomative cycloaddition reaction, which is possible with naphthols (Fig. 180) and indoles, and proceeds via an energy transfer mechanism.365 TADF compounds 2CzPN, 3DPA2FBN and 3DPADIPN were all considered as PCs in the dearomatisation of an indole substrate, providing a reactant to product ratio of 13:1, 1.5:1 and 3:1, respectively, under the same reaction conditions. Despite producing the worst outcome of these three organic PCs, 2CzPN was chosen as the PC for the remainder of the study. With naphthols, 2CzPN was shown to outperform [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, the PC previously identified as the best for this reaction.366 For example, with 1-(1-(but-3-en-1-yloxy)naphthalen-2-yl)ethan-1-one, 2CzPN produced 94% yield of product while the Ir PC yielded 86%. However, it should be noted that slightly different reaction conditions were used by König et al. for 2CzPN (CHCl3, 0.1 M, 14 h) in comparison to those of Glorius et al. for the iridium PC (1,4-dioxane, 0.04 M, 18 h). Similar superiority of 2CzPN is observed when using indoles. Recyclability of the PC was also investigated in this study, with 88% of 2CzPN being recovered. This recycled PC was then used in the cycloaddition of the aforementioned naphthol, yielding 90% of product, similar to the 94% yield obtained when using the PC for the first time, thereby demonstrating that there is no loss of activity.
 | ||
| Fig. 180 Reaction scheme for the dearomative cycloaddition. | ||
No mechanistic investigations were undertaken in this study, however, König et al. suggested that 2CzPN mimics quite strongly the success of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 on account of their similar ET values [2.63 eV (254 kJ mol−1) and 2.61 eV (252 kJ mol−1), respectively]. The mechanism in operation was assumed to be the same as that proposed by each of Glorius, Bochet and Ohkuma, depending on the substrate in question.366–369 Therefore, this reaction is described by analogy as a triplet sensitisation process, although there is no direct evidence that 2CzPN operates from the T1 state.
6. CDCB-based molecules used in dual catalysis
As previously documented, 4CzIPN has been extensively used as a component in dual catalysis, hence it follows that photocatalysts of similar structure would also be compatible in this class of reactions.Photocatalysis/nickel cross-coupling
In photoredox/Ni C(sp3)–C(sp2) cross-coupling reactions, both carboxylic acids and alkyltrifluoroborates have been used as the radical precursors, which upon single electron oxidation by the photocatalyst form alkyl radicals. These can then be cross-coupled with aryl halides (Fig. 181a and b). Zhang et al. investigated a wide number of CDCB-based photocatalysts, including 4CzIPN, 2CzIPN, 4CzPN, 2CzPN, 4CzTPN, 2CzTPN and 4DPAIPN.120 In the reaction with carboxylic acids as the radical precursors (Fig. 181a), all of the TADF compounds investigated should be able to participate in SET from the carboxylic acid to the excited photocatalyst (Ered* ranging from 1.10 V–1.40 V), owing to a low oxidation potential Eox = 0.93 V of N-Boc-Pro. Almost all should also be able to reduce the Ni(II) complex (Ered ≈ −1.1 V for the Ni(II) species and varies from −1.16 V to −1.50 V for the TADF compounds). Only 4CzTPN, with Ered = −1.02 V, may not be able to complete the second SET. However, only 4CzIPN and 4DPAIPN gave yields of greater than 80%, with 2CzIPN providing a yield of 56% and the other TADF photocatalysts, yielding between 5–20% of product. Zhang et al. tentatively hypothesised that the low activities of most of these photocatalysts was attributed to their photoinstability in the required reaction conditions where the solvent was DMF. Evidence for the photodecomposition of the PC came from the observed large blue-shift (∼100 nm) of the emission maximum in reaction mixtures containing the low-yielding CDCBs; HPLC analysis corroborated the contention that the PCs had degraded under these conditions whereas use of 4CzIPN, 4DPAIPN and 2CzIPN led to reasonable recovery of photocatalyst. High yields were obtained using [Ir(dF(CF3)ppy)2(dtbbpy)]+ and [Ir(dF(CF3)ppy)2(bpy)]+ (83% and 82%, respectively), owing to their appropriate redox potentials (Ered* = 1.21 V and 1.32 V and Ered = −1.37 V and −1.37 V, respectively) while neither the Ru(II) nor other organic photocatalysts tested were able to photocatalyse the reaction. For the cross-coupling with alkyltrifluoroborates (Fig. 181b), yields ranging from 72–91% were achieved for 4CzIPN, 4DPAIPN, 2CzIPN and 4CzPN. High yields were also obtained for [Ir(dF(CF3)ppy)2(dtbbpy)]+ and [Ir(dF(CF3)ppy)2(bpy)]+ (89% and 83%, respectively). For both cross-coupling reactions, 4DPAIPN was shown to provide the highest yields, which may be due to its large ground state reduction potential, making the reduction of the Ni(II) species more facile. | ||
| Fig. 181 Reaction schemes for the coupling of aryl halides with (a) carboxylic acids or (b) trifluoroborate salts in dual catalysis. | ||
The formation of two C(sp3)–C(sp2) bonds is also possible using this form of dual catalysis, as is illustrated by the reaction of aryl halides with dichloromethane (Fig. 182).370 The proposed mechanism essentially follows that shown for the generic mechanism shown in Fig. 84, but proceeds through this process twice to form the two C–C bonds. Four photocatalytic cycles and two nickel catalytic cycles are proposed, all of which involve reductive quenching of the PC by sacrificial electron donor TEA, supported by Stern–Volmer experiments. The reduced PC is invoked to reduce dichloromethane, forming the chloromethyl radical. The resultant C(sp3)–C(sp2) coupling then occurs as in Fig. 84. Once the first coupling is complete, the reduced PC is proposed to reduce the benzyl chloride derivate, delivering a benzyl radical that then enters into the nickel catalytic cycle (again, as in Fig. 84), alongside another equivalent of the aryl halide, to yield the final product. Addition of TEMPO supressed the reaction, with an adduct being formed with the chloromethyl radical. The presence of the chloromethyl radical was corroborated by the products formed upon addition of 1,1-diphenylethylene to the reaction mixture as a radical trapping agent.
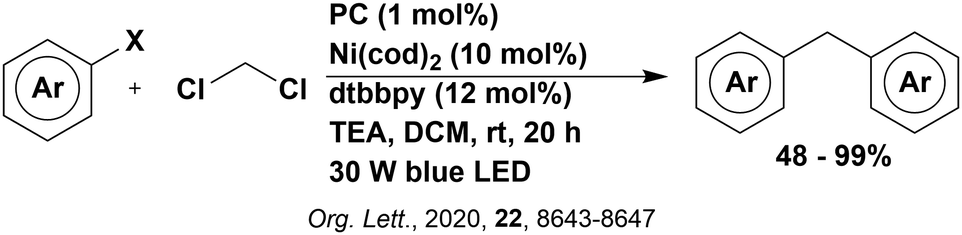 | ||
| Fig. 182 Reaction scheme for the cross coupling of aryl halides with dichloromethane. | ||
Both 4CzIPN and 4CzTPN proved successful in this reaction (63% and 35%, respectively), while [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3]Cl2 provided no product. The transition metal PCs may have failed due to their lower photooxidising ability (Ered* = 1.35 V, 1.41 V, 0.77 V and 1.21 V for 4CzIPN, 4CzTPN, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3]Cl2, respectively), while the difference in yield for the TADF compounds correlates with their Ered values (Ered = −1.21 V and −1.02 V for 4CzIPN and 4CzTPN, respectively).
Additionally, C(sp2)–C(sp) cross-coupling is possible using metallaphotocatalysis, as can be seen in the carboxylation of styrenes (Fig. 183a and b).371 Reductive quenching of the PC by a Hantzsch ester is observed before a SET transmetallation pathway occurs for the carboxylation in which the photocatalyst is used to reduce a nickel(II) species. Carboxylation of the in situ-formed Ni(I) complex, followed by product dissociation and regeneration of the Ni(II) catalyst encompasses the proposed mechanism (Fig. 184). The nickel catalyst used was NiBr2.glyme in combination with neocuproine or 1,4-bis(diphenylphosphino)butane (dppb), depending on the regioselectivity required for the cross-coupling with styrenes. The three TADF photocatalysts considered, 4CzIPN, 4CzTPN and 4CzPN, provided yields of 66%, 3% and 0%, respectively, of the Markovnikov hydrocarboxylation product. Iridium photocatalyst [Ir(dF(Me)ppy)2)(dtbbpy)]PF6 provided a similar yield to 4CzIPN of 67%. The difference in yield is likely due to differing reduction capacity of the reduced photocatalyst (Ered = −1.21 V, −1.02 V, −1.16 V and −1.44 V for 4CzIPN, 4CzTPN, 4CzPN and [Ir(dF(Me)ppy)2)(dtbbpy)]PF6, respectively), suggesting a strong ground state reductant is necessary to achieve high yields.
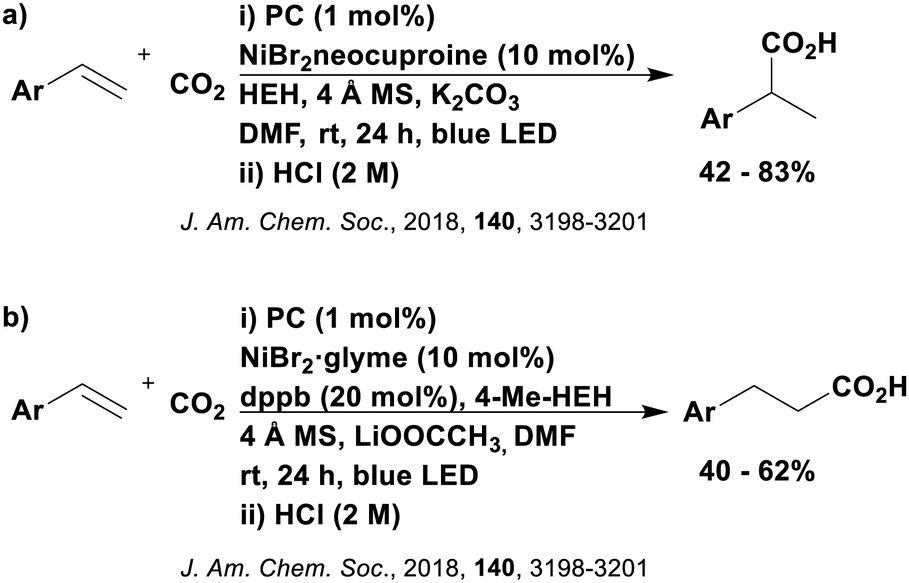 | ||
| Fig. 183 Reaction schemes for the carboxylation of styrene. | ||
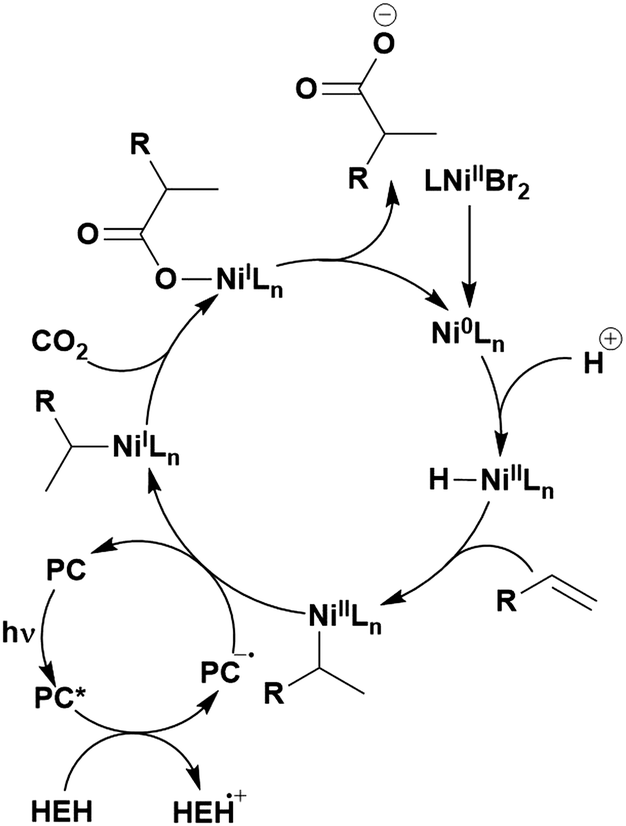 | ||
| Fig. 184 Proposed mechanism for the carboxylation of styrene. | ||
Moreover, photoredox/Ni cross-coupling reactions can be used to form C(sp2)–O bonds. This approach was applied towards the formation of C–O bonds within peptides post synthesis (Fig. 185) whereby the photocatalyst, in its excited state, is required to oxidise the nickel catalyst in order to facilitate reductive elimination, and the reduced photocatalyst is required to reduce the nickel catalyst (Fig. 186).372 In the photocatalyst screen, TADF molecules 4CzIPN and 4DPAIPN were considered, of which 4DPAIPN gave higher yields than all the other photocatalysts tested. A comparison between 4DPAIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which produced the second highest product yield (81% and 61%, respectively), reveals that the most significant difference in thermodynamic parameters arises in the ground state reduction potentials (Ered = −1.52 V for 4DPAIPNvs. Ered = −1.37 V for [Ir(dF(CF3) ppy)2(dtbbpy)]PF6), suggesting the stronger ground state reducing power of 4DPAIPN contributes to the higher yields obtained when this photocatalyst is in use.
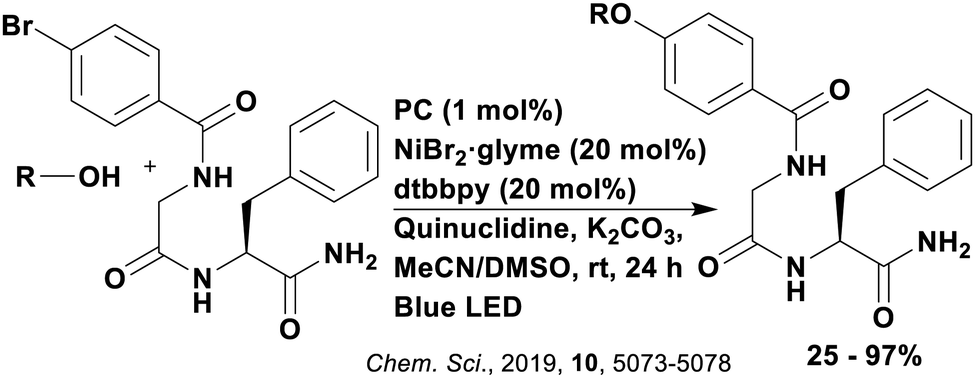 | ||
| Fig. 185 Reaction scheme for photoredox Ni-catalysed peptide C(sp2)–O cross coupling. | ||
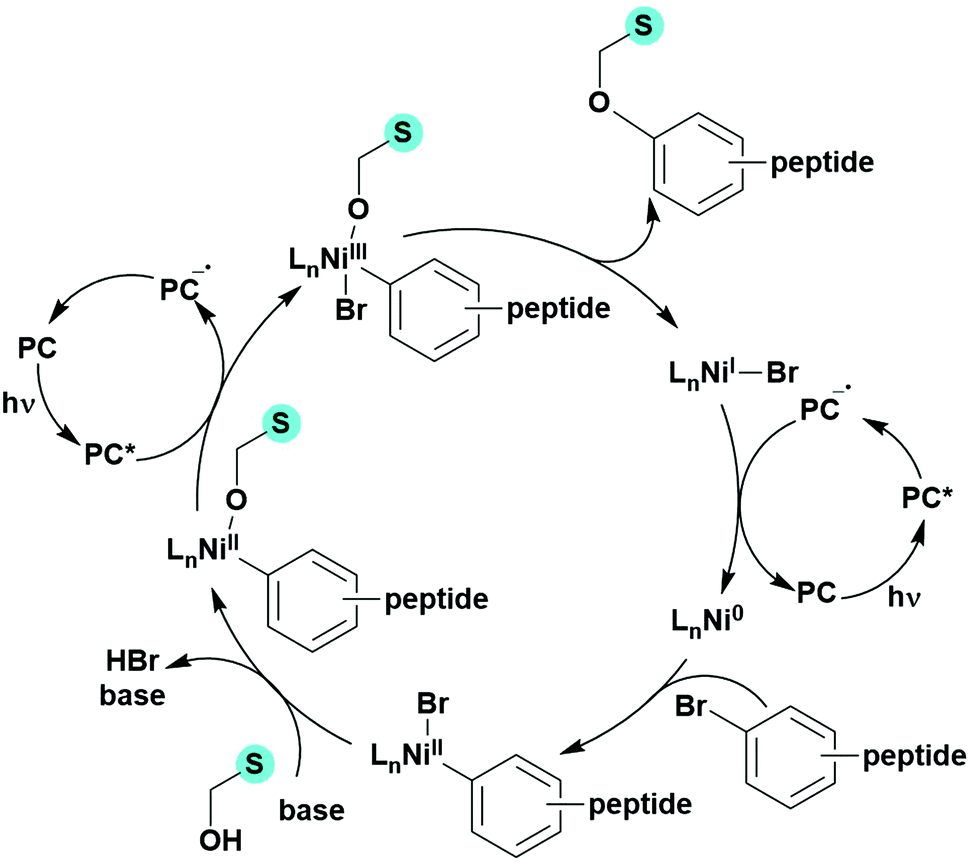 | ||
| Fig. 186 Proposed mechanism for C(sp2)–O cross coupling using photoredox/Ni dual catalysis where S = serine. | ||
Typically, dual catalysis for cross-coupling occurs through a photoredox-based mechanism; however, the same bonding-forming reaction can proceed via an energy transfer mechanism, such as the cross-coupling of carboxylic acids with aryl halides (Fig. 187a).325 Oxidative addition of an aryl halide to the Ni(0) catalyst followed by addition of the carboxylate radical, results in a Ni(II) complex. The excited photocatalyst can then transfer energy to this species (Fig. 188),373 which then undergoes reductive elimination to close the catalytic cycle and eject the product. Organic photocatalysts were investigated, including 4CzIPN, 4DPAIPN and 4DPAPN.325 The authors showed that both energy and electron transfer mechanisms were occurring competitively, producing two different products (Fig. 187b). Photocatalysts with strong excited state oxidation potentials are capable of oxidising a Ni(II) complex, generating a Ni(III) complex that can then release an imine; the presence of this imine was confirmed by GC-MS. Reductive elimination of the arene produces a Ni(I) complex that is reduced by the reduced PC, closing the photocatalytic cycle. The resultant Ni(0) complex partakes in oxidative addition with the aryl halide to continue the nickel catalytic cycle (Fig. 189). In order to suppress the SET required to generate the photoredox product, a weaker photooxidant must be used so that it is no longer thermodynamically feasible to undergo electron transfer. For example, for the aforementioned photocatalysts, the yields of the PEnT product are shown to increase with decreasing photooxidising ability (trace, 17% and 51% yields for 4CzIPN, 4DPAIPN and 4DPAPN, respectively, which have Ered* = 1.35 V, 1.1 V and 0.93 V, respectively). 4DPAPN proved the most efficient out of the organic compounds tested; however, it still produced inferior yields in comparison to fac-Ir(ppy)3 (70%) under the same reaction conditions. This is likely to be as a result of the even smaller photooxidising capacity of fac-Ir(ppy)3 which has Ered* = 0.31 V.
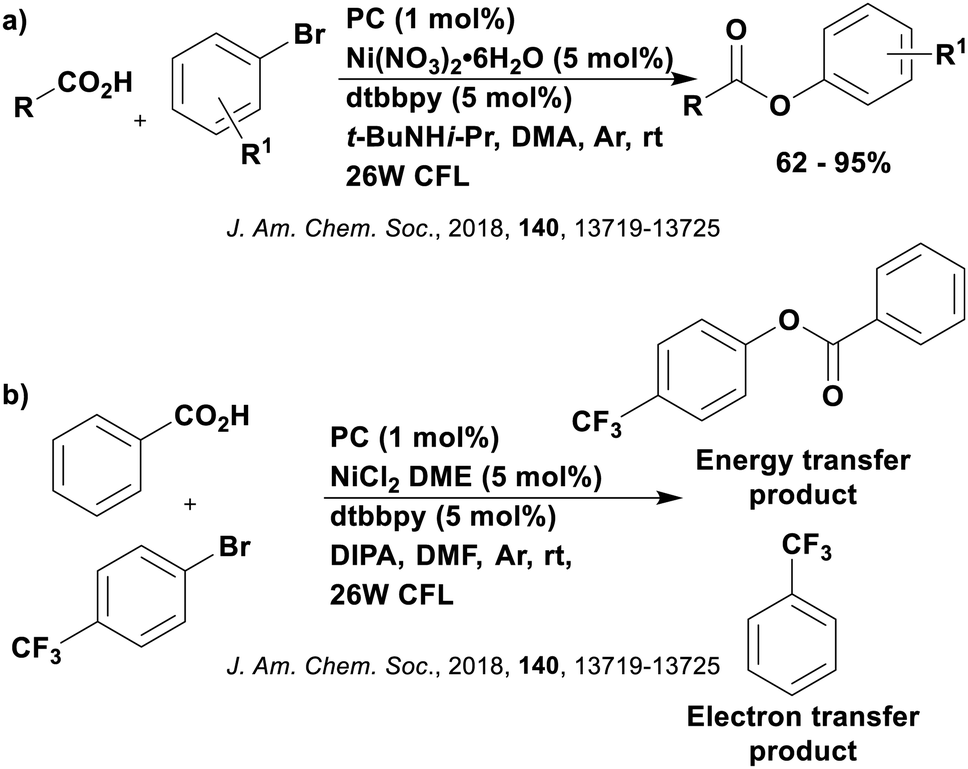 | ||
| Fig. 187 Reaction scheme showing (a) the cross coupling of carboxylic acids with aryl halides and (b) the two potential products for this reaction. | ||
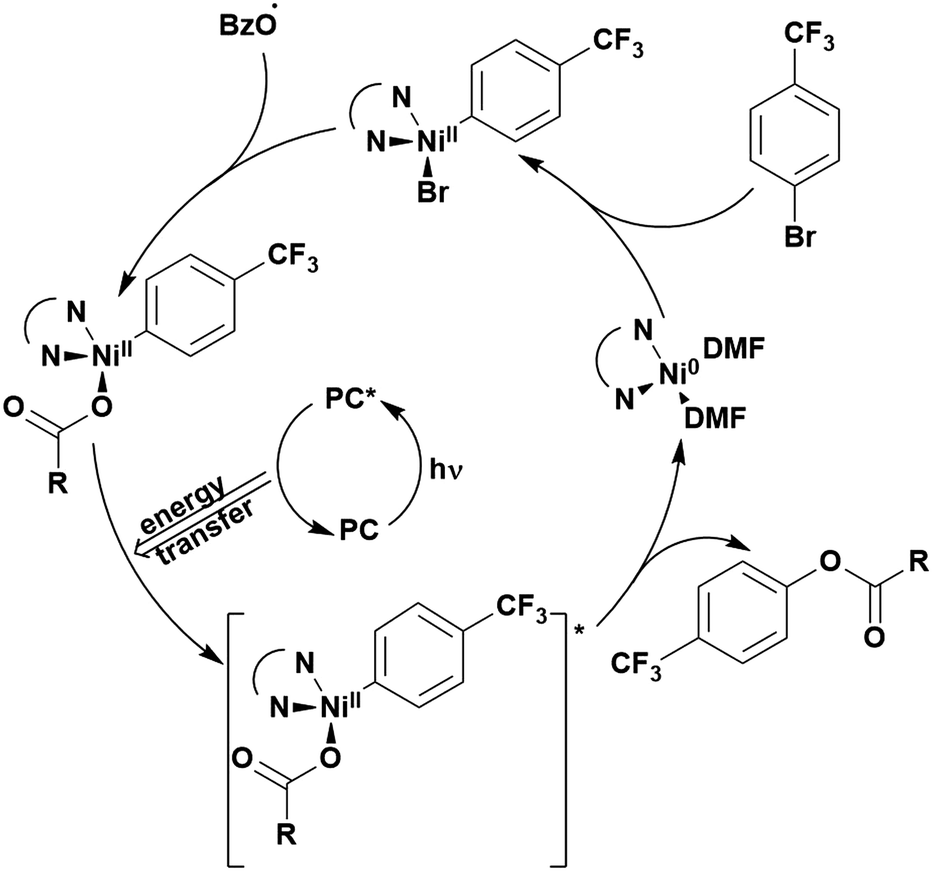 | ||
| Fig. 188 Proposed photocatalytic energy transfer mechanism in the cross coupling of carboxylic acids with aryl halides. | ||
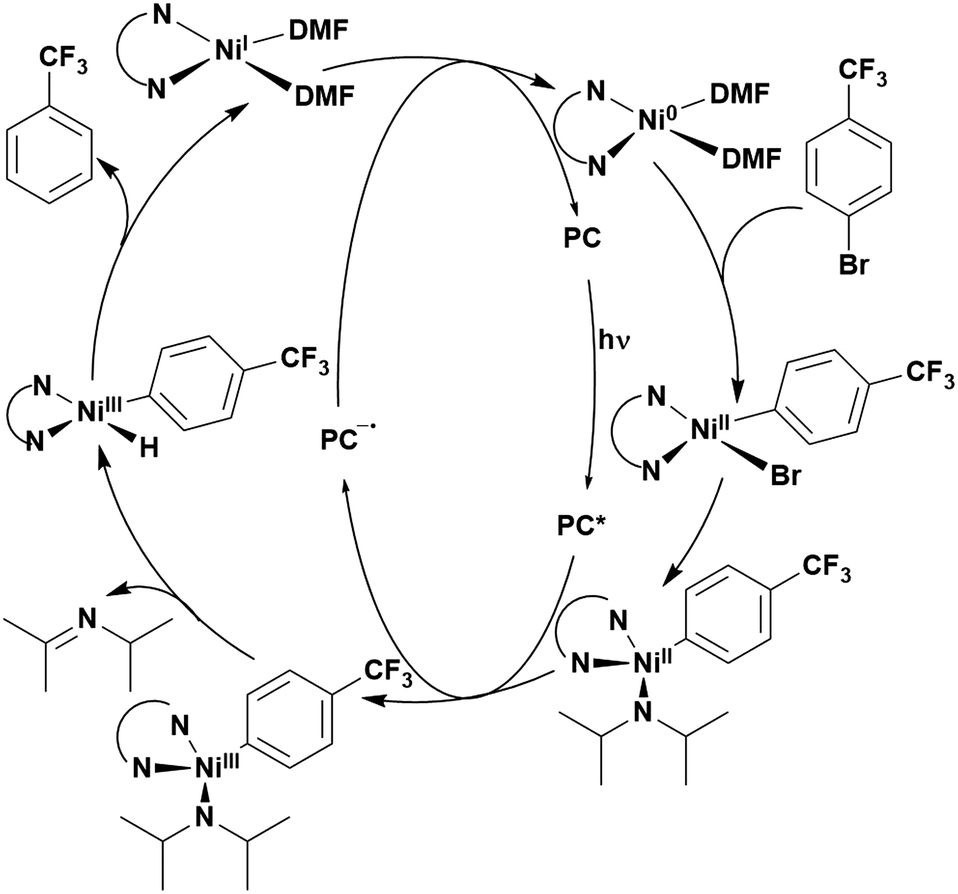 | ||
| Fig. 189 Proposed mechanism for the electron transfer route in the coupling of aryl halides with carboxylic acids. | ||
Photoredox/Pd cross coupling
In addition to Ni, CDCB-based organic PCs have been shown to be effective in dual catalysis with Pd catalysts. An example is the decarboxylative C(sp3)–C(sp3) coupling of carboxylic acids with π-electrophiles (Fig. 190),374 which uses Pd(OAc)2, in the presence of BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthalene), as the Pd catalyst. The proposed dominant catalytic pathway involves oxidative addition of the allyl carbonate to Pd(0) to generate a π-allyl-Pd carboxylate species, which reductively quenches the excited PC (Fig. 191). Decarboxylation followed by reductive elimination ensues, releasing the product. Both catalytic cycles are closed concurrently by SET transfer from the reduced PC to the Pd(I) species, regenerating the initial two catalysts.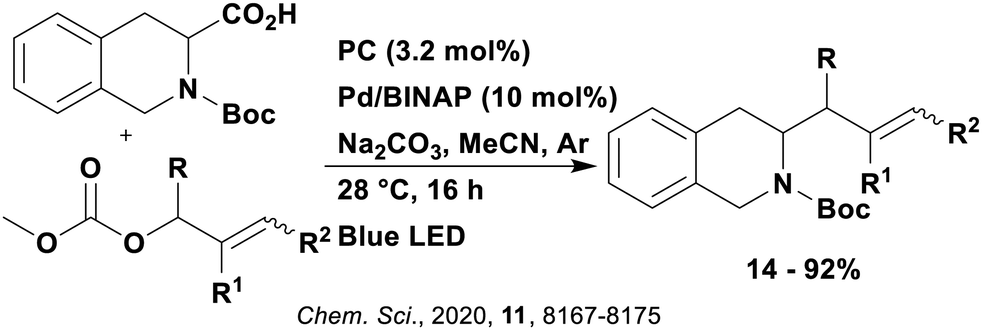 | ||
| Fig. 190 Reaction scheme for the decarboxylative C(sp3)–C(sp3) coupling of carboxylic acids and π-electrophiles. | ||
 | ||
| Fig. 191 Viable mechanism for the decarboxylative C(sp3)–C(sp3) cross coupling of carboxylic acids and π-electrophiles. | ||
In the presence of TEMPO, the reaction is inhibited, suggesting this is indeed a radical process while Stern–Volmer quenching experiments indicated the most efficient quenching of the excited PC occurred by the π-allyl-Pd carboxylate species. Four organic TADF compounds were tested as PCs in this reaction: 4CzIPN, 4CzPN, 2CzTPN and 4DPAIPN, providing yields of 82%, 48%, 5% and 0%, respectively. Additionally, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 gave 84% yield of product; 4CzIPN was used as the PC for the remainder of the study due to the lower cost of the organic PC.
The PC must be capable of reducing the Pd(I) species, which is estimated to have Ered = −1.26 V. This difficult reduction may explain why lower yields were obtained for 4CzPN in comparison to 4CzIPN (Ered = −1.21 V, −1.16 V, −1.30 V, −1.52 V and −1.37 V for 4CzIPN, 4CzPN, 2CzTPN, 4DPAIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively). The photooxidising ability of the PCs are relatively similar, save for 4DPAIPN, which may explain why this compound could not photocatalyze reaction (Ered* = 1.35 V, 1.40 V, 1.34 V, 1.1 V and 1.21 V for 4CzIPN, 4CzPN, 2CzTPN, 4DPAIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively). However, it seems as though something other than these thermodynamics considerations is responsible for the difference in yields as 2CzTPN for example, performed very poorly despite having the appropriate redox potentials required for the mechanism proposed.
Photoredox/Ti cross coupling
Synergistic catalysis involving TADF PCs and a titanium catalyst, Cp2TiCl2, can be seen in the allylation of aromatic and aliphatic aldehydes (Fig. 192).375 The proposed mechanism involves oxidative quenching of the excited PC by the titanium catalyst, before regeneration of the PC through oxidation of the Hantzsch ester (Fig. 193). Stern–Volmer quenching experiments indicated both the titanium catalyst and the Hantzsch ester can quench the excited 3DPAFIPN emission, although the former does so at a faster rate (kq = 5.2 × 108 M−1 s−1 and 1.5 × 107 M−1 s−1, respectively). A quantum yield of 0.013 is suggestive that this is not a radical chain process. Both 4CzIPN and 3DPAFIPN were studied as organic photocatalysts, providing respective yields of 45% and 99% at 5 mol% loading. Transition metal complex [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 also proved very successful, giving 96% yield at 1 mol% loading. The Hantzsch ester has an oxidation potential Eox = 1.0 V, which makes oxidation facile for all three photocatalysts (Eox = 1.3 V, 1.52 V and 1.69 V for 3DPAFIPN, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively). Therefore the difference in yield may be related more to the photoreducing capacity of the photocatalyst (Eox* = −1.38 V, −1.04 V and −0.89 V for 3DPAFIPN, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively), although this does not fully explain the poor yield obtained by 4CzIPN. One possible explanation may be that for this PC there is a change in mechanism to favour a reductive quenching pathway as 4CzIPN is the most photooxidising of the three (Ered* = 1.09 V, 1.35 V and 1.21 V for 3DPAFIPN, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively) and it was shown by Stern–Volmer quenching experiments that the Hantzsch ester does quench the photocatalyst emission. | ||
| Fig. 192 Reaction scheme for the allylation of aromatic aldehydes. | ||
 | ||
| Fig. 193 Proposed mechanism for the dual catalytic allylation of aldehydes. | ||
Photoredox/Cu cross coupling
Metallaphotoredox catalysis involving copper complexes has been investigated with a variety of CDCB type photocatalysts in the context of the decarboxylative C(sp3)–N coupling of anilines (Fig. 194a) and imines (Fig. 194b).376 Reductive quenching of the excited PC by NEt3 is proposed to occur (Fig. 195), with the reduced PC then being invoked to reduce the N-hydroxyphthalimide (NHPI) ester. Resultant decarboxylation and fragmentation produce an alkyl radical. Meanwhile, the aniline reagent coordinates to the Cu(I) catalyst, with the resultant complex undergoing HAT to the NEt3 radical cation. The alkyl radical is then trapped by the Cu(II)-anilido complex followed by reductive elimination to close the copper catalytic cycle and release the final product. Fluorescence quenching experiments showed that the aniline, CuCl and NEt3 could each quench the excited PC (KSV = 375.47, 278.27 and 205.25, respectively), while the NHPI could not. A combination of CuCl:aniline and CuCl:NEt3 were likewise considered as quenchers, but provided no significant luminescence quenching. The authors suggested that due to binding with aniline or NEt3, there is no free CuCl to quench the PC. Since the coupling was only successful when using NEt3 or DIPEA, rather than other bases, the authors contended that these amines act as more than a simple base and are serving to reductively quench the PC, despite having a lower Stern–Volmer quenching constant than the aniline. A low quantum yield of 0.054 was obtained, implying no radical chain process is in operation.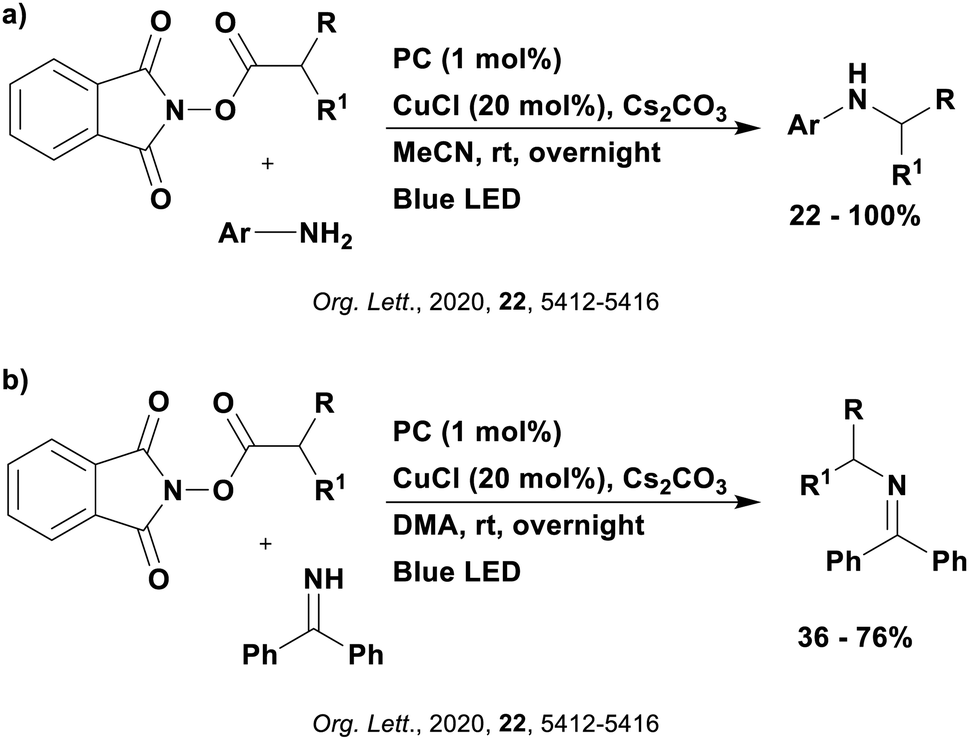 | ||
| Fig. 194 Reaction schemes for the decarboxylative C(sp3)–N coupling of (a) anilines and (b) imines. | ||
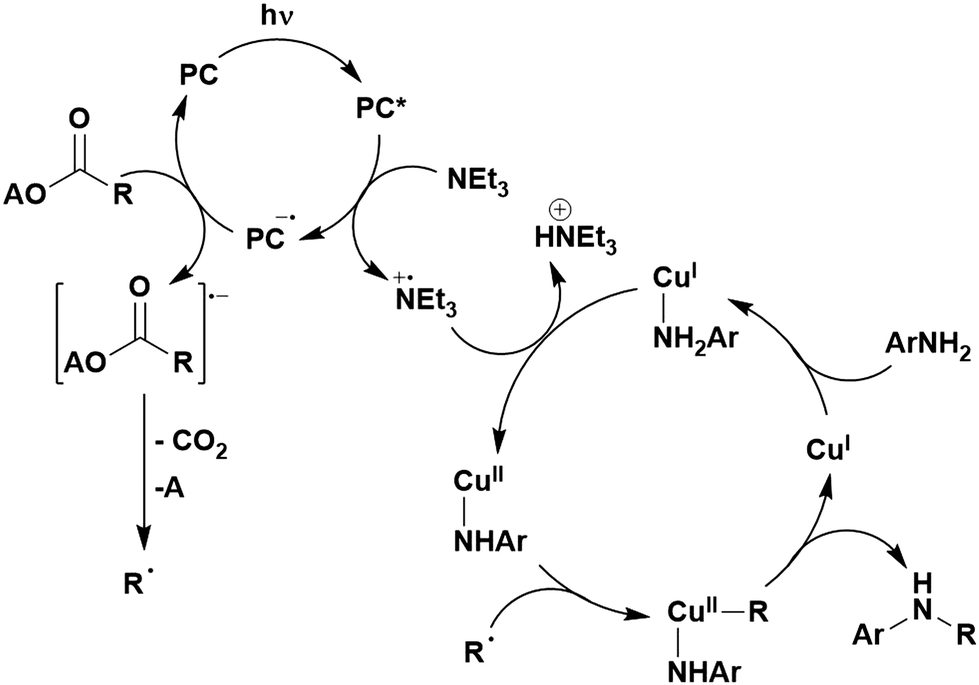 | ||
| Fig. 195 Proposed mechanism for the decarboxylative C(sp3)–N coupling of anilines where A represents phthalimide and R is an alkyl group. | ||
In the photocatalyst screen, 4CzIPN (100%) 4DPAIPN-Br (85%), 4DPAIPN-Cl (75%) 4DPAIPN (72%), 4CzIPN-tBu (59%) and 4CzIPN-Cl (10%) were all considered. It should be noted that the mechanism proposed differs from that suggested for an analogous coupling with [Ru(bpy)3](PF6)2, which proceeds via an oxidative quenching.377 The authors suggested the different mechanism proceeding with 4CzIPN may be on account of it being a stronger photooxidant (Ered* = 1.35 V and 0.77 V for 4CzIPN and [Ru(bpy)3](PF6)2, respectively). Hence, with this is mind, it is possible some of the other organic TADF compounds, which are weaker photooxidants, may also operate via an oxidative quenching mechanism, which may explain the differences in yields (Ered* = 0.98 V, 1.09 V, 1.1 V, 1.21 V for 4DPAIPN-Br, 4DPAIPN-Cl, 4DPAIPN and 4CzIPN-tBu, respectively). Although 4CzIPN-Cl is a strong photooxidant (Ered* = 1.71 V), it has a much weaker reducing capacity in ground state than 4CzIPN (Ered = −0.71 V and −1.21 V for 4CzIPN-Cl and 4CzIPN, respectively), which may explain why it only afforded a 10% product yield.
Dual photocatalytic hydrogen production
Photocatalytic water reduction has received significant attention as a potential renewable hydrogen energy vector, accessed through benign and sustainable methods.378 TADF-based photocatalysts have been considered as the photosensitiser component for this process, such as 4CzPN-R, 4CzIPN-R and 4CzTPN-R where R = H, Ph or tBu.316 A sacrificial reductant, triethylamine (TEA), as well as in situ generated water reduction catalyst (WRC) PdCl2(PPh3)2 are also required (Fig. 196). Stern–Volmer experiments suggested both oxidative and reductive quenching mechanisms may be possible, since both PdCl2(PPh3)2 and TEA can quench the emission of the PC; however, the large excess of TEA compared to the WRC implies that the reductive quenching cycle dominates. The success of the photocatalyst was assessed by the turnover number (TON), where TON = moles (H2)/moles (PC) and the volume of H2 produced. The photocatalytic activity increases following 4CzTPN-R < 4CzPN-R < 4CzIPN-R, whereby the phthalonitrile cyano groups are positioned para, ortho and meta, respectively. For 4CzIPN-R and 4CzPN-R, the photocatalytic activity also increases following R = Ph < tBu < H, while for 4CzTPN-R, R = H < Ph < tBu. The best TADF photocatalyst 4CzIPN-H produced higher TON than the known photosensitiser [Ru(bpy)3]Cl2 (2023 and 144, respectively), although the reaction was considerably more sluggish, proceeding in 60 hours compared to 5 hours when using the ruthenium complex. The authors concluded that the delayed fluorescence component (τd = 1–2 μs for 4CzPN and 4CzIPN in THF) was not essential, since the reductive quenching necessary for the photocatalytic cycle, took place on a much faster timescale (0.2–0.3 ns) than the intersystem crossing/reverse intersystem crossing cycle. | ||
| Fig. 196 Photocatalytic cycle for the water reduction reaction. | ||
Photocatalytic hydrogen production is also possible through dehydrogenation of amines (Fig. 197a). A 4CzPN motif (Fig. 197b) has been incorporated into a supramolecular assembly consisting of four pendant CoIII cobaloxime moieties. This system serves to promote the catalytic acceptorless dehydrogenation (CAD) of secondary amine to imines.379 The cobaloxime Co(dmgH)2PyCl is a well-known hydrogen evolution catalyst,380 which has been used in conjunction with a range of photosensitisers. For example, with a platinum(II) terpyridyl acetylide chromophore381 or a rhodamine based photosensitiser,382 water reduction is possible, whilst aromatic C–H thiolation could occur when the Co co-catalyst was used in combination with [Ru(bpy)3]Cl2.383 The proposed mechanism for the CAD procedure is shown in Fig. 198. Upon excitation of the photocatalyst, the Cobalt co-catalyst is reduced following a SET from the excited 4CzPN motif to CoIII. The oxidised photocatalyst then promotes oxidation of the secondary amine to the α-amino radical and concomitant regeneration of the 4CzPN moiety. This carbon-based radical is then further oxidized by the cobalt(II) species to afford the iminium ion, which is deprotonated by the as-formed cobalt(I) to afford the final imine product. The success of this photocatalyst was shown through the TON rather than yield of the imine product. Using a photoactive 4CzPN-based moiety and Co(dmgH)2PyCl as two separate entities only yielded a TON of 53 in comparison to 305 obtained for the combined supramolecular assembly, highlighting the advantages of covalently linking the two species together.
 | ||
| Fig. 197 (a) Reaction scheme for the photocatalytic dehydrogenation of secondary amines and (b) photoactive component (1) of the supramolecular catalyst. | ||
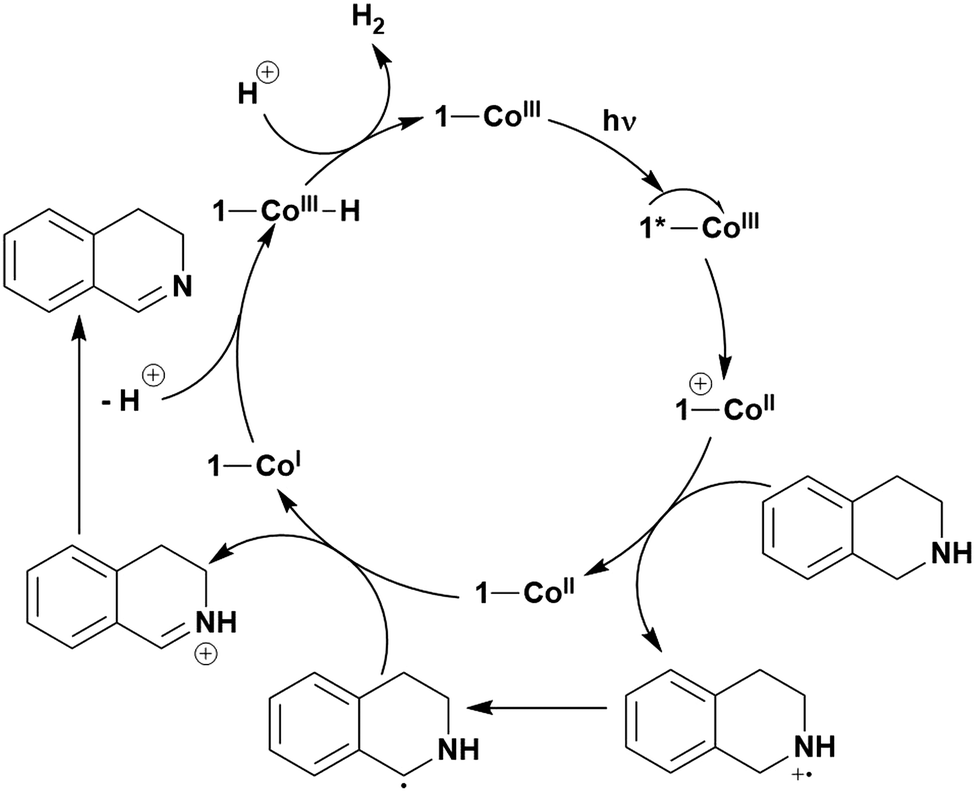 | ||
| Fig. 198 Mechanism for the catalytic acceptorless dehydrogenation of secondary amines to imines using a supramolecular assembly. | ||
HAT catalysis
Finally, dual catalysis using a CDCB-based TADF molecule as the photocatalyst has been reported to work effectively alongside a HAT catalyst in the trifluoromethylthiolation of tertiary ethers (Fig. 199).384 The excited photocatalyst undergoes reductive quenching from the thiolate anion, generating the thiyl radical. This electrophilic thiyl radical then undergoes HAT with the most hydridic α-C–H group to the ether, generating an alkoxyl radical. Cleavage of the C–O bond then results in the required alkyl radical. This part of the mechanism was confirmed by Stern–Volmer quenching experiments where emission of the PC was quenched by the presence of thiolate anions, and radical trapping experiments with TEMPO demonstrated the existence of the tertiary alkyl radical.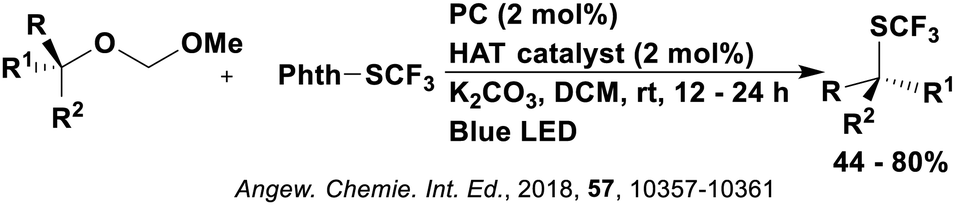 | ||
| Fig. 199 Reaction scheme for the trifluoromethylthiolation of tertiary ethers. | ||
Two potential mechanistic pathways were proposed for the regeneration of the photocatalyst: pathway A (Fig. 200a) involves the reduced PC acting to reduce Phth-SCF3 with the resultant radical coupling generating the product, with loss of the Phth anion; alternatively, pathway B (Fig. 200b) implicates the addition of the alkyl radical to Phth-SCF3, followed by release of the Phth radical, which is reduced to the anion by the reduced PC, regenerating the PC. The quantum yield was calculated to be 0.024, ruling out a radical chain pathway. 4CzIPN gave the highest product yield of 76% while 4CzPN gave the second highest product yield of 63% with other organic photocatalysts, such as eosin Y, proving unsuccessful. Both 4CzIPN and 4CzPN have similar ground and excited state reduction potentials (Ered = −1.21 V and −1.16 V and Ered* = 1.35 V and 1.40 V, respectively), which explains why they have similar yields whereas eosin Y is much weaker photooxidant and ground state reductant (Ered* = 0.83 V and Ered = −1.06 V); hence may not be able to complete the required SET.
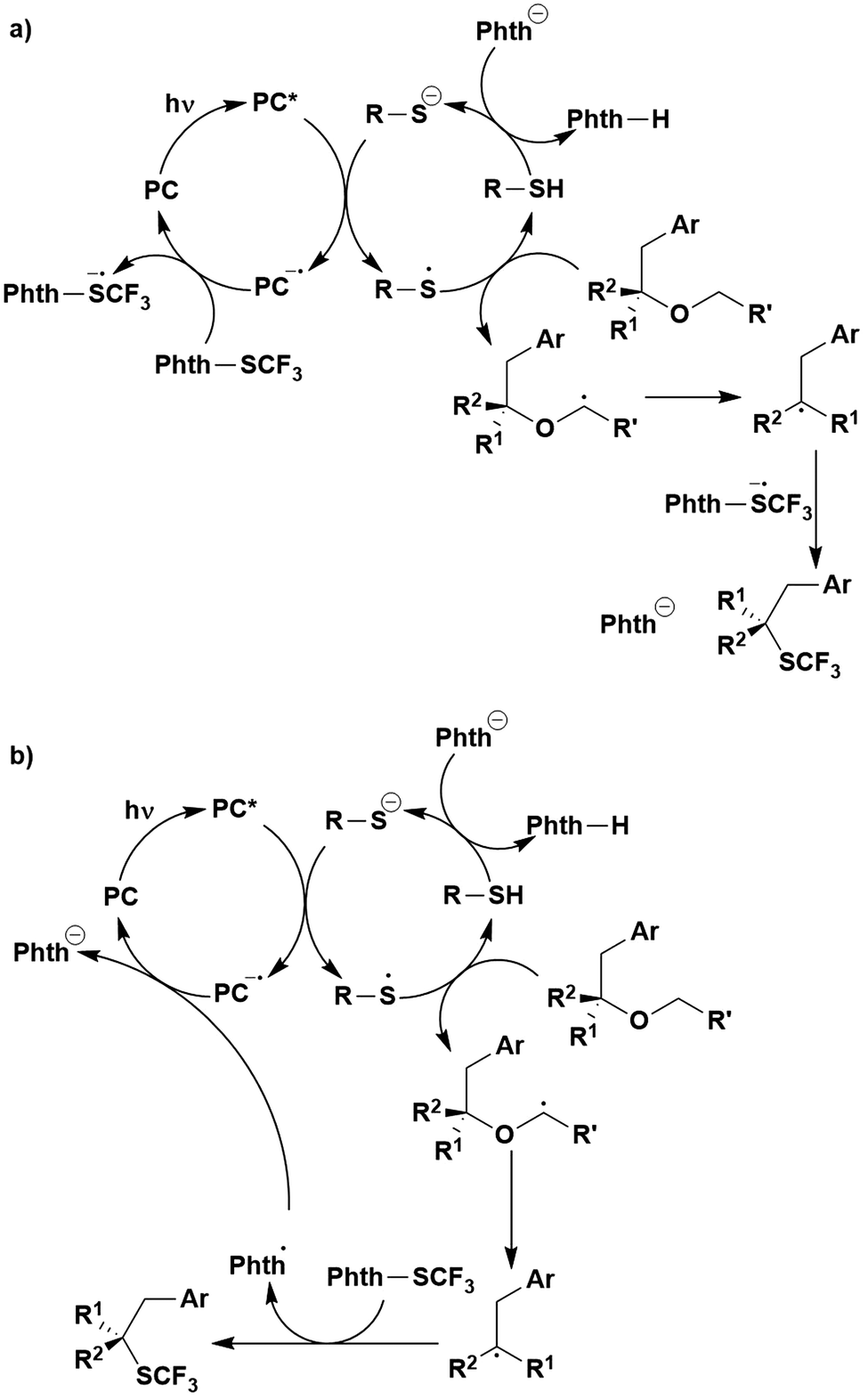 | ||
| Fig. 200 Two proposed mechanisms for the carbotrifluoromethylthiolation of tertiary ethers using dual catalysis. | ||
An additional example of dual photocatalysis with HAT catalysis can be seen in the carboxylation of benzylic C–H bonds (Fig. 201).324 The proposed mechanism involves reductive quenching of the excited photocatalyst by the triisopropylsilanethiol HAT catalyst (Fig. 202). This is unusual, as typically the thiol anion is suggested to reductively quench the photoexcited state of the PC. Regardless, the electrophilic thiyl radical that is generated after deprotonation of the oxidised HAT catalyst can abstract a H atom from the aryl substrate, closing the HAT catalytic cycle. The benzylic radical is then reduced by the reduced PC, closing the photocatalytic cycle. Carboxylation and protonation of the benzylic anion forms the required product. This proposed mechanism is supported by radical trapping experiments with TEMPO, confirming the presence of radical intermediates. The C–H bond cleavage step is postulated to be the rate-determining step from the observed kinetic isotope effect. In the photocatalyst screen, five TADF PCs investigated, 4CzIPN, 5CzBN, 3DPA2FBN, 3DPAFIPN, and 4CzBN, afforded similar product yields (14%, 7%, 11%, 14% and 23%, respectively) whereas the three iridium photocatalysts considered, fac-Ir(ppy)3, [Ir(ppy)2(dtbbpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, all yielded no product under the same reaction conditions. 4CzIPN was selected as the photocatalyst for this reaction, and further optimisation resulted in an increase in yield from 14% to 57%. König et al. rationalized the success of the TADF compounds compared to the iridium complexes by suggesting the organic compounds can generate an in situ photocatalyst, which cannot be formed with the transition metal PCs. When ethylbenzene was used as the starting material, the formation of 2,3,4,6-tetra(9-H-carbazol-9-yl)-5-(1-phenylethyl)benzonitrile (4CzPEBN) was detected, which was believed to be the active photocatalyst in the reaction (Fig. 203). The phenylethyl radical has a very negative reduction potential (Ered = −1.60 V), hence in terms of thermodynamics, the ground state reduction potential of 4CzIPN is not sufficiently reducing (since Ered = −1.21 V) whereas 4CzPEBN can complete this transformation (Ered = −1.69 V). This in situ-generated photocatalyst allows for challenging reductive transformations to occur, which are not possible with the iridium photocatalysts.
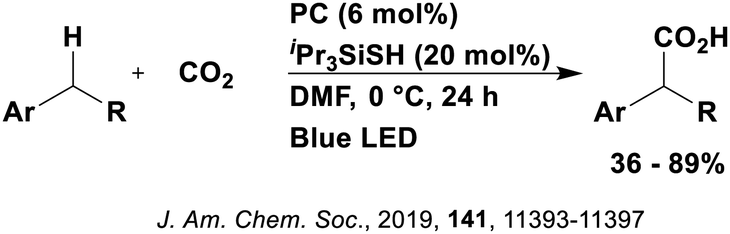 | ||
| Fig. 201 Reaction scheme for the photocarboxylation of benzylic C–H bonds. | ||
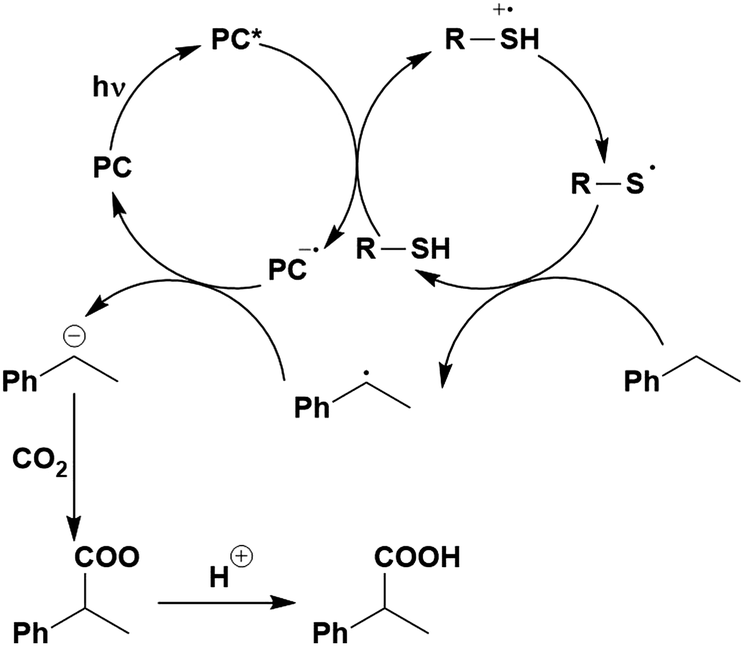 | ||
| Fig. 202 Proposed mechanism for the photocarboxylation of benzylic C–H bonds. | ||
 | ||
| Fig. 203 Proposed generation of 4CzPEBN from 4CzIPN, where Cz = carbazole. | ||
This mode of dual catalysis has been applied to the generation of benzylic carbanions, which were used to produce homobenzylic alcohols when reacted with electrophiles such as ketones and aldehydes (Fig. 204a and b).385 The mechanism proposed is similar to the one in Fig. 202, except that in this case the HAT catalyst, (iPr)3SiSH, is first deprotonated before undergoing SET to the excited PC (Fig. 205). The resultant thiyl radical abstracts a proton from ethylbenzene to form the benzylic radical, closing the HAT catalytic cycle. This benzylic radical is subsequently reduced to the benzylic carbanion by the reduced PC, completing the photocatalytic cycle. Reaction between the benzylic carbanion and a carbonyl results in the desired product. Mechanistic investigations were undertaken to confirm the presence of the carbanion as well as Stern–Volmer quenching experiments, which corroborated quenching of the excited PC by the thiolate. Optimisation of the reaction involving ketones was first conducted, with 4CzIPN, 3DPA2FBN and 3DPAFIPN being the TADF compounds under investigation and indeed being the only successful photocatalysts, providing yields of 30%, 50% and 28%, respectively. Their success seems to be a reflection of their suitably reducing ground state reduction potentials (Ered = −1.21 V, −1.92 V and −1.59 V, for 4CzIPN, 3DPA2FBN and 3DPAFIPN, respectively). Other PCs considered, including eosin Y, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and [Ru(bpy)3](PF6)2, all yielded no product, despite having appropriate redox potentials. Perhaps this may be related to the formation of 4CzPEBN as suggested by the earlier study of König et al.,324 although there is no mention of this in the follow up study by the same group. When using aldehydes, 4CzIPN, 3DPA2FBN, 3DPAFIPN and 4CzBN provided yields of 20%, 10%, 32% and 20%, respectively. The reaction was shown to plateau after a few hours, and it was uncovered that the presence of alcohols poisons the reaction, with König et al. proposing deleterious protonation of the carbanion.
 | ||
| Fig. 204 Reaction scheme for the synthesis of secondary and tertiary homobenzylic alcohols from unfunctionalised starting materials and (a) ketones or (b) aldehydes. | ||
 | ||
| Fig. 205 Possible mechanism for the dual catalytic generation of benzylic carbanions. | ||
Amide synthesis, through coupling of an alcohol and an amine (Fig. 206),329 is additionally possible. The mechanism was proposed to involve two photocatalytic cycles and can be thought of as a two-step process. The first cycle (Fig. 207a) involves oxidative quenching of the excited PC by oxygen, followed by oxidation of the HAT catalyst, quinuclidine, to close the photocatalytic cycle. The quinuclidine radical cation can then abstract a H atom from the α-hydroxy position of the H-bonded alcohol, resulting in the α-hydroxy radical. Oxidation of this radical produces the aldehyde. The second photocatalytic cycle by comparison depends on the nature of the amine. For both primary and secondary amines, the excited PC is reductively quenched by the presence of sacrificial electron donor DIPEA, to generate the reduced PC. Although DIPEA is an amine, its role in this reaction is purely sacrificial. An additional amine is present, which is used as the coupling partner. When this is a secondary amine (Fig. 207b), regeneration of the PC occurs by oxidation from oxygen, to form a superoxide radical. The aldehyde previously formed then reacts with amine, producing a hemiaminal, which can be oxidised by the superoxide radical to form the amide. By contrast, the proposed mechanism for a primary amine suggests that the reduced PC is used to reduce an in situ-formed imine, which following oxidation and protonation, yields the amide product (Fig. 207c). Radical trapping experiments confirmed the presence of the superoxide radical and Stern–Volmer quenching experiments indicated that DIPEA was acting as the quencher of the excited PC; however, no additional mechanistic evidence was provided, causing Singh et al. to propose five different mechanisms for the second photocatalytic cycle.
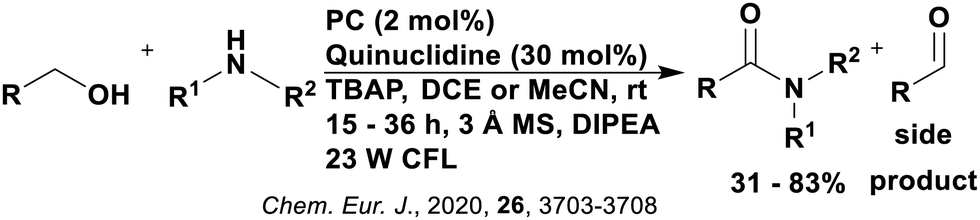 | ||
| Fig. 206 Reaction scheme for the synthesis of amides from alcohols and amines. | ||
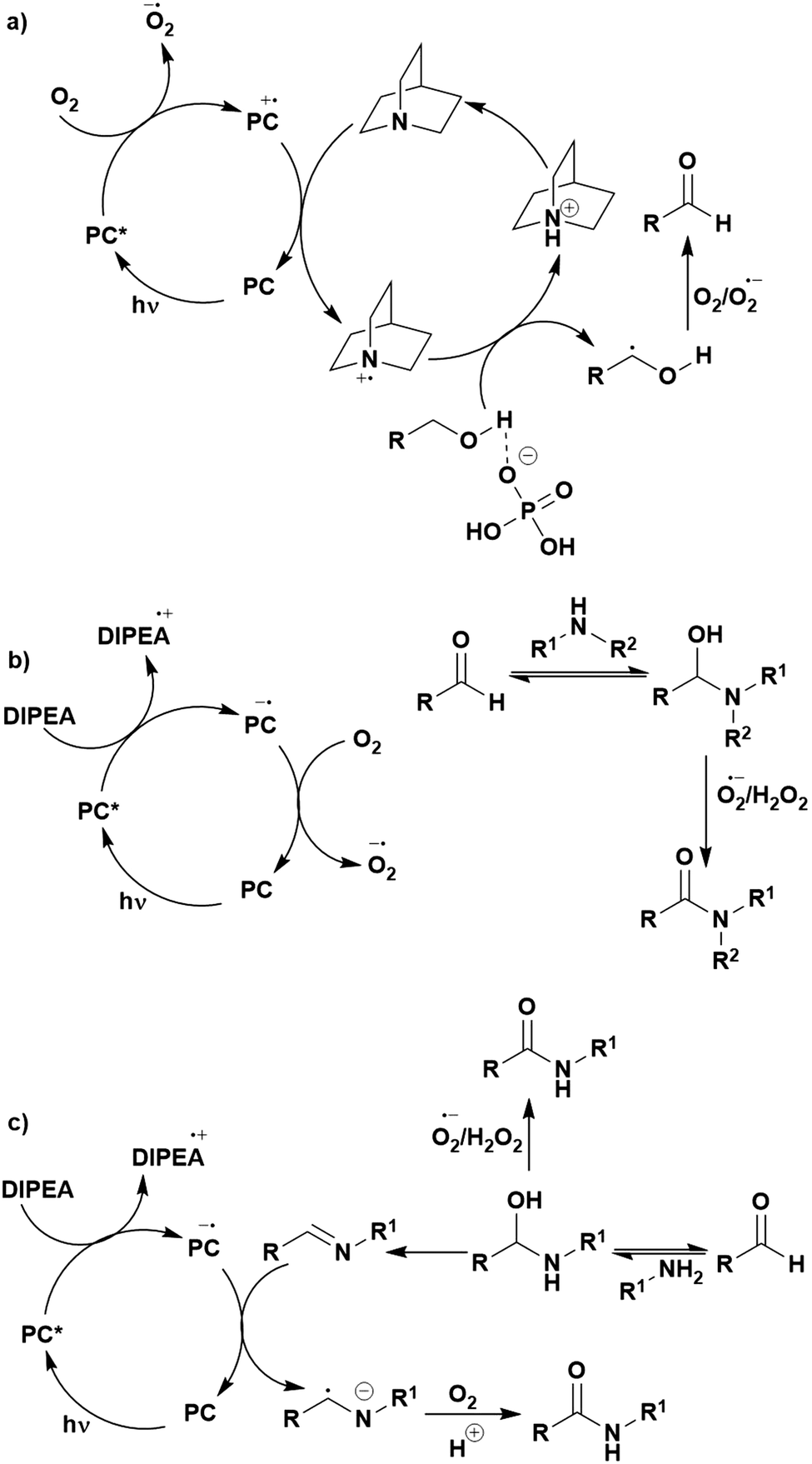 | ||
| Fig. 207 Plausible mechanism for the dual catalytic synthesis of amides from alcohols and amines: (a) formation of the aldehyde, (b) reaction with secondary amine and (c) reaction with a primary amine. | ||
Initial optimisation of the reaction conditions focused on the reaction of a benzylic alcohol with piperidine. Reference photocatalysts [Ru(bpy)3](PF6)2 and eosin Y produced no product while 4CzIPN and 4CzIPN-Br yielded 68% and 61% of amide, respectively, with additional aldehyde side product of 29% and 33%, respectively. The oxidation potential of quinuclidine is moderate (Eox = 1.1 V), hence the photocatalyst must be sufficiently oxidising in the ground state, which the TADF PCs most certainly are (Eox = 1.52 V and 1.76 V for 4CzIPN and 4CzIPN-Br, respectively) while eosin Y is not (Eox = 0.78 V). Although [Ru(bpy)3](PF6)2 is sufficiently oxidising in the ground state (Eox = 1.29 V), the problem here is its photoreducing ability (Eox* = −0.81 V), which isn’t sufficient to reduce oxygen (Ered = −0.86 V).269 When primary amines were investigated, 4CzIPN yielded only 15% of the amide product, hence 5CzBN-OMe was considered, which proved more efficient, affording a product yield of 67%. Singh et al. hypothesised that the change in performance is due to the in situ formation of an imine (Ered < −1.5 V),386 which cannot be reduced by the reduced 4CzIPN (Ered = −1.21 V) but can be reduced with 5CzBN-OMe (Ered = −1.79 V). The success of 5CzBN-OMe in this reaction seems to contradict the hypothesis of Zeitler et al. who suggested this TADF compound suffered from problems of BET, making it an inefficient PC.114
Finally, a base-free Corey Seebach reaction has been developed using this combination of dual catalysis (Fig. 208).387 In the proposed mechanism, the excited PC is reductively quenched by the HAT catalyst iPr3SiSH, which after deprotonation, yields the iPr3SiS radical. This radical can abstract a proton from the dithiane, with the resultant dithiyl radical being reduced by the reduced PC, closing the photocatalytic cycle. The carbanion nucleophile generated can attack non-activated ketones, with the final product formed after protonation. Support for this mechanism was provided in terms of Stern–Volmer quenching experiments which indicated the HAT catalyst was the cause of the luminescence quenching of the PC. The radical–radical homo-coupled side product, occurring from coupling of the dithiyl radical species, was detected by HRMS, with increasing yield of this species observed in the absence of the ketone electrophile. Deuterium labelling studies also acted in support of the mechanism; with deuterated tert-butanol as the electrophile, the deuterated dithiane product could be isolated.
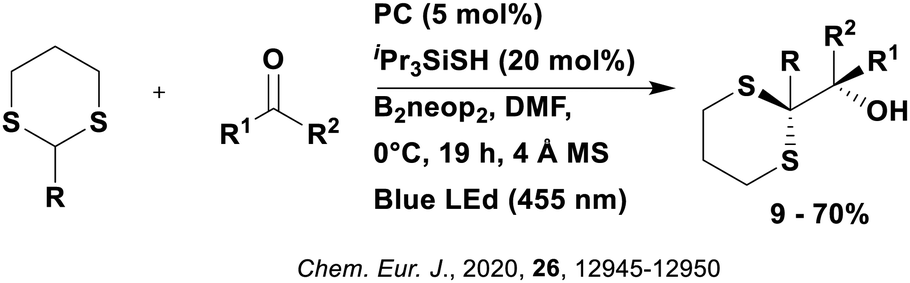 | ||
| Fig. 208 Reaction scheme for the base-free Corey-Seebach reaction. | ||
In the PC screen, four TADF compounds and two iridium PCs were tested, with both iridium species (fac-Ir(ppy)3 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6) producing no product. Three of the TADF compounds did not fair much better; 4CzIPN, 3DPAFIPN and 4CzBN all yielded between 2–7% while 3DPA2FBN managed to afford 30% of product. The dithiyl radical is difficult to reduce (Ered = −1.87 V vs. SCE), which hence explains why the majority of PCs struggled with this reaction (Ered = −1.37 V, −1.21 V, −1.59 V and −1.63 V for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 4CzIPN, 3DPAFIPN and 4CzBN, respectively) in comparison to 3DPAF2BN (Ered = −1.92 V). Despite fac-Ir(ppy)3 being suitably reducing in the ground state (Ered = −2.19 V), its poor photooxidising ability may have prevented the complex from turning over the reaction (Ered* = 0.31 V compared to 0.92 V for 3DPA2FBN).
7. Other organic TADF photocatalysts
While most of the examples in the literature have focused on 4CzIPN as well as structurally related CDCB family of compounds as organic TADF photocatalysts, there are three reports thus far that exists for evaluating another class of TADF molecules as PCs, all in polymerisation reactions. In the first study, four organic compounds based on carbazole/sulfone-based structures (Fig. 209),58 di(4-(4-(carbazole-9-yl)phenyl)sulfone) (CzS1), di(4-(4-(9-phenylcarbazol-3-yl)phenyl)sulfone) (CzS2), 9,9′-(sulfonylbis(4,1phenylene))bis(9H-carbazole) (2Cz-DPS) and 9,9′-(sulfonylbis(4,1phenylene))bis(3,6-di-tert-butyl-9H-carbazole) (2TCz-DPS), were tested as photocatalysts in the free radical polymerisation of methacrylates. FRP of methacrylates can proceed via both a reductive or oxidative quenching cycle, depending on whether a sacrificial reductant is present or oxidants such as iodonium salts are used.229 Since iodonium salts are employed in combination with the TADF compounds, an oxidative quenching mechanism is proposed to be in operation. Of these four sulfones, only two have an experimentally determined ΔEST sufficiently small to be considered TADF molecules: 2Cz-DPS and 2TCz-DPS. While all four compounds were effective in the photopolymerisation reaction, 2TCz-DPS, which displayed the longest emission lifetime (Table 3), was the most efficient in terms of both the rate of polymerisation and conversion. The ground state oxidation potentials of CzS1 and CzS2 are considerably less positive than 2Cz-DPS and 2TC-DPS (Eox = 0.81 V, 0.82 V, 1.32 V and 1.26 V, respectively), which may explain their poorer performance as photocatalysts in this reaction. This study by Lalevée et al. focused predominately on copper complexes as PCs, with only a scant mention of these organic compounds as photocatalysts. It is difficult to ascertain how potentially useful these PCs are as there is no comparison to reference photocatalysts, making it challenging to put these results into context.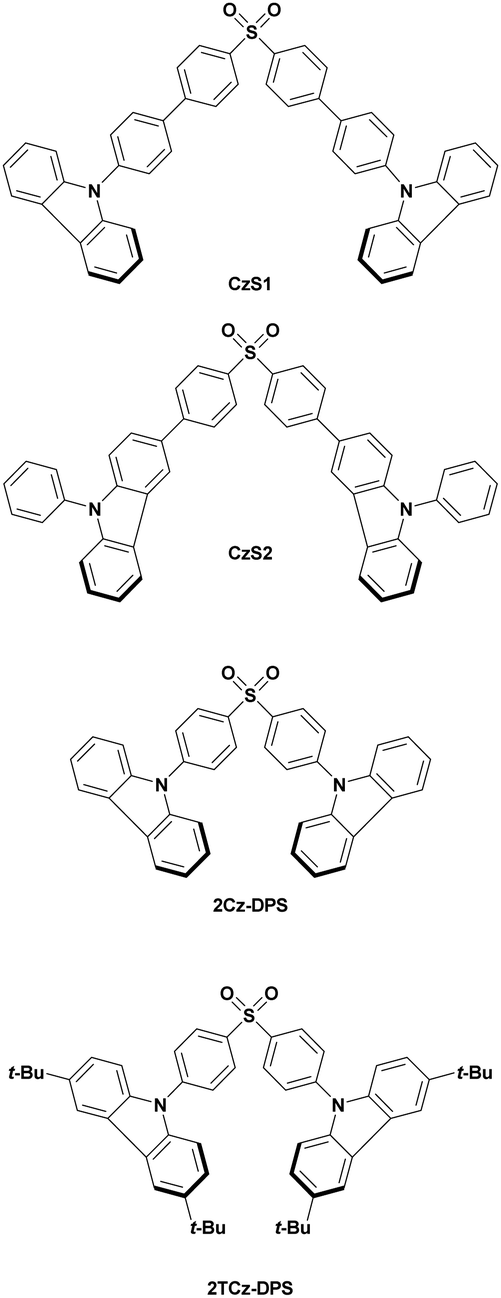 | ||
| Fig. 209 Carbazole-sulfone based molecules tested as photocatalysts. | ||
| PC | λ abs/nm | λ PL/nm | E 0,0/eV (kJ mol−1) | ΔEST/eV | E ox/V | E red/V | E ox*/V | E red */V | τ pf/ns | τ df/μs | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All potentials are given in volts versus SCE. Eox* = Eox − E0,0 and Ered* = Ered + E0,0. 1 eV = (1.602 × 10−22 kJ) × NA where NA = Avogradro's constant. λabs refers to the absorption maximum of the CT band. λPL refers to the photoluminescence maximum. Data reported in MeCN at room temperature unless otherwise noted. E0,0 determined from the intersection point of the normalised absorption and emission spectra unless otherwise noted. b Determined in toluene. c Estimated from the difference between fluorescence and phosphorescence maxima at 77 K. d Determined in CH2Cl2. e Determined in chlorobenzene. f Determined using powder sample. g Determined from the onset of the gated PL spectra at 77 K (delay time: 2 μs and gating time: 200 μs). h E 0,0 estimated using the medium wavelengths between the lowest fluorescence excitation peak (excitation λmax) and the fluorescence peak (emission λmax).120 | |||||||||||
| 4CzIPN | 435 | 535 | 2.67h (258) | 0.08b | 1.52 | −1.21 | −1.04 | 1.35 | 18.7 | 1.39 | 60, 114, 120 and 121 |
| CzS1 | 293b | 399b | 3.16 (305) | 0.62c | 0.81d | −2.35 | 2.55b | — | 388 | ||
| CzS2 | 300b | 381b | 3.16 (305) | 0.65c | 0.82d | −2.34 | 1.51b | — | 388 | ||
| 2Cz-DPS | 345f | 407e | 3.12g (301) | 0.28e | 1.32 | −1.96 | −1.80 | 1.16 | 113 and 389–391 | ||
| 2TCz-DPS | 342f | 404b | 3.08g (297) | 0.32b | 1.26 | −2.06 | −1.82 | 1.02 | 5.3b | 270b | 113, 389, 391 and 392 |
In the second example of other organic TADF photocatalysts considered as PCs, a computer-aided design strategy was implemented to identify donor–acceptor organic compounds that may have appropriate photophysical and electrochemical properties to act as a PC in the atom transfer radical polymerisation (ATRP) of methyl methacrylates, using diethyl 2-bromo-2-methylmalonate (DMB) as the initiator.113 The reaction is similarly proposed to proceed via an oxidative quenching mechanism, so significant focus was placed upon computing Eox* and Eox. A large combination of donors and acceptors were considered with the structures tested as PCs in the polymerisation reaction shown in Fig. 210 and their respective measured properties reported in Table 4.
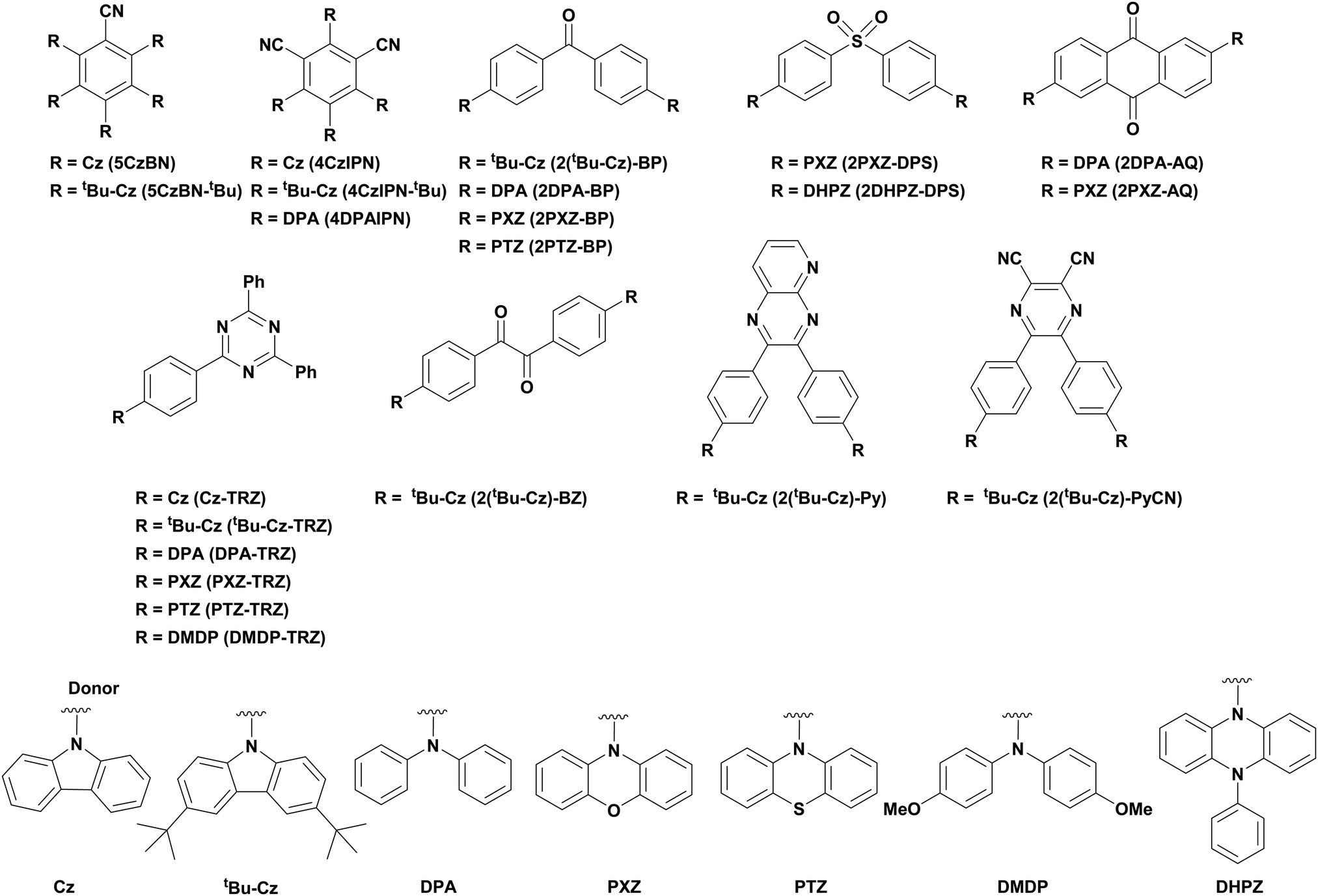 | ||
| Fig. 210 Donor–acceptor compounds considered as photocatalysts for the polymerisation of methyl acrylates. | ||
| PC | λ abs/nm | λ PL/nm | E 0,0/eV (kJ mol−1) | ΔEST/eV | E ox/V | E red/V | E ox*/V | E red */V | τ pf/ns | τ df/μs | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All potentials are given in volts versus SCE. Eox* = Eox − E0,0 and Ered* = Ered + E0,0. 1 eV = (1.602 × 10−22 kJ) × NA where NA = Avogradro's constant. λabs refers to the absorption maximum of the CT band. λPL refers to the photoluminescence maximum. Data reported in MeCN at room temperature unless otherwise noted. E0,0 determined from the intersection point of the normalised absorption and emission spectra unless otherwise noted. b Measured in DCM. c Measured in toluene. d Measured in 6 wt%-doped film in a host matrix, where the host is 1,3-bis(carbazole-9-yl)benzene (mCP), measured at 300 K under vacuum. e Measured in DMF. f E 0,0 (T1) was evaluated from the onset of gated PL spectrum at 77 K (delay time: 2 μs and gating time: 200 μs.113 g E 0,0 estimated using the medium wavelengths between the lowest fluorescence excitation peak (excitation λmax) and the fluorescence peak (emission λmax).120 h Estimated by time-dependent (TD)DFT at the M06-2X/6-31+G(d) level. i Estimated using onset wavelengths of the emission spectra using doped mCP films (6 wt%-doped), with Es measured at 300 K and ET at 5 K. j Determined from the onset of prompt and delayed spectra of 10 wt% doped films in bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO), measured at 77 K. k Measured in doped DPEPO films (10 wt%). l Estimated by TD-DFT at the the PBE0/6-31G level. m Obtained for the quasi-axial and quasi-equatorial conformers, respectively. n Estimated used TD-DFT at the CAMB3LYP/cc-pVDZ level. o Estimated from the absorption edge.401 p Measured using doped 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CPB) films (7 wt%). q Average lifetime calculated by τav = ΣAiτi2/ΣAiτi where Ai is the pre-exponential for lifetime τi. r Estimated from the emission onset.394 | |||||||||||
| 5CzBN | 384e | 2.86e,f (276) | 0.07e | 1.41 | −1.50 | −1.45 | 1.36 | 113 | |||
| 512 | 2.83 (273) | 0.07 | 1.41 | −1.52 | −1.42 | 1.31 | 16.2 | 7.8 | 114, 308 and 309 | ||
| 5CzBN-tBu | 400e | 2.78e,f (268) | 0.06e | 1.12 | −1.61 | −1.66 | 1.17 | 113 | |||
| 480c | 0.17c | 15.0c | 3.4c | 312 | |||||||
| 4CzIPN | 431e | 2.75e,f (265) | −0.03e | 1.52 | −1.21 | −1.23 | 1.54 | 113 | |||
| 435 | 535 | 2.67 g (258) | 0.08c | 1.52 | −1.21 | −1.04 | 1.35 | 18.7 | 1.39 | 60, 114, 120 and 121 | |
| 4CzIPN-tBu | 451e | 2.62e,f (253) | 0.00e | 1.30 | −1.31 | −1.32 | 1.31 | 113 | |||
| 380 | 588 | 2.53 (244) | 0.308h | 1.22 | −1.32 | −1.31 | 1.21 | 10 | 1.4 | 316, 317, 319 and 320 | |
| 4DPAIPN | 2.42e,f (233) | 0.17e | 1.01 | −1.66 | −1.41 | 0.76 | 113 | ||||
| 425 | 523 | 2.62g (253) | 1.34 | −1.52 | −1.28 | 1.1 | 120 | ||||
| 2(tBu-Cz)-BP | 364e | 2.93e,f (283) | 0.13e | 1.05 | −1.66 | −1.88 | 1.27 | 113 | |||
| 2DPA-BP | 375e | 2.62e,f (253) | 0.42e | 1.02 | −1.84 | −1.60 | 0.78 | 113 | |||
| 2PXZ-BP | 401e | 2.52e,f (243) | 0.19e | 0.79 | −1.61 | −1.73 | 0.91 | 113 | |||
| 413c | 509c | 0.3i | 23d | 12d | 393 | ||||||
| 2PTZ-BP | 343e | 2.93e,f (283) | 0.28e | 0.74 | −1.73 | −2.19 | 1.2 | 113 | |||
| 2PXZ-DPS | 383e | 2.81e,f (271) | 0.02e | 0.84 | −1.87 | −1.97 | 0.94 | 113 | |||
| 507c | 2.73c,r (263) | 0.08c | 15c | 2.5c | 394 | ||||||
| 2DHPZ-DPS | 431e | 2.56e,f (247) | 0.03e | 0.26 | −1.98 | −2.30 | 0.58 | 113 | |||
| 577c | 2.40c,r (232) | 5.6c | 0.28c | 394 | |||||||
| 2DPA-AQ | 445e | n.d. | n.d. | 1.20b | −0.83b | n.d. | n.d. | 113 | |||
| 449c | 601c | 2.42c (233) | 0.27c | 5.3c | 377c | 395 | |||||
| 2PXZ-AQ | 529e | n.d. | n.d. | 0.79 | −0.74 | n.d. | n.d. | 113 | |||
| Cz-TRZ | 359e | 2.88e,f (278) | 0.39e | 1.30 | −1.63 | −1.58 | 1.25 | 113 | |||
| 363c | 446c | 0.32j | 1.43b | −1.78b | 4.8k | 396 | |||||
| t Bu-Cz-TRZ | 373e | 2.85e,f (275) | 0.25e | 1.20 | −1.65 | −1.65 | 1.20 | 113 | |||
| 380c | 439c | 0.30c | 7.8k | 40.6k | 397 | ||||||
| DPA-TRZ | 388e | 2.59e,f (250) | 0.37e | 1.00 | −1.73 | −1.59 | 0.86 | 113 | |||
| 389b | 451b | 0.59c,i | 398 | ||||||||
| PXZ-TRZ | 411e | 2.43e,f (234) | 0.22e | 0.73 | −1.63 | −1.70 | 0.80 | 113 | |||
| 420c | 545c | 0.07l | 19c | 0.676c | 399 | ||||||
| PTZ-TRZ | 363e | 2.82e,f (272) | 0.48e | 0.73 | −1.62 | −2.09 | 1.20 | 113 | |||
| 409, 562c,m | 1.14, 0.18m,n | 400 | |||||||||
| DMDP-TRZ | 390e | 2.52e,f (243) | 0.14e | 0.77 | −1.76 | −1.75 | 0.76 | 113 | |||
| 2(tBu-Cz)-BZ | 387e | 2.58e,f (249) | 0.35e | 1.36 | −1.07 | −1.32 | 1.51 | 113 | |||
| 389c | 553c | 2.58c,o (249) | 0.13p | 12.6p | 24.9p,q | 401 | |||||
| 2(tBu-Cz)-Py | 396e | 2.35e,f (227) | 0.49e | 1.20 | −1.37 | −1.15 | 0.98 | 113 | |||
| 2(tBu-Cz)-PyCN | 386e | 2.49e,f (240) | 0.26e | 1.21 | −1.06 | −1.28 | 1.43 | 113 | |||
| 429c | 532c | 2.30c,o (222) | 0.43p | 25.2p | 210.5p,q | 401 | |||||
In the polymerisation of methyl methacrylates, 2DPA-BP, 2DHPZ-DPS, DMDP-TRZ and 4DPAIPN and provided the highest product yield (82%, 78%, 77%, respectively in DMF and 75% in DMSO for 4DPAIPN), which is an improvement over fac-Ir(ppy)3 (50% in DMF). The poorest yields were obtained using 2PXZ-BP, PTZ-TRZ and 2PTZ-BP (3%, 4% and 7%, respectively). The reduced yields obtained by some of the organic PCs are hypothesised by Kwon et al. to be due to inefficient triplet generation, weak visible light absorption, redox potential mismatch or a combination of these factors. The criteria for a good PC in this reaction is suggested to be linked to efficient generation of long-lived triplet excited states, strong reducing power of T1, high stability of radical cations and broad, strong visible light absorption. This was suggested due to the results obtained with 4DPAIPN as a PC, which performed particularly well, especially at a loading of 0.5 ppm, giving a yield of 75% in DMSO and 60% in DMF, in comparison to fac-Ir(ppy)3, which performed second best at these low loadings, giving 49% yield in DMF. The properties of these two PCs were then compared, where it was found that the molar absorptivity of the CT band of 4DPAIPN is roughly one order of magnitude larger than that of fac-Ir(ppy)3 (ε = 13900 M−1 cm−1 and 2450 M−1 cm−1 at 458 nm for 4DPAIPN and fac-Ir(ppy)3, respectively). Moreover, the excited state lifetime is considerably longer for 4DPAIPN (28 μs for τdf for 4DPAIPN and 1 μs for fac-Ir(ppy)3). 4DPAIPN was also shown to form a highly stable radical cation as demonstrated by the reversible oxidation waves observed by CV. Finally, the redox potentials indicate that while fac-Ir(ppy)3 is the better photoreductant (Eox* = −1.28 V and −1.73 V for 4DPAIPN and fac-Ir(ppy)3, respectively), both have sufficient photoreducing capacity to reduce MMA-Br (Ered ∼ −0.9 V). The higher performance of 4DPAIPN may be due to it being a stronger ground state oxidant (Eox = 1.34 V and 0.77 V for 4DPAIPN and fac-Ir(ppy)3, respectively).
The triplet state was suggested as being the state that participates in SET although no evidence is provided to support this assertion. Triplet exciton generation was confirmed with most of the molecules by the observed phosphorescence at 77 K using gated photoluminescence spectroscopy; the small calculated ΔEST values imply that these compounds are TADF. No further spectroscopic investigations were undertaken to prove the participation of the triplet state in this reaction mechanism.
A follow up study from the same group investigated the same subset of compounds in photoinduced electron/energy-transfer reversible addition–fragmentation chain-transfer (PET-RAFT) of methyl methacrylate.402 Since both electron and energy transfer processes are feasible photocatalytic pathways for this reaction, both the redox potentials and the energy of the triplet state were considered when choosing which organic PCs to investigate. Based on these considerations, 5CzBN, 4CzIPN, 4DPAIPN, 2DPA-BP, 2DHPZ-DPS and DMDP-TRZ were chosen for investigation, which provided monomer conversion of 35%, 22%, 70%, 10%, 21% and 43%, respectively. 4DPAIPN was thus clearly identified as the best PC for this reaction, providing comparable monomer conversion to fac-Ir(ppy)3 (72%). Again, the success of 4DPAIPN was assigned to its strong visible light absorption, suitable redox potentials, stability of the radical cation formed and long-lived excited state lifetimes. However, since the mechanism in operation was not clarified, it becomes difficult to assign which of these factors is most important.
These studies from Kwon et al. have identified a wide range of donor–acceptor compounds that may have potential as photocatalysts. While 4DPAIPN was identified in both cases as one of the most promising PCs, these investigations illustrate the value of a wider search into TADF compounds as photocatalysts.
8. Conclusions
Undoubtedly, photocatalysis has already proven to be a powerful tool for a wide range of organic transformations. These reactions proceed under much milder reaction conditions in comparison to alternative synthetic techniques. To further enhance the scope and diversity of reactions that can be photocatalyzed, new photocatalysts must be discovered. Organic photocatalysts provide an alluring class of catalysts as they are relatively inexpensive to synthesize, have toxicity profiles that are superior to heavy transition metal photocatalysts and can easily be structurally diversified. This review has documented why one organic photocatalyst, 4CzIPN, has become so popular. 4CzIPN has often been used on account of it being a “greener”, less toxic and cheaper photocatalyst, which can broadly undergo the same organic transformations as cationic iridium photocatalysts, without compromising on yields.The intrinsic photophysical properties of 4CzIPN, associated with the property of TADF, mean that this compound exhibits comparable excited state lifetimes to heavy metal photocatalysts. Factors such as similar ground and excited state redox properties, as well as enhanced molar absorptivity in the 400–450 nm region in comparison to [Ir(dF(CF3)ppy)2(dtbbpy)]+, contribute to 4CzIPN being a formidable photocatalyst. However, what is less clear is whether having accessible singlet and triplet excited states is intrinsically beneficial to enhancing reactivity in either PET or PEnT based reactions. On the other hand, the small ΔEST in TADF compounds implies that significantly less energy is lost during ISC to the triplet state than what is typically found in transition metal PCs, leading to a wider accessible excited state redox window given the higher energy optical gaps that are typically available to TADF compounds. A more in depth photophysical study would be required to adequately address this key point. What is, however, clear is that the donor–acceptor nature of these compounds allows for facile modification of the structure and a corresponding broad range of accessible ground and excited state redox potentials. These qualities alone explain the exponentially increasing popularity of this class of PCs.
From a mechanistic point of view, noteworthy is that TADF compounds, unlike heavy metal phosphorescent PCs, can participate in both Förster and Dexter energy transfer processes, which may be advantageous in PEnT reactions. However, the efficiency with which TADF compounds can participate in these mechanisms requires further investigation. When considering a photoredox catalysis mechanism, SET can potentially occur from the both the S1 and T1 states. Accessing SET from the S1 is advantageous in that this state is a more potent oxidant and reductant than the corresponding triplet state, conferring to TADF compounds yet another advantage to transition metal complexes, which participate in SET purely from their T1 state.
Despite the great potential of TADF PCs, their development is only in its infancy, lagging far behind the volume of TADF compounds designed for OLEDs. A more concerted and robust investigation of other TADF compounds seems certainly warranted.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Syngenta and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT) for financial support [PhD studentship to “M.A.B.”; Grant code: EP/L016419/1]. We wish to thank Prof. Dr. Andreas Steffen for helpful discussions. We thank Francis Millward, Callum Prentice, Lea Hämmerling and Bryony Hockin for proof reading of this document.Notes and references
- D. A. Nicewicz and D. W. C. Macmillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, ACS Cent. Sci., 2019, 5, 109–115 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 10075–10166 CrossRef CAS PubMed.
- R. A. Marcus, J. Phys. Chem., 1989, 93, 3078–3086 CrossRef CAS.
- R. A. Marcus, Discuss. Faraday Soc., 1960, 29, 21–31 RSC.
- D. Rehm and A. Weller, Ber. Bunsenges., 1969, 73, 834–839 CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- A. Juris and V. Balzani, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- J. Breu, P. Sto, S. Schrader, A. Starukhin, W. J. Finkenzeller, H. Yersin, P. Chemie, D. Regensburg, R. V. August, V. Re, M. Recei and V. No, Chem. Mater., 2005, 1745–1752 CrossRef CAS.
- G. St-Pierre, S. Ladouceur, D. Fortin and E. Zysman-Colman, Dalton Trans., 2011, 40, 11726–11731 RSC.
- Metals Daily, https://www.metalsdaily.com/live-prices/pgms/, (accessed 3 June 2020).
- A. Lebeau, in Hamilton and Hardy's Industrial Toxicology, ed. R. D. Harbidon, M. M. Bourgeois and G. T. Johnson, Wiley-Blackwell, New Jersey, 6th edn, 2015, pp. 187–192 Search PubMed.
- J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CAS.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- N. Robertson, B. M. Hockin, C. Li and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- H. Huang, X. Li, C. Yu, Y. Zhang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- H. G. Roth, S. F. Oliver, L. Campeau, D. Nicewicz and D. A. Dirocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef PubMed.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- Z. Li, H. Song, R. Guo, M. Zuo, C. Hou, S. Sun, X. He, Z. Sun and W. Chu, Green Chem., 2019, 21, 3602–3605 RSC.
- M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794–2800 CrossRef CAS.
- S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC.
- Z. Y. Yu, J. N. Zhao, F. Yang, X. F. Tang, Y. F. Wu, C. F. Ma, B. Song, L. Yun and Q. W. Meng, RSC Adv., 2020, 10, 4825–4831 RSC.
- M. A. Miranda and H. García, Chem. Rev., 1994, 94, 1063–1089 CrossRef CAS.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169–171 CrossRef CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- J. Luo, X. Zhang, J. Lu and J. Zhang, ACS Catal., 2017, 7, 5062–5070 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry – An Introduction, University Science Books, Sausalito, 2009 Search PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- O. A. El Seoud, W. J. Baader and E. L. Bastos, Practical Chemical Kinetics in Solution, John Wiley & Sons, Amsterdam, First Edit., 2016 Search PubMed.
- P. A. M. Dirac, Proc. R. Soc. A, 1928, 117, 610–624 Search PubMed.
- D. G. Fedorov, S. Koseki, M. W. Schmidt and M. S. Gordon, Int. Rev. Phys. Chem., 2003, 22, 551–592 Search PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1927, 114, 243–265 CAS.
- E. Fermi, Z. Phys., 1934, 88, 161–177 CrossRef CAS.
- J. Tanaka, Bull. Chem. Soc. Jpn., 1965, 38, 86–102 CrossRef CAS.
- I. S. K. Kerkines, I. D. Petsalakis, G. Theodorakopoulos and W. Klopper, J. Chem. Phys., 2009, 131, 224315 CrossRef PubMed.
- F. Gaida, A. G. Griesbeck and M. Vollmer, in Science of Synthesis: Photocatalysis in Organic Synthesis, ed. B. Konig, Thieme Verlagsgruppe, New York, 2019 Search PubMed.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- J. Widengren, Ü. Mets and R. Rigler, J. Phys. Chem., 1995, 99, 13368–13379 CrossRef CAS.
- N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639–5643 CrossRef CAS.
- N. R. Verhart, P. Navarro, S. Faez and M. Orrit, Phys. Chem. Chem. Phys., 2016, 18, 17655–17659 RSC.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- V. Balzani and S. Campagna, Top. Curr. Chem., 2007, 280, 143–203 CrossRef.
- E. Yu-Tzu, Li, T. Y. Jiang, Y. Chi and P. T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC.
- D. S. M. Ravinson and M. E. Thompson, Mater. Horizons, 2020, 7, 1210–1217 RSC.
- A. Juris and V. Balzani, Coord. Chem. Rev., 1998, 84, 85–277 CrossRef.
- M. Bouzrati-zerelli, N. Guillaume, F. Goubard, D. Gigmes, J. P. Fouassier and J. Laleve, New J. Chem., 2018, 42, 8261–8270 RSC.
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano and C. Milsmann, Nat. Chem., 2020, 12, 345–352 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- J. W. Verhoeven, Pure Appl. Chem., 1996, 68, 2223–2286 CAS.
- F. Strieth-kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- H. Sahoo, J. Photochem. Photobiol., C, 2011, 12, 20–30 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- A. D. McNaught and A. Wilkinson, IUPAC Compendium of Chemical Terminology, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- S. Bai, P. Zhang, P. Antoniou, S. S. Skourtis and D. N. Beratan, Faraday Discuss., 2019, 216, 301–318 RSC.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, Germany, 2018 Search PubMed.
- E. P. Wigner, Nachr. Akad. Wiss. Goettingen, Math.-Phys. Kl., 1927, 375–381 Search PubMed.
- H. Maudera and K. Petersb, Chem. Ber., 1991, 124, 407–410 CrossRef.
- A. Sinicropi, F. Barbosa, R. Basosi, B. Giese and M. Olivucci, Angew. Chem., Int. Ed., 2005, 44, 2390–2393 CrossRef CAS.
- B. Giese, P. Wettstein, C. Stähelin, F. Barbosa, M. Neuburger, M. Zehnder and P. Wessig, Angew. Chem., Int. Ed., 1999, 38, 2586–2587 CrossRef CAS PubMed.
- L. J. Johnston and J. C. Scaiano, Chem. Rev., 1989, 89, 521–547 CrossRef CAS.
- W. Adam, H. Platsch and J. Wirz, J. Am. Chem. Soc., 1989, 111, 6896–6898 CrossRef CAS.
- B. Giese, F. Barbosa, C. Stähelin, S. Sauer, P. Wettstein and C. Wyss, Pure Appl. Chem., 2000, 72, 1623–1629 CAS.
- W. H. Kramer and A. G. Griesbeck, J. Chem. Educ., 2008, 85, 701–709 CrossRef CAS.
- V. Alezra and T. Kawabata, Synthesis, 2016, 2997–3016 CrossRef CAS.
- T. Šumanovac Ramljak, M. Sohora, I. Antol, D. Kontrec, N. Basarić and K. Mlinarić-Majerski, Tetrahedron Lett., 2014, 55, 4078–4081 CrossRef.
- A. G. Griesbeck, W. Kramer and J. Lex, Angew. Chem., Int. Ed., 2001, 40, 577–579 CrossRef CAS PubMed.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- N. J. Turro, Pure Appl. Chem., 1977, 49, 405–429 CAS.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- R. A. Marcus and N. Sutin, Biochem. Biophys. Acta, 1985, 811, 265–322 CAS.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CrossRef CAS PubMed.
- J. R. Miller, J. Am. Chem. Soc., 1984, 106, 3047–3049 CrossRef CAS.
- T. Asahi and N. Mataga, J. Phys. Chem., 1989, 93, 6575–6578 CrossRef CAS.
- I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290–4301 CrossRef CAS.
- I. R. Gould and S. Farid, Acc. Chem. Res., 1996, 29, 522–528 CrossRef CAS.
- G. McLendon, Acc. Chem. Res., 1988, 21, 160–167 CrossRef CAS.
- J. R. Winkler and H. B. Gray, Chem. Rev., 1992, 92, 369–379 CrossRef CAS.
- G. Mclendon and R. Hake, Chem. Rev., 1992, 92, 481–490 CrossRef CAS.
- H. Rau, R. Frank and G. Greiner, J. Phys. Chem., 1986, 90, 2476–2481 CrossRef CAS.
- K. Kikuchi, Y. Takahashi, T. Katagiri, T. Niwa, M. Hoshi and T. Miyashi, Chem. Phys. Lett., 1991, 180, 403–408 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS PubMed.
- U. Megerle, M. Wenninger, R. J. Kutta, R. Lechner, B. König, B. Dick and E. Riedle, Phys. Chem. Chem. Phys., 2011, 13, 8869–8880 RSC.
- U. Steiner, G. Winter and H. E. A. Kramer, J. Phys. Chem., 1977, 81, 1104–1110 CrossRef CAS.
- L. Bergmann, New Emitters for OLEDs: The Coordination- and Photo-Chemistry of Mononuclear Neutral Copper(I) Complexes, Logos Verlag, Berlin, 2016 Search PubMed.
- K. Ohkubo and S. Fukuzumi, Org. Lett., 2000, 2, 3647–3650 CrossRef CAS PubMed.
- M. Fujita, A. Ishida, S. Takamuku and S. Fukuzumi, J. Am. Chem. Soc., 1996, 118, 8566–8574 CrossRef CAS.
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- J. Hore and R. W. Broadhurst, Prog. Nucl. Magn. Reson. Spectrosc., 1993, 25, 345–402 CrossRef.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS.
- J. Haimerl, I. Ghosh, B. König, J. Vogelsang and J. M. Lupton, Chem. Sci., 2019, 10, 681–687 RSC.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- M. Streiter, T. Fischer, C. Wiebeler, S. Reichert, J. Langenickel, K. Zeitler and C. Deibel, Impact of chlorine on the internal transition rates and excited states of the thermally delayed activated fluorescence molecule 3CzClIPN, http://arxiv.org/abs/2001.11317, (accessed 20 May 2020).
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607–5612 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, J. Organomet. Chem., 2015, 776, 51–59 CrossRef CAS.
- Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 143–203 Search PubMed.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 2763–2767 CrossRef CAS PubMed.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, H. J. Van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 16054–16064 CrossRef CAS.
- D. P. Hari and B. Ko, Chem. Commun., 2014, 50, 6688–6699 RSC.
- X. Zhang, I. Zhang and L. Liu, Photochem. Photobiol., 2010, 86, 492–498 CrossRef CAS.
- T. Shen, Z. G. Zhao, Q. Yu and H. J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203–212 CrossRef CAS.
- E. M. Arbeloa, C. M. Previtali and S. G. Bertolotti, ChemPhysChem, 2018, 19, 934–942 CrossRef CAS PubMed.
- A. Seret, E. Gandin and A. Van De Vorst, Chem. Phys. Lett., 1987, 135, 427–431 CrossRef CAS.
- L. Ludvíková, P. Friš, D. Heger, P. Šebej, J. Wirz and P. Klán, Phys. Chem. Chem. Phys., 2016, 18, 16266–16273 RSC.
- A. Penzkofer, M. Simmel and D. Riedl, J. Lumin., 2012, 132, 1055–1062 CrossRef CAS.
- L. E. Cramer and K. G. Spears, J. Am. Chem. Soc., 1978, 100, 221–227 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- E. G. Moschetta, T. Knauber, F. Susanne, F. Lévesque and L. J. Edwards, Nat. Commun., 2020, 11, 804–807 CrossRef.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS.
- J. R. Ochola and M. O. Wolf, Org. Biomol. Chem., 2016, 14, 9088–9092 RSC.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320–1330 CrossRef CAS PubMed.
- Z. Zhang, W. Xiao, J. Xuan, J. Xuan, Z. Zhang and W. Xiao, Angew. Chem., Int. Ed., 2015, 15632–15641 Search PubMed.
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustainable Energy Fuels, 2018, 2, 1905–1927 RSC.
- W. Wang, H. Huang, C. Yu, Y. Zhang, Y. Zhang and P. S. Mariano, J. Am. Chem. Soc., 2017, 139, 9799–9802 CrossRef PubMed.
- J. Davies, L. Angelini, M. A. Alkhalifah, L. M. Sanz, N. S. Sheikh and D. Leonori, Synthesis, 2018, 821–830 CAS.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS.
- H. Jiang and A. Studer, Chem. – Eur. J., 2019, 25, 516–520 CAS.
- O. Zhang and J. W. Schubert, J. Org. Chem., 2020, 85, 6225–6232 CrossRef CAS.
- N. X. Xu, B. X. Li, C. Wang and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 10639–10644 CrossRef CAS PubMed.
- S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed.
- X. L. Lyu, S. S. Huang, H. J. Song, Y. X. Liu and Q. M. Wang, RSC Adv., 2019, 9, 36213–36216 RSC.
- C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS PubMed.
- T. C. Sherwood, H. Xiao, R. G. Bhaskar, E. M. Simmons, S. Zaretsky, M. P. Rauch, R. R. Knowles and T. G. M. Dhar, J. Org. Chem., 2019, 84, 8360–8379 CrossRef CAS.
- R. Gueret, L. Pelinski, T. Bousquet, M. Sauthier, V. Ferey and A. Bigot, Org. Lett., 2020, 22, 5157–5162 CrossRef CAS PubMed.
- N. Bortolamei, A. A. Isse and A. Gennaro, Electrochim. Acta, 2010, 55, 8312–8318 CrossRef CAS.
- D. D. M. Wayner, B. A. Sim and D. Griller, J. Am. Chem. Soc., 1989, 755–757 Search PubMed.
- J. Guo, Q. Wu, Y. Xie, J. Weng and G. Lu, J. Org. Chem., 2018, 83, 12559–12567 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714–723 CAS.
- N. R. Patel, C. B. Kelly, A. P. Siegenfeld and G. A. Molander, ACS Catal., 2017, 7, 1766–1770 CrossRef CAS PubMed.
- C. Zheng, Y. Wang, Y. Xu, Z. Chen, G. Chen and S. H. Liang, Org. Lett., 2018, 20, 4824–4827 CrossRef CAS PubMed.
- Z. Sun, B. Tang, K. K. C. Liu and H. Y. Zhu, Chem. Commun., 2020, 56, 1294–1297 RSC.
- H. Cramail, Y. Landais and E. Grau, Chem. Commun., 2018, 54, 9337–9340 RSC.
- A. H. Jatoi, G. G. Pawar and Y. Landais, Chem. Commun., 2019, 55, 466–469 RSC.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. G. M. Dhar, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493–9500 CrossRef CAS PubMed.
- S. He, X. Chen, F. Zeng, P. Lu, Y. Peng, L. Qu and B. Yu, Chin. Chem. Lett., 2020, 31, 1863–1867 CrossRef CAS.
- A. N. Herron, D. Liu, G. Xia and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 2766–2770 CrossRef CAS PubMed.
- X. L. Huang, Y. Z. Cheng, X. Zhang and S. L. You, Org. Lett., 2020, 22, 9699–9705 CrossRef CAS PubMed.
- Y. Z. Cheng, X. L. Huang, W. H. Zhuang, Q. R. Zhao, X. Zhang, T. S. Mei and S. L. You, Angew. Chem., Int. Ed., 2020, 59, 18062–18067 CrossRef CAS PubMed.
- Q.-F. Bao, M. Li, Y. Xia, Y.-Z. Wang, Z.-Z. Zhou and Y.-M. Liang, Org. Lett., 2021, 23(3), 1107–1112 CrossRef CAS.
- L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS.
- A. Cartier, E. Levernier, V. Corcé, T. Fukuyama, A. Dhimane, C. Ollivier, I. Ryu and L. Fensterbank, Angew. Chem., Int. Ed., 2019, 58, 1789–1793 CrossRef CAS.
- Z. Wang, S. Zheng and Z. Lu, Chem. Sci., 2019, 10, 4389–4393 RSC.
- S. T. J. Cullen and G. K. Friestad, Org. Lett., 2019, 21, 8290–8294 CrossRef CAS PubMed.
- A. Cartier, E. Levernier, A. L. Dhimane, T. Fukuyama, C. Ollivier, I. Ryu and L. Fensterbank, Adv. Synth. Catal., 2020, 362, 2254–2259 CrossRef CAS.
- G. Ikarashi, T. Morofuji and N. Kano, Chem. Commun., 2020, 56, 10006–10009 RSC.
- X. Wang, M. Yang, W. Xie and J. Wu, Chem. Commun., 2019, 55, 6010–6013 RSC.
- T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 2, 21297–21300 RSC.
- J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, ACS Catal., 2016, 6, 8004–8008 CrossRef PubMed.
- M. Jou, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef.
- B. Zhang, Y. Yi, Z. Q. Wu, C. Chen and C. Xi, Green Chem., 2020, 22, 5961–5965 RSC.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- D. Kaiser, A. Noble, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 14104–14109 CrossRef CAS PubMed.
- J. Wu, Z. Wang, X. Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China: Chem., 2020, 63, 336–340 CrossRef CAS.
- S. Cai, Y. Tian, J. Zhang, Z. Liu, M. Lu, W. Weng and M. Huang, Adv. Synth. Catal., 2018, 360, 4084–4088 CrossRef CAS.
- M. Lu, Z. Liu, J. Zhang, Y. Tian, H. Qin, M. Huang, S. Hu and S. Cai, Org. Biomol. Chem., 2018, 16, 6564–6568 RSC.
- N. Meng, Y. Lv, Q. Liu, R. Liu, X. Zhao and W. Wei, Chin. Chem. Lett., 2021, 32, 258–262 CrossRef CAS.
- Q. F. Bao, Y. Xia, M. Li, Y. Z. Wang and Y. M. Liang, Org. Lett., 2020, 22, 7757–7761 CrossRef CAS PubMed.
- H. Wang, J. Zhang, J. Shi, F. Li, S. Zhang and K. Xu, Org. Lett., 2019, 21, 5116–5120 CrossRef CAS PubMed.
- J. Li, L. Li, M. Zhou and H. Wang, Org. Chem. Front., 2018, 5, 1003–1007 RSC.
- V. I. Supranovich, G. N. Chernov, V. V. Levin and A. D. Dilman, Eur. J. Org. Chem., 2020, 393–396 CrossRef CAS.
- M. Zheng, J. Hou, L. Zhan, Y. Huang, L. Chen, L. Hua, Y. Li, W. Tang and B. Li, ACS Catal., 2021, 11, 542–553 CrossRef CAS.
- Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512–5516 RSC.
- X. Ji, Q. Liu, Z. Wang, P. Wang, G.-J. Deng and H. Huang, Green Chem., 2020, 22, 8233–8237 RSC.
- Z. Wang, Q. Liu, X. Ji, G. J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- R. Bevernaegie, S. A. M. Wehlin, E. J. Piechota, M. Abraham, C. Philouze, G. J. Meyer, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2020, 142, 2732–2737 CrossRef CAS PubMed.
- J. J. Perkins, J. W. Schubert, E. C. Streckfuss, J. Balsells and A. ElMarrouni, Eur. J. Org. Chem., 2020, 1515–1522 CrossRef CAS.
- M. Lu, H. Qin, Z. Lin, M. Huang, W. Weng and S. Cai, Org. Lett., 2018, 20, 7611–7615 CrossRef CAS PubMed.
- M. Lu, T. Zhang, D. Tan, C. Chen, Y. Zhang, M. Huang and S. Cai, Adv. Synth. Catal., 2019, 361, 4237–4242 CrossRef CAS.
- Z. Lin, M. Lu, B. Liu, J. Gao, M. Huang, Z. Gan and S. Cai, New J. Chem., 2020, 44, 16031–16035 RSC.
- J. A. Milligan, J. P. Phelan, V. C. Polites, C. B. Kelly and G. A. Molander, Org. Lett., 2018, 20, 6840–6844 CrossRef CAS PubMed.
- J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037–8047 CrossRef CAS.
- C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS PubMed.
- N. B. Bissonnette, J. M. Ellis, L. G. Hamann and F. Romanov-Michailidis, Chem. Sci., 2019, 10, 9591–9596 RSC.
- Z. Song, C. Zhang and S. Ye, Org. Biomol. Chem., 2019, 17, 181–185 RSC.
- H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709–7715 CrossRef CAS PubMed.
- J. Li, X.-E. Yang, S.-L. Wang, L.-L. Zhang, X.-Z. Zhou, S.-Y. Wang and S.-J. Ji, Org. Lett., 2020, 22, 4908–4913 CrossRef CAS PubMed.
- T. Ju, Q. Fu, J. H. Ye, Z. Zhang, L. L. Liao, S. S. Yan, X. Y. Tian, S. P. Luo, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2018, 57, 13897–13901 CrossRef CAS PubMed.
- R. Alam and G. A. Molander, Org. Lett., 2018, 20, 2680–2684 CrossRef CAS PubMed.
- M. Graglia, N. Kanna and D. Esposito, ChemBioEng Rev., 2015, 2, 377–392 CrossRef.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- K. Walsh, H. F. Sneddon and C. J. Moody, Org. Lett., 2014, 16, 5224–5227 CrossRef CAS.
- L. J. Mitchell and C. J. Moody, J. Org. Chem., 2014, 79, 11091–11100 CrossRef CAS.
- V. Photocatalysts, D. Liu, M. Jiao, Z. Feng, X. Wang, G. Xu and P. Xu, Org. Lett., 2018, 20, 5700–5704 CrossRef.
- L. Wu and Q. Liu, Org. Lett., 2019, 4, 885–889 Search PubMed.
- X. C. Liu, X. L. Chen, Y. Liu, K. Sun, Y. Y. Peng, L. B. Qu and B. Yu, ChemSusChem, 2020, 13, 298–303 CrossRef CAS PubMed.
- X. Y. Yuan, F. L. Zeng, H. L. Zhu, Y. Liu, Q. Y. Lv, X. L. Chen, L. Peng and B. Yu, Org. Chem. Front., 2020, 7, 1884–1889 RSC.
- J. Y. Gu, W. Zhang, S. R. Jackson, Y. H. He and Z. Guan, Chem. Commun., 2020, 56, 13441–13444 RSC.
- W. J. Zhou, Z. H. Wang, L. L. Liao, Y. X. Jiang, K. G. Cao, T. Ju, Y. Li, G. M. Cao and D. G. Yu, Nat. Commun., 2020, 11, 3263 CrossRef CAS PubMed.
- W. J. Yoo and S. Kobayashi, Green Chem., 2013, 15, 1844–1848 RSC.
- F.-L. Zeng, X.-L. Chen, K. Sun, H.-L. Zhu, X.-Y. Yuan, Y. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Chem. Front., 2021, 8, 760–766 RSC.
- Z. Huang, Y. Gu, X. Liu, L. Zhang and Z. Cheng, Macromol. Rapid Commun., 2017, 38, 1600461 CrossRef PubMed.
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS.
- Z. Huang, L. Zhang, Z. Cheng and X. Zhu, Polymers, 2017, 9, 4–11 CrossRef.
- S. J. S. Düsel and B. König, Eur. J. Org. Chem., 2020, 1491–1495 CrossRef.
- J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. Macmillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- O. S. Wenger, Chem. – Eur. J., 2021, 27, 2270–2278 CrossRef CAS PubMed.
- A. Dumoulin, J. K. Matsui, Á. Gutiérrez-Bonet and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef CAS.
- S. Parisien-collette and S. K. Collins, ChemPhotoChem, 2018, 2, 855–859 CrossRef CAS.
- P. N. D. Cross-coupling, R. Alam and G. A. Molander, J. Org. Chem., 2017, 82, 13728–13734 CrossRef PubMed.
- E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679–3683 CrossRef CAS PubMed.
- J. K. Matsui and G. A. Molander, Org. Lett., 2017, 19, 436–439 CrossRef CAS PubMed.
- S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS PubMed.
- H. Hsieh, C. W. Coley, L. M. Baumgartner, K. F. Jensen and R. I. Robinson, Org. Process Res. Dev., 2018, 22, 542–550 CrossRef CAS.
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Chem. Commun., 2019, 55, 14657–14660 RSC.
- R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 CrossRef CAS PubMed.
- C. Ollivier, L. Fensterbank, C. Le and C. Ollivier, Chem. Commun., 2016, 52, 9877–9880 RSC.
- J. Yi, S. O. Badir, L. M. Kammer and G. A. Molander, Org. Lett., 2019, 21, 3346–3351 CrossRef CAS PubMed.
- T. J. Steiman, J. Liu, A. Mengiste and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7598–7605 CrossRef CAS PubMed.
- L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532–3539 CrossRef CAS.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172–5177 CrossRef CAS PubMed.
- S. Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370–4374 CrossRef CAS PubMed.
- M. Garbacz and S. Stecko, Adv. Synth. Catal., 2020, 362, 3213–3222 CrossRef CAS.
- E. M. Dauncey, S. U. Dighe, J. J. Douglas and D. Leonori, Chem. Sci., 2019, 10, 7728–7733 RSC.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS.
- M. Durandetti, M. Devaud and K. Perichon, New J. Chem., 1996, 20, 659–667 CAS.
- C. Lévêque, V. Corcé, L. Chenneberg, C. Ollivier and L. Fensterbank, Eur. J. Org. Chem., 2017, 2118–2121 CrossRef.
- M. S. Santos, A. G. Corrêa, M. W. Paixão and B. König, Adv. Synth. Catal., 2020, 362, 2367–2372 CrossRef CAS.
- Z. J. Wang, S. Zheng, E. Romero, J. K. Matsui and G. A. Molander, Org. Lett., 2019, 21, 6543–6547 CrossRef CAS.
- A. G. Diallo, D. Roy, S. Gaillard, M. Lautens and J. L. Renaud, Org. Lett., 2020, 22, 2442–2447 CrossRef CAS.
- F. Cong, X.-Y. Lv, C. S. Day and R. Martin, J. Am. Chem. Soc., 2020, 142, 20594–20603 CrossRef CAS PubMed.
- Q. Y. Meng, S. Wang and B. König, Angew. Chem., Int. Ed., 2017, 56, 13426–13430 CrossRef CAS PubMed.
- X. Q. Zhu, H. R. Li, Q. Li, T. Ai, J. Y. Lu, Y. Yang and J. P. Cheng, Chem. – Eur. J., 2003, 9, 871–880 CrossRef CAS PubMed.
- A. Klein, A. Kaiser, B. Sarkar, M. Wanner and J. Fiedler, Eur. J. Inorg. Chem., 2007, 965–976 CrossRef CAS.
- J. Santandrea, C. Minozzi, C. Cruché and S. K. Collins, Angew. Chem., Int. Ed., 2017, 56, 12255–12259 CrossRef CAS PubMed.
- L. Huang, C. Zhu, L. Yi, H. Yue, R. Kancherla and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 457–464 CrossRef CAS PubMed.
- B. Persson, Acta Chem. Scand., Ser. B, 1977, 31, 88–89 CrossRef.
- B. Zhao, R. Shang, W.-M. Cheng and Y. Fu, Org. Chem. Front., 2018, 5, 1782–1786 RSC.
- S. S. Shah, M. Shee, A. K. Singh, A. Paul and N. D. P. Singh, J. Org. Chem., 2020, 85, 3426–3439 CrossRef CAS PubMed.
- S. Sheri, A. Paul, M. Bera, Y. Venkatesh and N. D. P. Singh, Org. Lett., 2018, 20, 5533–5536 CrossRef PubMed.
- Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, A. I. Tufatullin, O. N. Kataeva, D. A. Vicic and Y. H. Budnikova, Organometallics, 2013, 32, 4785–4792 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- C. Zheng, W. Cheng, H. Li, R. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 CrossRef CAS PubMed.
- J.-C. Grenier-Petel and S. K. Collins, ACS Catal., 2019, 9, 3213–3218 CrossRef CAS.
- Q. Meng, T. E. Schirmer, K. Katou and B. Kçnig, Angew. Chem., Int. Ed., 2019, 58, 5723–5728 CrossRef CAS PubMed.
- K. Takizawa, T. Sekino, S. Sato, T. Yoshino, M. Kojima and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 9199–9203 CrossRef CAS PubMed.
- M. Kojima and S. Matsunaga, Synthesis, 2020, 1934–1946 CrossRef.
- H. Zhang, Q. Xiao, X. K. Qi, X. W. Gao, Q. X. Tong and J. J. Zhong, Chem. Commun., 2020, 56, 12530–12533 RSC.
- K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2016, 138, 15527–15530 CrossRef CAS.
- S. Lin, Y. Chen, F. Li, C. Shi and L. Shi, Chem. Sci., 2020, 11, 839–844 RSC.
- F. S. Li, Y. Q. Chen, S. J. Lin, C. Z. Shi, X. Y. Li, Y. C. Sun, Z. W. Guo and L. Shi, Org. Chem. Front., 2020, 7, 3434–3438 RSC.
- F. Li, S. Lin, Y. Chen, C. Shi, H. Yan, C. Li, C. Wu, L. Lin, C. Duan and L. Shi, Angew. Chem., Int. Ed., 2021, 60, 1561–1566 CrossRef CAS PubMed.
- Y. Li, F. Ying, T. Fu, R. Yang, Y. Dong, L. Lin, Y. Han, D. Liang and X. Long, Org. Biomol. Chem., 2020, 18, 5660–5665 RSC.
- Y. Wang, X. W. Gao, J. Li and D. Chao, Chem. Commun., 2020, 56, 12170–12173 RSC.
- J. L. Schwarz, H. M. Huang, T. O. Paulisch and F. Glorius, ACS Catal., 2020, 10, 1621–1627 CrossRef CAS.
- C. T. Buse and C. H. Heathcock, Tetrahedron Lett., 1978, 19, 1685–1688 CrossRef.
- T. Hiyama, K. Kimura and H. Nozaki, Tetrahedron Lett., 1981, 22, 1037–1040 CrossRef CAS.
- J. P. Cheng, Y. Lu, X. Q. Zhu, Y. Sun, F. Bi and J. He, J. Org. Chem., 2000, 65, 3853–3857 CrossRef CAS.
- J. He, G. Chen, B. Zhang, Y. Li, J. R. Chen, W. J. Xiao, F. Liu and C. Li, Chem, 2020, 6, 1149–1159 CAS.
- M. M. Mastandrea, S. Cañellas, X. Caldentey and M. A. Pericàs, ACS Catal., 2020, 10, 6402–6408 CrossRef CAS.
- R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621–16625 CrossRef CAS PubMed.
- J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- C. Dai, Y. Zhan, P. Liu and P. Sun, Green Chem., 2021, 23, 314–319 RSC.
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS.
- A. Ryder, W. Cunningham, G. Ballantyne, T. Mules, A. Kinsella, J. Turner-Dore, C. Alder, L. Edwards, B. McKay, M. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS.
- W. Xu, H. Jiang, J. Leng, H. W. Ong and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 4009–4016 CrossRef CAS PubMed.
- M. Shimoi, T. Watanabe, K. Maeda, D. P. Curran and T. Taniguchi, Angew. Chem., Int. Ed., 2018, 57, 9485–9490 CrossRef CAS.
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- K. Chen, J. Schwarz, T. A. Karl, A. Chatterjee and B. König, Chem. Commun., 2019, 55, 13144–13147 RSC.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. Macmillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- Y. Zhang, P. Ji, Y. Dong, Y. Wei and W. Wang, ACS Catal., 2020, 10, 2226–2230 CrossRef CAS.
- F. Legros, P. Fernandez-Rodriguez, A. Mishra, R. Weck, A. Bauer, M. Sandvoss, S. Ruf, M. Méndez, H. Mora-Radó, N. Rackelmann, C. Pöverlein and V. Derdau, Chem. – Eur. J., 2020, 26, 12738–12742 CrossRef CAS.
- M.-S. Liu and W. Shu, ACS Catal., 2020, 10, 12960–12966 CrossRef CAS.
- A. V. Davies, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef PubMed.
- L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed.
- D. F. Chen, C. H. Chrisman and G. M. Miyake, ACS Catal., 2020, 10, 2609–2614 CrossRef CAS PubMed.
- T. Shang, L. Lu, Z. Cao, Y. Liu, W. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- P. P. Singh and V. Srivastava, Org. Biomol. Chem., 2021, 19, 313–321 RSC.
- D. Zhang, M. Cai, Z. Bin, Y. Zhang and D. Zhang, Chem. Sci., 2016, 7, 3355–3363 RSC.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- T. Hosokai, J. Photonics Energy, 2018, 8, 032102 Search PubMed.
- W. Ou, R. Zou, M. Han, L. Yu and C. Su, Chin. Chem. Lett., 2020, 31, 1899–1902 CrossRef CAS.
- A. Al Mousawi, D. M. Lara, G. Noirbent, F. Dumur, J. Toufaily, T. Hamieh, T. Bui, F. Goubard, B. Gra, D. Gigmes, J. P. Fouassier and J. Laleve, Macromolecules, 2017, 50, 4913–4926 CrossRef CAS.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- Y. J. Cho, S. K. Jeon, B. D. Chin, E. Yu and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 5201–5204 CrossRef CAS.
- T. Nishimoto, T. Yasuda, S. Y. Lee, R. Kondo and C. Adachi, Mater. Horiz., 2014, 1, 264–269 RSC.
- C. Wang, C. Deng, D. Wang and Q. Zhang, J. Phys. Chem. C, 2018, 122, 7816–7823 CrossRef CAS.
- Z. Yu, W. Lou, H. Junge, A. Päpcke, H. Chen, L. Xia, B. Xu, M. Wang, X. Wang, Q. Wu, B. Lou, S. Lochbrunner, M. Beller, S. Luo, S. Key and G. C. Technology, Catal. Commun., 2019, 119, 11–15 CrossRef CAS.
- R. Ishimatsu, T. Edura, C. Adachi, K. Nakano and T. Imato, Chem. – Eur. J., 2016, 22, 4889–4898 CrossRef CAS PubMed.
- Y. Liu, X. Chen, X. Li, S. Zhu, S. Li, Y. Song, L. Qu and B. Yu, J. Am. Chem. Soc., 2021, 143(2), 964–972 CrossRef CAS PubMed.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 6642–6646 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn’Ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 RSC.
- A. Kretzschmar, C. Patze, S. T. Schwaebel and U. H. F. Bunz, J. Org. Chem., 2015, 80, 9126–9131 CrossRef CAS.
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 RSC.
- Q. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. Konig, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS PubMed.
- R. A. Scott and C. M. Lukehart, Applications of physical methods to inorganic and bioinorganic chemistry, John Wiley & Sons, 2007, vol. 2 Search PubMed.
- Y. He, D. Anand, Z. Sun and L. Zhou, Org. Lett., 2019, 21, 3769–3773 CrossRef CAS.
- F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790–1800 RSC.
- S. S. Shah, M. Shee, Y. Venkatesh, A. K. Singh, S. Samanta and N. D. P. Singh, Chem. – Eur. J., 2020, 26, 3703–3708 CrossRef CAS.
- B. A. Sim, D. Griller and D. D. M. Wayner, J. Am. Chem. Soc., 1989, 111, 754–755 CrossRef CAS.
- D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132–137 CrossRef CAS.
- K. Donabauer and B. Konig, Acc. Chem. Res., 2021, 54, 242–252 CrossRef CAS PubMed.
- K. P. L. Kuijpers, C. Bottecchia, D. Cambié, K. Drummen, N. J. König and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 11278–11282 CrossRef CAS PubMed.
- M. A. Graham, G. Noonan, J. H. Cherryman, J. J. Douglas, M. Gonzalez, L. V. Jackson, K. Leslie, Z. Q. Liu, D. McKinney, R. H. Munday, C. D. Parsons, D. T. E. Whittaker, E. X. Zhang and J. W. Zhang, Org. Process Res. Dev., 2021, 25, 57–67 CrossRef CAS.
- W. Cheng, R. Shang, M. Fu and Y. Fu, Chem. – Eur. J., 2017, 23, 2537–2541 CrossRef CAS PubMed.
- B. W. Hadrys and R. J. Phipps, Synlett, 2021, 179–184 CAS.
- W. M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 CrossRef CAS.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS.
- R. C. Betori and K. A. Scheidt, ACS Catal., 2019, 9, 10350–10357 CrossRef CAS.
- S. Bas, Y. Yamashita and S. Kobayashi, ACS Catal., 2020, 10, 10546–10550 CrossRef CAS.
- M. C. Nicastri, D. Lehnherr, Y. H. Lam, D. A. Dirocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 987–998 CrossRef CAS PubMed.
- C. Zhou, T. Lei, X.-Z. Wei, C. Ye, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142, 16805–16813 CrossRef CAS.
- J. A. Milligan, K. L. Burns, A. V. Le, V. C. Polites, Z. J. Wang, G. A. Molander and C. B. Kelly, Adv. Synth. Catal., 2020, 362, 242–247 CrossRef CAS.
- L. Cardinale, M. O. Konev and A. Jacobi von Wangelin, Chem. – Eur. J., 2020, 26, 8239–8243 CrossRef CAS.
- R. Abrams and J. Clayden, Angew. Chem., Int. Ed., 2020, 59, 11600–11606 CrossRef CAS PubMed.
- M. Shee, S. S. Shah and N. D. P. Singh, Chem. Commun., 2020, 56, 4240–4243 RSC.
- D. H. R. Barton, J. C. Jaszberenyi and E. A. Theodorakis, Tetrahedron Lett., 1991, 32, 3321–3324 CrossRef CAS.
- D. H. R. Barton, J. C. Jaszberenyi and E. A. Theodorakis, Tetrahedron, 1992, 48, 2613–2626 CrossRef CAS.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- V. Pirenne, G. Kurtay, S. Voci, L. Bouffier, N. Sojic, F. Robert, D. M. Bassani and Y. Landais, Org. Lett., 2018, 20, 4521–4525 CrossRef CAS.
- S. G. E. Amos, S. Nicolai and J. Waser, Chem. Sci., 2020, 11, 11274–11279 RSC.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël, C. W. Am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS.
- M. Shee, S. S. Shah and N. D. P. Singh, Chem. – Eur. J., 2020, 26, 14070–14074 CrossRef CAS.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS.
- I. Ghosh, R. S. Shaikh and B. Kçnig, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS PubMed.
- A. Jutand and S. Ne, Eur. J. Org. Chem., 1998, 1811–1821 CrossRef CAS.
- T. Ohtani, Y. Tsuchiya, D. Uraguchi and T. Ooi, Org. Chem. Front., 2019, 6, 1734–1737 RSC.
- H. J. Timpe, K. P. Kronfeld, U. Lammel, J. P. Fouassier and D. J. Lougnot, J. Photochem. Photobiol., A, 1990, 52, 111–122 CrossRef CAS.
- M. Sayes, G. Benoit and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 13514–13518 CrossRef CAS PubMed.
- A. R. Flynn, K. A. McDaniel, M. E. Hughes, D. B. Vogt and N. T. Jui, J. Am. Chem. Soc., 2020, 142, 9163–9168 CrossRef CAS PubMed.
- W. G. Herkstroeter and D. S. McClure, J. Am. Chem. Soc., 1968, 90, 4522–4527 CrossRef CAS.
- Q. A. Wu, F. Chen, C. C. Ren, X. F. Liu, H. Chen, L. X. Xu, X. C. Yu and S. P. Luo, Org. Biomol. Chem., 2020, 18, 3707–3716 RSC.
- T. Lei, C. Zhou, M. Y. Huang, L. M. Zhao, B. Yang, C. Ye, H. Xiao, Q. Y. Meng, V. Ramamurthy, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 15407–15410 CrossRef CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- A. B. Rolka and B. Koenig, Org. Lett., 2020, 22(13), 5035–5040 CrossRef CAS PubMed.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed.
- U. Streit, F. Birbaum, A. Quattropani and C. G. Bochet, J. Org. Chem., 2013, 78, 6890–6910 CrossRef CAS PubMed.
- N. Arai and T. Ohkuma, Tetrahedron Lett., 2019, 60, 151252 CrossRef CAS.
- F. Birbaum, A. Neels and C. G. Bochet, Org. Lett., 2008, 10, 3175–3178 CrossRef CAS.
- W. Xu, P. Zheng and T. Xu, Org. Lett., 2020, 22, 8643–8647 CrossRef CAS.
- Q. Meng and S. Wang, J. Am. Chem. Soc., 2018, 140, 3198–3201 CrossRef CAS.
- T. K. Sawyer and N. Sciammetta, Chem. Sci., 2019, 10, 5073–5078 RSC.
- E. R. Welin, C. Le, D. M. Arias-rotondo, J. K. Mccusker and D. W. C. Macmillan, Science, 2017, 385, 380–385 CrossRef PubMed.
- K. C. Cartwright and J. A. Tunge, Chem. Sci., 2020, 11, 8167–8175 RSC.
- A. Gualandi, F. Calogero, M. Mazzarini, S. Guazzi, A. Fermi, G. Bergamini and P. G. Cozzi, ACS Catal., 2020, 10, 3857–3863 CrossRef CAS.
- G. Barzanò, R. Mao, M. Garreau, J. Waser and X. Hu, Org. Lett., 2020, 22, 5412–5416 CrossRef PubMed.
- R. Mao, A. Frey, J. Balon and X. Hu, Nat. Catal., 2018, 1, 120–126 CrossRef CAS.
- S. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- D. Chao and M. Zhao, Dalton Trans., 2019, 48, 5444–5449 RSC.
- S. Roy, A. Bhunia, N. Schuth, M. Haumann and S. Ott, Sustainable Energy Fuels, 2018, 2, 1148–1152 RSC.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J. X. Jian, L. Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed.
- W. Xu, J. Ma, X. Yuan, J. Dai, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 10357–10361 CrossRef CAS.
- A. L. Berger, K. Donabauer and B. König, Chem. Sci., 2019, 10, 10991–10996 RSC.
- J. L. Jeffrey, F. R. Petronijević and D. W. C. Macmillan, J. Am. Chem. Soc., 2015, 137, 8404–8407 CrossRef CAS PubMed.
- K. Donabauer, K. Murugesan, U. Rozman, S. Crespi and B. König, Chem. – Eur. J., 2020, 26, 12945–12950 CrossRef CAS.
- J. Ye, Z. Chen, M. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C. Lee, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- J. Fan, L. Lin and C. Wang, Chem. Phys. Lett., 2016, 652, 16–21 CrossRef CAS.
- M. Aydemir, S. Xu, C. Chen, M. R. Bryce, Z. Chi and A. P. Monkman, J. Phys. Chem. C, 2017, 121, 17764–17772 CrossRef CAS.
- S. Xu, T. Liu, Y. Mu, Y. Wang, Z. Chi, C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS.
- N. Sharma, E. Spuling, C. M. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Bräse, I. D. W. Samuel and E. Zysman-Colman, Chem. Sci., 2019, 10, 6689–6696 RSC.
- X. Li, J. Li, D. Liu, D. Li and R. Dong, New J. Chem., 2020, 9743–9753 RSC.
- Y. Zhang, Y. Zheng, B. Wang, H. Ran, X. Wang, J. Y. Hu and Q. Wang, J. Mater. Chem. C, 2020, 8, 4461–4468 RSC.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- X. Cai, X. Li, G. Xie, Z. He, K. Gao, K. Liu, D. Chen, Y. Cao and S. J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner, L. Lüer and M. S. Kwon, Macromolecules, 2019, 52, 5538–5545 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |