
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA heteroepitaxial interface of Pt//CeO2 nanoparticles for enhanced catalysis of the oxygen reduction reaction (ORR)†
Nasrat Hannah
Shudin
ab,
Ryuto
Eguchi
ac,
Shigenori
Ueda
 a,
Ankit
Singh
a,
Ayako
Hashimoto
*ac and
Hideki
Abe
*ab
a,
Ankit
Singh
a,
Ayako
Hashimoto
*ac and
Hideki
Abe
*ab
aNational Institute for Materials Science, Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ABE.Hideki@nims.go.jp; Hashimoto.Ayako@nims.go.jp
bGraduate School of Science and Engineering, Saitama University, 255 Shimo-Okubo, Saitama 338-8570, Japan
cUniversity of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
First published on 21st February 2025
Abstract
Metal-oxide nanocomposites (MONs) have garnered significant interest in catalysis due to their excellent performance in various chemical reactions. A key focus of research on MONs is the heteroepitaxial metal-oxide interface, which is known to serve as a highly active catalytic center. In this report, we demonstrate that nanometer-sized MONs with heteroepitaxial interfaces can be engineered to exhibit enhanced catalytic performance owing to their strong interfacial effects. Specifically, a MON material composed of platinum (Pt) and cerium dioxide (CeO2), denoted as Pt//CeO2, can be obtained by exposing graphene-supported precursor Pt5Ce alloy nanocrystals (Pt5Ce/graphene), which are synthesized by the pyrolytic dissociation of chloroplatinic acid (H2PtCl6) and cerium trichloride (CeCl3) in a hydrogen-containing atmosphere, to a gas mixture of carbon monoxide (CO) and oxygen (O2) at elevated temperatures. Transmission electron microscopy (TEM) observations revealed a sharp heteroepitaxial interface between Pt(110) and CeO2(110) planes within the Pt//CeO2 material. This nanometer-sized heteroepitaxial interface showed superior catalytic activity of Pt//CeO2 compared to carbon-supported Pt and large-grained Pt//CeO2 bulk catalysts for the oxygen reduction reaction (ORR) in basic media.
1 Introduction
Catalysts play a crucial role in the economy, with approximately 90% of chemical manufacturing processes worldwide relying on high-performance catalysts. Among them, heterogeneous nanocatalysts comprising metal-oxide nanocomposites (MONs) are widely used due to their enhanced performance compared to their bulk counterparts.1–4 The enhanced catalytic performance of MONs is widely attributed to strong interfacial effects that improve electronic transfer and atomic exchanges at the metal-oxide interface.5–9 The interfacial effects are particularly strengthened at the heteroepitaxial interface, where a pair of the metal-oxide phases is conjugated sharing one or more crystal axes. This heteroepitaxial interface with fewer atomic defects and lattice-mismatch distortions facilitates charge and atomic transfers across the interface, further improving their catalytic performance.10–12Heterogeneous catalysts comprising MONs are typically synthesized as nanoparticles smaller than 100 nanometers to maximize the exposure of active sites for catalysis, using conventional wet chemistry processes involving impregnation methods or co-precipitation methods.13,14 In both methods, the building blocks for the desired MON nanoparticles, consisting of small molecules and/or nanoparticles, are initially dissolved or highly dispersed in a solvent to form a solution. The solution is then dried, and the resulting solid sediment is typically heated in a controlled atmosphere to produce the desired MON nanoparticles.15–17 However, a technical challenge arises in synthesizing MON nanoparticles with a catalytically active metal-oxide heteroepitaxial interface via wet chemistry approaches where these methods often yield randomly oriented heterointerfaces rather than the desired atomically oriented heteroepitaxial interfaces.
We propose here a hybrid approach for synthesizing MON nanoparticles with metal-oxide heteroepitaxial interfaces. Graphene nanoplatelets are dispersed in an aqueous solution containing chloroplatinic acid (H2PtCl6) and cerium trichloride (CeCl3), and subsequently dried to form a solid-state sediment. This sediment is then heated in a stream of hydrogen (H2) and argon (Ar) gases at 900 °C to precipitate Pt5Ce alloy nanocrystals on the surface of graphene nanoplatelets, yielding graphene-supported Pt5Ce nanocrystals. The Pt5Ce nanocrystals are further treated in a gas mixture of carbon monoxide (CO) and oxygen (O2) at 600 °C to selectively oxidize Ce atoms, forming the desired Pt–CeO2 MON nanoparticles with a heteroepitaxial interface (Pt//CeO2). Transmission electron microscope (TEM) observations reveal that the Pt metal and CeO2 phases of the Pt//CeO2 nanoparticles contact at a heteroepitaxial interface. Catalytic tests for the oxygen reduction reaction (ORR) in aqueous electrolytes have further demonstrated that the Pt//CeO2 nanomaterial exhibits superior ORR activity compared to the bulk counterpart, Pt//CeO2 bulk catalyst. This enhancement is attributed to strengthened interfacial effects at the heteroepitaxial interface. The findings of this study highlight the simple approach in obtaining a heteroepitaxial interface in nanomaterials via oxidation-triggered nanophase separation.
2 Materials and methods
2.1 Synthesis of Pt5Ce/graphene nanoplatelets
Pt5Ce nanocrystals supported on graphene nanoplatelets have been synthesized via a two-step approach: impregnation followed by hydrogen reduction. In the first step, chloroplatinic acid hexahydrate (H2PtCl6·6H2O; Sigma-Aldrich) and cerium(III) chloride heptahydrate (CeCl3·7H2O; Kishida Chemical) were used as precursors. An aliquot of 100 mg graphene nanoplatelets (Sigma-Aldrich) and 50 ml ultrapure water were added as the support material. The mixture was placed in a sealed 100 ml round bottom flask and sonicated with a magnetic stirrer overnight. This was followed by sonication in a bath sonicator for one hour. The mixture was then transferred to a beaker and placed in an oil bath to dry using the double bowl method. In the second step, the dried powder was placed on a graphite boat and transferred into a glass tube for hydrogen reduction. A 5% H2/Ar gas mixture was supplied, and the reaction was conducted at 900 °C to obtain Pt5Ce nanocrystals supported on graphene nanoplatelets.2.2 Synthesis of Pt5Ce bulk
The Pt5Ce bulk sample was prepared to explore the effect of dimensionality on catalytic performance. Pt5Ce alloy was prepared by melting Pt and Ce metals at a mole ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 using an arc torch in an argon environment.5 The obtained ingot was subsequently crushed and sieved to collect particles with an average size of approximately ∼50 μm.
:1 using an arc torch in an argon environment.5 The obtained ingot was subsequently crushed and sieved to collect particles with an average size of approximately ∼50 μm.
2.3 Phase separation in a CO–O2 gas environment
The graphene-supported Pt5Ce nanocrystals were placed in a ceramic crucible and heated at 600 °C in a stream of a mixed gas of CO and O2 (CO:O2 = 2:1 by volume ratio) for 1 minute to induce phase separation of Pt5Ce, forming the desired Pt//CeO2 nanoparticles. A similar procedure was applied to the bulk Pt5Ce alloy but extended to 12 hours, ensuring complete phase separation across its large grain size of 50 μm to produce the Pt//CeO2 bulk catalyst. Fig. 1 shows the schematic of the synthetic route for Pt//CeO2 nanoparticles.
 | ||
| Fig. 1 Schematic of the synthesis route. (a) The materials, (b) impregnation of molecular precursors over graphene nanoplatelets, (c) development of precursor alloy Pt5Ce nanocrystals, and (d) formation of Pt//CeO2 nanoparticles through promoted nanophase separation of Pt5Ce nanoparticles. The (110) planes of Pt and CeO2 are epitaxially oriented. | ||
2.4 Characterization
The synthesized Pt5Ce nanocrystals and Pt//CeO2 nanoparticles supported on graphene nanoplatelets were characterized using powder X-ray diffraction (pXRD; PANalytical X'Pert Pro (Cu Kα, 45 kV, 30 mA)) over a 2θ range of 0° to 90°. The elemental composition and structure were observed using an aberration-corrected TEM (JEM-ARM200F, JEOL Ltd, Japan) with an energy-dispersive X-ray spectrometer (EDS, JED-2300, JEOL Ltd, Japan). Hard X-ray photoemission spectroscopy (HAXPES) with synchrotron radiation X-rays (5.95 keV) was performed at BL09XU of SPring-8 (Super Photon Ring 8, Hyōgo Prefecture, Japan).2.5 Electrochemical measurements
Electrochemical measurements were conducted using a three-electrode cell connected to a potentiostat (CHI 842B). A 5 mm diameter glassy carbon electrode was used as the working electrode, an Ag/AgCl electrode was used as the reference electrode and a 1 mm diameter Pt electrode was used as the counter electrode. A mixture of ultrapure water (370 μL), isopropanol (100 μL), Nafion solution (5 μL), and the catalyst powder (2.5 mg Pt content) was prepared and sonicated to obtain ink, which was subsequently dropped onto the working electrode. The loading weight of the catalysts was approximately 0.0316 mg mm−2 over the glassy carbon electrode. Electrochemical tests were carried out in 0.1 M KOH solution at room temperature. The electrolyte was first purged with Ar gas for 30 minutes, and cyclic voltammetry (CV) was conducted at a scanning rate of 20 mV s−1. The electrolyte was later purged with O2 gas for 30 minutes, and linear sweep voltammetry (LSV) for the ORR was conducted at a scanning rate of 1 mV s−1.3 Results and discussion
3.1 Nanoparticles of Pt5Ce and Pt//CeO2 supported on graphene nanoplatelets
The pXRD pattern in Fig. 2a(i) represents Pt5Ce nanocrystals supported on graphene nanoplatelets, which show peaks that match closely with those of the atomically ordered Pt5Ce phase (ICDD 01-071-7052), indicating a successful synthesis of the desired phase. After the post-treatment in a CO–O2 atmosphere for 1 minute, all the peaks corresponding to Pt5Ce diminished (Fig. 2a(ii)). Instead, the peaks for Pt are clearly observed, suggesting a phase transformation from Pt5Ce to Pt//CeO2 has completed within the exposure time. | ||
| Fig. 2 (a) pXRD of Pt5Ce nanocrystals and Pt//CeO2 nanoparticles supported on graphene nanoplatelets. (b) HAXPES spectra of the Pt 4f region (blue, red, green, and black curves represent Pt nanoparticles, Pt//CeO2 nanoparticles, Pt5Ce nanocrystals, and Pt5Ce bulk, respectively). (c) HAXPES spectra of the Ce 3d region (blue, red, green, and black curves represent CeO2, Pt//CeO2 nanoparticles, Pt5Ce nanocrystals, and Pt5Ce bulk respectively). | ||
The HAXPES spectrum in the Pt 4f region for the Pt5Ce nanocrystals is similar to that for Pt5Ce bulk in peak positions and intensities (Fig. 2b). The Pt 4f emission peaks for the Pt//CeO2 nanoparticles show shifts towards lower binding energy compared to the nanocrystals and bulk of Pt5Ce. The Pt 4f spectrum for the Pt//CeO2 nanoparticles is similar to that for pure Pt bulk instead of the Pt5Ce bulk, indicating the chemical environment for Pt in the Pt//CeO2 nanoparticles is close to that in pure Pt instead of Pt5Ce. The HAXPES spectrum in the Ce 3d region for the Pt//CeO2 is similar to that for pure CeO2, unlike any of the spectra for the nanocrystals and bulk Pt5Ce, indicating that the Ce atoms in Pt//CeO2 are situated in a similar environment in pure CeO2 instead of Pt5Ce (Fig. 2c). There are recognized Ce3+ 3d emissions from the nanocrystals and bulk Pt5Ce at 884.5 and 903.8 eV, which are absent in the Ce 3d spectrum for the Pt//CeO2 nanoparticles and pure CeO2. Ce in Pt5Ce bulk and nanocrystals primarily exhibits photoelectron peaks at binding energy positions corresponding to Ce3+ species, whereas the Ce in Pt//CeO2 and CeO2 primarily shows peaks at binding energy positions corresponding to Ce4+ species. However, in the case of Pt5Ce nanocrystals, photoelectron peaks corresponding to Ce4+ species are also observed. These peaks most likely originated from an impurity containing Ce4+ within the Pt5Ce nanocrystals. (see ESI Table S1†). Summarizing the results of pXRD and HAXPES, we conclude that the Pt5Ce nanocrystals were fully phase-separated into the Pt//CeO2 nanoparticles by the atmosphere treatment.
Pt5Ce nanocrystals with a size of approximately 10 nm were observed using a TEM and a high-angle annular dark-field (HAADF) scanning TEM (STEM), as shown in Fig. 3a and b, respectively. Elemental mapping with an EDS in Fig. 3c shows spatially overlapped regions of Pt and Ce, confirming the successful synthesis of the desired Pt5Ce nanocrystals. ESI Fig. S1† shows the corresponding Fast Fourier Transformation (FFT) patterns and the calculated interplanar distance further confirmed Pt5Ce nanocrystals. STEM-EDS in Fig. 3d show the full phase separation of Pt5Ce nanocrystals into Pt//CeO2 nanoparticles, showing a clear contrast between Pt and Ce regions after exposure to an oxidative atmosphere containing O2. FFT patterns and EDS spectra in ESI Fig. S2† show areas corresponding to Pt and CeO2 phases further proving that phase separation has completed. Note that a longer duration of 12 hours was required for the bulk Pt5Ce to achieve complete phase separation (see a cross-sectional STEM image of the fully nanophase-separated Pt//CeO2 bulk material, ESI Fig. S3†). Oxygen species from the atmospheric O2 likely need more time to diffuse into the inner regions of the Pt5Ce bulk alloy, which explains the extended exposure time to ensure full phase separation.
 | ||
| Fig. 3 (a) TEM image of a Pt5Ce nanocrystal. Inset shows a structural model. The red and black spheres in the inset correspond to the Ce and Pt atoms, respectively. (b) HAADF-STEM image of a Pt5Ce nanocrystal. Inset shows a filtered STEM image, clearly showing crystal structures. (c) STEM image and EDS mapping of the Pt5Ce nanocrystal. (d) STEM image and EDS mapping of Pt//CeO2 nanoparticles after phase separation. The red and green regions in the mapping correspond to Pt and Ce, respectively. | ||
3.2 Heteroepitaxial interface of nanosized Pt//CeO2 supported on graphene nanoplatelets
An epitaxial interface was observed in the heterostructure of the Pt//CeO2 nanoparticles, as shown in Fig. 4a. All the Pt//CeO2 nanoparticles exhibited an asymmetrical structure with sharp facets (see ESI Fig. S4†). Phase separation initiated by the spontaneous oxidation of the oxyphilic component, in this case, Ce, resulted in distinct regions of Pt and CeO2.18 The white lines in Fig. 4a highlight the crystal facets and lattice planes of the Pt and CeO2 phases. The configurational relationship between the two phases was verified from the FFT patterns (Fig. 4b–d). The Pt- and CeO2 phases are both on the [01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] zone axis, where the Pt(111) plane is parallel to the CeO2(002) plane (see Fig. 1d). Fig. 4e depicts a configurational model of the Pt//CeO2 nanoparticle along the [01] direction at the heterointerface. The Pt- and CeO2 phases are conjugated at their (01) planes to form an epitaxial interface, perpendicular to their [01] axes.
] zone axis, where the Pt(111) plane is parallel to the CeO2(002) plane (see Fig. 1d). Fig. 4e depicts a configurational model of the Pt//CeO2 nanoparticle along the [01] direction at the heterointerface. The Pt- and CeO2 phases are conjugated at their (01) planes to form an epitaxial interface, perpendicular to their [01] axes.
 | ||
| Fig. 4 (a) TEM image of a Pt//CeO2 particle showing a heteroepitaxial interface. (b–d) FFT patterns from the blue and red squares, and the interface in (a), respectively, demonstrating the configurational relationship between the Pt- and CeO2 phases. (e) Schematic illustrating the epitaxial (110) interface between the Pt- and CeO2 phases. | ||
Another configurational relationship, with a parallel relation between the Pt(111) plane and the CeO2(111) plane as a minor phase, was observed in other nanoparticles having larger grain sizes than 10 nm (ESI Fig. S4†). Note that, in contrast to the nanoparticle system, the epitaxial interface observed in the Pt//CeO2 bulk system solely showed a Pt(111)//CeO2(111) relationship.19 We attribute this difference in configuration to dimension effects, where a larger rotational angle is necessary to accommodate larger particle sizes, resulting in the Pt (111)//CeO2 (111) relationship.
3.3 ORR catalytic activity
Fig. 5a shows the result of CV in Ar-saturated 0.1 M KOH. The electrochemical surface areas (ECSA) are evaluated as 9.0 m2 per g Pt for the Pt//CeO2 nanoparticles and 29.0 m2 per g Pt for the Pt//CeO2 bulk catalyst from the area of the hydrogen desorption valley in CV curves (see ESI Fig. S5† for details on the ECSA evaluation). Fig. 5b shows the LSV profiles for the ORR over the different catalysts and Fig. 5c shows the LSV normalized ECSA. CV and LSV for the Pt5Ce nanocrystals and graphene nanoplatelets are provided in ESI Fig. S6a–c.† The onset potential for the Pt//CeO2 nanoparticles, +1.01 V vs. a reversible hydrogen electrode (RHE), is more positive than those of the Pt//CeO2 bulk catalyst, +0.91 V vs. RHE, and Pt5Ce nanocrystal, +0.93 V vs. RHE (Fig. 5c and S6c ESI†). This positive shift indicates enhanced ORR catalytic activity of the Pt//CeO2 nanoparticles compared to the bulk Pt//CeO2 catalyst and Pt5Ce nanocrystals. The related parameters are compared to electrochemical data of a typically prepared CeO2-supported Pt catalyst from the literature and provided in ESI Table. S2.† The onset potential was observed to be more positive for Pt(110)//CeO2(110) nanoparticles compared to other CeO2-supported Pt catalysts which is likely due to the stronger interfacial effect. We further conducted an electrochemistry test in 1.0 M KOH to evaluate the performance of Pt(110)//CeO2(110) nanoparticles at different electrolyte concentrations (see ESI Fig. S6d and e†). LSV recorded in 1.0 M KOH in ESI Fig. S6e† shows that the onset potential was observed at +0.88 V vs. RHE. There is a shift from +1.01 V vs. RHE recorded in 0.1 M KOH. This is expected as by increasing the molar concentration of KOH, the increased OH− concentration will disrupt oxygen absorption on the active surface. The stability test performed in 0.1 M KOH (ESI Fig. S6f†) shows that the CV curve did not exhibit significant changes after repeated cycles. This suggest that the heterointerface of Pt(110)//CeO2(110) remains stable throughout the cycle.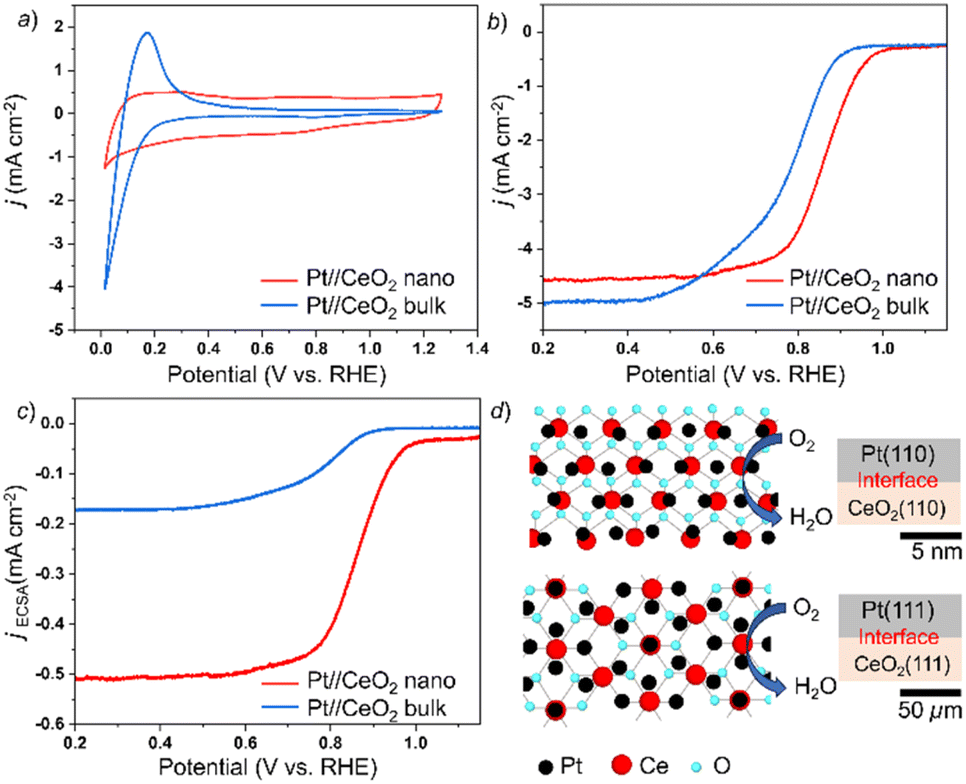 | ||
| Fig. 5 (a) CV and (b) LSV measurements performed at 1200 rpm in a 0.1 M KOH solution, (c) LSV normalized ECSA and (d) schematic illustrations of the top view and side view of the Pt//CeO2 interface. The potential was referenced against an Ag/AgCl electrode and then converted to the values for the reversible hydrogen electrode (RHE) for better readability (see the ESI†). | ||
Kinetics analysis of the ORR was performed using rotating disk electrode (RDE) voltammetry in O2 saturated 0.1 M KOH at a different rotating speed with a scanning rate of 1 mV s−1. The current density increases as the rotating speed increases from 100 rpm to 1200 rpm due to the enhanced diffusion of reactants (see ESI Fig. S7a, c and e†). The corresponding K–L plots in Fig. S7b, d and f† indicate the four-electron pathway for all the Pt//CeO2 nanoparticles, Pt//CeO2 bulk catalyst and a carbon-supported Pt catalyst (Pt/C, Pt loading: 20 wt%; Fuel Cell Store, Inc).
The HAXPES spectrum in the Pt 4f region for the Pt//CeO2 nanoparticles exhibits a 74 meV deep-level shift compared to that of pure Pt bulk and the Pt//CeO2 bulk catalyst (see ESI Fig. S8†). This shift is attributed to a strengthened interfacial effect at the Pt(110)//CeO2(110) interface in the nanoparticles, as opposed to the Pt(111)//CeO2(111) interface in the bulk catalyst as shown in the schematic image in Fig. 5d. This enhanced interfacial effect causes a deep-level shift in the Pt core levels, along with the center of gravity of the Pt d-band in the nanoparticles. Consequently, this deep-level shift in the Pt d-band can weaken the adsorption of intermediate species, such as OH− anions, near the Pt(110)//CeO2(110) interface.20,21 This weakening in the adsorption of reaction intermediates can optimize the balance between mass transfer and electron transfer, leading to the improved ORR activity of the Pt//CeO2 nanoparticles.22
4 Conclusions
A nanometer-sized MON with an epitaxial interface, specifically Pt//CeO2 nanoparticles, has been successfully synthesized by utilizing the selective oxidation of precursor Pt5Ce nanocrystals supported on graphene nanoplatelets.23 The spontaneous nanophase separation of Pt5Ce nanocrystals results in a (110) epitaxial interface between single-crystalline Pt and CeO2 phases. The catalytic Pt//CeO2 nanoparticles exhibit superior ORR activity compared to a carbon-supported Pt catalyst and bulk Pt//CeO2 catalysts, due to strengthened interfacial effects at the epitaxial Pt(110)//CeO2(110) interface.Data availability
Electrochemistry datasets for the catalytic performance test are available at https://doi.org/10.5281/zenodo.13888952.Author contributions
N. H. Shudin synthesized and characterized the nanoparticles. Microscopic observations were conducted by A. Hashimoto, R. Eguchi, and A. Singh. N. H. Shudin, H. Abe, and A. Hashimoto wrote and edited the manuscript. All authors have read and approved the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The HAXPES experiments were performed at BL09XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2024A1536). AH acknowledges the JST FOREST Program (Grant Number JPMJFR213U) and JST PRESTO (Grant Number JPMJPR17S7) for financial support. HA acknowledges the JSPS Grant-in-Aid for Scientific Research (B) (KAKENHI) (Grant number 22H01799 and 23K23067). This work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Grant number JPMXP1224NM5122, JPMXP1224NM5255).References
- T. W. Van Deelen, C. H. Mejia and K. P. Jong, Nat. Catal., 2019, 2, 955–970 CAS.
- M. A. Memon, Y. Jang, M. A. Hassan, M. Ajmal, H. Wang and Y. Liu, Catalysts, 2023, 13, 1514 CAS.
- J. Zhang, J. Lian, Q. Jiang and G. Wang, Chem. Eng. J., 2022, 439, 135634 CrossRef CAS.
- X. Zhang, Y. Chen, X. Li, Y. Zhang, M. Li, Y. Yang, X. Sun, H. Wang and Y. Xiong, Catal. Small, 2021, 17(42), 2101573 Search PubMed.
- Y. Wen, H. Abe, A. Hirata and A. Hashimoto, ACS Appl. Nano Mater., 2021, 4, 13602–13611 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. F. Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- S. Lee, A. Moysiadou, Y. Chu, H. M. Chen and X. Hu, Energy Environ. Sci., 2022, 15, 206–214 RSC.
- J. Zhang, T. H. M. Pham, Y. Ko, M. Li, S. Yang, C. D. Koolen, L. Zhong, W. Luo and A. Zuttel, Cell Rep. Phys. Sci., 2022, 3(7), 100949 CrossRef CAS.
- C. Xu, Y. Wu, S. Li, J. Zhou, J. Chen, M. Jiang, M. Zhao and G. Qin, J. Mater. Sci. Technol., 2020, 40, 39–46 CAS.
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2021, 34, 2108133 CrossRef PubMed.
- C. Zhu, A. Wang, W. Xiao, D. Chao, X. Zhang, N. H. Tiep, S. Chen, J. Kang, X. Wang, J. Ding, J. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1705516 Search PubMed.
- E. Serena, Materials, 2019, 12(4), 668 Search PubMed.
- J. C. Vedrine, ChemSusChem, 2019, 12, 577–588 CrossRef CAS PubMed.
- J. Song, Y. Yang, S. Liu, L. Li, N. Yu, Y. Fan, Z. Chen, L. Kuai and B. Geng, Nano Res., 2021, 14(12), 4841–4847 CrossRef CAS.
- S. B. T. Tran, H. S. Choi, S. Y. Oh, S. Y. Moon and J. Y. Park, RSC Adv., 2018, 8, 21528–21533 RSC.
- X. Xu, Y. Zhong, M. Wajrak, T. Bhatelia, S. P. Jiang and Z. Shao, InfoMat, 2024, 6, e12608 CrossRef CAS.
- N. H. Shudin, R. Eguchi, T. Fujita, T. Tokunaga, A. Hashimoto and H. Abe, Phys. Chem. Chem. Phys., 2024, 26, 14103–14107 RSC.
- Y. Wen, H. Abe, K. Mitsuishi and A. Hashimoto, Nanoscale, 2021, 13, 18987–18995 RSC.
- N. Han, W. Zhang, W. Guo, H. Pan, B. Jiang, L. Xing, H. Tian, G. Wang, X. Zhang and J. Fransaer, Nano–Micro Lett., 2023, 15, 185 CrossRef CAS PubMed.
- C. Lim, A. R. Fairhurst, B. J. Ransom, D. Haering and V. R. Stamenkovic, ACS Catal., 2023, 13, 14874–14893 CAS.
- A. Velázquez-Palenzuela, F. Masini, A. Pedersen, M. Escudero-Escribano, D. Deiana, P. Malacrida, T. Hansen, D. Friebel, A. Nilsson, I. Stephens and I. Chorkendorff, J. Catal., 2015, 328, 297–307 Search PubMed.
- W. Wang, Y. Dong, Y. Yang, D. Chai, Y. Kang and Z. Lei, J. Hydrogen, 2018, 43, 12119–12128 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07380k |
| This journal is © The Royal Society of Chemistry 2025 |