
Leukocyte-mimicking nanovesicles for effective doxorubicin delivery to treat breast cancer and melanoma†
Roberto
Molinaro
 ab,
Jonathan O.
Martinez
a,
Assaf
Zinger
a,
Alessandro
De Vita
ac,
Gianluca
Storci
ad,
Noemi
Arrighetti
ae,
Enrica
De Rosa
a,
Kelly A.
Hartman
a,
Nupur
Basu
a,
Nima
Taghipour
a,
Claudia
Corbo
*af and
Ennio
Tasciotti
*ag
ab,
Jonathan O.
Martinez
a,
Assaf
Zinger
a,
Alessandro
De Vita
ac,
Gianluca
Storci
ad,
Noemi
Arrighetti
ae,
Enrica
De Rosa
a,
Kelly A.
Hartman
a,
Nupur
Basu
a,
Nima
Taghipour
a,
Claudia
Corbo
*af and
Ennio
Tasciotti
*ag
aRegenerative Medicine Program, Houston Methodist Research Institute, Houston, TX, USA
bSchool of Pharmacy, Department of Biomolecular Sciences, University of Urbino Carlo Bo, Urbino, Italy
cOsteoncology and Rare Tumors Center, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola, Italy
dDepartmentof Experimental, Diagnostic and Specialty Medicine, University of Bologna, Bologna, Italy
eDepartment of Applied Research and Technological Development, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy
fSchool of Medicine and Surgery, Nanomedicine Center NANOMIB, University of Milano-Bicocca, Milano, Italy. E-mail: claudia.corbo@unimib.it
gHouston Methodist Orthopedics and Sports Medicine, Houston Methodist Hospital, Houston, TX, USA. E-mail: etasciotti@houstonmethodist.org
First published on 7th November 2019
Abstract
In the last decades, several approaches were developed to design drug delivery systems to address the multiple biological barriers encountered after administration while safely delivering a payload. In this scenario, bio-inspired and bio-mimetic approaches have emerged as promising solutions to evade the mononuclear phagocytic system while simultaneously negotiating the sequential transport across the various biological barriers. Leukocytes freely circulate in the bloodstream and selectively target the inflamed vasculature in response to injury, infection, and cancer. Recently we have shown the use of biomimetic nanovesicles, called leukosomes, which combine both the physical and biological properties of liposomes and leukocytes, respectively, to selectively deliver drugs to the inflamed vasculature. Here we report the use of leukosomes to target and deliver doxorubicin, a model chemotherapeutic, to tumors in syngeneic murine models of breast cancer and melanoma. Exploiting the inflammatory pathway responsible for recruiting immune cells to the site of injury, leukosomes exhibited increased targeting of cancer vasculature and stroma. Furthermore, delivery of doxorubicin with leukosomes enabled significant tumor growth inhibition compared with free doxorubicin in both breast and melanoma tumors. This study demonstrates the promise of using biomimetic nanovesicles for effective cancer management in solid tumors.
Introduction
Researchers have developed several approaches to bestow nanoparticles with superior pharmaceutical properties for targeting tumor tissues. After several iterations and discoveries a consensus was reached that the ideal drug delivery platform for tumors should: (i) modulate drug bioavailability and pharmacokinetics,1 (ii) protect the payload from degradation/clearance in biological environments,2,3 and (iii) favor the biodistribution and targeting at the site of interest.4 These properties are particularly relevant for the delivery of payloads characterized by high toxicity (chemotherapeutics)5 or poor stability in harsh biological environments (siRNA).2 Nanomedicine provides several tools to increase the therapeutic index of currently used therapeutics and has gained much interest in the cancer field due to the concerns raised by the limits of chemotherapeutics, i.e., (a) improving drug physical and chemical properties (evasion of mononuclear phagocytic system (MPS) and prolonged blood circulation), enhancing their pharmacokinetics (PK),6 (b) selective drug delivery to the cancer lesion through passive (enhanced permeation and retention (EPR) effect) or active (i.e., targeting with antibodies) mechanisms reducing off-target toxicity,7 and (c) enhancing cell internalization of the payload, thus overcoming multidrug resistance (MDR).8Current FDA approved therapies rely on passive nanoparticles that exploit the anti-opsonizing properties of hydrophilic polymers (e.g., polyethylene glycol (PEG)) conjugated on their surface.9 These stealth liposomes showed reduced MPS-associated uptake and prolonged circulation in the blood stream, allowing increased accumulation in cancer lesions through the EPR effect.10 However, initial praise of PEG's stealth properties has been reconsidered after several studies showed the production of anti-PEG antibodies following repeated injections of PEGylated liposomes. This accelerated blood clearance phenomenon increased the clearance of PEGylated liposomes and poses serious concerns about the long-term efficacy of PEGylated formulations.11
Recently, biomimetic approaches have emerged as novel approaches for the design of drug delivery platforms.12–14 The transfer of the membrane protein signature of a source cell (e.g., leukocytes,15 platelets,16 or cancer cells17) to a synthetic nanoparticle (e.g., liposomes or polymeric) resulted in the transfer of biological functions to the synthetic entity. For example, using this strategy, biomimetic nanoparticles have demonstrated promise for inflammatory bowel disease,18 rheumatoid arthritis,19 infection,16 sepsis,20 and cancer immunotherapy.21 Here, we describe the biological performance of leukocyte-mimicking biomimetic nanovesicles, called leukosomes, for the delivery of chemotherapeutics to solid tumors. Leukosomes exploited their intrinsic targeting properties due to the presence of the leukocyte membrane proteins responsible for cellular adhesion molecule ligation, such as lymphocyte function-associated antigen 1 (LFA-1), macrophage-1 antigen (Mac-1), and P-selectin glycoprotein ligand-1 (PSGL-1) to deliver doxorubicin to both tumor-associated vasculature and parenchyma. This strategy enabled significant survival advantage to mice with breast and melanoma tumors compared to conventional free drug treatments.
Results
Physicochemical properties of doxorubicin-loaded leukosomes
We recently reported the physicochemical features of leukosomes and their ability to retain similar characteristics of conventional liposomes, including loading and release and versatility for encapsulating compounds with various chemical features.15 Here, we encapsulated doxorubicin (DOX) using the established remote loading method.22 DOX encapsulation did not significantly affect the size and polydispersity of DOX-loaded leukosomes (Leuko DOX) (Fig. 1A). Indeed, compared to empty leukosomes (hydrodynamic size = 122 ± 1.2 nm; PDI = 0.06 ± 0.009), DOX encapsulation did not alter both nanovesicle's diameter (hydrodynamic size = 120 ± 2 nm), while slightly increased size distribution (PDI = 0.1 ± 0.01) to values that are still below 0.2, thus indicating a very high size homogeneity (Fig. 1). A substantial increase, instead, in the surface charge was observed (Fig. 1B). This increase may be attributed to the DOX that was naturally adsorbed on the surface, as previously shown by others.23 Leuko DOX retained similar encapsulation of DOX compared to liposomes (Lipo DOX), confirming the versatility of this platform (Fig. 1C). In particular, DOX encapsulation efficiency into liposomes and leukosomes was 75% and 60% of the initial amount (equivalent to a loading capacity of 7.5% and 6% by weight), respectively. The resulting loading efficiency values were above 94% for both formulations. DOX encapsulation in both Lipo and Leuko conferred them the typical coffee-bean shape, as previously reported by others22 (ESI Fig. S1†). The release kinetics of DOX from Lipo DOX and Leuko DOX followed a first-order release profile with leukosomes exhibiting a delayed release (Fig. 1D). This slight delay in the full discharge of DOX from leukosomes was likely attributed to the presence of membrane proteins and to the increased bilayer thickness and rigidity, previously demonstrated with dexamethasone, caffeine and paclitaxel.15 Furthermore, to evaluate the release at the intracellular and extracellular pH typical of solid tumors,24 we tested DOX release from Leuko DOX at both physiological (7.4) and acidic (6.5) pH at 37 °C. We observed a faster release of DOX at lower pH, consistent with published reports for liposomal formulations25 (ESI Fig. S2†).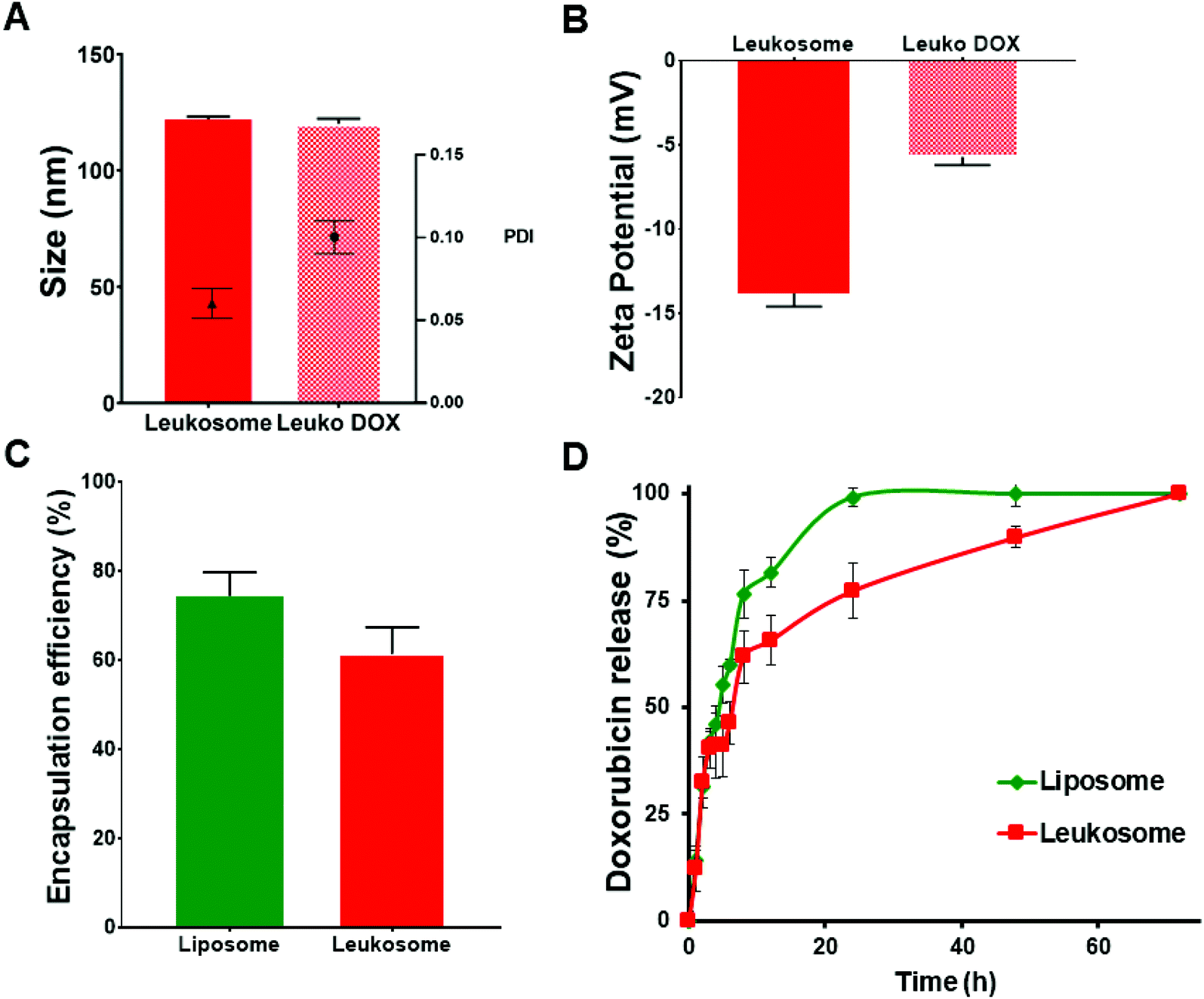 | ||
| Fig. 1 Physical and pharmaceutical characterization of empty and doxorubicin-loaded leukosomes. (A) Dynamic light scattering and (B) zeta potential measurements comparing empty leukosomes and Leuko DOX. (C) Encapsulation efficiency and (D) release profiles of Lipo DOX and Leuko DOX in PBS at 37 °C. | ||
To gain insights into the physical stability of Leuko DOX once injected in the blood stream, they were incubated with a mixture of FBS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBS (50:50 v/v). Leuko DOX initially displayed a slight reduction of their average size, which then remained constant up to 19 h of incubation (ESI Fig. S3†). This result is in line with what previously reported by Wolfram and coworkers.26 Leuko DOX surface charge, instead, gradually decreased from −6 mV at time zero to −14 mV at 19 h, most likely as consequence of serum proteins absorbance on leukosomes surface.
:PBS (50:50 v/v). Leuko DOX initially displayed a slight reduction of their average size, which then remained constant up to 19 h of incubation (ESI Fig. S3†). This result is in line with what previously reported by Wolfram and coworkers.26 Leuko DOX surface charge, instead, gradually decreased from −6 mV at time zero to −14 mV at 19 h, most likely as consequence of serum proteins absorbance on leukosomes surface.
Targeting properties of Leukosomes towards inflamed tumor-associated vasculature
We previously assessed the ability of leukosomes to avoid MPS uptake and increase circulation time in healthy mice;27 and to selectively adhere to the inflamed vasculature in breast tumors, local inflammation, and atherosclerosis plaques.15,28,29 Leukosomes were characterized by a significant reduction in phagocytosis by macrophages and reduced accumulation in the liver and spleen, confirming the stealth properties of the biomimetic proteolipid material in healthy mice.27 In a cancer setting, we evaluated the bio-distribution of liposomes and leukosomes to accumulate in MPS-associated organs and tumors in a syngeneic 4T1 murine triple negative breast cancer model. This model was selected as it grows in immune competent mice and permitted testing in a normal immune system. Spectrofluorometric analysis of the distribution of fluorescently labeled liposomes and leukosomes into MPS organs (spleen and liver) and other organs (kidney, lung, heart, pancreas) validated previous results in healthy mice (ESI Fig. S4†). In particular, the accumulation of control liposomes remained higher in the main MPS organs at 1, 6 and 24 h (respectively 7, 1.7, and 1.6-fold increases in the spleen and 7.5, 3.1, and 4.4-fold increases in the liver; ESI Fig. S4†) compared to leukosomes. Furthermore, spectrofluorometric analysis on homogenized tumor tissues confirmed significant tumor accumulation of leukosomes at 1, 6, and 24 h relative to liposomes (ESI Fig. S5†). This analysis confirmed that leukosomes avoided sequestration in MPS-associated organs while exhibiting a significant increase in tumor targeting in breast cancer.Our previous work demonstrated that leukosomes exhibited superior targeting of 4T1 breast tumors with significant accumulation observed at the vessel wall.28 However, the targeting of leukosomes in other solid tumors is not known. Intravital microscopy (IVM) analysis on immune competent mice bearing B16 melanoma tumors revealed the adhesion dynamics of leukosomes in tumor-associated inflamed vasculature. Compared to control liposomes, leukosomes exhibited increased adhesion toward tumor-associated vasculature in melanoma tumors at 1 h after systemic injection (Fig. 2A and B, respectively). Particularly, compared to liposomes, leukosomes showed a 9-fold (p < 0.0001) increased accumulation in B16 tumors. IVM analysis of the adhesion dynamics of leukosomes to inflamed tumor endothelium in melanoma tumors revealed that while liposomes uniformly distributed across the vessel section (ESI Fig. S6a†), leukosomes exhibited substantial accumulation at the vessel walls (ESI Fig. S6b†). This behavior remarkably mimics the innate ability of leukocytes to selectively adhere to inflamed vessels prior to trans-endothelial migration.30–32 Furthermore, this same behavior was observed in breast tumors and provides confirmation of the universal targeting of leukosomes for solid tumors.
 | ||
| Fig. 2 Targeting properties of liposomes and leukosomes towards tumor-associated vasculature. (A) Representative images of intravital microscopy analysis of melanoma-associated vasculature at 1 hour after intravenous injection of rhodamine-labeled vesicles. (B) End point biodistribution of liposomes and leukosomes. ****p < 0.0001. Scale bar = 20 μm. | ||
In vitro and in vivo cytotoxic effect of doxorubicin-loaded leukosomes
The ability of Leuko DOX to deliver DOX to cancer cells was tested in vitro for dose–response and in vivo for tumor growth inhibition and survival analysis. The in vitro cytotoxicity of free DOX and Leuko DOX was tested against 4T1 and B16 cancer cell lines. We observed a significantly increased activity for the encapsulated DOX at 0.05, 0.1 and 0.5 μM against 4T1 breast cancer cells. Specifically, compared to untreated cells, free DOX resulted in 27, 23 and 29% of reduction of cell viability, while Leuko DOX produced a reduction of cell viability of 11, 13 and 12% at a DOX concentration of 0.05, 0.1 and 0.5 μM, respectively (Fig. 3A).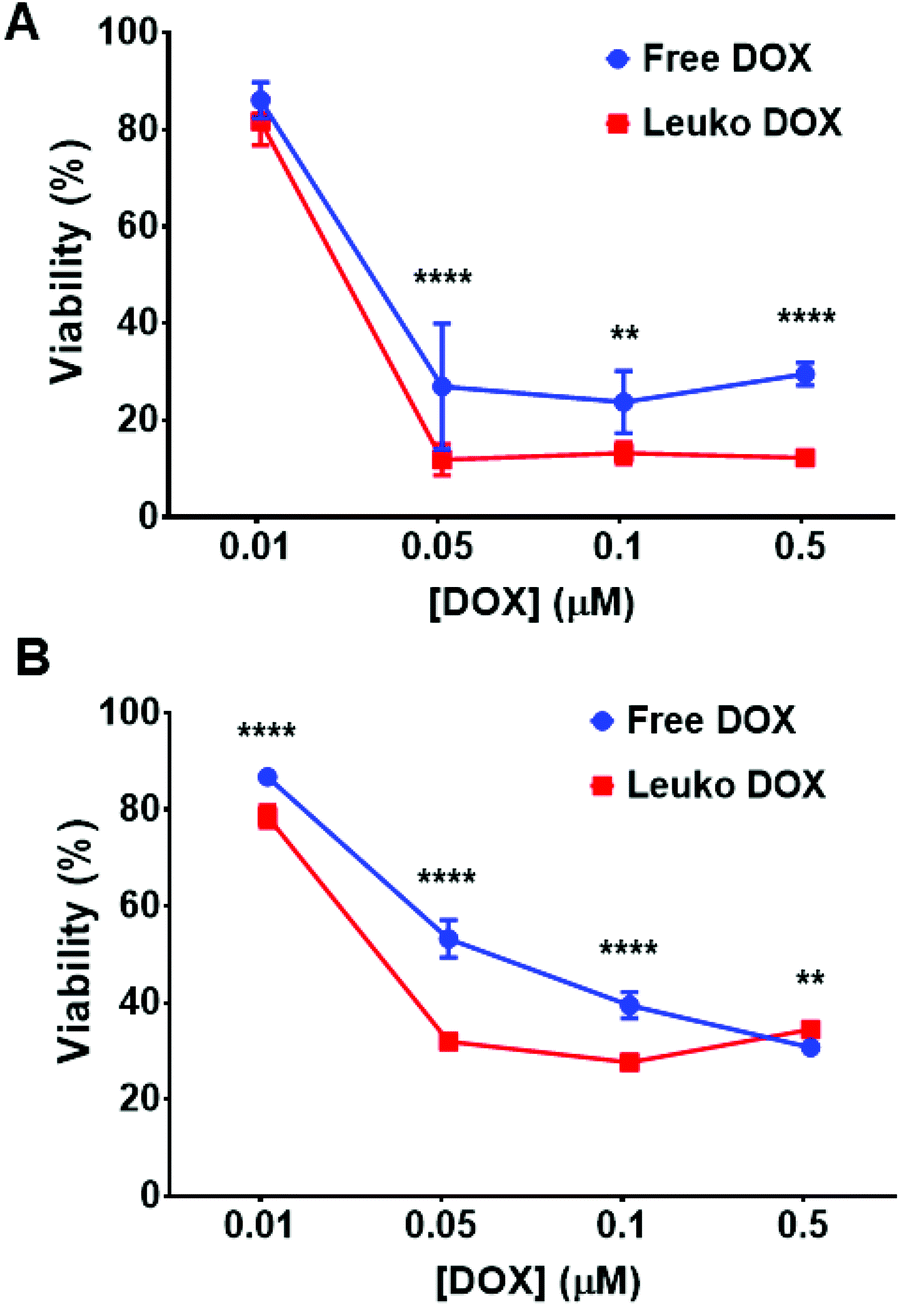 | ||
| Fig. 3 In vitro cytotoxicity of free and encapsulated DOX. A, 4T1 and (B) B16 cell viability after incubation with growing concentrations (e.g., 0.01, 0.05, 0.1 and 0.5 μM) of DOX either free DOX or Leuko DOX. Treatment efficacy is expressed as survival percentage compared to untreated control (100% survival, not shown). Each bar shows the mean ± SD. Experiments were performed in triplicate. **p < 0.01; ****p < 0.0001. | ||
For B16 melanoma cells, Leuko DOX demonstrated a more potent response at 0.01, 0.05 and 0.1 μM compared to free DOX (Fig. 3B). Particularly, Leuko DOX yielded a reduction in cell viability of 78, 32 and 27% vs. 86, 53 and 39% of free DOX at DOX concentrations of 0.01, 0.05 and 0.1 μM, respectively. Empty leukosomes had minimal effect on the proliferation of both cancer cell lines (data not shown).
These findings prompted the testing of the in vivo anti-tumor activity of Leuko DOX in both 4T1 and B16 models. In these experiments, Leuko DOX was compared with no treatment and free DOX. Tumor volume measurements and survival analysis in both tumor models revealed that Leuko DOX displayed a substantial reduction in tumor growth compared to the free DOX allowing for an extended median survival (Fig. 4 and 5).
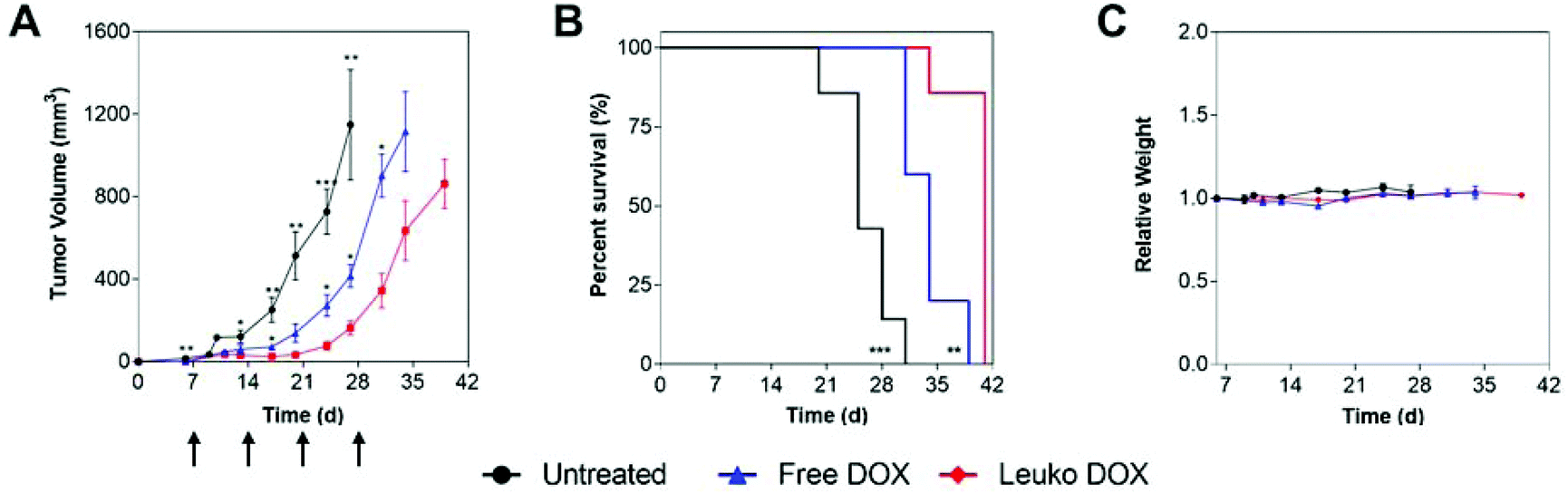 | ||
| Fig. 4 Breast cancer efficacy of Leuko DOX. (A) Tumor volume measurements of mice with 4T1 triple negative breast tumors implanted on Day 0. (B), Kaplan–Meier curves comparing untreated, free DOX, and Leuko DOX monitoring survival for 6 weeks after tumor implantation. (C) Relative weights of mice during treatment with free DOX, Leuko DOX, or left untreated. Data is shown as means with standard error of the mean. Mice were treated at 3 mg kg−1 of DOX starting at Day 7 and continued once a week for 4 total treatments (i.e., Day 7, Day 14, Day 21, and Day 28). *p < 0.05, **p < 0.01, ***p < 0.001. | ||
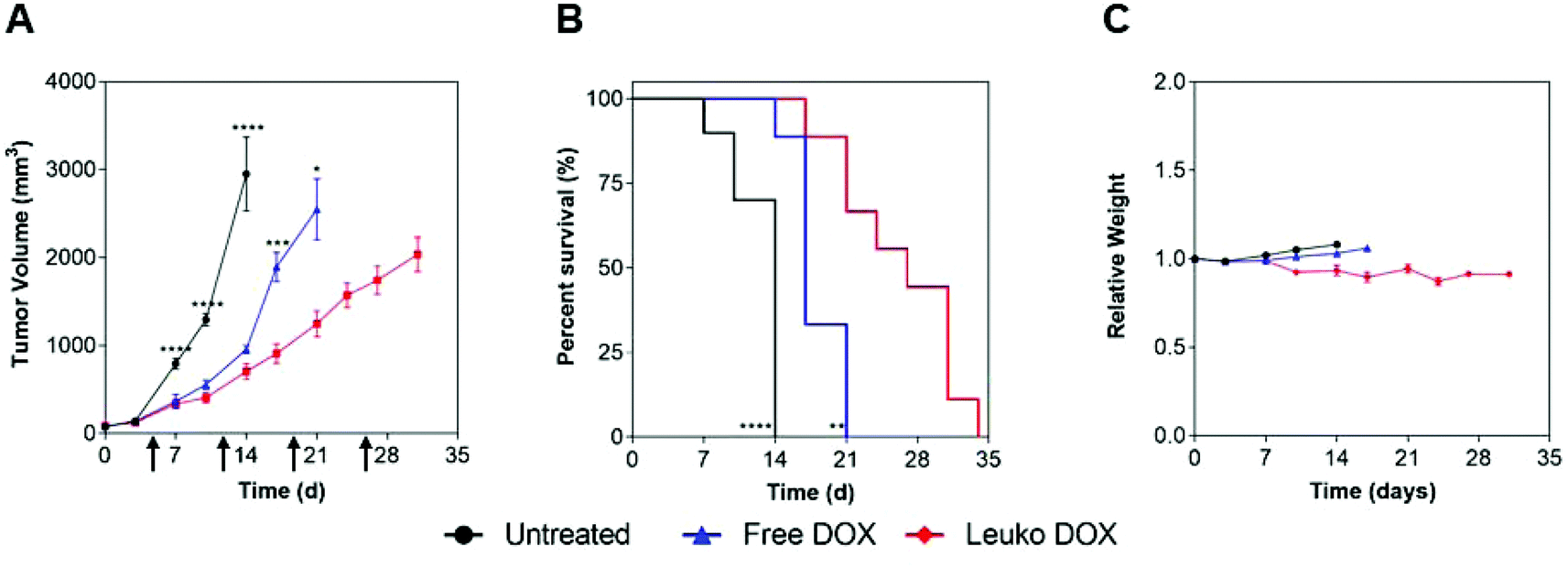 | ||
| Fig. 5 Melanoma efficacy of Leuko DOX. (A) Tumor volume measurements of mice with melanoma tumors implanted on Day 0. (B) Kaplan–Meier curves comparing untreated, free DOX, and Leuko DOX monitoring survival for 6 weeks after tumor implantation. (C) Relative weights of mice during treatment with free DOX, Leuko DOX, or left untreated. Data is shown as means with standard error of the mean. Mice were treated at 7 mg kg−1 of DOX starting at Day 5 and continued once a week until tumors reached any dimension greater than 2.0 cm2 (i.e., Day 5, Day 12, Day 19, and Day 26). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001. | ||
In 4T1 tumors, tumor volume measurements demonstrated that mice treated with Leuko DOX exhibited significant inhibition of breast cancer growth from 17, 24, 27, and 31 days after tumor implantation relative to free DOX therapy and from day 6 compared to untreated (Fig. 4A). Tumors left untreated or treated with free DOX displayed sizes larger than 9-fold (728 vs. 77 mm3) and 3.5-fold (273 vs. 77 mm3) at 24 days compared to tumors treated with Leuko DOX, respectively. This inhibition in tumor growth translated to significant increases in the overall survival of mice treated with Leuko DOX (Fig. 4B). Mice treated with Leuko DOX extended median survival by 64% (25 vs. 41 days) and 21% (34 vs. 41 days) compared to untreated and free DOX, respectively. Furthermore, monitoring of mouse weight allowed for evaluation of the overall safety of Leuko DOX treatments (Fig. 4C). No substantial differences were observed in untreated and Leuko DOX mice indicating that treatments with Leuko DOX in mice with breast cancer were well-tolerated.
In melanoma tumors, Leuko DOX group displayed the most significant reduction in tumor volume from 7, 10, and 14 days after tumor implantation compared to untreated and from day 17 and 21 relative to free DOX treatment (Fig. 5). Particularly, Leuko DOX significantly reduced tumor growth of 4.2-fold (705 vs. 2949 mm3) compared to untreated control (p < 0.0001) and 2-fold compared to free DOX treatment (1248 vs. 2548 mm3, p < 0.05), respectively (Fig. 5A). Tumor growth inhibition by Leuko DOX treatment resulted in extension of median survival by 142% (34 vs. 14 days, p < 0.0001) and 62% (34 vs. 21 days, p < 0.01) compared to untreated and free DOX, respectively (Fig. 5B). Monitoring of mouse weight throughout the treatment revealed a slight reduction in the group treated with Leuko DOX, which was, however, lower than 10% of initial weight (Fig. 5C).
Discussion
Among all nanoscale drug delivery platforms developed to date, liposomes are the most investigated and the first to gain approval for treatment in humans.1 The engineering of lipid materials allows the reproducible synthesis of biocompatible and biodegradable nanocarriers with tunable loading and release properties.33 The immune system and the physiologic compartmentalization of our body are extremely effective in recognizing, sequestering, and clearing foreign objects, including the synthetic materials used for drug delivery applications.34 Bio-mimetic approaches represent a viable and sustainable alternative for the development of carriers able to interact with and negotiate the complexity of the biological barriers protecting our body.35–37 In particular, leukocyte physiology represents an extraordinary source of inspiration for the synthesis of injectable biomimetic carriers. Here we exploit the targeting properties of leukosomes towards the inflamed, tumor-associated vasculature for effective delivery of doxorubicin to solid tumors in syngeneic mouse models of breast cancer and melanoma. In this study, we demonstrated that leukosomes retained liposomal pharmaceutical properties to load and release DOX, while displaying a delayed release effect due a more densely packed bilayer. In vivo targeting experiments revealed a significantly higher tumor accumulation for leukosomes in B16 tumors, thus highlighting the contribution of leukocyte membrane proteins to target tumor-associated vasculature. In vitro cytotoxic studies revealed a similar effect for both free and DOX-loaded leukosomes on 4T1 and B16 cancer cells. In tumor efficacy studies for 4T1 and B16 tumors, DOX-loaded leukosomes showed a more potent anti-cancer activity in terms of reduction of tumor volume and prolonged survival.These findings confirm the potentiality and versatility of leukosomes for cancer treatment among the other biomedical applications seen so far.18,20 The similarity of leukosomes with the established liposomal technology simplifies their clinical translation since they share the same physicochemical and pharmaceutical background. The biomimetic nature of leukosomes, and other similar nanovesicles, which enable the use of cells derived directly from the patient could transform the landscape of nano-based therapies for precision medicine.
Methods
Assembly and physical characterization of leukosomes
Leukosomes were prepared using the thin layer evaporation (TLE) method as previously reported.15 Briefly, choline-based phospholipids, i.e., 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, 1,2-distearoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphocholine, and cholesterol (Avanti Polar Lipids), 5:1:3:1 molar ratio, respectively, were dissolved in a mixture of chloroform:methanol (3:1 v/v). The solvent was then evaporated through a rotary evaporator (BÜCHI Labortechnik AG, Switzerland) to form a thin film, which was hydrated with membrane proteins (1:300 protein-to-lipid ratio) dispersed in PBS or PBS alone to assemble leukosomes or conventional liposomes, respectively. Lipid suspension was then extruded ten times through 200 nm pore-size cellulose acetate membranes at 45 °C and the obtained unilamellar vesicles were then dialyzed overnight through 1000 kDa membranes (Spectrum Laboratories, Inc.) to eliminate raw material and unincorporated proteins. Membrane proteins were obtained by whole leukocytes deriving from peripheral blood of Balb/c (for breast cancer studies) and C57BL/6J (for melanoma studies) mice. Briefly, blood was collected by a heart puncture procedure. After lysis of erythrocytes using proper buffers, leukocytes were collected, and membrane proteins extracted as previously reported.15 Purified membrane proteins were then quantified using Bradford assay and stored at −80 °C until usage. For intravital microscopy studies, liposomes and leukosomes were labeled with a rhodamine-marked phospholipid added in the organic phase containing the lipid mixture. Physical characterization was performed with a Nanosizer ZS (Malvern Instruments).
Evaluation of doxorubicin encapsulation efficiency
Doxorubicin hydrochloride (DOX) was encapsulated in both liposomes and leukosomes using the remote loading method through the creation of a transmembrane ammonium sulfate gradient.22 Briefly, the lipid film was hydrated with a 250 mM ammonium sulfate solution by warming-vortex cycles as described above in the presence or absence of membrane proteins. Multi-lamellar liposomes or leukosomes (nanovesicles) were then extruded 10 times through 200 nm pore-size cellulose acetate membranes at 45 °C. The unentrapped ammonium sulfate solution was removed by dialysis in a 0.9% NaCl solution, thus obtaining an acidic pH gradient between the vesicle aqueous compartment and the medium. The resulting formulation was then incubated with 1 ml of a solution of DOX (2 mg ml−1) and kept at 37 °C for 1 hour. Unencapsulated DOX was separated from the encapsulated one through ultracentrifugation at 130000g for 1 h at 20 °C, the supernatant was removed, and the pellet resuspended in HBS pH 7.4 for further experiments. DOX concentration was determined spectrofluorometrically (λexc 480 nm, λem 590 nm) after lysing nanovesicles with Triton X-100 (0.5% v/v final concentration38) according to the following equation:| EE% = (DU/DT) × 100 |
| LC% = (DU/W) × 100 |
Doxorubicin release experiments
The release of DOX from liposomes and leukosomes was investigated using a fluorescence dequenching assay.39,40 DOX-loaded nanovesicles (500 μl) were incubated in PBS:FBS (50:50 v/v) at 37 °C. The pH of PBS was adjusted to pH values of 6.5 and 7.4 with sodium hydroxide to mimic acidic and physiologic pH, respectively. At various time points, the percentage of DOX released from the nanovesicles was determined by measuring the increase in fluorescence intensity at the above-mentioned wavelength. Total dequenched DOX was measured after addition of Triton X-100 (0.5% v/v). The experiment was repeated three times for each formulation and the results are expressed as the mean ± standard deviation.
Evaluation of physical stability of doxorubicin-loaded leukosomes in simulated serum
To gain insights into the stability of Leuko DOX once injected in vivo, we incubated these nanovesicles in a mixture of FBS:PBS (50:50 v/v) at 37 °C under stirring to mimic in vivo conditions. At determined time points (e.g., 0, 20, 40, 60, 120 minutes, 17 and 19 hours), 20 μl of samples were withdrawn and physically analyzed through DLS as above described. Results are expressed as the mean of seven measurements ± standard deviation.
In vitro experiments
In vitro studies were performed with the use of murine breast cancer cell line 4T1 and melanoma B16-F10 purchased from the American Type Culture Collection (Rockville, Maryland, USA). Cells were cultured either in Dulbecco's modified Eagle's medium (DMEM) (for 4T1 cultures) or RPMI-1640 (for B16-F10) supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin and 1% glutamine (PAA, Piscataway, NJ, USA) at 37 °C in a 5% CO2 atmosphere. 1 × 103 4T1 per 100 μl per well and 0.6 × 105 B16-F10 cells per 100 μl per well were seeded in 96-well plates. Cells were treated with either free doxorubicin (free DOX) or leukosome-loaded DOX (Leuko DOX) at DOX concentration of 0.01, 0.05, 0.1 and 0.5 μM. Cell viability percentage was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MMT) assay (Sigma Aldrich, St Louis, MO, USA) after a 24-hour drug exposure. Results were compared to untreated cells. Experiments were performed in triplicate.In vivo experiments
All animal experiments were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals approved by The Houston Methodist Institutional Animal Care and Use Committee guidelines (Houston, TX).Intravital microscopy (IVM)
All animal experiments were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals approved by The Houston Methodist Institutional Animal Care and Use Committee guidelines (Houston, TX) and were performed in accordance to IACUC-approved protocols AUP-0714-0043 and IVM imaging performed under protocol AUP-0611-0032. Before imaging, mice were anesthetized with isoflurane. Rhodamine-labeled nanovesicles were injected intravenously via retro orbital injection. Upon systemic administration, we monitored accumulation real-time dynamic flow for up to 60 min post-injection, comparing extent of uptake versus conventional liposomes. Fluorescein isothiocyanate (FITC)-labeled dextran dye (70 kDa) (Invitrogen, Carlsbad, CA) was used as vessel tracer. Tumor imaging was performed by firstly exposing tumor using a skin-flap procedure. Before imaging, a bolus injection of 70 kDa FITC-dextran (50 μL in PBS) was used to delineate the vasculature. Images were obtained with a three-channel setup in which fluorescence was collected at 488/525 nm for FITC dextran and at 561/579 nm for rhodamine-labeled particles. Image acquisition was performed over selected field of views (FOVs) with resolution of 512 × 256 pixels with an optical slice thickness of 7.1 μm.Biodistribution profile of leukosomes
Tumor bearing Balb/c mice were i.v. injected with 2 mg of rhodamine-labeled liposomes and leukosomes (n = 5 for each group) in order to evaluate particle biodistribution and pharmacokinetic profile. After 1, 6 and 24 h mice were sacrificed and major organs (kidney, lung, heart, pancreas, spleen and liver) and tumor tissue were collected, washed twice with PBS, weighted and transferred in a falcon tube. Tubes were then filled with formamide (1 ml per 100 mg of tissue weight) and tissues were homogenized. After 2 h of incubation at room temperature, samples were centrifuged at 5000g per 10 min and supernatant was collected and spectrofluorometrically analyzed for rhodamine detection (excitation/emission 561/579; slit width, 5 nm). Results are represented as percentage of injected dose per gram of tissue based on a standard curve to calibrate rhodamine-labeled phospholipid.Generation of tumor models
4T1 breast cancer tumors were implanted into Balb/c mice at 2 × 104 cells per mouse in their mammary fat pad. Treatment with free DOX (n = 5), Leuko DOX (n = 7), and no treatment (n = 7) began 7 days after tumor implantation and continued once a week for 4 total treatments at a dose of 3 mg kg−1 of DOX. Mice were monitored with calipers for tumor growth and changes in weight twice a week. Mice were sacrificed when tumors reached any dimension greater than 1.0 cm2 or if any substantial degree of ulceration was observed.For the melanoma model, C57BL/6 mice were subcutaneously injected with 5 × 105 B16-F10 cells. Mice were monitored twice a week with calipers for tumor growth and changes in weight for treatment safety. At 5 days after tumor implantation, when tumor volume reached about 150–200 mm3, mice were randomly divided into three groups (n = 10), the same as above, and treated at a dose of 7 mg kg−1 of DOX once per week until tumors reached any dimension greater than 2.0 cm2 or if any substantial degree of ulceration was observed.
Statistical analysis
All data were presented as means and standard error of the mean (SEM). Intravital microscopy data were presented as means ± standard error of the mean obtained from at least 8 field of views (FOVs) for n = 4. GraphPad statistical software (La Jolla, CA) was used to compute statistical significance between groups and control using student's test and one-way ANOVA test to compare differences between groups. Statistics for tumor volume growth and weight measurements were assessed using multiple t-tests using unpaired t-tests assuming unequal scatter with correction for multiple comparisons (Holm-Sidak method). Survival data was analyzed for significance with survival curve analysis with curve comparison using a Log-rank (Mantel-Cox) test. Outliers for tumor volume measurement were identified using ROUT with Q set at 0.1%. A value of p = 0.05 was considered statistically significant.Author contributions
R.M conceived and designed the experiments; E.T., R.M., J.O.M. and C.C. wrote the paper and interpreted the data; R.M., C.C. and A.D.V. performed survival experiments, R.M. and G.S. performed biodistribution experiments. R.M. and E.D.R. performed ex vivo confocal experiments; R.M. and K.A.H. synthesized and characterized the nanoparticles; N.T., N.B. and N.A. performed in vitro experiments, A.Z. performed stability and cryoEM experiments, E.T. provided mentoring and contributed the funding support. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants to ET from The Cullen Trust for Health Care and the William Randolph Hearst Foundation. N. A. was supported by Associazione Bianca Garavaglia Onlus A/15/01O (Zaffaroni). This manuscript is dedicated to the memory of Salvatore Montesanti (12/24/1956–01/23/2019).References
- S. Svenson, Clinical translation of nanomedicines, Curr. Opin. Solid State Mater. Sci., 2012, 16, 287–294 CrossRef CAS.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, Endosomal escape pathways for delivery of biologicals, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- A. Zinger, et al., Collagenase Nanoparticles Enhance the Penetration of Drugs Into Pancreatic Tumors, ACS Nano, 2019, 13, 11008–11021 CrossRef CAS.
- M. E. Davis, Nanoparticle therapeutics: an emerging treatment modality for cancer, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- T. Lammers, W. E. Hennink and G. Storm, Tumour-targeted nanomedicines: principles and practice, Br. J. Cancer, 2008, 99, 392–397 CrossRef CAS.
- E. K. Chow and D. Ho, Cancer nanomedicine: from drug delivery to imaging, Sci. Transl. Med., 2013, 5, 216rv214 Search PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS.
- M. S. Kim, et al., Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells, Nanomedicine, 2016, 12, 655–664 CrossRef CAS.
- P. Milla, F. Dosio and L. Cattel, PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery, Curr. Drug Metab., 2012, 13, 105–119 CrossRef CAS.
- A. A. Gabizon, Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet, Clin. Cancer Res., 2001, 7, 223–225 CAS.
- A. Rajput, et al., Characterization of HCT116 human colon cancer cells in an orthotopic model, J. Surg. Res., 2008, 147, 276–281 CrossRef CAS.
- A. Parodi, et al., Bio-inspired engineering of cell- and virus-like nanoparticles for drug delivery, Biomaterials, 2017, 147, 155–168 CrossRef CAS.
- M. R. Mohammadi, C. Corbo, R. Molinaro and J. R. T. Lakey, Biohybrid Nanoparticles to Negotiate with Biological Barriers, Small, 2019, e1902333 CrossRef.
- R. Li, Y. He, S. Zhang, J. Qin and J. Wang, Cell membrane-based nanoparticles: a new biomimetic platform for tumor diagnosis and treatment, Acta Pharm. Sin. B, 2018, 8, 14–22 CrossRef PubMed.
- R. Molinaro, et al., Biomimetic proteolipid vesicles for targeting inflamed tissues, Nat. Mater., 2016, 15, 1037–1046 CrossRef CAS.
- C. M. Hu, et al., Nanoparticle biointerfacing by platelet membrane cloaking, Nature, 2015, 526, 118–121 CrossRef CAS PubMed.
- J. Jin, et al., Human Cancer Cell Membrane-Coated Biomimetic Nanoparticles Reduce Fibroblast-Mediated Invasion and Metastasis and Induce T-Cells, ACS Appl. Mater. Interfaces, 2019, 11, 7850–7861 CrossRef CAS PubMed.
- C. Corbo, et al., Engineered biomimetic nanovesicles show intrinsic anti-inflammatory properties for the treatment of inflammatory bowel diseases, Nanoscale, 2017, 9, 14581–14591 RSC.
- Q. Zhang, et al., Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis, Nat. Nanotechnol., 2018, 13, 1182–1190 CrossRef CAS.
- R. Molinaro, et al., Macrophage-derived nanovesicles exert intrinsic anti-inflammatory properties and prolong survival in sepsis through a direct interaction with macrophages, Nanoscale, 2019, 11, 13576–13586 RSC.
- A. V. Kroll, et al., Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity, Adv. Mater., 2017, 29, 1703969 CrossRef.
- Y. Barenholz, Doxil®—The first FDA-approved nano-drug: Lessons learned, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS.
- H. C. Arora, et al., Nanocarriers Enhance Doxorubicin Uptake in Drug-Resistant Ovarian Cancer Cells, Cancer Res., 2012, 72, 769–778 CrossRef CAS.
- X. Zhang, Y. Lin and R. J. Gillies, Tumor pH and its measurement, J. Nucl. Med., 2010, 51, 1167–1170 CrossRef CAS.
- H. Shibata, K. Izutsu, C. Yomota, H. Okuda and Y. Goda, Investigation of factors affecting in vitro doxorubicin release from PEGylated liposomal doxorubicin for the development of in vitro release testing conditions, Drug Dev. Ind. Pharm., 2015, 41, 1376–1386 CrossRef CAS.
- J. Wolfram, et al., Shrinkage of pegylated and non-pegylated liposomes in serum, Colloids Surf., B, 2014, 114, 294–300 CrossRef CAS.
- C. Corbo, et al., Unveiling the in Vivo Protein Corona of Circulating Leukocyte-like Carriers, ACS Nano, 2017, 11, 3262–3273 CrossRef CAS.
- J. O. Martinez, et al., Biomimetic nanoparticles with enhanced affinity towards activated endothelium as versatile tools for theranostic drug delivery, Theranostics, 2018, 8, 1131–1145 CrossRef CAS PubMed.
- R. Molinaro, et al., Design and Development of Biomimetic Nanovesicles Using a Microfluidic Approach, Adv. Mater., 2018, 30, e1702749 CrossRef.
- K. E. de Visser, A. Eichten and L. M. Coussens, Paradoxical roles of the immune system during cancer development, Nat. Rev. Cancer, 2006, 6, 24–37 CrossRef CAS.
- L. M. Coussens and Z. Werb, Inflammation and cancer, Nature, 2002, 420, 860–867 CrossRef CAS.
- R. A. Worthylake and K. Burridge, Leukocyte transendothelial migration: orchestrating the underlying molecular machinery, Curr. Opin. Cell Biol., 2001, 13, 569–577 CrossRef CAS.
- S. K. Sriraman and V. P. Torchilin, Recent Advances with Liposomes as Drug Carriers, Adv. Biomater. Biodevices, 2014, 79–119 CAS.
- P. P. Karmali and D. Simberg, Interactions of nanoparticles with plasma proteins: implication on clearance and toxicity of drug delivery systems, Expert Opin. Drug Delivery, 2011, 8, 343–357 CrossRef CAS.
- J.-W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Bio-inspired, bioengineered and biomimetic drug delivery carriers, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS.
- R. Molinaro, et al., Inflammation and Cancer: In Medio Stat Nano, Curr. Med. Chem., 2018, 25, 4208–4223 CrossRef CAS PubMed.
- R. Molinaro, et al., Vascular Inflammation: A Novel Access Route for Nanomedicine, Methodist Debakey Cardiovasc. J., 2016, 12, 169–174 CrossRef.
- T. Ishida, M. Kirchmeier, E. Moase, S. Zalipsky and T. Allen, Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells, Biochim. Biophys. Acta, Biomembr., 2001, 1515, 144–158 CrossRef CAS.
- A. Fritze, F. Hens, A. Kimpfler, R. Schubert and R. Peschka-Süss, Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1633–1640 CrossRef CAS.
- G. Pasut, et al., Polyethylene glycol (PEG)-dendron phospholipids as innovative constructs for the preparation of super stealth liposomes for anticancer therapy, J. Controlled Release, 2015, 199, 106–113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01766f |
| This journal is © The Royal Society of Chemistry 2020 |