Photophysics of pyrene-labelled compounds of biophysical interest†
Chunxiang
Yao
,
Heinz-Bernhard
Kraatz
and
Ronald P.
Steer
*
Department of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, SK, Canada S7N 5C9. E-mail: ron.steer@usask.ca; Fax: 306-966-4730; Tel: 306-966-4667
First published on 6th December 2004
Abstract
The effects of the chemical constitution and structure of the substituent on the excited state dynamics of several model fluorescent pyrene-labelled molecules of biophysical interest have been examined. Nine new 1-substituted pyrenyl compounds, Py–NH–CO–C2H5, Py–NH–CO–Leu–Boc, Py–CH2–NH–CO–C2H5, Py–CH2–NH–CO–Leu–Boc, Py–CO–NH–C3H7, Py–CO–NH–Leu–OMe, Py–CH2–CO–NH–C3H7, Py–CH2–CO–NH–Leu–OMe and Py–C3H6–CO–NH–Leu–OMe, have been synthesized and their electronic spectra, fluorescence quantum yields and excited state lifetimes measured. These data have been used to calculate the radiative, kr, and non-radiative decay constants of their S1 states and the values of these constants correlated with the structures of the tethers. Non-radiative S1 decay rates (mainly intersystem crossing to T1) vary in parallel with the radiative rates so that the excited state lifetimes and radiative rate constants change considerably with the structure of the substituent whereas the quantum yields of fluorescence do not. An excellent correlation between εmax of the S1–S0 transition and either kr or the excited state lifetime is observed as long as no additional intermolecular or intramolecular excited state decay process of significant rate competes with the ‘normal’ radiative and non-radiative (ISC) decay processes of the pyrenyl chromophore. This correlation may have predictive value. Rates of bimolecular quenching of the S1 states of these molecules by molecular oxygen have been measured. The quenching process is diffusion-controlled with a spin statistical factor of 1, indicating that the S1–T1 electronic energy spacings of all the derivatives exceed the O2 (1Δg–3Σ−g) electronic excitation energy of ca. 1 eV.
Introduction
One of the most commonly used approaches to studying the dynamics of electron transfer (ET) and electronic energy transfer (EET) involves time-resolved fluorescence measurements on molecules that contain an electron or energy donor linked to an electron or energy acceptor. In biomolecules, such studies make use of either naturally fluorescent amino acid residues (intrinsic fluorescence) or, more frequently, extrinsic fluorescent probes covalently tethered to the molecule under investigation.1 Pyrene, 1, is conveniently substituted at the 1-position, and has often been employed as an extrinsic fluorescent label in such studies.Pyrene's electronic spectrum and state assignments2 are well established and it has several photophysical characteristics that make it useful as a fluorescent probe. The latter include a high quantum yield of fluorescence and a relatively long S1 lifetime (e.g.ϕf = 0.6; τ = 450 ns in deaerated cyclohexane at room temperature)3 that are readily measurable using straightforward techniques. In addition, the relative intensities of the vibrational structure of pyrene's fluorescence spectrum (the Ham bands) are sensitive to the polarity of the surrounding medium, and this molecule forms spectroscopically distinct, fluorescent excimers that can be used to detect aggregation. These characteristics of pyrene and its derivatives have been employed widely in the investigation of multimolecular aggregates of micelles and membranes,4 in studies of molecular optical memory and optical switching,5 and in the inspection of the active sites of enzymes.6 Recently, pyrene has also been used widely as a fluorescent label in investigations of the distance dependence,7,8 electric field effects9,10 and conformation dependence11 on the ET rates of polypeptides, DNA oligomers and duplexes.
Surprisingly, relatively little attention has been focused in these studies on understanding the electronic communication between the pyrene fluorophore and the rest of the molecule through its tethering group. The latter information is critically important for establishing a full understanding of the mechanisms of complex biological processes, especially ET in pyrene-labelled peptides or DNA compounds where it has often been assumed that electron transfer occurs through the molecular backbone of the tether between the electron donor/acceptor pair.12,13 This paper seeks to address this deficiency by systematically studying the effects of the nature of the tether on the fluorescence spectra and photophysical properties of several pyrene-labelled molecules of biophysical interest.
Experimental
Synthesis and characterization of pyrenyl compounds
The structures of the compounds synthesized for this study are shown below. The procedures used for their synthesis and the data obtained for characterizing their structures are provided as ESI.†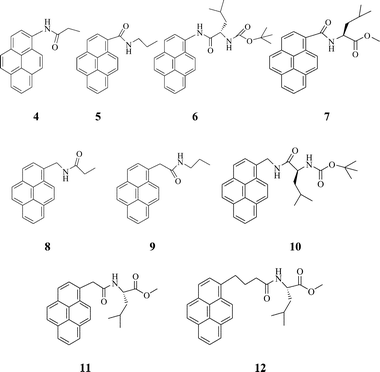
General methods
The starting materials: 1-pyreneanime (2), 1-pyrenecarboxylic acid (3), 1-pyreneacetic acid (14), 1-pyrenebutyric acid (15), 1-aminopyrene (16), and 1-pyrenemethylamine hydrochloride (17), di-tert-butyl dicarbonate (Boc2) (18), H–L-Leu·HCl (19), Boc–Leu–OH (20) and H–Leu–OMe·HCl (21) were obtained from Aldrich. The coupling reagents N-hydroxybenzotriazole·H2O (HOBt·H2O) (22) and ethyl-3(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) (23) were commercially available from Quantum Biotechnologies. Thionyl chloride, 1,4-dioxane, triethylamine, n-propylamine, propionic acid, and Na2SO4 were used as received from BDH. For synthetic purposes, CH2Cl2 (BDH) was dried over CaH2 under an atmosphere of N2 prior to use. All reactions with pyrenyl complexes were carried out in the absence of light. TLC was carried out on aluminium plates coated with silica gel (EM Science).The pyrenyl compounds were purified by flash column chromatography using 230–400 mesh silica gel (Silicycle Chemical Division). 1H and 13C{1H} NMR spectra were recorded on a Brucker AMX-300 instrument at 300.133 MHz and 75.478 MHz, respectively. Unless otherwise noted, NMR spectra were recorded in CDCl3, which was stored over molecular sieves. All chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz, relative to tetramethylsilane (δ = 0), and are referenced to the residual signal of CHCl3 (δH = 7.26, δC = 77.23). Chemical shifts and coupling constants from 1H NMR were determined assuming first-order behavior. Abbreviations for multiplicities are as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). All assignments were made on the basis of chemical shift, multiplicity and by comparison with related structures. If necessary, assignments were confirmed by 1H–1H COSY (correlation spectroscopy). The multiplicity (peak intensity) of decoupled 13C NMR signals refers to the number of attached protons and was determined by J-modulation. High-resolution mass spectra (HRMS) were obtained on a VG 70E mass spectrometer. Electron impact (EI) ionization at 70 eV was employed, and fast-atom bombardment (FAB) was carried out in the positive ion mode from a glycerol and methanol matrix. Elemental analyses were carried out using a Perkin-Elmer 2400 CHN elemental analyzer.
For spectroscopic measurements, CH2Cl2 was dried over CaH2 under a N2 atmosphere prior to use and was checked for purity by measuring its emission, which was negligible when excited at λex < 250 nm. Samples were deoxygenated either by bubbling dry, oxygen-free N2 through dried CH2Cl2 and preparing solutions inside a glove box or by three or four freeze–pump–thaw cycles on a high vacuum system. All measurements were carried out at room temperature of 22 °C.
Spectra
UV-visible absorption spectra were recorded with a Varian Cary Model 500 spectrophotometer in double beam mode at a constant spectral bandwidth of 1.0 nm using a matched pair of 1 × 1 cm quartz cells. Pure solvent was used as a reference. Values of the molar extinction coefficients, εmax, of the compounds of interest were obtained from the slopes of linear plots of their absorbance at λmaxvs. concentration.Fluorescence emission and excitation spectra were obtained using a Spex Fluorolog Model 222 spectrofluorometer (dispersion of 1.8 nm mm−1) using 1 × 1 cm cells and right angle observation geometry, acceptable for the optically dilute solutions employed. The emission signal was detected by an RCA C31034-02 photomultiplier tube, operating in the single photon counting mode. The excitation spectra were corrected by employing a rhodamine B reference quantum counter whereas the emission spectra were corrected using the manufacturer's pre-installed relative instrument response routine.
Quantum yields
Fluorescence quantum yields were measured by the relative method, using recrystallized quinine sulfate in 0.5M aqueous H2SO4 (ϕf = 0.546 14) as the reference standard. In these measurements, spectrometer slit widths were adjusted so that the spectral bandwidth of the absorption and emission instruments were identical at 1.0 nm, and the absorbances of the sample and the reference standard were chosen so they were in the 0.1–0.2 range and nearly identical at the same excitation wavelength. Emission quantum yields were then calculated according to the method described by Demas and Crosby,14 taking differences in the refractive indices of the sample and reference solutions into account. Errors in the absolute values of quantum yields in the ϕf > 0.10 range are estimated to be no greater than 10%, but are larger for smaller yields.Fluorescence lifetimes
The excitation light source used in the fluorescence lifetime measurements was a Coherent diode-pumped Mira, Model 900-P Ti:sapphire laser, which provided mode-locked pulses in the 700 to 1000 nm range. The 76 MHz pulse train was sampled using a Coherent Model 9200 pulse picker to provide excitation pulses at an acceptable repetition rate and was frequency doubled using an INRAD model 5-050 second harmonic generator. An excitation wavelength of 361 nm was chosen for most samples. Fluorescence was collected using a convex lens and was dispersed using a Carl Zeiss M4QIIId quartz prism monochromator (which broadens the instrument response function minimally) with a variable slit that was set to give a constant 20 nm bandwidth.Fluorescence lifetimes were recorded using the time correlated single photon counting (TCSPC) method,15 as previously described in detail. Briefly, the signal from a cooled Hammamatsu microchannel plate photomultiplier tube was fed into a preamplifier (HP Model 6216A) and then to a constant fraction discriminator (CFD, Tennelec TC 453). The output from the CFD was used as the start pulse in a time-to-amplitude converter (TAC, Tennelec TC 684). The stop signal was obtained by diverting a small fraction of the laser output from the pulse picker onto a fast photodiode (Spectra-Physics, Model 403B). The output signal of the TAC was fed into 1024 channels of a multichannel analyzer (MCA, Tracor Northern, Model 7200), the contents of which were transferred to and stored in a computer. Excitation repetition rates were chosen so that the intervals between excitation pulses were greater than 5 exponential fluorescence lifetimes, and photon count rates were adjusted to be less than 2% of the excitation rates, to avoid pulse pile-up.
Data analysis was performed using custom-developed software. This program performs an iterative deconvolution of the instrument response from the observed fluorescence decay functions using non-linear least-squares error minimization based on the Leavenberg–Marquardt16 and Nelder–Mead–Simples algorithms.17 Normal, reduced ‘chi-squared’ values (χr2), Durbin–Watson parameters, and weighted residual distributions were calculated to assess the goodness-of-fit between the deconvoluted data and trial single, double or triple exponential decay functions, 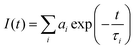 . The fractional intensity, Fi, of the ith fluorescent component was calculated from
. The fractional intensity, Fi, of the ith fluorescent component was calculated from 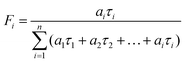 .
.
Results
Spectra
The UV-visible absorption spectra of all the pyrenyl compounds investigated (Figs. 1, 2, 3) show some similarities to pyrene itself. All exhibit two strong, vibrationally structured transitions in the λ < 365 nm region, which are assigned to the S2–S0 and S3–S0 transitions. By amplifying and expanding the spectrum on the red edge of the S2–S0 0–0 band, another weak absorption band system, convoluted with the low energy tail of the S2–S0 absorption band, may be clearly observed. Based on previous spectroscopic analyses of pyrene,18 this weak absorption is assigned to the lowest energy singlet-singlet transition, S1–S0. The resolved vibronic feature of this band system having substantial intensity and lying at lowest energy is taken to be the 0–0 transition. The absorbance due to the S1–S0 band system alone was obtained using standard curve-fitting software, by subtracting the absorbance due to the tail of the S2–S0 absorption from the recorded absorbance in the region of overlap when the spectrum was plotted in wavenumbers. Assuming that all vibrational bands of the S2–S0 and S1–S0 absorption systems could be adequately represented by Gaussian functions, values of εmax of the S1–S0 transitions obtained by deconvolution in this way were found to correlate well with measured radiative lifetimes (vide infra).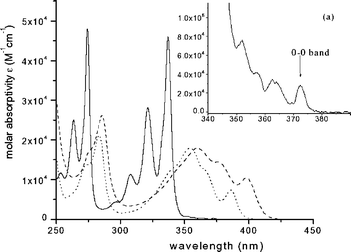 | ||
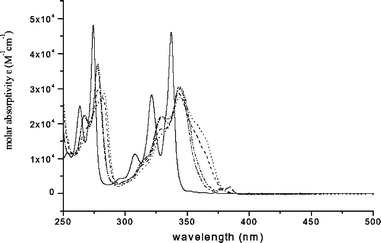
Fig. 1 Absorption spectra of compounds 1 (——), 2 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and 3 (⋯⋯⋯) in the range of 10−5–10−3 M in CH2Cl2 at room temperature; (a) the amplified spectrum of 1 (×100) in the range of 340–390 nm. The 0–0 band of Py 1 is at 372.5 nm. ) and 3 (⋯⋯⋯) in the range of 10−5–10−3 M in CH2Cl2 at room temperature; (a) the amplified spectrum of 1 (×100) in the range of 340–390 nm. The 0–0 band of Py 1 is at 372.5 nm. | ||
 | ||
| Fig. 2 Absorption spectra of compounds 4 (), 5 (–·
–·
–), 6 (⋯⋯), 7 (–··
–··
–) and 1 (——) in the range of 10−5–10−3 M in CH2Cl2 at room temperature. | ||
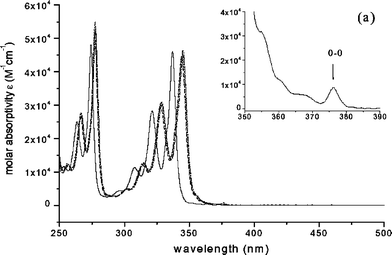 | ||
| Fig. 3 Absorption spectra of 1 (——), 8 (⋯·), 9 (–·
–·
–), 10 (-), 11 (–··
–··
–) and 12 () in the range of 10−5–10−3 M in CH2Cl2 at room temperature. (a) the ×10 amplified spectrum of 12 in the range of 360–390 nm, with 0–0 band at 377 nm. | ||
The UV-visible absorption spectra of 1 (pyrene), and the parents of its nine synthesized derivatives, 2 (1-pyrene amine) and 3 (1-pyrene carboxylic acid) in CH2Cl2 at room temperature are shown in Fig. 1. The UV-visible absorptions of compounds 2 and 3 are substantially red-shifted and the S2–S0 and S3–S0 absorption systems lose much of their vibrational structure, compared to pyrene itself. Note, however, that the shift in the absorption spectrum is quite uniform over the near-UV region, so that the electronic energy gaps between the S1 and S2 states, ΔE(S2–S1), are not very different from pyrene itself. Note also that the weak absorptions due to the S1–S0 transitions of 2 and 3 are intensified by substitution of amino and carboxyl groups at the 1-position of pyrene, at the cost of absorption intensity in the S2–S0 and S3–S0 transitions. As shown in Table 1, the values of the maximum molar extinction coefficient, εmax, of the S1–S0 transitions for compounds 3 and 2, as recovered by numerical curve-fitting, are a factor of ca. 20 larger than that of pyrene, while the values of εmax of the S2–S0 transitions for these two compounds decrease to almost half compared with 1.
| Compound | λ max/nm (εmax/M−1 cm−1)a | ΔE(S2–S1)/cm−1c | ||
|---|---|---|---|---|
| S1–S0b | S2–S0 | S3–S0 | ||
| a Wavelength error: ±0.5 nm. b From curve fitting. c Ca. ±50 cm−1. | ||||
| Py (1) | 372.5 (2.05 × 102) | 337.0 (4.60 × 104) | 274.5 (4.80 × 104) | 2800 |
| Py–NH2 (2) | 398.0 (4.51 × 103) | 360.0 (1.80 × 104) | 286.0 (2.62 × 104) | 2650 |
| Py–COOH (3) | 386.0 (4.90 × 103) | 355.0 (1.79 × 104) | 283.0 (2.11 × 104) | 2250 |
| Py–NH–CO–C2H5 (4) | 384.0 (1.44 × 103) | 343.0 (2.83 × 104) | 278.5 (2.93 × 104) | 3100 |
| Py–CO–NH–C3H7 (5) | 377.0 (8.56 × 102) | 344.0 (3.01 × 104) | 278.0 (3.58 × 104) | 2550 |
| Py–NH–CO–Leu–Boc (6) | 384.0 (1.55 × 103) | 344.0 (2.72 × 104) | 283.0 (2.83 × 104) | 3000 |
| Py–CO–NH–Leu–OMe (7) | 378.0 (1.36 × 103) | 344.0 (3.06 × 104) | 277.5 (3.70 × 104) | 2600 |
| Py–CH2–NH–CO–C2H5 (8) | 375.0 (3.40 × 102) | 344.0 (4.76 × 104) | 277.0 (5.55 × 104) | 2400 |
| Py–CH2–CO–NH–C3H7 (9) | 376.0 (3.54 × 102) | 345.0 (4.50 × 104) | 278.0 (5.17 × 104) | 2400 |
| Py–CH2–NH–CO–Leu–Boc (10) | 375.5 (2.87 × 102) | 344.0 (4.34 × 104) | 277.0 (5.04 × 104) | 2450 |
| Py–CH2–CO–NH–Leu–OMe (11) | 376.0 (2.62 × 102) | 345.0 (4.59 × 104) | 277.5 (5.24 × 104) | 2400 |
| Py–C3H6–CO–NH–Leu–OMe (12) | 376.0 (7.86 × 102) | 344.5 (4.51 × 104) | 277.5 (5.34 × 104) | 2450 |
Compounds 4 and 6 are derived from 2 and have an N-amido group in the 1-position whereas 5 and 7 can be considered as being derived from 3, linked by a C-amido group; their UV-visible spectra are shown in Fig. 2. Not surprisingly, the spectrum of compound 4 is similar to that of 6 whereas the spectrum of 5 is similar to that of 7. Compared to pyrene itself, the absorptions of 4–7 are shifted to the red (but lie to the blue of 2 and 3), the S3–S0 and S2–S0 absorptions are weaker by about 1/3 and exhibit less vibrational structure, and the S1–S0 transitions increase in intensity by a factor of ca. 5.
Fig. 3 presents the UV-visible absorption spectra of compounds 8–12, all of which have at least one methylene group interposed between the pyrene fluorophore and the amido group. These spectra are all similar and exhibit only small red-shifts compared with pyrene. Consequently, they all exhibit nearly identical S2–S1 electronic energy gaps of ca. 2400–2600 cm−1. The values of εmax for these compounds show that the S1–S0 transitions are somewhat enhanced in intensity compared to pyrene, but not nearly as much as when an N-amido or C-amido group is attached directly to the pyrene ring. The S2–S0 transitions carry slightly smaller oscillator strengths than pyrene whereas the S3–S0 transitions are somewhat more intense. The data are summarized in Table 1.
Representative S1–S0 emission spectra of pyrene and its derivatives in CH2Cl2 are shown in Figs. 4, 5 and 6. These spectra were all measured within the 10−6–10−4 M concentration range in CH2Cl2; no features which might be unequivocally assigned to an excimer11 were observed at the low end of this concentration range. The corrected, normalized excitation spectra are each identical, within calibration error limits, to their respective UV-visible absorption spectra, indicating that the quantum yield of S1–S0 fluorescence is constant for excitation within the whole of this near-UV range and that the quantum yields of Sn (n = 2, 3)–S1 internal conversion are unity. The emission spectra are all qualitatively similar and differ mainly in the relative intensities of the vibrational bands. Deconvolution of the vibrational features in the emission and excitation spectra (presented in wavenumbers) was achieved by simulating the structure as a sum of Gaussian bands after normalization of both spectra at the S1–S0 origin. Table 2 presents the data obtained from these spectra, from which the wavenumbers of the prominent vibrational spacings and the Stokes shifts may be determined.
| Compound | λ/nm | Stokes shift, Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) /cm−1c /cm−1c |
|
|---|---|---|---|
| S1→S0a | S1↔S0b | ||
|
a
The wavelengths in nm [wavenumbers] of the vibrational band maxima (shoulders) recorded in the fluorescence emission spectra.
b
The wavelengths in nm [wavenumbers] of the origin bands of the S1–S0 transitions in the excitation spectra.
c
The Stokes shift, Δ, is calculated from the difference (in cm−1) of the S1–S0 origin bands in the emission and excitation spectra. The uncertainty is ca.
±50 cm−1.
d
See text.
|
|||
| Py (1) | 371.5, 377.5, 382.5, 392.5, (413.0) [26918, 26490, 26414, 25478] | 371.5 [26918] | 0 |
| Py–NH2 (2) | 413.0, (428.5) [24213] | 397.0 [25189] | 980d |
| Py–NH–CO–C2H5 (4) | 384.5, 342.0, (329.0) [26008, 29240] | 382.5 [26144] | 140 |
| Py–NH–CO–Leu–Boc (6) | 385.5, 405.5, 427.5 [25940, 24661, 23392] | 382.0 [26178] | 200 |
| Py–CH2–NH–CO–C2H5 (8) | 375.0, 381.0, 386.0, 395.0, (413.0) [26667, 26247, 25907, 25316] | 374.0 [26738] | 70 |
| Py–CH2–NH–CO–Leu–Boc (10) | 375.0, 381.0, 386.0, 395.0, (413.0) [26667, 26247, 25907, 25316] | 374.0 [26738] | 70 |
| Py–COOH (3) | 392.0, 413.0, (431.0) [25510, 24213] | 385.5 [25940] | 430d |
| Py–CO–NH–C3H7 (5) | 381.0, 400.0, (419.0) [26247, 25000] | 376.0 [26596] | 350 |
| 7: Py–CO–NH–Leu–OMe | 383.0, 402.5, (420.5) [26110, 24845] | 377.0 [26525] | 420 |
| Py–CH2–CO–NH–C3H7 (9) | 375.5, 381.5, 387.0, 396.5, (416.0) [26631, 26212, 25840, 25221] | 375.0 [26667] | 0 |
| Py–CH2–CO–NH–Leu–OMe (11) | 376.0, 381.5, 387.0, 396.5, (415.5) [26596, 26212, 25840, 25221] | 376.0 [26667] | 0 |
| Py–C3H6–CO–NH–Leu–OMe (12) | 375.5, 381.5, 387.5, 396.5, 416.0 [26631, 26212, 25806, 25220, 24038] | 374.5 [26702] | 70 |
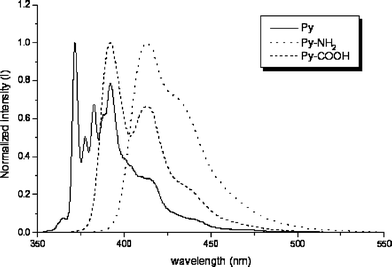 | ||
| Fig. 4 Normalized and corrected emission spectra of 1 (——), 2 (⋯⋯) and 3 () in CH2Cl2. | ||
 | ||
| Fig. 5 Normalized and corrected emission spectra of 1 (——), 4 (—■—), 5 (—●—), 6 (—□—) and 7 (—△—) in CH2Cl2. | ||
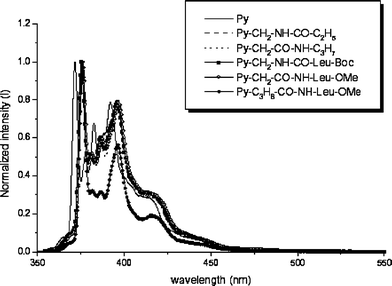 | ||
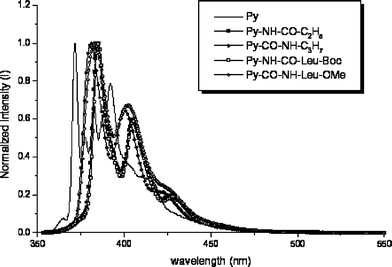
| Fig. 6 Normalized and corrected emission spectra of 1 (——), 8 (⋯⋯), 9 (–), 10 (—■—), 11 (—△—) and 12 (—●—) in CH2Cl2. | ||
For 8–12, the excited state and ground state vibrations were compared by measuring the relative intensities and wavenumbers of the vibrational bands in the respective excitation and emission spectra. The wavenumbers of the five resolved emission bands, designated I–V, and the four observable bands in the excitation spectrum, designated I′ and III′–V′ (band II′ is weak and sometimes lost in deconvolution from the S2 absorption) have been measured for all five of these compounds. The S1–S0 emission and excitation spectra are distorted mirror-images and the Stokes shift is small. The vibrational data for compound 12, of particular interest (vide infra), are compiled in Table 3.
| Peak | Emission | Excitation | ||
|---|---|---|---|---|
|
/cm−1a |
Displacement from 0–0 line/cm−1 |
/cm−1a |
Displacement from 0–0 line/cm−1 | |
| a Wavenumber (cm−1) of the vibrational band maxima with labels shown in Fig. 4; the uncertainty is about 50 cm−1 for each vibrational band. | ||||
| * | 27100 | −484 | — | — |
| I | 26616 | 0 | 26690 | 0 |
| II | 26223 | 393 | — | — |
| III | 25824 | 792 | 27373 | 653 |
| IV | 25541 | 1075 | 27789 | 1099 |
| V | 25258 | 1358 | 28259 | 1569 |
Although their absorption spectra are similar, compounds 8–12 exhibit differences in the relative intensity ratios of their vibrational bands in the emission spectra (Figs. 4–6). The emission intensity ratio of peaks I and III in the Ham bands (labelled I/III, below) has been taken as an indicator of the polarity of the surrounding medium. The value of I/III = 0.68 for pyrene in CH2Cl2, similar to that observed when pyrene is dissolved in other solvents of similar polarity.4,19 The data are summarized in Table 4. Note that the value of I/III = 0.32 for 12 is unusually small compared with pyrene and its other similar derivatives 8–11.
| Compound | λ max I/nm | λ max III/nm | I/IIIa |
|---|---|---|---|
| a Emission intensity ratio of vibrational peaks I and III in the normalized and corrected emission spectrum. | |||
| Py (1) | 371.5 | 382.5 | 0.68 |
| Py–CH2–NH–CO–C2H5 (8) | 375.0 | 386.0 | 0.61 |
| Py–CH2–CO–NH–C3H7 (9) | 375.5 | 387.0 | 0.57 |
| Py–CH2–NH–CO–Leu–Boc (10) | 375.0 | 386.0 | 0.60 |
| Py–CH2–CO–NH–Leu–OMe (11) | 376.0 | 387.0 | 0.59 |
| Py–C3H6–CO–NH–Leu–OMe (12) | 375.5 | 387.0 | 0.32 |
The emission spectra of compounds 2–7 lie within the 350–500 nm region, but exhibit fewer vibrational features. Compound 2, Fig. 4, is least structured and the location of the origin band is uncertain, making deconvolution of its vibrational structure impossible. Measured as the difference between the wavenumbers at the maximum of the emission spectrum and the maximum of the first discernible band in the excitation spectrum, 2 exhibits a large nominal Stokes shift (ca. 1000 cm−1). However this should certainly not be compared with the Stokes shifts observed in the structured spectra of pyrene itself and its derivatives 8–12. Compounds 4–7 possess very similar emission spectra, with better resolution of the vibrational structure and almost the same λmax. Stokes shifts measured in the spectra of these four compounds are smaller, in the <200 cm−1 range, similar to that observed in the spectrum of 3. The data are compiled in Table 2.
Quantum yields and excited state lifetimes
The temporal fluorescence decay profiles of pyrene and its derivatives were all measured by exciting at a wavelength of 361 nm and employing a non-linear least-squares fitting procedure to extract the exponential fluorescence lifetimes of the emitting species. Some of the pyrenyl compounds, such as 2, exhibit a fluorescence decay profile that can be well-represented by a single mono-exponential decay function. In most cases, however, a small fraction of the fluorescence was observed to decay by one or more faster processes, leading to a second or third exponential component of the decay. In such cases, the major, slower component could be attributed to the fluorescence of the major conformer of the pyrenyl fluorophore, whereas any faster components could be attributed to conformers of minor fractional populations (vide infra).The fluorescence quantum yields and lifetimes of compounds 2–12 in deoxygenated CH2Cl2 solution are collected in Table 5. Pyrene exhibits a nearly monoexponential decay in this solvent with a fluorescence lifetime (τf = 167 ns), in agreement with previous results for pyrene dissolved in solvents with similar solvatochromic properties.20 Note also that the fluorescence quantum yields of pyrene's derivatives are relatively insensitive to substitution whereas the lifetimes vary over thirty-fold. Substitution at the 1-position by a tether that starts with even a single methylene group results in only a modest change in the lifetime, whereas substitution by -NH2, -COOH or any C-amido or N-amido group results in a very substantial decrease in the S1 lifetime of the major fluorescent component of the compound. The temporal fluorescence profile of compound 7, Fig. 7, is shown as an example.
| Compound | Quantum yield, Φfa | Lifetime, τ/nsb |
|---|---|---|
| a Estimated maximum errors in quantum yields are ± 10% of the quoted value. b Excited at 361 nm and observed at 400 nm. Numbers in brackets are the values of Fi of the major component in %. Errors are ±1 ns for lifetimes > 100 ns, and ±0.2 ns for lifetimes < 100 ns. | ||
| Py (1) | 0.41 | 167 (98) |
| Py–NH2 (2) | 0.61 | 4.9 (100) |
| Py–NH–CO–C2H5 (4) | 0.35 | 10.8 (84) |
| Py–NH–CO–Leu–Boc (6) | 0.34 | 9.3 (92) |
| Py–CH2–NH–CO–C2H5 (8) | 0.47 | 133 (78) |
| Py–CH2–NH–CO–Leu–Boc (10) | 0.46 | 145 (64) |
| Py–COOH (3) | 0.75 | 5.3 (100) |
| Py–CO–NH–C3H7 (5) | 0.64 | 32.4 (97) |
| Py–CO–NH–Leu–OMe (7) | 0.70 | 20.3 (97) |
| Py–CH2–CO–NH–C3H7 (9) | 0.52 | 131 (92) |
| Py–CH2–CO–NH–Leu–OMe (11) | 0.51 | 141 (95) |
| Py–C3H6–CO–NH–Leu–OMe (12) | 0.48 | 104 (96) |
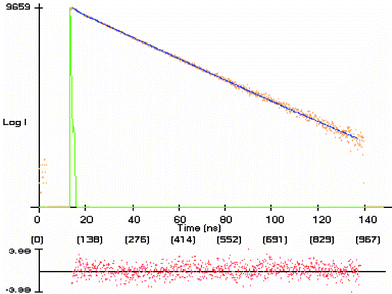 | ||
Fig. 7 Fluorescence decay curve of Py–CO–NH–Leu–OMe, 7, in degassed CH2Cl2 at room temperature: λex![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =361 nm, λem=400 nm. a1=0.86, τ1=20.3 ns, a2=0.14, τ2=2.6 ns, χ2=1.09. The bottom panel shows the distribution of weighted residuals. =361 nm, λem=400 nm. a1=0.86, τ1=20.3 ns, a2=0.14, τ2=2.6 ns, χ2=1.09. The bottom panel shows the distribution of weighted residuals. | ||
Py–CH2–NH–CO–C2H5, 8, was used to investigate further the nature of the multi-exponent decays of the compounds with flexible tethers by making fluorescence decay measurements as a function of solute concentration and emission wavelength in degassed solution. Table 6 presents the results of analyzing these fluorescence decays using trial biexponential or triexponential functions. At least two decay components were detected. The longer-lived component (τ ≈ 130 ns) is dominant when observed in the 375 < λem < 420 nm range, but progressively loses relative emission intensity as the observation wavelength shifts to the red. A second component with a shorter lifetime (τ ≈ 6 ns) exhibits a progressive increase in its emission intensity as the observation wavelength moves towards the red. When observed at 460 nm, the fluorescence decays are mainly due to the component with the shorter lifetime. Similar results were observed at both concentrations, and the distribution of the emission intensity between the two components did not change much within the (0.4–1.9) × 10−5 M concentration range, suggesting that the red-shifted one is not due to an excimer. Note, however that the values of χ2 are relatively large for several of these fits, so these data may only be of semi-quantitative value.
| Wavelengtha/nm | Conc. = 3.69 × 10−6 M | Conc. = 1.85 × 10−5 M | ||
|---|---|---|---|---|
| Decayb/ns | χ 2 | Decayb/ns | χ 2 | |
| a The observation wavelength for emission (bandwidth is 20 nm). b [Preexponential factor] fluorescence decay (fractional intensity in percent) for a two or three exponential fit. | ||||
| 375 | [0.74] 137 ± 2 (99) | 1.34 | [0.77] 136 ± 2 (98) | 1.24 |
| [0.26] 5 ± 1 (1) | [0.23] 7 ± 1 (2) | |||
| 385 | [0.48] 130 ± 2 (93) | 1.65 | [0.57] 126 ± 7 (91) | 1.25 |
| [0.52] 9 ± 1 (7) | [0.44] 16 ± 5 (9) | |||
| 395 | [0.17] 130 ± 3 (83) | 1.41 | [0.30] 132 ± 3 (89) | 1.26 |
| [0.83] 5 ± 2 (17) | [0.70] 7 ± 2 (11) | |||
| 400 | [0.14] 133 ± 3 (78) | 1.31 | [0.28] 129 ± 3 (88) | 1.35 |
| [0.86] 6 ± 2 (22) | [072] 6 ± 2 (12) | |||
| 420 | [0.05] 123 ± 5 (52) | 1.50 | [0.13] 121 ± 5 (72) | 1.24 |
| [0.95] 6 ± 3 (48) | [0.87] 6 ± 3 (28) | |||
| 440 | [0.02] 96 ± 7 (26) | 1.57 | [0.04] 106 ± 7 (46) | 1.27 |
| [0.98] 5 ± 2 (64) | [0.08] 17 ± 3 (17) | |||
| [0.88] 3 ± 1 (37) | ||||
| 460 | [0.03] 73 ± 10 (27) | 1.56 | [0.06] 64 ± 15 (54) | 1.57 |
| [0.97] 6 ± 3 (73) | [0.20] 8 ± 3 (22) | |||
| [0.74] 2 ± 1 (24) | ||||
Calculations
Semiempirical PM3 calculations were carried out for each pyrenyl derivative to search for its ground-state conformation of minimum energy and for other conformations of similar energy that could be reached by torsion around single bonds of the tether's backbone. The results are unremarkable, except to note first that a wide range of orientations of the plane of the amido >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group with respect to the plane of the pyrene ring are found in conformations of lowest energy in compounds 4–7 in which the amido group is bound directly to the pyrene ring via either the N or C atom. Second, compounds 8–12 in which the amido group is at least one methylene group removed from the ring, exhibit several different conformations of similar energy, separated by relatively low torsional barriers, as expected. Since barriers between conformational minima are often lower in the electronic excited state than the ground state, it appears that several different conformations could be significantly thermally populated in the excited states of these molecules.
O group with respect to the plane of the pyrene ring are found in conformations of lowest energy in compounds 4–7 in which the amido group is bound directly to the pyrene ring via either the N or C atom. Second, compounds 8–12 in which the amido group is at least one methylene group removed from the ring, exhibit several different conformations of similar energy, separated by relatively low torsional barriers, as expected. Since barriers between conformational minima are often lower in the electronic excited state than the ground state, it appears that several different conformations could be significantly thermally populated in the excited states of these molecules.
Quenching by oxygen
Molecular oxygen is an effective quencher of pyrene in its S1 electronically excited state, and the same is observed for its longer-lived 1-alkylpyrene derivatives, 8–12. On the other hand, aeration produces little change in the quantum yield or excited state lifetime of those compounds with shorter-lived excited states, 2–7, suggesting that the quenching process could be diffusion limited. Careful examination shows that the fluorescent quantum yields and excited state lifetimes decrease proportionately on aeration of the solutions, suggesting that it may be possible to extract the rate constants for the quenching process from these data.Discussion
With the S1 excited state lifetimes, τ, and S1–S0 fluorescence quantum yields, ϕf, in hand, it is possible to calculate both the radiative rate constant, kr, and the sum of the rate constants of all parallel non-radiative processes, Σknr, by which the S1 state decays. The τ and ϕf data for deoxygenated solutions were used to determine the decay parameters for these excited molecules in the absence of intermolecular quenching. For species that exhibited multi-exponential temporal fluorescence decay profiles, the quantum yield of fluorescence attributable to each species was obtained by multiplying the measured value of ϕf by the appropriate fractional contribution, Fi, of the species to the total emission, all measured at the same emission observation wavelength. The values of the decay constants are then obtained for each species, i, using kr,i = ϕfFi/τi and Σknr,i = (1 − ϕfFi)/τi. In each case, the long-lived major component of a bi- or tri-exponential decay in dilute solution was assigned to the most stable excited state conformer of the monomer. The decay parameters for these species are given in Table 7.| Compound | Σknr/s−1a | k r/s−1b |
|---|---|---|
| a Sum of the rate constants of non-radiative decay of the S1 state (major component). b Radiative rate constant of the S1 state (major component). | ||
| Py (1) | 3.6 × 106 | 2.4 × 106 |
| Py–NH2 (2) | 8.0 × 107 | 1.2 × 108 |
| Py–NH–CO–C2H5 (4) | 6.5 × 107 | 2.7 × 107 |
| Py–NH–CO–Leu–Boc (6) | 7.4 × 107 | 3.4 × 107 |
| Py–CH2–NH–CO–C2H5 (8) | 4.8 × 107 | 2.8 × 106 |
| Py–CH2–NH–CO–Leu–Boc (10) | 4.9 × 106 | 2.0 × 106 |
| Py–COOH (3) | 4.7 × 107 | 1.4 × 108 |
| Py–CO–NH–C3H7 (5) | 1.2 × 107 | 1.9 × 107 |
| Py–CO–NH–Leu–OMe (7) | 1.6 × 107 | 3.3 × 107 |
| Py–CH2–CO–NH–C3H7 (9) | 4.0 × 106 | 3.7 × 106 |
| Py–CH2–CO–NH–Leu–OMe (11) | 3.7 × 106 | 3.4 × 106 |
| Py–C3H6–CO–NH–Leu–OMe (12) | 5.2 × 106 | 4.4 × 106 |
The most remarkable characteristic of the data for this group of pyrenyl compounds is that the quantum yields are all large and lie within a small range (0.3–0.7), despite the fact that the excited state lifetimes vary by a factor of more than 30. As shown by the calculations presented in Table 7, this is due to the fact that the S1 radiative and non-radiative decay rates vary in an approximately proportionate fashion; the ratio Σknr/kr is roughly constant and equal to 1.3 ± 0.6 for the twelve compounds under study.
The radiative decay constants, kr, exhibit the expected direct proportionality to the values of εmax, as shown in Fig. 8. The good linear relationship observed provides direct evidence that both the experimental quantum yield and lifetime data from which the values of kr are derived and the spectral deconvolution procedure used to extract the values of εmax are reasonably accurate. Moreover, this correlation and the relative invariance of the S2–S1 electronic energy spacings (Table 1) suggest that the substantial change in radiative rate (oscillator strength) in the C- or N-coupled derivatives is not due to an inversion of states or the appearance of an altogether different state.
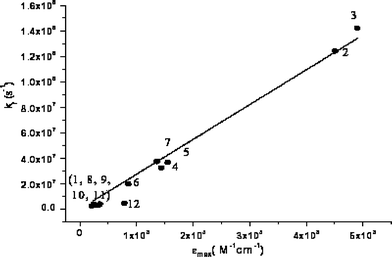 | ||
| Fig. 8 Radiative rate constant of the S1–S0 transition as a function of the molar absorptivity, εmax, in degassed CH2Cl2. The equation of the straight line is kf=2.73 × 104εmax, R2=0.98. 1, Py; 2, Py–NH2; 3, Py–COOH; 4, Py–NH–CO–C2H5; 5, Py–CO–NH–C3H7; 6, Py–NH–CO–Leu–Boc; 7, Py–CO–NH–Leu–OMe; 8, Py–CH2–NH–CO–C2H5; 9, Py–CH2–CO–NH–C3H7; 10, Py–CH2–NH–CO–Leu–Boc; 11, Py–CH2–CO–NH–Leu–OMe and 12, Py–C3H6–CO–NH–Leu–OMe. | ||
One compound, 12, appears to be an outlier in Fig. 8; its measured value of kr is similar to those of pyrene and the other methylene-tethered derivatives, but its value of εmax appears to be about a factor of 3 or 4 too large. This compound is the only one of the 12 compounds investigated that has a –(CH2)3– tethering chain that is long enough to permit the polar free end of the substituent to fold back upon and interact with the pyrene ring. (Note that 9 is of similar length but does not have a polar head group.) Moreover, the S1 lifetime of 12 in deoxygenated solution is long enough to permit such a torsional folding process to occur prior to emission. The I/III vibronic band intensity ratio of compound 12 (cf.Table 4) suggests that its pyrenyl chromophore experiences a more polar microenvironment that those in compounds of similar structure (8–11) tethered by a single methylene group. Although these observations provide empirical evidence in favour of a folded major conformer in the excited state, further work is required to clarify this point.
A clue to the nature of the mechanism by which the S1–S0 transition gains oscillator strength may be obtained by considering the spectroscopic characteristics of the substituted pyrenyl compounds listed in Table 1, as compared with pyrene itself. Note that all derivatives are substituted at the 1-position, so their effective electronic state symmetries are the same. Those compounds that acquire substantially greater oscillator strength by substitution of an amido, amine or carboxyl group directly on the pyrene ring also experience a concomitant substantial decrease in the oscillator strengths of the accompanying S2–S0 and S3–S0 transitions. This is not seen in those derivatives in which the tether is linked via a methylene group, and for which the effect of substitution on the photophysics is rather small. Note also that the S2–S1 electronic energy spacing varies somewhat from compound to compound, but not in a fashion that is correlated with the S1 radiative decay constants. These observations suggest that the S1–S0 transition borrows intensity by a Herzberg–Teller mechanism that involves vibronic mixing of the S1 state with S2 and S3, nearby states that are strongly radiatively coupled to the ground state.
State mixing must also be responsible for the proportionate change in the rate constant(s) of S1's parallel radiationless decay processes. We have uncovered no evidence of hidden electronic states. We therefore assume that the only possible intramolecular radiationless processes by which S1 could decay are intersystem crossing (ISC) to the triplet manifold and internal conversion (IC) to the ground state, i.e. Σknr = kIC + kISC. The quantum yields of triplet states resulting from S1–T1 intersystem crossing were not measured. Nevertheless, it is expected that these yields will be substantial based on the known behaviour of pyrene itself and the fact that the energies of states low in the triplet manifold appear to be similar in all 12 compounds (cf. the oxygen quenching results, following). If the derivatives are similar to pyrene itself, the sum of the quantum yield of fluorescence and the quantum yield of T1 formation is expected to be unity. In the absence of contrary information we therefore assume that the only significant competing radiationless decay process depopulating the S1 states of these pyrenyl derivatives is intersystem crossing to the triplet manifold.
The quantum yield and lifetime data taken on air-saturated solutions can be used in conjunction with the data on de-oxygenated solutions to extract the second order rate constants, kQ, for the bimolecular quenching of the S1 states by ground state molecular oxygen. The intermolecular oxygen quenching process exhibits pseudo-first-order kinetics so that the difference between kobs = kr + kIC + kISC +kQ[O2] = 1/τ obtained from measurements on air-saturated solutions, and kobs = kr + kIC + kISC from measurements of the same compound in deoxygenated solutions will be equal to kQ[O2]. Assuming Henry's law applies, the equilibrium concentration of oxygen in CH2Cl2 solutions subject to 1 bar air pressure at 20 °C is calculated to be 2.14 × 10−3 M (where Henry's law constant is calculated using standard methods21). The values of kQ obtained are given in Table 8. Only data for those compounds exhibiting longer-lived fluorescence and significant oxygen quenching are listed; the shorter-lived S1 states are only slightly quenched under these conditions making it impossible to extract accurate data from the small differences in the fluorescence decay constants.
| Compound | Undegassed | Degassed | k q[O2]/s−1 | k q/M−1 s−1 |
|---|---|---|---|---|
| Σknf/s−1 | k nf/s−1 | |||
| Py (1) | 3.1 × 107 | 3.6 × 106 | 2.7 × 107 | 1.3 × 1010 |
| Py–CH2–NH–CO–C2H5 (8) | 2.4 × 107 | 3.7 × 106 | 2. × 107 | 9.6 × 109 |
| Py–CH2–CO–NH–C3H7 (9) | 2.6 × 107 | 3.7 × 106 | 2.3 × 107 | 1.1 × 1010 |
| Py–CH2–NH–CO–Leu–Boc (10) | 2.6 × 107 | 3.7 × 106 | 2. × 107 | 1.0 × 1010 |
| Py–CH2–CO–NH–Leu–OMe (11) | 2.6 × 107 | 3.5 × 106 | 2.2 × 107 | 1.0 × 1010 |
| Py–C3H6–CO–NH–Leu–OMe (12) | 2.9 × 107 | 5.0 × 106 | 2.4 × 107 | 1.1 × 1010 |
These accurate values of kQ are all near 1 × 1010 M−1s−1, and although the data from the compounds with shorter-lived excited states are not as accurate, the approximate values of kQ obtained for them are also of the same order of magnitude. These data indicate that the quenching of the S1 states of all 11 pyrenyl derivatives by oxygen is diffusion controlled, with a spin-statistical factor of 1, just as it is for pyrene itself. Consequently, the same spin-conservative oxygen quenching mechanism should hold, i.e.,
| Py(S1) + O2(3Σ−g) → Py(T1) + O2(1Δg), |
The minor, shorter-lived contributor(s) to the emission intensity remain unidentified. Nevertheless, the small amount of information available from the measurement of the decay parameters of compound 8 as a function of emission wavelength suggests that this short-lived emission is due to an excited state conformer of significantly different structure than the corresponding ground state. Measuring Fi for this emitter as a function of emission wavelength allows one to construct its crude emission spectrum, as shown in Fig. 9. Note that this reconstructed spectrum is substantially red-shifted compared with that of the major emitter, consistent with an excited state structure that is rather different from that of the most stable ground state conformation. Note also that this spectrum has vibronic structure and this structure exhibits about the same spacing (ca. 1350 cm−1) as the major peaks of the emission spectrum of the major emitter. The latter observation and the fact that the spectrum does not change significantly at somewhat higher concentrations suggest that the short-lived component is not a pyrenyl excimer, whose fluorescence emission spectrum would be expected to be structureless and to increase in relative intensity at higher solute concentration.
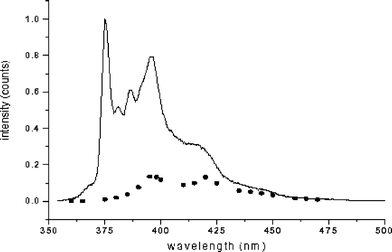 | ||
| Fig. 9 Simulated emission spectra of the unstable conformations of Py–CH2–NH–CO–C2H5, 8, in degassed CH2Cl2. (——) the emission spectra of the principal conformation; (⋯⋯) the simulated emission spectra of the unstable conformations. | ||
Finally we note that the data presented in this paper suggest that care should be taken in interpreting fluorescence lifetime data when pyrenyl chromophores tethered to a polypeptide backbone are photoexcited and used as acceptors in electron transfer experiments. A number of different tethers of different length and chemical constitution have been used in previous reports. A tetramethylene tether22 will minimize ground state perturbation of the pyrene by the backbone, but will permit the chromophore to fold onto the polypeptide chain, with effects that are seen in compound 12. A monomethylene tether, such as that used by Fox and Galoppini,10 may serve as the ideal compromise between minimal chromophore perturbation and fixed proximity to the backbone. A pyrenyl ketone such as that used by Jones and Vullev23 will, however, exhibit photophysical behaviour much different from that of pyrene and its alkylated derivatives.
Conclusions
The effects of the chemical constitution and structure of the substituent on the excited state dynamics of several model fluorescent pyrene-labelled molecules of biophysical interest have been examined. Nine new 1-substituted compounds, Py–NH–CO–C2H5, Py–NH–CO–Leu–Boc, Py–CH2–NH–CO–C2H5, Py–CH2–NH–CO–Leu–Boc, Py–CO–NH–C3H7, Py–CO–NH–Leu–OMe, Py–CH2–CO–NH–C3H7, Py–CH2–CO–NH–Leu–OMe and Py–C3H6–CO–NH–Leu–OMe, have been synthesized and their electronic spectra, fluorescence quantum yields and excited state lifetimes measured. These data have been used to calculate the radiative and non-radiative decay constants of the S1 states and correlate the results with the structures of the tethers.The observed increase in the oscillator strength of the S1–S0 transitions in the derivatives at the expense of the intensities of nearby strongly allowed transitions is qualitatively consistent with a Herzberg–Teller model of excited state vibronic mixing. A linear relationship is observed between the values of εmax for the S1–S0 transition and the measured rate constants for S1 radiative decay. Non-radiative S1 decay rates (mainly intersystem crossing to T1) vary in parallel with the radiative rates so that the excited state lifetimes change considerably with substituent whereas the quantum yields of fluorescence do not. The correlation between εmax and either kr or the excited state lifetime is excellent as long as no additional intermolecular or intramolecular excited state decay process of significant rate competes with the ‘normal’ radiative and non-radiative (ISC) decay processes of the pyrene chromophore. With this information in hand, it should be possible to estimate the photophysical properties of similar pyrenyl-tethered biomolecules under conditions (that may be impossible to achieve experimentally) where excited state intramolecular or intermolecular quenching does not take place. That is, simply by measuring the compound's UV-visible absorption spectrum and obtaining its εmax for the S1–S0 transition it should be possible to estimate the lifetime (or quantum yield) of the derivative closely for the (perhaps hypothetical) situation in which no excited state intermolecular or intramolecular ET or EET is taking place. The difference between the measured, shorter lifetime of a pyrenyl fluorophore tethered to a biomolecule and the unperturbed lifetime calculated from the absorption spectrum may then be attributed to a competing excited state decay process whose rate can also be estimated. The results presented here suggest that care should be taken in interpreting fluorescence lifetime data when pyrenyl chromophores tethered to a polypeptide backbone are photoexcited and used as acceptors in electron transfer experiments.
Rates of bimolecular quenching of the S1 states of the derivatives by molecular oxygen have been measured. The quenching process is diffusion-controlled with a spin statistical factor of 1, indicating that the S1–T1 electronic energy spacings of all the derivatives exceed the O2 (1Δg–3Σ−g) electronic excitation energy of 7882 cm−1.
Acknowledgements
The authors are pleased to acknowledge the support of this research by the Natural Sciences and Engineering Research Council of Canada. CY is grateful for the award of a University of Saskatchewan graduate scholarship.References
- G. Erhard; J. Meienhofer, The Peptides: Analysis, Synthesis, Biology, Academic Press, New York, 1979, p. 66 Search PubMed.
- A. Bree and V. V. B. Vilkos, Singlet and triplet electronic states of pyrene, Spectrochim. Acta, A, 1971, 27(11), 2333–2354 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, New York, London, 1970 Search PubMed.
- K. Kalyanasundaram and J. K. Thomas, Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems, J. Am. Chem. Soc., 1977, 99(7), 2039–2044 CrossRef CAS.
- T. Saika, T. Iyoda, K. Honda and T. Shimidzu, Emission control of a pyrene-thioindigo compound, J. Chem. Soc., Chem. Commun., 1992, 591–592 RSC.
- T. Sakurai, K. Watanabe, K. Nojima and H. Inoue, The role of papain in the association process of a 1-pyrenoyl pendant attached to its active site, J. Chem. Soc., Chem. Commun., 1995, 1729–1730 RSC.
- M. Sisido, S. Hoshino, H. Kusano, M. Kuragaki, M. Makino, H. Sasaki, T. A. Smith and K. P. Ghiggino, Distance dependence of photoinduced electron transfer along a-helical polypeptides, J. Phys. Chem. B, 2001, 105(42), 10407–10415 CrossRef CAS.
- S. S. Isied, M. Y. Ogawa and J. F. Wishart, Peptide-mediated intramolecular electron transfer: long-range distance dependence, Chem. Rev., 1992, 92(3), 381–394 CrossRef CAS.
- P. Piotrowiak, Photoinduced electron transfer in molecular systems: recent developments, Chem. Soc. Rev., 1999, 28(2), 143–150 RSC.
- M. A. Fox and E. Galoppini, Electric field effects on electron transfer rates in dichromophoric peptides: the effect of helix unfolding, J. Am. Chem. Soc., 1997, 119(23), 5277–5285 CrossRef.
- M. Manoharan, K. L. Tivel, M. Zhao, K. Nafisi and T. L. Netzel, Base-sequence dependence of emission lifetimes for DNA oligomers and duplexes covalently labeled with pyrene: relative electron-transfer quenching efficiencies of A, G, C, T nucleosides toward pyrene, J. Phys. Chem., 1995, 99(48), 17461–17472 CrossRef CAS.
- S. Aich and H.-B. Kraatz, Quenching studies of pyrenoyl glutanmic acid derivatives, J. Chem. Soc., Perkin Trans. 2, 1999, 2, 301–308 RSC.
- T. Hayashita, S. Taniguchi, Y. Tanamura, T. Uchida, S. Nishizawa, N. Teramae, Y. S. Jin, J. C. Lee and R. A. Bartsch, A dibenzo-16-crown-5 fluoroionophore for selective emission ratio sensing of Na+ in basic aqueous dioxane solution, J. Chem. Soc., Perkin Trans. 2, 2000, 5, 1003–1006 RSC.
- G. A. Crosby and J. N. Demas, Measurement of photoluminescence quantum yields. Review, J. Phys. Chem., 1971, 75(8), 991–1024 CrossRef.
- D. V. O' Connor and D. Phillips, Time-correlated Single Photon Counting; Academic Press, London, Orlando, 1984 Search PubMed.
- D. W. Marquardt, An algorithm for least-squares estimation of nonlinear parameters, J. Soc. Ind. Appl. Math., 1963, 11, 431–441 Search PubMed.
- M. Ameloot and H. Hendrickx, Extension of the performance of Laplace deconvoluntion in the analysis of fluorescence decay curves, Biophys. J., 1983, 44(1), 27–38 CrossRef CAS.
- C. Armbruster, M. Knapp, K. Rechthaler, R. Schamschule, A. B. J. Parusel, G. Köhler and W. Wehrmann, Fluorescence properties of 1-heptanoylpyrene: a probe for hydrogen bonding in microaggregates and biological membranes, J. Photochem. Photobiol., A: Chem., 1999, 125(1–3), 29–38 CrossRef CAS.
- D. S. Karpovich and G. J. Blanchard, Relating the polarity-dependent fluorescence response of pyrene to vibronic coupling. Achieving a fundamental understanding of the py polarity scale, J. Phy. Chem., 1995, 99(12), 3951–3958 Search PubMed.
- F. W. Langkilde, E. W. Thulstrup and J. Michl, The effect of solvent environment on molecular electronic transition moment directions: symmetry lowering in pyrene, J. Chem. Phys., 1983, 78(6), 3372–3381 CrossRef CAS.
- R. Battino, Solubility Data Series: Oxygen and Ozone, Pergamon, Oxford, New York, 1981 Search PubMed.
- G. II. Jones, X. Zhou and V. I. Vullev, Photoinduced electron transfer in α-helical polypeptides: dependence on conformation and electron donor–acceptor distance, Photochem. Photobiol. Sci., 2003, 2(11), 1080–1087 RSC.
- G. II. Jones and V. I. Vullev, Photoinduced electron transfer between non-native donor-acceptor moieties incorporated in synthetic polypeptide aggregates, Org. Lett., 2002, 4(23), 4001–4004 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of compounds 4–12. See http://www.rsc.org/suppdata/pp/b4/b414577c/ |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |