The stereoselective synthesis of aziridine analogues of diaminopimelic acid (DAP) and their interaction with dap epimerase†
Christopher M.
Diaper
a,
Andrew
Sutherland
b,
Bindu
Pillai
c,
Michael N. G.
James
c,
Paul
Semchuk
d,
John S.
Blanchard
e and
John C.
Vederas
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2. E-mail: john.vederas@ualberta.ca; Fax: (780) 4922134; Tel: (780) 4924575
bWestChem, Department of Chemistry, University of Glasgow, Glasgow, United Kingdom G12 8QQ
cDepartment of Biochemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2H6
dInstitute for Biomolecular Design, University of Alberta, Edmonton, Alberta, Canada T6G 2H7
eDepartment of Biochemistry, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York, 10461
First published on 15th November 2005
Abstract
Aziridine analogues of diaminopimelic acid (DAP) have been prepared stereoselectively for the first time and evaluated as inhibitors of DAP epimerase. (2R,3S,3′S)-3-(3′-Aminopropane)aziridine-2,3′-dicarboxylate 4 was synthesised and shown to be a reversible inhibitor of DAP epimerase with an IC50 value of 2.88 mM. (2S,4S)- and (2S,4R)-2-(4-Amino-4-carboxybutyl)aziridine-2-carboxylic acid (LL-azi-DAP 14 and DL-azi-DAP 29) were made as pure diastereomers, and both were shown to be irreversible inhibitors of DAP epimerase. LL-Azi-DAP 14 selectively binds to Cys-73 of the enzyme active site whereas DL-azi-DAP 29 binds to Cys-217 via attack of sulfhydryl on the methylene of the inhibitor aziridine ring. These observations are consistent with the two base mechanism proposed for the epimerisation of LL-DAP 1 and meso-DAP 2 by DAP epimerase.
Introduction
The worldwide increase in bacterial resistance has led to renewed interest in the development of new antibiotics,1,2 including methods of inhibiting microbial cell wall biosynthesis.3 As meso-diaminopimelic acid (meso-DAP 2) is a precursor to L-lysine and an important component of the peptidoglycan layer of Gram-negative bacteria,4 it has attracted considerable interest for the possible development of new antibiotics.5 Crucially, the biosynthesis of meso-DAP is restricted to bacteria5 and plants and is absent in mammals, which require L-lysine in their diet.6 Therefore specific inhibitors of DAP enzymes are likely to be antimicrobial agents with low mammalian toxicity.One of the key enzymes in the DAP pathway is DAP epimerase, an unusual enzyme that interconverts LL-DAP 1 and meso-DAP 2 without the use of co-factors, metals, or reducible keto or imino functionality (Scheme 1).7
 | ||
| Scheme 1 The biosynthetic pathways to meso-DAP and proposed transition state for the inter-conversion of LL-DAP 1 and meso-DAP 2 by DAP epimerase. | ||
This process is mechanistically analogous to the other PLP-independent amino acid racemases.8,9 It is proposed to involve two active site cysteine residues, one of which as a thiolate, acts as a general base while the other, as a thiol, acts as a general acid.10 Initial tritium labeling studies showed that the α-proton of DAP is exchanged during epimerisation.7 Further investigations using 3-fluoro-DAP isomers demonstrated that the mechanism involves development of anionic character at the α-carbon and that the substrate is rigidly held in the enzyme active site.11 In 1990, the irreversible inactivation of DAP epimerase was reported12 using a crude mixture of all possible diastereomers of 2-(4-amino-4-carboxybutyl)aziridine-2-carboxylic acid (azi-DAP) 3. Enzymatic digestion studies determined that Cys-73 was alkylated by the aziridino moiety, thereby demonstrating its presence in the active site (Scheme 2).13 Recently a diastereomeric mixture of oxa analogues of azi-DAP was found to irreversibly inhibit DAP epimerase, presumably due to thiol opening of the epoxide moiety.14
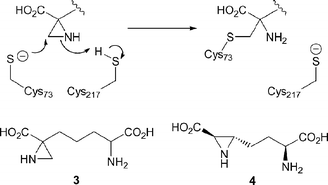 | ||
| Scheme 2 Proposed mechanism for the irreversible inhibition of DAP epimerase by azi-DAP 3. | ||
The crystal structure of inactive DAP epimerase from Haemophilus influenzae has been solved15 and subsequently refined16 to reveal a disulfide linkage between Cys-73 and Cys-217 at the interface of two structurally superimposable domains. Based on this observation it was proposed that in the active reduced form of the enzyme, these conserved residues are the key catalytic units required for activity. This led to detailed kinetic studies of DAP epimerase.10 Assignment of the specific roles of Cys-217 and Cys-73 in the active site could be confirmed by the interaction of pure 3-fluoro-DAP isomers with the single DAP epimerase mutants C73A, C73S, C217A and C217S.17
The substrates LL-DAP 1 and meso-DAP 2 for DAP epimerase differ from the other PLP-independent amino acid racemases8,18 (e.g. glutamate racemase,19–21 aspartate racemase,22 proline racemase23,24) in that they contain two stereocentres. DAP epimerase has very strict requirements for its substrates,5 and only accepts DAP isomers with the L configuration at the distal site.7,25 Neither D nor L isomers of lysine or α-aminopimelic acid are substrates or good inhibitors, thereby demonstrating that both carboxyl and amino groups must be present.26
Unfortunately, the crystal structures of the inactive disulfide form of DAP epimerase15,16 do not allow reliable modeling of the substrate in the collapsed active site. Interestingly, no crystal structures of other PLP-independent amino acid racemases have been obtained thus far with a substrate analog correctly bound in the active site to illustrate catalytic function. It therefore appeared that formation of a complex of DAP epimerase with pure diastereomers of the aziridine analogues 3 and 4 could provide further insight into the organisation of the enzyme active site. In particular, crystal structures of the DAP epimerase with stereochemically pure isomers of azi-DAP 14 and 29 could afford a detailed picture of the active site complex for each substrate isomer and the arrangement of residues for catalysis of this unusual reaction. In this report we describe the first stereoselective synthesis of highly reactive aziridine analogues 4, 14 and 29 and analysis of their interaction with DAP epimerase.
Results
Stereoselective synthesis of DAP aziridine analogue 4
It seemed that aziridine 4 could be prepared by aziridination of a suitable α,β-unsaturated ester or amide bearing a chiral auxiliary.27 In 1993 we reported the aziridination of N-enoylbornane[10,2]sultams by the oxidative addition of N-aminophthalimide mediated by lead tetraacetate with excellent diastereoselectivity.28 This approach was subsequently found to be compatible with other auxiliaries such as oxoalkenyloxazolidinone29 and camphor.30 It appeared that a similar approach using the 3-amino-2-ethyl-3,4-dihydroquinazolin-4-one 1231 would lead to the stereoselective synthesis of the internal N-quinazolinyl aziridine 7. The desired aziridine 4 could then be accessed by N–N bond reduction using dissolved metal in ammonia (Scheme 3).32 | ||
Scheme 3 The synthesis of internal aziridine DAP analogues. Reagents and conditions: (i) XcCOCH2PO(OEt)2, DBU, LiCl, MeCN (85%); (ii) TFA, CH2Cl2 (94%); (iii) 12, Pb(OAc)4, HMDS, CH2Cl2 (72%); (iv) TFA, CH2Cl2 (99%); (v) LiOH, MeOH–H2O (85%); (vi) Li, NH3 (48%); (vii) MeO2CCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3, THF (88%); (viii) TFA, CH2Cl2 (88%); (ix) 12, Pb(OAc)4, HMDS, CH2Cl2 (83%); (x) TFA, CH2Cl2 (quant.); (xi) LiOH, MeOH–H2O, (84%); (xii) Li, NH3 (48%). PPh3, THF (88%); (viii) TFA, CH2Cl2 (88%); (ix) 12, Pb(OAc)4, HMDS, CH2Cl2 (83%); (x) TFA, CH2Cl2 (quant.); (xi) LiOH, MeOH–H2O, (84%); (xii) Li, NH3 (48%). | ||
Reaction of phosphonate ester XcCOCH2PO(OEt)233 with N,N-di-Boc glutamate semialdehyde 534 using a Horner–Wadsworth–Emmons type coupling35 gives exclusively the trans-alkene in 85% yield. One of the Boc protecting groups could then be removed by TFA to generate the aziridine precursor 6 in 94% yield. Aziridination of 6 with quinazolin-4-one 12 proceeds with optimum selectivity at a temperature of −40 °C to give a diastereomeric ratio of 9 : 1. The major isomer 7 can be isolated by recrystallisation in 72% yield. Efforts to prove the absolute stereochemistry of aziridine 7 by X-ray crystallography were unsuccessful. However, it has been demonstrated that addition to N-enoylbornane[10,2]sultams occurs by syn attack from the re face of the α-carbon.28 Moreover, syn attack would give an aziridine with trans geometry which is consistent with the observed 1H NMR coupling constant of 4.8 Hz for the 2′-H proton.36 Removal of the Boc group followed by base hydrolysis and reductive cleavage of the N–N bond with Li–NH3 affords the target molecule 4 in 40% yield over three steps (Scheme 3).
In addition to testing the single diastereomer 4 with DAP epimerase, it was desirable to examine interaction of the enzyme with another diastereomer to provide insight into stereochemical preferences for the active site. Hence, a mixture of diastereomers 4 and 11 was prepared from N,N-di-Boc glutamate semialdehyde 5via the trans-alkene 837 in an analogous manner (Scheme 3).
Inhibition study of aziridines 4 and 11 with DAP epimerase
Although several assays for DAP epimerase have been developed,38 the activity of DAP epimerase is easily monitored using a coupled enzyme assay. At pH 7.8, meso-DAP 2 is generated from LL-DAP 1 by DAP epimerase and is then transformed by DAP dehydrogenase to produce L-THDP and NADPH (Scheme 1), which can be measured spectrophotometrically at 340 nm.7,39 The enantiopure LL-DAP 1 is prepared by the photolysis of a suitably functionalised diacyl peroxide,40 which is then subjected to hydrolysis to remove the protecting groups.Aziridine 4 was initially tested as a substrate and as an inhibitor of DAP dehydrogenase, the enzyme used for analysis. As expected, this compound is neither a substrate nor an inhibitor of the dehydrogenase at a concentration of 1 mM. Disappointingly, aziridine 4 is also a very weak inhibitor of DAP epimerase with an IC50 value of only 2.88 mM. A time dependence study showed 4 to be a fully reversible inhibitor. These results show that unlike azi-DAP 3, which rapidly inactivates DAP epimerase, presumably by cysteine attack at the methylene position of the aziridine ring, both secondary carbons at the aziridine ring positions of 4 are more stable towards the enzyme, resulting in poor reversible binding. The mixture of trans-stereoisomers 4 and 11 was also tested against DAP epimerase. Inhibition by this mixture is similar to the results obtained for the pure diastereomer, suggesting analogous modes of ineffective binding for both compounds.
Stereoselective synthesis of azi-DAP
Although a number of synthetic methods have been developed for the asymmetric synthesis of disubstituted aziridines,41,42 examples of their application in the synthesis of the 1-carboxy-1-alkyl aziridine functionality required for the synthesis of azi-DAP are limited.43–47 A number of synthetic strategies were explored in the stereospecific synthesis of azi-DAP. Wenker ring closure of (2S,6S)-2,6-diamino-6-(hydroxymethyl)pimelic acid 13,48,49 was unsuccessful (Scheme 4). Attempts to utilise the chiral auxiliary methodology used in the construction of the internal aziridine 4 were frustrated by problems encountered in the synthesis of the required N-enoylbornane[10,2]sultam precursor. This led us to investigate the use of chiral 3-acetoxyaminoquinazolinones,31 which exert reagent controlled diastereoselectivity in the aziridination of prochiral alkenes such as 16 (Scheme 4). Boc protected unsaturated α-aminopimelic acids have been reported,37,50 and it was proposed that aziridination of the Cbz protected analogue with chiral aminoquinazolinones 19–21 would allow the rapid preparation of a single diastereomer of the fully protected azi-DAP derivative 17.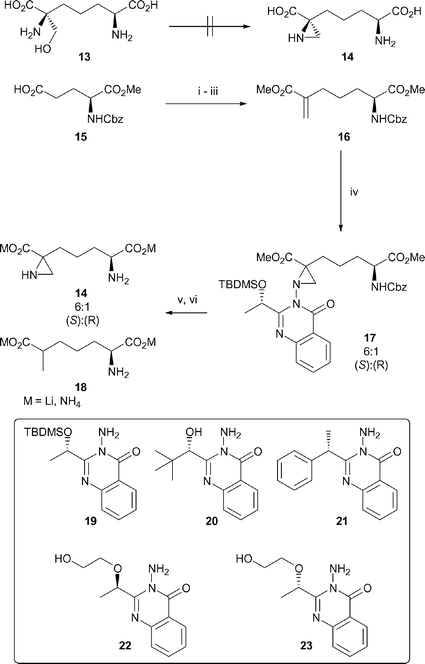 | ||
| Scheme 4 The synthesis of azi-DAP. Reagents and conditions: (i) i-BuOCOCl, NMM, THF; (ii) NEt3, 2-mercaptopyridine N-oxide, THF; (iii) methyl α-thiophenol methylacrylate, hυ (tungsten lamp), THF (68% over 3 steps); (iv) 19, Pb(OAc)4, HMDS, CH2Cl2 (49%); (v) LiOH, MeOH–H2O (quant.); (vi) Li, NH3. | ||
The required unsaturated α-aminopimelic ester 16 was constructed in one-pot by photolysis of the hydroxamate ester of N-Cbz-L-glutamic acid α-methyl ester 15 with methyl α-thiophenol methylacrylate using a tungsten lamp at room temperature to give a 68% yield over three steps (Scheme 4).50
The 3-acetoxy derivatives of the known aminoquinazolin-4(3H)-ones 19,4720,51 and the previously unreported 21 were investigated as aziridinating agents. No reaction is observed during titanium mediated aziridination31 using the hydroxy aminoquinazolin-4(3H)-one 20. Aziridination using 21 and TFA31 gives low conversion (22%) and poor selectivity (3 : 1 dr), whereas the reaction of 19 in the presence of HMDS31 proceeds with moderate yield and selectivity (49%, dr 6 : 1). The mixture of diastereomers 17 can be saponified with base and then reduced using lithium in ammonia at −78 °C, to produce azi-DAP 14 as a 6 : 1 mixture of diastereomers at the quaternary carbon along with the unexpected fully saturated ε-methyl-α-aminopimelic acid 18 (1 : 1 dr at the carbon bearing methyl).52 The structure of 18 was confirmed by independent synthesis (see ESI†). Separation of 14 and 18 is possible by careful preparative TLC under basic conditions (azi-DAP undergoes very rapid decomposition on exposure to acid). To our chagrin, extensive attempts to separate the minor azi-DAP diastereomer from 14, or to purify their corresponding diacid salts or the fully protected intermediates 17 using flash chromatography, HPLC or recrystallisation all failed.
To improve the selectivity of the aziridination and thus obviate the problematic isomer separation, we adopted an intramolecular variant of the aminoquinazolin-4(3H)-one methodology that had been reported to give complete diastereoselectivity during the aziridination of O-cinnamoyl esters.53 This necessitated the modification of our synthetic strategy to couple (R)-aminoquinazolin-4(3H)-one 22 to the 1,2-disubstituted α,β-unsaturated acid 24 (Scheme 5).
 | ||
| Scheme 5 The stereoselective synthesis of LL- and DL-azi-DAP. Reagents and conditions: (i) LiOH, MeOH–H2O (82%); (ii) PS-PTSA, MeOH (95%); (iii) 22, DCC, DMAP, CH2Cl2 (62%); (iv) 23, PS-DCC, DMAP, CH2Cl2 (51%); (v) Pb(OAc)4, HMDS, CH2Cl2 (26 55%, 28 87%); (vi) Na2CO3, MeOH–H2O (from 28 69%); (vii) Li, NH3 (14 30% over 2 steps, 29 23%). | ||
The acid 24 could be synthesised in two steps from the diester 16 by basic hydrolysis and regioselective esterification of the diacid using catalytic p-toluene sulfonic acid (PTSA) in anhydrous methanol.54 Although the selective esterification proceeds in >95% yield (1H NMR), the isolated yields after purification by flash chromatography are significantly lower (30–60%). Use of polymer bound PTSA55 circumvents this problem and allows the quantitative recovery of the product 24. The coupling of acid 24 to (R)-aminoquinazolin-4(3H)-one 22 proceeds in satisfactory 62% yield using DCC–DMAP. However, several cycles of flash chromatography are required to achieve the complete removal of the unwanted urea by-product. Switching to resin bound N-benzyl-N′-cyclohexylcarbodiimide reagent (PS-DCC)56 permits facile purification without significant loss of yield, as demonstrated by the coupling of acid 24 to the (S)-aminoquinazolin-4(3H)-one 23 to give 27 in 51% after a single pass through a plug of silica.
Intramolecular aziridination of 27 progresses rapidly at 0 °C to give the aziridine 28 with complete diastereoselectivity (>99 : 1 dr, see ESI†). The stereochemical outcome of this reaction can be determined by comparison of the observed NOE interactions with energy minimised models of the two possible reaction products (see ESI†). Preparation of the aziridine 26 under analogous conditions is slightly less selective (98 : 2 dr). Fortunately, the minor diastereomer could be removed by careful purification using flash column chromatography to give a >99 : 1 dr.
Hydrolysis of the fully protected azi-DAP 28 using lithium hydroxide or sodium hydroxide results in decomposition. However, sodium carbonate57 in methanol–water is sufficiently mild to permit quantitative conversion to the corresponding salt of the diacid (1H NMR). Final deprotection using lithium or sodium in ammonia at −78 °C results in the formation of both the desired azi-DAP isomer 29 as the major product and the fully saturated ε-methyl-α-aminopimelic acid 18 in a 5.6 : 1 ratio (1H NMR). The undesired 18 is again formed as a 1 : 1 mixture of diastereomers at the carbon bearing the methyl. Reductions with sub-stoichiometric quantities of lithium on carefully purified (RP-HPLC) carboxylate salts of the diacid intermediate demonstrate that the fully saturated ε-methyl-α-aminopimelic acid 18 is formed concomitantly with azi-DAP 29. The temperature of the reaction has little effect on the product ratio. The reaction to form 18 may proceed via reductive aziridine ring cleavage to generate an anion α to the carboxylate followed by elimination of nitrogen and further reduction of the resulting conjugated olefin. Separation of the crude reaction mixture could be achieved using preparative TLC under basic conditions, albeit in low isolated yield (23% for azi-DAP 29). It was later found that HPLC purification of the diacid salt is not necessary prior to the reductive deprotection. Therefore pure azi-DAP 14 could be prepared directly from 26 with an improved yield of 30% over the final two steps. As expected, azi-DAP isomers 14 and 29 are extremely sensitive compounds that are stable in strong base, but degrade rapidly under neutral or acidic conditions. The presence of multiple internal nucleophiles (four carboxylate oxygens and the distal amino group) assists rapid intramolecular aziridine ring opening. In solid form as carboxylate salts, 14 and 29 decompose gradually at room temperature, with half lives of about 1 to 2 weeks. Although storage at −20 °C retards decomposition, they are only stable for prolonged periods under cryogenic (−80 °C) conditions. Unfortunately, lack of stability in aqueous media precludes evaluation of their antimicrobial activity.
Inhibition of DAP epimerase by azi-DAP isomers 14 and 29
Inhibition studies of DAP epimerase by Higgins et al. using a mixture of all azi-DAP diastereomers generated in situ from the corresponding α-(fluoromethyl)diaminopimelic acids demonstrated that azi-DAP is an irreversible inhibitor. They were unable to determine Ki or ki values due to the extremely fast conversion of the enzyme-inhibitor complex to the inactivated enzyme.12,58 Using the coupled enzyme assay we were able to confirm that both diastereomers of azi-DAP are irreversible inhibitors, and show both time and concentration dependence. Furthermore, the covalent attachment of both isomers to the enzyme can be demonstrated by electrospray mass spectroscopy (MS).The original studies by Higgins et al. using tryptic digests of inhibited DAP epimerase combined with Edman degradation of the purified fragments reported that the enzyme is attacked at Cys-73 by the crude mixture of all diastereomers of azi-DAP.13 Subsequent studies suggested17 that inhibition of DAP epimerase with pure LL-azi-DAP 14 should result in selective attachment to Cys-73, whereas DL-azi-DAP 29 would react with Cys-217. In order to test this proposal, a trypsin digestion was done on DAP epimerase inhibited with excess pure azi-DAP isomers. HPLC purification of the fragments followed by MS/MS analysis displays a surprisingly low level of sequence coverage (40% for inhibition with 14 and 32% with 29). In each case, a fragment incorporating the Cys-73 unit (residues 62–78) could be sequenced and shown to contain free Cys-73 or Cys-73 with azi-DAP covalently attached. No fragment containing Cys-217 could be identified after HPLC of the crude digest.
In order to study the interaction of Cys-217 with azi-DAP, an alternative digestion protocol was required. As mentioned above, covalent attachment of an azi-DAP molecule to DAP epimerase could be observed for both 14 and 29 by MS. Although treatment with cyanogen bromide followed by trypsin digestion allowed the detection of a suitable fragment (residues 210–230) of the parent (not inhibited) DAP epimerase, it was found that the reaction of cyanogen bromide resulted in the cleavage of azi-DAP from the enzyme inactivated by 29. Presumably a mechanism similar to the cleavage of the peptide chain adjacent to methionine residues occurs under these conditions. The inactivation of DAP epimerase by azi-DAP occurs by nucleophilic ring opening of the aziridine to generate 30, which has a sulfide linkage between the enzyme and the inhibitor.13 Nucleophilic attack of the sulfur on cyanogen bromide followed by intramolecular displacement of the resulting positively charged sulfonium species 31 from the nearby peptide bond affords the dealkylated peptide 33, which can subsequently undergo hydrolysis (Scheme 6).
 | ||
| Scheme 6 Proposed mechanism for the cleavage of covalently linked azi-DAP from cysteine residues of DAP epimerase on treatment with cyanogen bromide. | ||
An analogous process was previously observed during cyanogen bromide cleavage of a protease inhibited by N-iodoacetyl peptides.59 Fortunately, inactivation of DAP epimerase with azi-DAP 29 followed by treatment with thermolysin and trypsin provides excellent sequence coverage (89%) of the resulting fragments by MS/MS. More importantly, fragments incorporating both cysteines of interest could be analysed using this protocol (Table 1). Analysis of DAP epimerase inhibited with a large excess (10–100 fold) of azi-DAP 29 reveals that in addition to the expected alkylation of Cys-217, attack also occurs at Cys-73 to some extent. In principle, it is possible that a single diasteroisomer of azi-DAP (i.e.29) reacts with both cysteines. However, the excess of inhibitor could possibly be contaminated by very small amounts (<1.0%) of the other azi-DAP diastereomer 14, which is not easily detectable and could react with the DAP epimerase. The use of a such a large excess of inhibitor was initially prompted by the desire to completely and rapidly inactivate the enzyme for crystallographic studies prior to its facile aerobic oxidation to form the internal disulfide between Cys-73 and Cys-217.

Fortunately, preliminary X-ray crystallography studies of DAP epimerase separately inhibited with the individual azi-DAP isomers show LL-azi-DAP 14 bound exclusively to Cys-73, and DL-azi-DAP 29 attached only to Cys-217. In both cases, inactivation proceeds as predicted by attack of thiolate on the aziridine methylene. After complete refinement and structural analysis, these three dimensional structures will be the subject of a separate report.
In conclusion, we have described the first stereoselective synthesis of highly sensitive aziridine analogues of DAP 4, 14 and 29. The results show that internal aziridine analogue 4 is a reversible inhibitor of DAP epimerase. The LL-azi-DAP 14 and DL-azi-DAP 29 rapidly and irreversibly inactivate DAP epimerase, as can be seen from a coupled enzyme assay with DAP dehydrogenase as well as by MALDI-TOF MS analysis. Proteolytic digestion of the enzyme inactivated by each inhibitor with thermolysin/trypsin followed by MS/MS provides excellent sequence coverage (89%) and allows analysis of fragments incorporating both cysteines of interest. MS/MS analysis of the digest of each inactivated DAP epimerase shows that the individual azi-DAP isomers bind irreversibly to Cys-73 and Cys-217. The use of excess inhibitor to assist preparation of samples for X-ray crystallographic analysis results in some unexpected alkylation of the cysteine residue that is expected to act as the protonating thiol. This may be due to the presence of very small quantities (<1.0%; undetectable by NMR) of the “wrong” isomer of azi-DAP. Preliminary X-ray analysis of inhibited DAP epimerase crystals showed that LL-azi-DAP 14 binds selectively to Cys-73 and DL-azi-DAP 29 to Cys-217. The covalent attachment of azi-DAP is consistent with the proposed mechanism of inhibition i.e. attack of thiolate at the methylene position of the aziridine ring. Furthermore, the observed selectivity of azi-DAP inhibition is consistent with the two base mechanism proposed for the epimerisation of LL-DAP 1 and DL-DAP 2 by DAP epimerase, more specifically that Cys-73 acts as a thiolate base to deprotonate LL-DAP 1, and Cys-217 in the case of meso-DAP 2.17
Experimental
General methods
All reactions were performed under dry argon. All solvents were purified and distilled according to Perrin et al.60 Flash chromatography employed silica gel 60 (Silicycle, 230–420 mesh) and was performed according to the Still procedure.61 One or more of the following methods were used for visualisation: UV fluorescence, iodine staining, phosphomolybdic acid/ceric sulfate, potassium permangante or ninhydrin. Melting points were determined on a Büchi apparatus using open-end capillary tubes and are uncorrected. NMR spectra were recorded on Inova Varian 300, 400, 500 and 600 MHz instruments. IR spectra were determined with a Nicolet Magna 750 FT-IR spectrometer. Mass spectra (MS) were recorded with a Micromass ZabSpec Hybrid Sector-TOF instrument (electrospray ionization (ES)). Optical rotations were measured on a Perkin-Elmer 241 polarimeter at 26 °C with a micro cell (100 mm; 0.9 mL) or a standard cell (100 mm, 8 mL) respectively. UV spectroscopy was performed on a GBC Cintra 40 UV spectrometer. [α]D are given in units of 10−1 deg cm2 g−1. Microanalyses were completed at the University of Alberta Microanalytical Laboratory. All literature compounds had IR, 1H NMR, 13C NMR and mass spectra consistent with assigned structures.CHH), 6.12 (1H, s, CCHH), 7.36 (5H, m, Ph); δC (125 MHz, CDCl3) 24.1, 31.4, 32.3, 51.9, 52.4, 53.7, 67.0, 125.3, 128.0, 128.1, 128.4, 136.2, 139.7, 155.7, 167.3, 172.7; m/z (ES) 372.1421 (MNa+). C18H23NO6Na requires 372.1423.
CHH), 6.25 (1H, s, CCHH), 7.28–7.34 (5H, m, Ph); δC (125 MHz, CDCl3)23.7, 31.5, 53.6, 67.1, 128.1, 128.2, 128.5, 136.1, 139.1, 155.8, 172.2, 177.7; m/z (ES) calcd. for C16H19NO6Na 344.1105 (MNa+), found 344.1105.
CHH), 6.27 (1H, s, CCHH), 7.28–7.34 (5H, m, Ph); δC (125 MHz, CDCl3) 24.0, 30.9, 32.1, 52.4, 53.6, 67.0, 127.4, 128.1, 128.2, 128.5, 136.2, 139.2, 155.9, 171.7, 172.9; m/z (ES) calcd. for C17H21NO6Na 358.1263 (MNa+), found 358.1261.
CHH), 6.11 (1H, s, CCHH), 7.28–7.34 (5H, m, Ph), 7.47–7.50 (1H, m, ArCH), 7.74–7.75 (2H, m, ArCH), 8.26–8.28 (1H, m, ArCH); δC (125 MHz, CDCl3) 16.8, 24.1, 29.8, 31.4, 32.2, 52.4, 53.8, 63.9, 67.1, 75.9, 120.3, 125.7, 126.5, 127.1, 127.9, 128.2, 128.6, 134.1, 136.3, 139.7, 146.4, 153.9, 156.0, 160.6, 166.8, 172.9; m/z (ES) calcd. for C29H35N4O8 567.2449 (M+), found 567.2446.
CHH), 6.10 (1H, s, CCHH), 7.28–7.34 (5H, m, Ph), 7.45–7.48 (1H, m, ArCH), 7.73–7.74 (2H, m, ArCH), 8.24–8.26 (1H, m, ArCH); δC (125 MHz, CDCl3) 16.8, 24.0, 31.3, 32.1, 33.9, 53.87, 63.8, 67.0, 75.8, 120.2, 125.6, 126.4, 127.0, 127.8, 128.1, 128.5, 134.1, 136.3, 139.6, 146.2, 153.9, 160.5, 166.7, 172.8; m/z (ES) calcd. for C29H35N4O8 567.2449 (M+), found 567.2449.
Acid 18 as a mixture of diastereomers: Rf 0.61; νmax(microscope)/cm−1 3050, 2940, 1693, 1583, 1513, 1441, 1408; δH (300 MHz, D2O) 1.10 (3H, d, J = 11.5 Hz, CH3), 1.32–1.51 (3H, m, CHH, CH2), 1.58–1.65 (1H, m, CHH), 1.81–1.92 (2H, m, CH2), 2.38–2.46 (1H, m, CHCH3), 3.73 (1H, t, J = 6.5 Hz, CHN), δC (100 MHz, D2O) 17.8 & 17.9, 23.2 & 23.3, 31.2 & 31.2, 33.9, 41.6 & 41.7, 55.5 & 55.6, 175.7, 185.1; m/z (ES) calcd. for C8H14NO4 188.0917 (M+), found 188.0918.
L,L-Diaminopimelic acid 1
A solution of (2S)-4-[(4S)-4-benzyloxycarbonylamino-4-methoxycarbonyl-butyrylperoxy)-2-tert-butoxycarbonylamino-4-oxo-butyric acid tert-butyl ester40 (92 mg, 0.19 mmol) in Et2O (5 mL) was added to 6 M HCl (5 mL) and the mixture was then heated to 90 °C for 2 h with stirring. The resulting aqueous solution was concentrated in vacuo and purified by ion-exchange chromatography (Biorad AG 50W-X8 hydrogen form resin), loading with distilled H2O, flushing for 6 column lengths with distilled H2O and then eluting with 10% NH4OH to give the title compound 1 (28 mg, 79%) as a white powder after lyophilisation. [α]D + 2.2 (c 2.0, H2O); νmax(microscope)/cm−1 2942, 1582, 1465, 1436, 1413, 1350, 1327, 1103; δH (500 MHz, D2O) 1.39–1.45 (2H, m, 4-CH2), 1.84–1.89 (4H, m, 3, 5-CH2), 4.06–4.08 (2H, t, J = 6.0 Hz, 2 × CHN), δC (125 MHz, D2O) 20.9, 30.9, 55.2, 175.4; m/z (ES) calcd. for C7H15N2O4 191.1026 (M+), found 191.1026.Inhibition Studies with DAP Epimerase Using Coupled Assay
DAP epimerase activity was monitored using a coupled assay with DAP dehydrogenase which detects the production of NADPH at 340 nm. The assay was performed in a 1 mL quartz cuvette filled with buffer solution (0.1 M tris-HCl, 1 mM EDTA, 1 mM DTT, pH 7.8), 0.4 mM L,L-DAP, 0.3 mM NADP+ and 0.06 units of DAP dehydrogenase. One unit of DAP epimerase activity corresponds to the production of one µmol of NADPH per minute. For inhibition studies with aziridine analogue 4, the assay buffer contained varying concentrations of the aziridine, 0.4 mM L,L-DAP, 0.3 mM NADP+, 50 mU of DAP dehydrogenase and 15 mU of DAP epimerase. These results were compared against a control performed simultaneously, where the Tris-HCl buffer replaced the inhibitor. The DAP epimerase used in the study of aziridines 4 and 11 was isolated from Escherichia coli mutant BL21(DE3) pLysS using a modified procedure7,39 and DAP dehydrogenase was purified from Bacillus sphaericus IFO 3525 as previously reported.62 The DAP epimerase used in the study of azi-DAP was isolated from Haemophilus influenza as described previously.15Labeling of DAP Epimerase with azi-DAP
A solution of azi-DAP in water (1–5 mg mL−1) was added in excess (10–100 equiv) to a solution of DAP epimerase (0.54 mg mL−1) in buffered solution (0.1 M tris-HCl, 1 mM EDTA, 1 mM DTT, pH 7.8) at room temperature and allowed to stand for 16 h. An aliquot (50 µL) was removed and added to a 10 mM solution of DTT in ammonium bicarbonate (50 µL, pH 8) and the mixture was heated at 37 °C for 30 min. An aliquot was removed and purified by RP-HPLC using a Jupiter™ reverse phase 1 mm Microbore column (5 µ, C18, 300 Å), eluting with a linear gradient over 50 min from 0.05% aqueous TFA to 0.05% TFA in MeCN, column temperature 70 °C, detection at 210 nm, injection volume 50 µL, flow rate 100 µL min−1, tR 29 min.Acknowledgements
Financial support was provided by the Natural Sciences and Engineering Research Council of Canada, the Canada Research Chair in Bioorganic and Medicinal Chemistry, Merck Frosst Canada and the Alberta Heritage Foundation for Medical Research (postdoctoral fellowship for A. S.).References
- F. R. Schmidt, Appl. Microbiol. Biotechnol., 2004, 63, 335 Search PubMed.
- I. Chopra, L. Hesse and A. J. O'Neill, J. Appl. Microbiol., 2002, 92, 4.
- A. H. Katz and C. E. Caufield, Curr. Pharm. Des., 2003, 9, 857 Search PubMed.
- J. van Heijenoort, Nat. Prod. Rep., 2001, 18, 503 RSC.
- R. J. Cox, A. Sutherland and J. C. Vederas, Bioorg. Med. Chem., 2000, 8, 843 CrossRef CAS.
- A. O. Hudson, C. Bless, P. Macedo, S. P. Chatterjee, B. K. Singh, C. Gilvarg and T. Leustek, Biochim. Biophys. Acta, 2005, 1721, 27 CrossRef CAS.
- J. S. Wiseman and J. S. Nichols, J. Biol. Chem., 1984, 259, 8907 CAS.
- M. E. Tanner, Acc. Chem. Res., 2002, 35, 237 CrossRef CAS.
- T. Yoshimura and N. Esaki, J. Biosci. Bioeng., 2003, 96, 103 Search PubMed.
- C. W. Koo and J. S. Blanchard, Biochemistry, 1999, 38, 4416 CrossRef CAS.
- M. H. Gelb, Y. Lin, M. A. Pickard, Y. Song and J. C. Vederas, J. Am. Chem. Soc., 1990, 112, 4932 CrossRef CAS.
- F. Gerhart, W. Higgins, C. Tardif and J. B. Ducep, J. Med. Chem., 1990, 33, 2157 CrossRef CAS.
- W. Higgins, C. Tardif, C. Richaud, M. A. Krivanek and A. Cardin, Eur. J. Biochem., 1989, 186, 137 CAS.
- R. J. Cox, J. Durston and D. I. Roper, J. Chem. Soc., Perkin Trans. 1, 2002, 1029 RSC.
- M. Cirilli, R. Zheng, G. Scapin and J. S. Blanchard, Biochemistry, 1998, 37, 16452 CrossRef CAS.
- A. J. Lloyd, T. Huyton, J. Turkenburg and D. I. Roper, Acta Crystallogr., 2004, D60, 397 CAS.
- C. W. Koo, A. Sutherland, J. C. Vederas and J. S. Blanchard, J. Am. Chem. Soc., 2000, 122, 6122 CrossRef CAS.
- J. P. Richard and T. L. Amyes, Bioorg. Chem., 2004, 32, 354 CrossRef CAS.
- M. Anderson, S. L. Fisher, R. H. A. Folmer, G. Kern, R. T. Lundqvist, D. T. Newton and Y. Xue, Crystal structure of bacterial glutamate racemase (MurI) and the design of antibacterial agents, WO, 2004/061097, pp. 1125 Search PubMed.
- K. Y. Hwang, C.-S. Cho, S. S. Kim, H.-C. Sung, Y. G. Yu and Y. Cho, Nat. Struct. Biol., 1999, 6, 422 CrossRef CAS.
- H. Mobitz and T. C. Bruice, Biochemistry, 2004, 43, 9685 CrossRef.
- L. Liu, K. Iwata, A. Kita, Y. Kawarabayasi, M. Yohda and K. Miki, J. Mol. Biol., 2002, 319, 479 CrossRef CAS.
- G. Williams, E. P. Maziarz, T. L. Amyes, T. D. Wood and J. P. Richard, Biochemistry, 2003, 42, 8354 CrossRef CAS.
- P. Minoprio, P. Alzari, A. Buschiazzo, W. Degrave, C. Gregoire, N. Chamond and A. Berneman, Crystallographic structure of Trypanosoma cruzi PRACA proline racemase and use of the structural information for drug discovery, WO, 2004//106506, pp. 119 Search PubMed.
- L. K. P. Lam, L. D. Arnold, T. H. Kalantar, J. G. Kelland, P. M. Lane-Bell, M. M. Palcic, M. P. Pickard and J. C. Vederas, J. Biol. Chem., 1988, 263, 11814 CAS.
- Y. Gao, P. Lane-Bell and J. C. Vederas, J. Org. Chem., 1998, 63, 2133 CrossRef CAS.
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC.
- J. T. Kapron, B. D. Santarsiero and J. C. Vederas, J. Chem. Soc., Chem. Commun., 1993, 1074 RSC.
- T. K. Chakraborty and A. Ghosh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2001, 40B, 895 CAS.
- K.-S. Yang and K. Chen, J. Org. Chem., 2001, 66, 1676 CrossRef CAS.
- R. S. Atkinson, Tetrahedron, 1999, 55, 1519 CrossRef.
- A. Koohang, C. L. Stanchina and R. M. Coates, Tetrahedron, 1999, 55, 9669 CrossRef CAS.
- W. Oppolzer, D. Dupuis, G. Poli, T. M. Raynham and G. Bernardinelli, Tetrahedron Lett., 1988, 29, 5885 CrossRef CAS.
- J. M. Padron, G. Kokotos, T. Martin, T. Markidis, W. A. Gibbons and V. S. Martin, Tetrahedron: Asymmetry, 1998, 9, 3381 CrossRef CAS.
- M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183 CrossRef CAS.
- Z. Chilmonczyk, M. Egli, C. Behringer and A. S. Dreiding, Helv. Chim. Acta, 1989, 72, 1095 CAS.
- A. Sutherland, J. F. Caplan and J. C. Vederas, Chem. Commun., 1999, 555 RSC.
- A. N. C. Weir, C. Bucke, G. Holt, M. D. Lilly and A. T. Bull, Anal. Biochem., 1989, 180, 298 CAS.
- Y. Song, D. Niederer, P. M. Lane-Bell, L. K. P. Lam, S. Crawley, M. M. Palcic, M. A. Pickard, D. L. Pruess and J. C. Vederas, J. Org. Chem., 1994, 59, 5784 CrossRef CAS.
- M. D. Spantulescu, R. P. Jain, D. J. Derksen and J. C. Vederas, Org. Lett., 2003, 5, 2963 CrossRef CAS.
- P. Muller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef CAS.
- H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693 CrossRef CAS.
- R. Haener, B. Olano and D. Seeback, Helv. Chim. Acta, 1987, 70, 1676 CrossRef.
- R. S. Atkinson and G. Tughan, J. Chem. Soc., Perkin Trans. 1, 1987, 2797 RSC.
- K.-S. Yang and K. Chen, Org. Lett., 2002, 4, 1107 CrossRef CAS.
- H. Shao, Q. Zhu and M. Goodman, J. Org. Chem., 1995, 60, 790 CrossRef CAS.
- R. S. Atkinson, A. P. Ayscough, W. T. Gattrell and T. M. Raynham, J. Chem. Soc., Perkin Trans. 1, 1998, 2783 RSC.
- J. E. Baldwin, V. Lee and C. J. Schofield, Synlett, 1992, 249 CrossRef CAS.
- R. M. Williams, M. N. Im and J. H. Cao, J. Am. Chem. Soc., 1991, 113, 6976 CrossRef CAS.
- D. H. R. Barton, S. D. Gero, B. Quiclet-Sire and M. Samadi, J. Chem. Soc., Perkin Trans. 1, 1991, 981 RSC.
- R. S. Atkinson and P. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 205 RSC.
- B. Cavaller, G. Volpe, G. Sartori, G. Carniti and R. J. White, Farmaco, Ed. Sci., 1974, 24, 257 Search PubMed.
- R. S. Atkinson, R. D. Draycott, D. J. Hirst, M. J. Parratt and T. M. Raynham, Tetrahedron Lett., 2002, 43, 2083 CrossRef CAS.
- A. R. Devi and S. Rajaram, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2000, 39B, 294 CAS.
- P. Gogoi, Synlett, 2005, 2263 CrossRef CAS.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815 RSC.
- K. L. Kaestle, M. K. Anwer, T. K. Audhya and G. Goldstein, Tetrahedron Lett., 1991, 32, 327 CrossRef CAS.
- C. Richaud, W. Higgins, D. Mengin-Lecreulx and P. Stragier, J. Bacteriol., 1987, 169, 1454 CAS.
- J. E. Mckendrick, S. Frormann, C. Luo, P. Semchuck, J. C. Vederas and B. A. Malcolm, Int. J. Mass Spectrom., 1998, 176, 113 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, UK, 1988 Search PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- S. D. Abbott, P. Lane-Bell, K. P. S. Sidhu and J. C. Vederas, J. Am. Chem. Soc., 1994, 116, 6513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details for the preparation of 7, 8, 18, 21, 22 and 23; 600 MHz NOE assignment of stereochemistry for 26; 600 MHz 1H NMR determination of dr's for 26 and 28; HPLC analysis of 14 precursor; MS/MS analysis enzymatic digests of DAP epimerase inhibited with either 14 or 29. See DOI: 10.1039/b513409a |
| This journal is © The Royal Society of Chemistry 2005 |