Design and synthesis of aromatic inhibitors of anthranilate synthase†
Richard J. Paynea, Esther M. M. Bullocha, Andrew D. Abellb and Chris Abell*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 336405
bDepartment of Chemistry, University of Canterbury, Christchurch, New Zealand
First published on 9th September 2005
Abstract
Anthranilate synthase catalyses the conversion of chorismate to anthranilate, a key step in tryptophan biosynthesis. A series of 3-(1-carboxy-ethoxy) benzoic acids were synthesised as chorismate analogues, with varying functionality at C-4, the position of the departing hydroxyl group in chorismate. Most of the compounds were moderate inhibitors of anthranilate synthase, with inhibition constants between 20–30 µM. The exception was 3-(1-carboxy-ethoxy) benzoic acid, (C-4 = H), for which KI = 2.4 µM. These results suggest that a hydrogen bonding interaction with the active site general acid (Glu309) is less important than previously assumed for inhibition of the enzyme by these aromatic chorismate analogues.
Introduction
The shikimate pathway is a major biosynthetic pathway utilised by plants, fungi, bacteria and protozoa to produce a range of important aromatic metabolites.1 This pathway has generated considerable interest as a target for potential herbicides2 and antimicrobial agents.3,4In E. coli the product of the pathway, chorismate (1), is a substrate for five separate enzymes that are responsible for the production of a range of aromatic compounds, including the aromatic amino acids phenylalanine, tyrosine and tryptophan (Fig. 1).5 Three of the branchpoint enzymes; anthranilate synthase, ADC synthase and isochorismate synthase, catalyse mechanistically-related reactions6,7 and probably diverged from a common ancestor.8–10
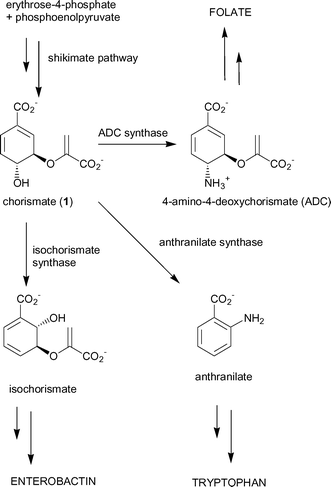 | ||
| Fig. 1 Chorismate-utilising enzymes. | ||
Anthranilate synthase represents the first committed step towards the biosynthesis of tryptophan.11 It is a multifunctional enzyme composed of a small TrpG and large TrpE subunit either as an αβ dimer or a α2β2 heterotetramer. TrpG belongs to the family of “triad” glutamine amidotransferases. The subunit hydrolyses the side chain amine of glutamine and transfers the nascent ammonia through an intramolecular channel to the synthase active site of TrpE. The TrpE subunit catalyses the production of anthranilate in two steps, (Scheme 1).12
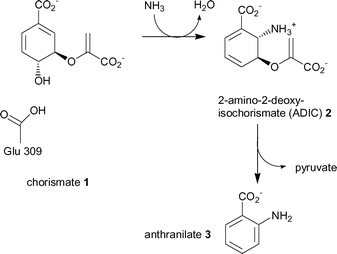 | ||
| Scheme 1 The two reactions catalysed by the TrpE subunit of anthranilate synthase, (ammonia is produced by the TrpG subunit). | ||
The first step is the reversible reaction of chorismate (1) with ammonia to give the intermediate, 2-amino-2-deoxyisochorismate (2, ADIC). The mechanism is thought to proceed by attack of ammonia at C-2 of chorismate, and concomitant loss of the C-4 hydroxyl as water via protonation by Glu309 (Scheme 1).6 The second reaction is the elimination of the enol-pyruvyl side chain from ADIC to produce the aromatic product anthranilate (3).
We recently described the synthesis of a range of aromatic inhibitors of anthranilate synthase.13 It was found that an aromatic ring was a reasonable mimic for the cyclohexadiene ring of the natural substrate, and that a lactyl side chain at the C-3 position was a good replacement for the enol-pyruvyl side chain. Racemic compounds 4–6 containing these features exhibited inhibition constants ranging from 3–43 µM (Fig. 2). Building on these findings we have developed a further series of aromatic chorismate analogues, where the substituent at C-3 is always the racemic lactyl side chain but where the substitution at C-4 is varied.
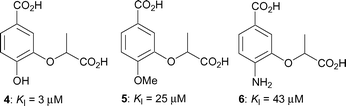 | ||
| Fig. 2 Inhibitors of S. marcescens anthranilate synthase.13 | ||
We now report a development of this study to systematically explore the effect of changing the substituent at C-4.
Results and discussion
Design
C-4 is the site of departure of the hydroxyl group from chorismate, and undergoes rehybridisation during the formation of ADIC (2). It seemed probable that the enzyme would be sensitive to the nature of the substituent at this position, and preliminary evidence for this is seen in the relative potency of the inhibitors 4–6, where there is a preference for a hydroxyl group over an amino group. The design strategy was to hold the main components (the C-1 carboxyl, the aromatic ring, the C-3 lactyl sidechain) constant, but introduce various functionality at C-4 by making compounds 7–12 (Fig. 3).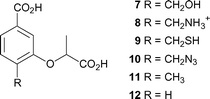 | ||
| Fig. 3 Proposed synthetic targets as anthranilate synthase inhibitors. | ||
As the C-4 hydroxyl departs, the C–O bond will lengthen and the hydroxyl will acquire a partial positive charge as it is protonated by Glu309. We sought to design compounds that would mimic these effects, e.g. by increasing the distance between C-4 and the hydroxyl (7, R = CH2OH), and having a positive charge near C-4 (8, R = CH2NH3+). To probe the space available and the importance of hydrogen bonding we also made the mercaptomethyl (9, R = CH2SH), azidomethyl (10, R = CH2N3), and methyl (11, R = CH3) analogues. Finally the C-4 hydroxyl was replaced with a hydrogen (12), to allow the binding due to the C-1 carboxylate and C-3 lactyl side chain alone to be assessed.
Modelling of chorismate and analogues 7–11 into the active site of S. marcescens anthranilate synthase using GOLD2.1,14 indicates that the C-4 substituents of the analogues are in close proximity to Glu309 in silico. Fig. 4 shows (R)-7 docked into the active site of anthranilate synthase and is overlayed with the corresponding chorismate docking. It is apparent that the C-4 hydroxyl of the analogue is within hydrogen-bonding distance from the carboxylate of Glu309. The hydroxyl of (R)-7 is in a similar orientation but is closer to Glu309 (2.3 Å cf. 2.5 Å for chorismate), suggesting a stronger hydrogen bond.
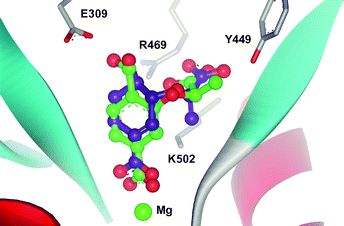 | ||
| Fig. 4 Chorismate (1) (purple) and (R)-7 (green) docked into the active site of anthranilate synthase from Serratia marcescens (PDB code: 1I7Q).12,15 | ||
Synthesis of 4-substituted analogues
The synthesis involved initial methyl ester formation from 3-hydroxy-4-methylbenzoic acid (13), followed by alkylation of the 3-hydroxy functionality with methyl-2-bromopropionate to introduce the lactyl side chain, (Scheme 2). The 4-methylbenzoic acid analogue 11 was produced by treatment of 15 with aqueous base.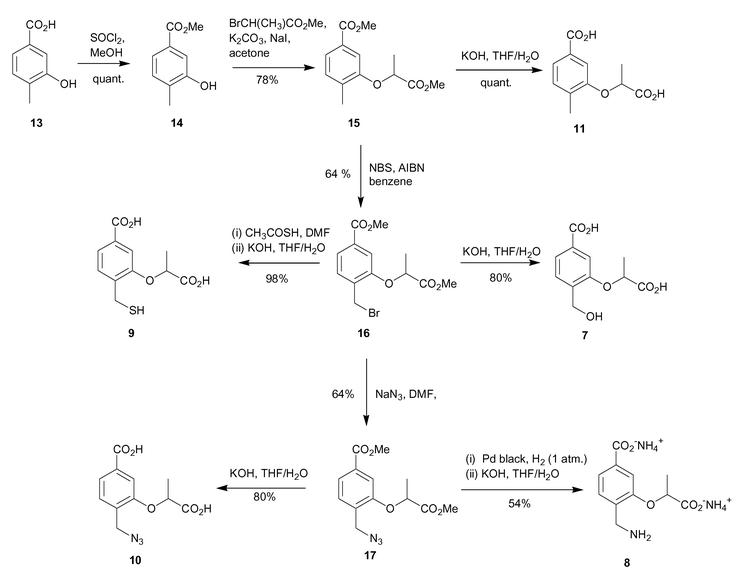 | ||
| Scheme 2 | ||
Bromination of the 4-methyl group in 15 was achieved using N-bromosuccinimide and AIBN in refluxing benzene, and produced the desired bromide 16 in 64% yield. SN2 displacement of the bromide with aqueous base occurred with concomitant hydrolysis of the methyl esters to afford the 4-hydroxymethyl diacid 7 in reasonable yield.
In order to synthesise mercaptomethyl analogue 9, bromide 16 was first treated with potassium thiolacetate in DMF, followed by hydrolysis of the methyl esters and thiolacetate protecting groups with aqueous potassium hydroxide, generating diacid 9 in 98% yield over the two steps.
The azide 10 was synthesised by SN2 displacement of the bromide of 16 using sodium azide in DMF in 64% yield. Methyl ester hydrolysis of 17 with aqueous potassium hydroxide gave the 4-azidomethyl diacid 10 in 80% yield. Synthesis of the aminomethyl analogue involved reduction of azide 17 to the corresponding amine, and finally methyl ester deprotection. The azide reduction was achieved by catalytic hydrogenation using Pd black catalyst under one atmosphere of hydrogen. A mixture of the desired 4-aminomethyl compound and the ring closed lactam were formed. The mixture was further treated with aqueous base to open up the lactam ring, and hydrolyse the methyl esters to yield the desired aminomethyl dicarboxylate 8 in 54% yield over the two steps. Purification of 8 was achieved by FPLC using a SourceQ anion exchange column.
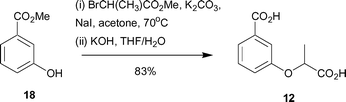
Synthesis of 12 began with methyl 3-hydroxybenzoate (18) and involved alkylation with methyl 2-bromopropionate, followed by ester hydrolysis to give the desired diacid 12 in 83% yield (Scheme 3).
 | ||
| Scheme 3 | ||
Inhibition assays
The aromatic chorismate analogues were assayed against S. marcescens anthranilate synthase. The production of anthranilate was detected fluorimetrically with excitation at 313 nm and detection at 390 nm as reported previously.13 The inhibition results are shown in Table 1. Intriguingly, analogues 7–11 exhibited very similar inhibition constants with KI values ranging from 21–26 µM. The only difference between these inhibitors is the substitution at C-4. This inhibition data suggests a lack of sensitivity of the enzyme for the C-4 substituent of these aromatic analogues.Analogue 12, which has a hydrogen atom at C-4, is ten-fold more potent than analogues 7–11, with a KI of 2.4 µM and represents the most potent aromatic anthranilate synthase analogue synthesised to date (Fig. 5). The 10-fold higher potency of 12 compared with 7–11 may be due to a decrease in steric bulk at C-4, or it may be that binding of 12 allows a water to bind between C-4 and Glu309. At this stage it is not possible to distinguish between these alternatives.
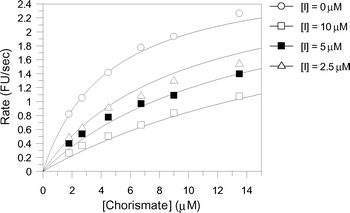 | ||
| Fig. 5 Least squares fitting for the reversible competitive inhibition of anthranilate synthase by 12 (KI = 2.4 ± 0.3 µM). | ||
In order to determine which enantiomer of 12 binds with higher affinity to anthranilate synthase, enantiopure forms of 12 were synthesised. Treatment of commercially available (R) and (S)-2-bromopropionic acids with diazomethane gave the corresponding methyl esters (Scheme 4). Alkylation of 18 with (R)-methyl 2-bromopropionate and (S)-methyl 2-bromopropionate, under identical conditions to those employed for the synthesis of racemic 12, gave the corresponding diesters. Subsequent hydrolysis of the esters with aqueous potassium hydroxide gave (R)-12 and (S)-12 in 50% and 72% yields respectively over the three steps.
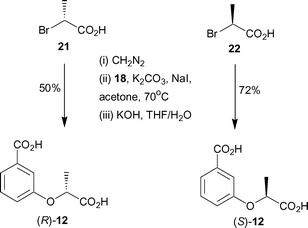 | ||
| Scheme 4 | ||
Analogues (R)- and (S)-12 were assayed against S. marcescens anthranilate synthase. (S)-12 was found to have a KI = 1.9 ± 0.2 µM, and was slightly more potent than the (R)-enantiomer (KI = 3.6 ± 0.5 µM). This result indicates a relatively modest sensitivity to the chirality of the side chain, but one that can be incorporated in the design of future inhibitors.
Conclusions
We have previously introduced aromatic chorismate analogues as competitive inhibitors of anthranilate synthase. That study established that a lactyl group bound well into the enol-pyruvyl side chain binding site. In this paper we have explored the importance of the substituent in the position (C-4) corresponding to the departing hydroxyl group in chorismate. It was assumed that there would be important binding interactions to this position and that these would be reflected in a sensitivity to the nature of the C-4 substituent on the aromatic inhibitor. It has been a major surprise to find that this is not the case, and that the inhibition constants are almost invariant for a range of different C-4 substituents. The biggest surprise was that removal of the C-4 substituent gave the most potent inhibitor (compound 12). It may be that having only hydrogen at C-4 allows a water molecule to bind between C-4 and Glu309.There are several possible explanations for the observed lack of sensitivity of the enzyme to the nature of the C-4 substituent. The C-4 substituent projects out in the same plane as the aromatic ring. This may be a poor mimic of the transition state conformation in the conversion of chorismate to ADIC (2) if chorismate adopts a conformation where the C-4 hydroxyl moves to a more axial orientation prior to departure. If this is the case, it may reflect a limitation in using aromatic analogues as inhibitors of anthranilate synthase. It is also possible that the inhibitor could be binding in a different orientation. Docking studies identify another binding mode where the binding of the C-1 and side chain carboxylates is reversed. However, this places the C-4 substituent close to the magnesium ion, and it would be surprising if variations of the C-4 substituent did not modulate the inhibition constant. Whatever the origin of the effect, the insensitivity to the C-4 substitution has important consequences to the development of aromatic inhibitors of anthranilate synthase.
Experimental
All organic solvents were freshly distilled prior to use and milli-Q deionised water was used for all biochemical work. Analytical thin layer chromatography was carried out on commercial silica gel 60 0.25 mm plates using either UV absorption or potassium permanganate stain, (3 g potassium permanganate, 20 g potassium carbonate, 5 ml of 5% sodium hydroxide, 300 ml water), for visualisation. RF values are quoted with respect to the solvent system used to develop the plate. Column chromatography was carried out using 230–400 mesh silica gel 60. Unless otherwise stated petroleum ether refers to the fraction collected between 40–60 °C. 1H NMR spectra were recorded on a Bruker AM-400 spectrometer in deuterated solvents, as indicated. 13C NMR spectra were recorded on a Bruker AM-400 spectrometer operating at 100 MHz. All chemical shifts are quoted in parts per million (ppm) δ Coupling constants for 1H NMR spectroscopy are assigned where possible and are given in Hz. Infrared spectra were recorded on a Perkin Elmer Spectrum One FTIR spectrometer using attenuated transmittance reflectance (ATR). High resolution mass spectrometry was carried out using a Micromass Quadrapole-Time of Flight (Q-Tof) spectrometer. FPLC purification was carried out on a SourceQ anion exchange resin column using a gradient from 10 mM ammonium bicarbonate to 1 M ammonium bicarbonate. Liquid-chromatography mass-spectrometry (LCMS) was carried out using an Alliance HT Waters 2795 Separations Module coupled to a Waters Micromass ZQ Quadrapole Mass Analyzer. Samples were detected using a photomultiplier detection system. Samples were run on a gradient from 10 mM ammonium acetate containing 0.1% formic acid to 95% acetonitrile over a period of 8 min. Purified anthranilate synthase from Serratia marcescens were stored in aliquots as concentrated solutions in buffer at −78 °C.Over-expression and purification of anthranilate synthase from S. marcescens was carried out as described previously.13
Gold docking
All ligands and the receptor were prepared using SYBYL6.5 and used as MOL2 files.16 The ligands were prepared as carboxylate anions and their structures were energy minimised using the Tripos force-field Gasteiger-Huckel charges calculated prior to docking. Each ligand was docked using GOLD2.1 in 25 independent genetic algorithm (GA) runs. For each of these, a maximum number of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GA operations was performed on a single population of 50 individuals.14 Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10 respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of the active site was introduced and the radius was set to 10 Å, with the automatic active-site detection on.
000 GA operations was performed on a single population of 50 individuals.14 Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10 respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of the active site was introduced and the radius was set to 10 Å, with the automatic active-site detection on.Enzyme assays
Kinetic parameters for S. marcescens anthranilate synthase were determined using the kinetic fluorescence assay developed previously.13 The assay was carried out in 100 mM potassium phosphate buffer (pH = 7); 10 mM magnesium chloride and 20 mM glutamine at 25 °C. Assays were initiated by the addition of S. marcescens anthranilate synthase (final concentration = 17.3 nM). The formation of anthranilate was detected by fluorescence (ex. 313 nm, em. 390 nm).13Grafit17 software was used to construct Michaelis–Menton plots of the kinetic data and carry out a least squares fitting for the reversible competitive inhibition of anthranilate synthase. The software was also utilised to calculate the inhibition constants and the standard errors of these values.
3-Hydroxy-4-methyl benzoic acid methyl ester 14
Thionyl chloride (20 drops) was added dropwise to a suspension of 3-hydroxy-4-methylbenzoic acid (2.4 g, 0.02 mol) in freshly distilled methanol (40 ml). The reaction mixture was heated to reflux under nitrogen for 16 h. The solution was allowed to cool to 22 °C before the solvent was removed in vacuo to give the crude product as a white solid. Purification by column chromatography eluting with 3 : 1 v/v petroleum ether–ethyl acetate gave 14 as a white solid. (2.66 g, quant.).RF
(3 : 1 petroleum ether–ethyl acetate)
= 0.39; νmax
(ATR): 3266 (br OH stretch), 2962 (Ar C–H stretch), 1695 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ar), 1596, 1513 (CC, ar), 1436 (CC) cm−1; 1H NMR (CDCl3)
δ 2.28 (3H, s, CH3), 3.89 (3H, s, CH3), 5.51 (1H, br s, OH), 7.17 (1H, d, J 8.0 Hz, ArH, C-5), 7.50 (2H, m, ArH, C-6 and C-2); 13C NMR (CDCl3)
δ 16.5, 52.5, 116.1, 122.3, 129.3, 130.5, 131.3, 154.5, 167.7; LCMS (M−
= 165.3, MH+
= 167.2), ret time = 3.7 min; HRMS calcd for C9H10O3Na: MNa+, 189.0530. Found: MNa+,189.0528.
O, ar), 1596, 1513 (CC, ar), 1436 (CC) cm−1; 1H NMR (CDCl3)
δ 2.28 (3H, s, CH3), 3.89 (3H, s, CH3), 5.51 (1H, br s, OH), 7.17 (1H, d, J 8.0 Hz, ArH, C-5), 7.50 (2H, m, ArH, C-6 and C-2); 13C NMR (CDCl3)
δ 16.5, 52.5, 116.1, 122.3, 129.3, 130.5, 131.3, 154.5, 167.7; LCMS (M−
= 165.3, MH+
= 167.2), ret time = 3.7 min; HRMS calcd for C9H10O3Na: MNa+, 189.0530. Found: MNa+,189.0528.
3-(1-Methoxycarbonyl-ethoxy)-4-methyl benzoic acid methyl ester 15
Methyl-2-bromopropionate (0.87 ml, 7.83 mmol) was added dropwise to a solution of 14 (1 g, 6.02 mmol), anhydrous potassium carbonate (1.66 g, 0.01 mol), sodium iodide (18 mol%, 0.16 g, 1.08 mmol) in acetone (50 ml) and the reaction heated to 70 °C for 16 h. The reaction was allowed to cool to 22 °C before the solvent was removed in vacuo. The residue was redissolved in ethyl acetate and washed with water (50 ml), brine (50 ml), dried (MgSO4) and the solvent removed in vacuo. The product was purified by column chromatography eluting with 4 : 1 v/v 50/70 petroleum ether–ethyl acetate to afford 15 as a white solid (1.19 g, 78%).RF
(4 : 1 petroleum ether/ethyl acetate)
= 0.66; νmax
(ATR): 1733, 1715 (CO, str), 1610, 1584, 1507 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.62 (3H, d, J 6.7 Hz), 2.30 (3H, s, CH3), 3.74 (3H, s, CO2Me), 3.85 (3H, s, CO2Me), 4.84 (1H, q, J 6.7 Hz, CH), 7.18 (1H, d, J 7.9 Hz, H-5), 7.39 (1H, d, J 1.2 Hz, H-2), 7.62 (1H, dd, J 7.9, 1.2 Hz, H-6); 13C NMR (CDCl3)
δ 16.5, 18.5, 52.0, 52.2, 72.6, 112.3, 122.7, 128.7, 130.8, 133.3, 155.6, 166.8, 172.3; HRMS calcd for C13H16O5Na: MNa+, 275.0895. Found: MNa+, 275.0893.
3-(1-Carboxy-ethoxy)-4-methyl benzoic acid 11
Potassium hydroxide (0.19 g, 3.30 mmol) in milliQ water (5 ml) was added dropwise to a solution of 15 (200 mg, 0.80 mmol) in THF (5 ml) and the reaction was heated to 40 °C for 2 h. The reaction was allowed to cool to 22 °C before dilution with water (10 ml). The aqueous fraction was washed with ethyl acetate (15 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 15 ml). The organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford 11 as a white solid (168 mg, quant).νmax
(ATR): 2845, 2631 (br, acid OH stretch), 1726, 1713, 1678 (CO, str), 1583, 1508 (CC, ar); 1H NMR (CD3OD)
δ 1.62 (3H, d, J 6.0 Hz, CH3), 2.30 (3H, s, CH3), 4.83 (1H, q, J 6.0 Hz, CH), 7.21 (1H, d, J 7.8 Hz, H-5), 7.39 (1H, d, J 1.5 Hz, H-2), 7.62 (1H, dd, J 7.8, 1.5 Hz, H-6); 13C NMR (CD3OD)
δ 17.0, 19.2, 73.8, 113.6, 124.1, 130.8, 132.0, 134.5, 157.4, 170.0, 175.9; HRMS calcd for C11H12O5Na: MNa+, 247.0582. Found: MNa+, 247.0578.
4-Bromomethyl-3-(1-methoxycarbonyl-ethoxy) benzoic acid methyl ester 16
A mixture of 15 (2.55 g, 0.01 mol), N-bromosuccinimide (1.78 g, 0.01 mol) and AIBN (5 mol%, 82 mg, 0.50 mmol) was dissolved in freshly distilled benzene (65 ml). The reaction was heated to 85 °C under argon for 24 h. The reaction was allowed to cool to 22 °C before dilution with dichloromethane (100 ml). The organic fraction was washed with saturated sodium carbonate solution (150 ml), brine (150 ml), dried (MgSO4) and the solvent removed in vacuo. The product was purified by column chromatography eluting with 9 : 1 v/v petroleum ether 50–70–ethyl acetate to afford bromide 16 as a yellow oil (2.12 g, 64%).RF
(9 : 1 petroleum ether–ethyl acetate)
= 0.12; νmax
(ATR): 1736, 1717 (CO, str), 1610, 1582 1505 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.72 (3H, d, J 6.7 Hz), 3.78 (3H, s, CO2Me), 3.91 (3H, s, CO2Me), 4.50 (1H, d, J 9.9 Hz, CHHBr), 4.75 (1H, d, J 9.9 Hz, CHHBr), 4.95 (1H, q, J 6.7 Hz, CH), 7.42 (2H, m, 2 × ArH), 7.64 (1H, dd, J 7.9, 1.6 Hz, H-6); 13C NMR (CDCl3)
δ 17.5, 26.5, 51.4, 51.5, 72.0, 112.2, 122.0, 130.1, 130.6, 131.0, 154.3, 165.2, 170.8; HRMS calcd for C13H15O5BrNa: MNa+, 353.0001. Found: MNa+, 352.9984.
3-(1-Carboxy-ethoxy)-4-hydroxymethyl benzoic acid 7
Potassium hydroxide (0.08 g, 1.36 mmol) in milliQ water (4 ml) was added dropwise to a solution of 16 (150 mg, 0.45 mmol) in THF (4 ml) and the reaction was stirred at 22 °C for 2 h. The reaction was diluted with water (10 ml). The aqueous fraction was washed with ethyl acetate (15 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 15 ml), the organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford 7 as an off white solid (110 mg, 80%).νmax
(ATR): 2927, 2542 (br, acid OH stretch), 1686 (CO, str), 1581, 1506 (CC, ar); 1H NMR (CD3OD)
δ 1.62 (3H, d, J 6.8 Hz, CH3), 4.68 (1H, d, J 14.6 Hz, CHH), 4.79 (1H, d, J 14.6 Hz, CHH), 4.90 (1H, q, J 6.8 Hz, CH), 7.43 (1H, d, J 1.3 Hz, H-2), 7.50 (1H, d, J 7.8 Hz, H-5), 7.65 (1H, dd, J 7.8, 1.3 Hz, H-6); 13C NMR (CD3OD)
δ 21.5, 62.8, 76.4, 116.1, 126.6, 131.2, 134.3, 139.6, 158.6, 172.1, 178.2; HRMS calcd for C11H11O6Na: MNa+, 263.0532. Found: MNa+, 263.0537.
3-(1-Carboxy-ethoxy)-4-mercaptomethyl benzoic acid 9
Potassium thiolacetate (99 mg, 0.87 mmol) was added to a 0 °C solution of bromide 16 (200 mg, 0.61 mmol) in dry DMF (5 ml) and the reaction stirred at 22 °C for 24 h. The reaction was diluted with ethyl acetate (100 ml) and washed with brine (5 × 50 ml). The organic fraction was dried (MgSO4) and the solvent removed in vacuo. Purification by column chromatography (eluent: 4 : 1 v/v petroleum ether–ethyl acetate) gave the diester as a pale yellow oil. (194 mg, 98%).RF
(4 : 1 v/v petroleum ether–ethyl acetate)
= 0.30; νmax
(ATR): 1756, 1719, 1687 (CO, str), 1621 (CC), 1580, 1503 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.63 (3H, d, J 6.8 Hz, CH3), 2.30 (3H, s, COCH3), 3.75 (3H, s, CO2Me), 3.86 (3H, s, CO2Me), 4.18 (1H, d, J 13.5 Hz, CHH), 4.22 (1H, d, J 13.5 Hz, CHH), 4.88 (1H, q, J 6.8 Hz, CH), 7.33 (1H, d, J 1.1 Hz, H-2), 7.39 (1H, d, J 7.9 Hz, H-5), 7.64 (1H, dd, J 7.9, 1.1 Hz, H-6); 13C NMR (CDCl3)
δ 18.4, 28.0, 30.2, 52.1, 52.3, 72.7, 112.5, 122.8, 130.4, 130.6, 132.2, 155.1, 166.5,171.9, 195.2; LCMS: ret. time = 4.04 min; MH+
= 327.1; HRMS calcd for C15H22O6NS: MNH4+, 344.1162. Found: MNH4+, 344.1159.
Potassium hydroxide (41 mg, 0.73 mmol) in milliQ water (2 ml) was added dropwise to a solution of the above diester (95 mg, 0.29 mmol) in THF (2 ml) and the reaction was stirred at 22 °C for 3 h. The reaction was diluted with milliQ water (10 ml). The aqueous fraction was washed with dichloromethane (10 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 15 ml), the organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford 9 as a white solid (75 mg, quant, 98% over the two steps).
νmax
(ATR): 2987, 2901 br. (O–H acid str.), 1727, 1693 (CO, str), 1583, 1507 (CC, ar) cm−1; 1H NMR (CD3OD)
δ 1.65 (3H, d, J 6.8 Hz, CH3), 3.72 (1H, d, J 13.8 Hz, CHH), 3.78 (1H, d, J 13.8 Hz, CHH), 4.92 (1H, q, J 6.8 Hz, CH), 5.02 (1H, s, SH), 7.36 (1H, d, J 7.8 Hz, H-5), 7.42 (1H, d, J 1.4 Hz, H-2), 7.58 (1H, dd, J 7.8, 1.4 Hz, H-6); 13C NMR (CD3OD)
δ 18.9, 24.0, 73.6, 113.8, 124.0, 130.6, 131.7, 137.4, 156.2, 169.3, 175.2; HRMS calcd for C11H16O5NS: MNH4+, 274.07443. Found: MNH4+, 274.0749.
4-Azidomethyl-3-(1-methoxycarbonyl-ethoxy) benzoic acid methyl ester 17
Sodium azide (193 mg, 2.97 mmol) was added to a solution of 16 (490 mg, 1.48 mmol) in DMF (10 ml) and the reaction was heated to 40 °C under argon for 16 h. The reaction was cooled to 22 °C before dilution with ethyl acetate (40 ml). The organic fraction was washed with water (5 × 40 ml), dried (MgSO4) and the solvent removed in vacuo. The product was purified by column chromatography eluting with 5 : 1 v/v petroleum ether 50–70–ethyl acetate to afford 17 as a colourless oil (0.30 g, 68%).RF
(5 : 1 v/v petroleum ether–ethyl acetate)
= 0.29; νmax
(ATR): 2110 (N3 stretch), 1745, 1698 (CO, str), 1612, 1583, 1502 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.67 (3H, d, J 6.8 Hz, CH3), 3.73 (3H, s, CO2Me), 3.88 (3H, s, CO2Me), 4.40 (1H, d, J 14.1 Hz, CHH), 4.56 (1H, d, J 14.1 Hz, CHH), 4.92 (1H, q, J 6.8 Hz, CH), 7.35 (1H, d, J 7.8 Hz, H-5), 7.41 (1H, d, J 1.4 Hz, H-2), 7.66 (1H, dd, J 7.8, 1.4 Hz, H-6); 13C NMR (CDCl3)
δ 20.8, 52.2, 54.7, 75.1, 115.0, 125.4, 132.1, 132.5, 133.7, 157.7, 168.8, 174.2; HRMS calcd for C13H15N3O5Na: MNa+, 316.0909. Found: MNa+, 316.0919.
4-Azidomethyl-3-(1-carboxy-ethoxy) benzoic acid 10
Potassium hydroxide (58 mg, 1.03 mmol) in milliQ water (2 ml) was added dropwise to a solution of 17 (76 mg, 0.26 mmol) in THF (2 ml) and the reaction was stirred at 22 °C for 2 h. The reaction was diluted with water (10 ml) and the aqueous fraction was washed with ethyl acetate (15 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 15 ml). The organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford 10 as a white solid (55 mg, 80%).νmax
(ATR): 2949, 2570 (br OH acid), 2093, 2068 (N3 stretch), 1703, 1692 (CO, str), 1584 (CC, ar) cm−1; 1H NMR (CD3OD)
δ 1.64 (3H, d, J 6.8 Hz, CH3), 4.39 (1H, d, J 14.0 Hz, CHH), 4.55 (1H, d, J 14.0 Hz, CHH), 4.92 (1H, q, J 6.8 Hz, CH), 7.37 (1H, d, J 7.8 Hz, H-5), 7.49 (1H, d, J 1.3 Hz, H-2), 7.62 (1H, dd, J 7.8, 1.3 Hz, H-6); 13C NMR (CD3OD)
δ 17.2, 49.2, 72.3, 112.3, 122.2, 129.2, 129.7, 131.5, 155.4, 167.5, 173.6; HRMS calcd for C11H11N3O5Na: MNa+, 288.0596. Found: MNa+, 288.0603.
4-Aminomethyl-3-(1-carboxy-ethoxy) benzoate 8
A solution of 17 (0.15 g, 0.52 mmol) in freshly distilled methanol (3 ml) was added to a suspension of palladium black (10 mg, 0.09 mmol) in freshly distilled methanol (2 ml) under a hydrogen atmosphere. The reaction was stirred at 22 °C for 3 h. The methanolic solution was passed through Celite and washed with a further portion of methanol (10 ml). The solution was dried (MgSO4) and the solvent removed in vacuo to afford the crude product. This was dissolved in THF (2 ml) and a solution of potassium hydroxide (87 mg, 1.55 mmol) in milliQ water (2 ml) added dropwise. The reaction was stirred at 22 °C for 2 h. The reaction was diluted with milliQ water (5 ml) and washed with ethyl acetate (10 ml). The aqueous fraction was acidified to pH of 8 with 0.1 M HCl and the solution lyophilised. Purification was achieved on a SourceQ anion exchange column using an FPLC system (gradient from 10 mM ammonium bicarbonate to 1 M ammonium bicarbonate) to afford 8 as the diammonium salt as a white solid. (0.28 mmol, 54%, the yield was determined by 1H NMR spectroscopy using 3-(trimethylsilyl)-2,2,3,3-d4-acid sodium salt as an internal standard).νmax (ATR): 3044, 2297, 1555, 1405 cm−1; 1H NMR (D2O) δ 1.64 (3H, d, J 6.8 Hz, CH3), 4.07 (1H, d, J 13.3 Hz, CHH), 4.32 (1H, d, J 13.3 Hz, CHH), 4.83 (1H, q, J 6.8 Hz, CH), 7.40 (1H, d, J 7.8 Hz, H–5), 7.49 (1H, d, J 1.4 Hz, H–2), 7.62 (1H, dd, J 7.8, 1.4 Hz, H–6); 13C NMR (D2O) δ 18.8, 39.6, 75.8, 113.3, 122.4, 124.9, 131.4, 138.4, 156.4, 173.9, 180.5; HRMS calcd for C11H12NO5Na2: [M + Na2 + H]+, 284.0511. Found: [M + Na2 + H]+, 284.0494.
3-(1-Carboxy-ethoxy) benzoic acid 12
Methyl 2-bromopropionate (478 µl, 4.28 mmol) was added dropwise to a solution of methyl 3-hydroxybenzoate (0.50 g, 3.29 mmol), anhydrous potassium carbonate (0.91 g, 6.57 mmol) and sodium iodide (0.09 g, 0.59 mmol) in distilled acetone (15 ml) and the reaction was heated at 70 °C under nitrogen for 24 h. The reaction was allowed to cool to 22 °C before dilution with ethyl acetate (30 ml). The organic fraction was washed with water (40 ml), brine (40 ml), dried (MgSO4) and the solvent removed in vacuo. Purification by column chromatography (eluent: 4 : 1 petroleum ether–ethyl acetate) gave the diester as a colourless oil. (0.65 g, 83%).RF
(4 : 1 petroleum ether–ethyl acetate)
= 0.33; νmax
(ATR): 1756, 1719 (CO, str), 1587, 1489 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.59 (3H, d, J 6.8 Hz, CH3), 3.71 (3H, s, CO2Me), 3.84 (3H, s, CO2Me), 4.79 (1H, q, J 6.8 Hz, CH), 7.04 (1H, ddd, J 1.3, 2.5, 8.0 Hz, ArH), 7.29 (1H, t, J 1.8 Hz, ArH), 7.48 (1H, dd, J 1.3, 2.5 Hz, ArH), 7.61 (1H, dt, J 1.3, 8.0 Hz, ArH); 13C NMR (CDCl3)
δ 18.3, 52.0, 52.2, 72.5, 115.4, 120.2, 122.7, 129.5, 157.4, 166.5, 172.1; LCMS: ret. time = 3.70 min; MH+
= 240.1; HRMS calcd for C12H18O5N: MNH4+, 256.1179. Found: MNH4+, 256.1182.
Potassium hydroxide (375 mg, 6.05 mmol) in milliQ water (4 ml) was added dropwise to a solution of the above diester (400 mg, 1.67 mmol) in THF (4 ml) and the reaction was stirred at 40 °C for 3 h. The reaction was diluted with water (10 ml) and the aqueous fraction was washed with dichloromethane (15 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 15 ml). The organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford 12 as a white solid (350 mg, quant).
νmax
(ATR): 2989, 2532 br. (O–H acid str.), 1682, 1610 (CO, str), 1583, 1492 (CC, ar) cm−1; 1H NMR (CD3OD)
δ 1.58 (3H, d, J 6.8 Hz, CH3), 4.82 (1H, q, J 6.8 Hz, CH), 7.08 (1H, ddd, J 0.8, 2.5, 8.0 Hz, ArH), 7.31 (1H, t, J 8.0 Hz, ArH), 7.51 (1H, dd, J 1.3, 2.5 Hz, ArH), 7.61 (1H, dt, J 1.3, 8.0 Hz, ArH); 13C NMR (CD3OD)
δ 18.7, 73.4, 116.6, 121.1, 123.7, 130.5, 133.0, 158.9, 169.5, 175.5; HRMS calcd for C10H14O5N: MNH4+, 228.0866. Found: MNH4+, 228.0869.
(R)-3-(1-Carboxy-ethoxy) benzoic acid (R)-12
An ethereal diazomethane (CAUTION) solution (generated from Diazald)18 was added dropwise to a solution of (R)-(+)-2-bromopropanoic acid (1 g, 6.54 mmol) at 0 °C until a bright yellow colour persisted. The reaction was stirred at 22 °C for 2 h. The solvent was removed in vacuo to give an oil, which was subsequently purified by kugelrohl distillation under reduced pressure to afford the desired methyl ester as a colourless liquid (1 g, 5.99 mmol). The resulting (R)-methyl 2-bromopropionate (0.71 g, 4.28 mmol) was added dropwise to a solution of methyl 3-hydroxybenzoate (0.25 g, 3.29 mmol), anhydrous potassium carbonate (0.45 g, 3.29 mmol) and sodium iodide (0.04 g, 0.30 mmol) in distilled acetone (7.5 ml) and the reaction was heated at 70 °C under nitrogen for 24 h. The reaction was allowed to cool to 22 °C before dilution with ethyl acetate (30 ml). The organic fraction was washed with water (40 ml), brine (40 ml), dried (MgSO4) and the solvent removed in vacuo. Purification by column chromatography (eluent: 4 : 1 petroleum ether–ethyl acetate) gave the diester as a colourless oil. (0.26 g, 67%).1H NMR (CDCl3) δ 1.59 (3H, d, J 6.8 Hz, CH3), 3.71 (3H, s, CO2Me), 3.84 (3H, s, CO2Me), 4.79 (1H, q, J 6.8 Hz, CH), 7.04 (1H, ddd, J 1.3, 2.5, 8.0 Hz, ArH), 7.29 (1H, t, J 1.8 Hz, ArH), 7.48 (1H, dd, J 1.3, 2.5 Hz, ArH), 7.61 (1H, dt, J 1.3, 8.0 Hz, ArH); 13C NMR (CDCl3) δ 18.3, 52.0, 52.2, 72.5, 115.4, 120.2, 122.7, 129.5, 157.4, 166.5, 172.1.
Potassium hydroxide (188 mg, 3.03 mmol) in milliQ water (2 ml) was added dropwise to a solution of the above diester (200 mg, 0.84 mmol) in THF (2 ml) and the reaction was stirred at 40 °C for 3 h. The reaction was diluted with water (5 ml) and the aqueous fraction was washed with dichloromethane (8 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 8 ml). The organic fractions were dried (MgSO4) before the solvent was removed in vacuo to afford (R)-12 as a white solid (135 mg, 74%, 50% over the three steps).
[α]25D −0.8 (c 1 in MeOH); 1H NMR (CD3OD) δ 1.58 (3H, d, J 6.8 Hz, CH3), 4.82 (1H, q, J 6.8 Hz, CH), 7.08 (1H, ddd, J 0.8, 2.5, 8.0 Hz, ArH), 7.31 (1H, t, J 8.0 Hz, ArH), 7.51 (1H, dd, J 1.3, 2.5 Hz, ArH), 7.61 (1H, dt, J 1.3, 8.0 Hz, ArH); 13C NMR (CD3OD) δ 18.7, 73.4, 116.6, 121.1, 123.7, 130.5, 133.0, 158.9, 169.5, 175.5; LCMS: ret. time = 4.12 min; MH+ = 209.1.
(S)-3-(1-Carboxy-ethoxy) benzoic acid (S)-12
(S)-12 was synthesised in an identical fashion to (R)-12 and was produced as a white solid (150 mg, 72% over three steps).[α]25D +1.0 (c 1 in MeOH); 1H NMR (CD3OD) δ 1.58 (3H, d, J 6.8 Hz, CH3), 4.82 (1H, q, J 6.8 Hz, CH), 7.08 (1H, ddd, J 0.8, 2.5, 8.0 Hz, ArH), 7.31 (1H, t, J 8.0 Hz, ArH), 7.51 (1H, dd, J 1.3, 2.5 Hz, ArH), 7.61 (1H, dt, J 1.3, 8.0 Hz, ArH); 13C NMR (CD3OD) δ 18.7, 73.4, 116.6, 121.1, 123.7, 130.5, 133.0, 158.9, 169.5, 175.5; LCMS: ret. time = 4.13 min; MH+ = 209.1.
Acknowledgements
RJP and EMMB would like to thank the Gates Cambridge Trust for PhD funding and the BBSRC for research support. We would also like to thank Glen Spraggon (Genomics Institute of the Novartis Research Foundation, San Diego, California) and Charles Yanofsky (Stanford University, Stanford, California) for kindly supplying the ptttSmEDC vector containing the Serratia marcescens trpEGDC operon.References
- C. Abell, in Comprehensive Natural Products Chemistry, ed. U. Sankawa, Elsevier, Amsterdam, 1999, vol. 1, pp. 573–607 Search PubMed.
- J. L. Rubin, C. G. Gaines and R. A. Jensen, Plant Physiol., 1984, 75, 839–845 CrossRef CAS.
- C. K. Matthews, in Biochemistry, Benjamin/Cummings Publishing, Redwood City, CA, 1990, pp. 693–699 Search PubMed.
- F. Roberts, C. W. Roberts, J. J. Johnson, D. E. Kyle, T. Krell, J. R. Coggins, G. H. Coombs, W. K. Milhous, S. Tzipori, D. J. P. Ferguson, D. Chakrabarti and R. McLeod, Nature, 1998, 393, 801–805 CrossRef CAS.
- C. T. Walsh, J. Liu, F. Rusnak and M. Sakaitani, Chem. Rev., 1990, 90(7), 1105–1129 CrossRef CAS.
- Z. He, K. D. Stigers Lavoie, P. A. Bartlett and M. D. Toney, J. Am. Chem. Soc., 2004, 126, 2378–2385 CrossRef CAS.
- E. M. M. Bulloch, M. A. Jones, E. J. Parker, A. P. Osbourne, E. Stephens, G. M. Davies, J. R. Coggins and C. Abell, J. Am. Chem. Soc., 2004, 126, 9912–9913 CrossRef CAS.
- P. Goncharoff and B. P. Nichols, J. Bacteriol., 1984, 159, 57–62 CAS.
- B. A. Ozenberger, T. J. Brickman and M. A. McIntosh, J. Bacteriol., 1989, 171, 775–783 CAS.
- M. F. Elkins and C. F. Earhart, FEMS Microbiol. Lett., 1988, 56, 35–40 CrossRef CAS.
- F. Dosselaere and J. Vanderleyden, Crit. Rev. Microbiol., 2001, 27(2), 75–131 CAS.
- G. Spraggon, C. Kim, X. Nguyen-Huu, M. Yee, C. Yanofsky and S. Mills, Proc. Nat. Acad. Sci. USA, 2001, 98, 6021–6026 CrossRef CAS.
- R. J. Payne, M. T. Toscano, E. M. Bulloch, A. D. Abell and C. Abell, Org. Biomol. Chem., 2005, 3, 2271–2281 RSC.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- Tripos SYBYL v6.5, Tripos, Inc, 1699, South Hanley Road, St. Louis, Missouri, 63144, USA Search PubMed.
- GraFit (version 5.0.6), 1989–2003, Erithacus Software Limited Search PubMed.
- J. S. Pizey, in Synthetic Reactions, Halsted, New York, 1974, vol. II, ch. 4 Search PubMed.
Footnote |
| † Part II. For Part I see ref. 13. |
| This journal is © The Royal Society of Chemistry 2005 |