Directing the secondary structure of polypeptides at will: from helices to amyloids and back again?
Kevin
Pagel
,
Toni
Vagt
and
Beate
Koksch
*
Freie Universität Berlin, Institut für Chemie-Organische Chemie, Takustrasse 3, 14195, Berlin, Germany. E-mail: koksch@chemie.fu-berlin.de; Fax: +49-30-838-55644; Tel: +49-30-838-55344
First published on 20th September 2005
Abstract
An ageing society faces an increasing number of neurodegenerative diseases such as Alzheimer's, Parkinson’s, and Creutzfeld–Jacob disease. The deposition of amyloid fibrils is a pathogenic factor causing the destruction of neuronal tissue. Amyloid-forming proteins are mainly α-helical in their native conformation, but undergo an α-helix to β-strand conversion before or during fibril formation. Partially unfolded or misfolded β-sheet fragments are discussed as direct precursors of amyloids. To potentially cure neurodegenerative diseases we need to understand the complex folding mechanisms that shift the equilibrium from the functional to the pathological isoform of the proteins involved. This paper describes a novel approach that allows us to study the interplay between peptide primary structure and environmental conditions for peptide and protein folding in its whole complexity on a molecular level. This de novo designed peptide system may achieve selective inhibition of fibril formation.
 Kevin Pagel Kevin Pagel | Kevin Pagel was born in Werdau (Germany) and studied chemistry at the University of Leipzig. After graduation under supervision of Beate Koksch in 2003 he started his PhD thesis in the same group. In 2004 the group moved to Berlin where he is currently working on the de novo design of coiled coil based model peptides that change their conformation under the influence of differing environmental conditions. |
 Toni Vagt Toni Vagt | Toni Vagt was born in Leipzig (Germany) and studied biochemistry at the University of Leipzig. In 2004 he graduated in the group of Beate Koksch, where he is currently working on his PhD thesis. His main research interests are the development of an in vitro expression system for the incorporation of fluorinated amino acids into proteins and the role of membranes in peptide folding. |
 Beate Koksch Beate Koksch | Beate Koksch received her Diploma in Chemistry from the University of Leipzig where she then earned her PhD in biochemistry under the supervision of Professor H.-D. Jakubke in 1995. She was a DFG research fellow in the laboratories of Professor M. R. Ghadiri and Professor C.F. Barbas III at The Scripps Research Institute, La Jolla. In 2000 she returned to the University of Leipzig to start her independent career. In June 2004 she assumed a position as Professor for Organic Chemistry and Natural Product Chemistry at the Free University of Berlin. Her research interests focus on the interface of chemistry and biology and in particular on the role of complementary interactions and cooperativity in peptide and protein folding as well as peptide and protein modification by non-natural amino acids. |
Introduction
The special feature of proteins involved in Alzheimer's or prion diseases is their ability to adopt at least two different (meta)stable conformations. Thus, amyloid-forming proteins that mainly contain α-helical structures in their native conformation must undergo an α-helix to β-strand conversion before or during fibril formation. Protein aggregation leads to the deposition of the insoluble protein forms in the tissue concerned.1,2 The conformational transition that shifts the equilibrium from the functional to the pathological isoform can happen sporadically. The conformational change can also be triggered by mutations in the primary structure as well as by changes of the environmental conditions such as pH, ionic strength, metal ions, protein concentration, oxidative stress, free radicals, and by physiological or pathological chaperons. Alternatively, a small quantity of a misfolded protein fragment may act as a structural template that initiates the conformational conversion causing the disease. While the spontaneous forms of such diseases are extremely common, inherited factors, even single mutations within the amino acid sequence, may be of paramount importance.How can we elucidate the complex folding mechanisms that occur during the transformation from α-helix to β-sheet and beyond that to the formation of amyloids? What would be a practical system for studying the consequences of the interplay between peptide primary structure and environmental factors for peptide/protein folding on a molecular level? What kind of intrinsic factors enable a peptide/protein structure to transfer its secondary structure motif onto an unfolded protein fragment? The aim not only to inhibit, but also to reverse β-sheet formation and aggregation is even more challenging. If aggregation can be inhibited, at what stage of this process do we have a chance to interfere? The understanding of how complementary interactions determine the structure of polypeptides holds the key to answering these questions.
The system we have developed is based on an antiparallel, 26 amino acid α-helical coiled coil peptide with unprotected N and C termini. It can be modified such that a switch from the α-helical state to a random coil and/or a β-sheet structure, and in reverse, can be induced by changing various environmental conditions. Thus, our strategy is different from those reported in the literature. The big advantages that we see in the coiled coil system are:
(1) The α-helical coiled coil protein folding motif is very well studied and the design principles are known in detail, implying a better predictability of the system compared to isolated helices.
(2) The driving force for the helix formation is the interaction of intrinsically complementary surfaces. This is a valuable property not only for the study of cooperative processes, but also for a systematic investigation of the influence of mutations in the primary structure on the stability of secondary structure motifs.
(3) No restrictions are exerted on the system, such as covalent modifications, non-natural building blocks, linkages, etc., allowing our investigations to be compared with naturally occurring scenarios.
(4) No peptide bonds are formed or rearranged during the folding processes, thereby laying the foundation for the reversibility of the system.
Before we introduce our concept in detail, we will first explain the basic features of the secondary structures that are involved, namely α-helices and β-sheets, as well as several known approaches for switching between these secondary structure motifs.
Switch peptides
The formation of amyloid fibrils in neurodegenerative diseases is caused by partially misfolded and/or unfolded intermediates of the proteins involved. Therefore, the investigation of peptides and proteins which can change their conformation under certain conditions is of increasing interest (Fig. 1).3–9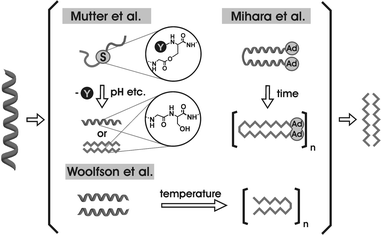 | ||
| Fig. 1 Several approaches for studying α-helix → β-strand switches. | ||
The de novo design of such “switch peptides” was first described by Mutter et al. in 1991.10 They could show that aggregated amphiphilic α-helices can be switched to double layer β-sheets by changing the pH. This system is based on an amphiphilic β-sheet peptide that is built of alternating hydrophobic and hydrophilic amino acids. The primary structure was modified to make a helical conformation more favorable. Therefore, some of these alternating amino acids were substituted by neutral amino acids resulting in a 16 amino acid peptide that forms amphiphilic helical aggregates with a hydrophobic and a hydrophilic side on each helical cylinder.
Another approach was presented by the group of Mihara,11–13 who generated a parallel 16 residue α-helical coiled coil dimer, covalently linked by a cysteine spacer at the C-terminus and equipped with a highly hydrophobic adamantane group at the N-terminus. This system forms β-sheets in buffered solution that rearrange to fibrils within two weeks. The conformational change is induced by the interaction of adamantane groups that serve as a “hydrophobic defect” and trigger a slow α to β transition. Capturing the adamantane groups by cyclodextrine prevents aggregation and the α-helical coiled coil structure remains stable. A similar system was described recently by Kammerer et al., where neither covalent linkages nor hydrophobic defects were necessary for the time-dependent structural change.14
In addition to time and pH dependent switches, the redox state also appears to be a factor that influences the secondary structure of peptides. Gellman et al. studied a methionine-rich peptide that changes its conformation from an antiparallel β-sheet structure at the oxidized sulfon and sulfoxide state of the methionine to an aggregated α-helical form in the reduced species.15 Thus, the secondary structure of an 18 amino acid peptide has been shown to be alterable just by converting four residues from a lipophilic to a hydrophilic character. These results lead to the assumption that the amphiphilic order of amino acids is a strong determinant of peptide and protein folding.
Woolfson et al. described a peptide system that alters the conformation from an α-helix to a β-hairpin motif under the influence of thermal denaturation.16 They conclude that a straight destabilization of the α-helical coiled coil structure does not necessarily result in a conformational change, but incorporation of β-sheet preferring amino acids into the f-position of an α-helical coiled coil peptide promotes the sensitivity of the structural switch. Furthermore, the results show that the formation of the β-sheet structure and the resulting amyloid aggregation proceeds with an unfolded intermediate as a direct precursor. A more recent study by Mutter et al. induces the secondary structure switch by the formation of a newly formed peptide bond between two fragments.17
In a theoretical approach, Peng and Hansmann applied multicanonical Monte Carlo simulations to investigate the nature of a secondary structure switch of a small peptide sequence which occurs in various proteins.18 The simulations showed that this seven-residue peptide adopts a helical conformation in solution as well as under gas phase conditions. However, the secondary structure switches to a β-sheet once two strands of this peptide get into close proximity to each other due to strong electrostatic effects and van der Waals interactions.
These results show that the α-helix to β-sheet secondary structure switch appears to be a highly sensitive process triggered by a huge diversity of environmental factors and parameters. Additionally, cooperativity effects seem to play a major role in the conformational evolution and switch properties of α-helical species as well as β-sheet motifs. Hence, it follows that the detailed investigation of the coherence between the switch properties of peptide structures and cooperativity is of paramount importance to understand disease related β-sheet and amyloid formation processes.
α-Helical coiled coil peptides
The α-helical coiled coil folding motif is one of the most widespread structural motifs in nature.19 Approximately 3% of amino acids in naturally occurring peptides and proteins are involved in the formation of coiled structures.20 α-Helical coiled coils typically consist of two to five right-handed α-helices which are wrapped around each other to form a left-handed superhelical twist.21 A schematic view of a dimeric antiparallel coiled coil is shown in Fig. 2. | ||
| Fig. 2 Schematic model of an antiparallel coiled coil dimer. | ||
The primary structure of each helix is characterized by a periodicity of seven residues, the so-called 4–3 heptad repeat which is commonly denoted (a-b-c-d-e-f-g)n (Fig. 3). Positions a and d are typically occupied by apolar residues (Leu, Ile, Val, Met) that form a special interaction surface at the interface of the helices by hydrophobic core packing (“knobs-into-holes”).22 In contrast, the positions e and g are frequently occupied by charged amino acids (most commonly Glu and Lys) that form inter-helical ionic interactions.23,24 Polar residues are often found in the remaining heptad repeat positions b, c, and f, which are solvent exposed, located at the opposite side of the motif.
 | ||
| Fig. 3 Helical wheel presentation of an α-helical coiled coil motif. Yellow: positions occupied by hydrophobic amino acids, red: positions occupied by charged amino acids. | ||
The hydrophobic core provides the major contribution to the structural stability of the α-helical coiled coil. The allocation of the two positions, a and d, of this interface in regard to the different types of hydrophobic residues, β- and γ-branched, controls the order of aggregate formation. Introducing specific, buried polar interactions within the hydrophobic core provides an efficient way to direct the helical alignment.25 In contrast, the inter-helical ionic pairing positions, e and g, mainly dictate the specificity of folding (parallel versus antiparallel) as well as promoting the preference for homo- or heterotypic α-helical coiled coil formation.26–30 Amino acids in these positions provide less overall stability for this folding motif and have less effect on the direction of the oligomerization state than hydrophobic core residues.
A third recognition domain is formed by intramolecular charge interactions between positions c/g and b/e, respectively, of the single helices. These interactions indirectly influence the stability of α-helical coiled coil folding by stabilizing or destabilizing the single helices.31 Position f of the heptad repeat is not part of any of the three recognition domains. Therefore, its contribution to helix stability has not yet been defined if it exists at all. Fig. 3 shows a simple model of an α-helical coiled coil peptide dimer.
β-Sheet peptides
β-Sheets are not as uniform as α-helices and their basic design principles and folding characteristics are much more complicated. Despite the importance of β-sheets as one of the most important secondary structure elements in proteins, the principles underlying their formation and stability are not understood in detail.32 In general, two major problems complicate detailed elucidation of the β-sheet folding motifs. The intrinsic tendency to aggregate usually results in solubility problems that make sample handling difficult. Furthermore, the absence of well determined cross-strand amino acid preferences in protein β-sheets constrains the use of long-range interactions in the de novo design.33 Thus, the design of β-sheets, that fold with high specificity, is a challenging topic in modern peptide chemistry.Various attempts have been made to design β-sheet peptides de novo. Due to the frequent occurrence of β-hairpin motifs in natural proteins, the majority of work in the early years focused on constructs that consist of two β-strands linked by a β-turn inducing element or a short loop.34 A schematic view of such a β-hairpin is shown in Fig. 4. Based on this work, many non aggregating multi-stranded peptides with up to eight β-strands have been synthesized, characterized, and applied in several studies to elucidate the general interaction pattern in β-sheets.33,35,36,37,38 Although the non-aggregating nature of these peptides facilitates detailed investigation of the β-sheet motif itself by high resolution methods such as NMR, these systems are usually unsuitable for use as models for studying peptide aggregation and fibril formation.
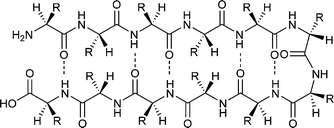 | ||
| Fig. 4 Schematic view of a β-hairpin peptide. | ||
Designing monostranded β-sheet peptides that are not linked by loops, turns, or other non-natural scaffolds is very complicated due to the absence of well-defined interaction patterns between peptide strands. Early studies revealed that an alternating pattern of hydrophobic and hydrophilic amino acids compared to those observed for amphipathic α-helices is required, especially if the peptide is associated with amyloid-like fibril formation.10 In 2000, Koide et al. proposed that β-sheet formation is not necessarily related to the formation of a hydrophobic core and showed that large amino acid side chains are able to stabilize the β-sheet by burying the non-polar surfaces.39,40 Interestingly, the interactions that force a peptide to fold into a β-sheet are in general not as different as those found for α-helical structures. A comparison of the major contributors to the stability of α-helices and β-sheets given in Table 1 shows only one main difference which is the pattern of hydrogen bond formations.
| α-Helical coiled coil | β-Sheet |
|---|---|
| Amino acid intrinsic secondary structure properties | |
| Sidechain–sidechain hydrophobic interactions | |
| Interstrand electrostatic interactions | |
| Intramolecular hydrogen bonds | Intermolecular hydrogen bonds |
The intrinsic secondary structure propensities of amino acids are an important factor. It is necessary, however, that these values are determined by calculating the total distribution in secondary structure elements of naturally occurring peptides and proteins.41 Thus, the suitability of these data for de novo design is restricted due to the partial incomparability of the structural data of large proteins and those obtained for small peptides. Hydrophobic effects are one of the major contributors to β-sheet stability. Numerous studies on β-hairpin peptides and strand-extended hairpins have shown that the burying of hydrophobic residues within the β-sheet strands is one of the major thermodynamic driving forces for the secondary structure formation.34,35,42,43 According to these findings, the general rule “the more hydrophobic, the more β” is at least partially true, although the incorporation of hydrophobic residues into a peptide does not necessarily result in the formation of a β-sheet secondary structure. Electrostatic interactions and salt bridges contribute significantly to β-sheet stability as well. As in case of α-helical coiled coil peptides, these electrostatic interactions are only minor contributors to overall stability, even though they assist the secondary structure formation and direct the specificity of folding.34,35 The last and perhaps most important contributor to β-sheet stability is hydrogen bonds. Unlike α-helical structures, where hydrogen bonds are formed intramolecularly within one helix, hydrogen bonds in β-sheets are formed between two different peptide strands. In conclusion, as in the case of α-helices, the important factors for β-sheet formation appear to be a balanced mixture of intrinsic amino acid propensities, hydrogen bonds, and hydrophobic effects. Thus, the general interaction patterns of α-helices and β-sheets are more similar to each other than expected. Indeed, the ability to form amyloid fibrils is not only limited to some proteins associated with known physical dysfunctions, but rather a widespread feature of many, perhaps all, polypeptide chains.44,45 Accordingly, amino acid side chains seem to be less important for fibril stability as they are stabilized by interactions involving the polypeptide main chain. However, the primary structure strongly affects the propensity of a peptide sequence to form amyloid fibrils, which can differ drastically between varying sequences under given circumstances.46
Inhibition of fibril formation
Whereas the soluble forms of proteins involved in amyloid diseases are highly various in sequence and structure, the aggregated forms have many structural characteristics in common, such as the binding of certain dye molecules e.g. Thioflavin T or Congo red. Many of the aggregates show typical fibrillar structures featuring very similar morphologies, namely unbranched and twisted structures with a diameter of a few nanometres and mostly several micrometres in length. In these aggregates β-sheets are found to be orientated perpendicularly to the fibril axis.46To develop therapies for amyloid-related diseases several strategies are currently being pursued. One of the most promising therapeutic strategy is the inhibition of peptide/protein aggregation using inhibitor molecules. To date a large number of diverse organic compounds are known to inhibit fibril formation. The ability to prevent aggregation of the β-amyloid peptide (Aβ), involved in Alzheimer's disease, has been reported for Congo red,47 the yellow curry pigment curcumin,48 β-cyclodextrin,49 aspirin,50 oligomeric aminopyrazoles51 as well as for nicotine which seems to prevent amyloid formation by binding either an α-helical or a β-sheet element within Aβ.52,53 Due to the lack of specificity and toxicity of most of these substances, their suitability as defined anti-agents of specific diseases is highly limited.
The design of peptide based inhibitors provides an attractive alternative to overcome such hurdles. Tjernberg et al. ascertained that the residues 16–20 within Aβ are essential for Aβ aggregation. The pentapeptide Aβ(16–20) is able to bind full length Aβ and prevents its polymerization into fibrils.54 Based on this finding, different peptide based inhibitors have been developed using the wild-type peptide as a lead structure.
Proline is well known as a β-sheet breaker amino acid. Soto and co-workers, one of the pioneers in the study of β-sheet breaker peptides, designed inhibitors for Aβ aggregation that incorporate proline into a short peptide sequence deduced from the recognition element of Aβ. This approach led to the generation of small peptides that inhibit amyloid formation in vitro as well as reduces cerebral Aβ deposition in rat brain.55 A similar strategy is the insertion of α-aminoisobutyric acid. This non-natural amino acid interrupts fibril aggregation due to its high tendency to induce helical conformations.56
The incorporation of N-methyl amino acids was shown to inhibit amyloidosis. These peptides bind to fibrils by presenting a complementary hydrogen-bonding face on one side, while the opposite side presents N-methyl instead of backbone NH groups. Thus, further fibril growth is disturbed by retarding hydrogen bond formation.57,58
The concept
The general interaction patterns of α-helices and β-sheets are very similar (see above). This led to the idea of developing a model of peptide sequences which contains structural elements for both stable α-helical folding as well as β-sheet formation as competing subunits. Ideally, this system could then be predicted to adopt one of these secondary structures at will by applying the appropriate environmental conditions.The structure as well as the design principles of the α-helical coiled coil folding motif have been subject to intense scrutiny.26 Therefore, this protein folding motif can serve as a perfect basis to start investigating the influence of a peptide primary structure and its modifications on the stability of the resulting secondary structure. The heptad repeat primary structure can be divided into two main domains; positions that form the dimerization domains (a, d, e, g) and positions that are solvent exposed (b, c, f).59 The fundamental idea of our design is that, although amino acids in positions b, c, f do not directly influence interactions between both helices, they still affect the stability of the helix monomer itself. Therefore, amino acids in these positions will have an impact on the cooperative interactions that induce helix dimerization. Furthermore, in the case where the same peptide would fold into a β-sheet, these amino acids will contribute to the stability of such an assembly equally to all other positions (Fig. 3).
Fig. 5 A shows the helical wheel presentation of the de novo designed parallel α-helical coiled coil basis peptide. Positions a and d are occupied by leucine residues, forming an ideal first recognition domain. In addition, inter- and intrahelical electrostatic interactions between position e, g, b, and c are exclusively optimized for attractions by the incorporation of oppositely charged lysine and glutamate residues. Thus, the observed secondary structure of this peptide is a perfectly folded α-helical coiled coil.
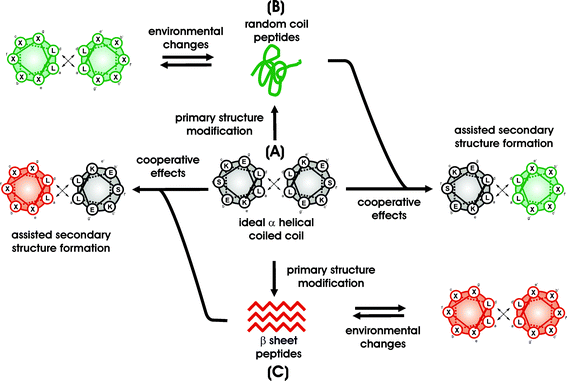 | ||
| Fig. 5 The general concept. | ||
Based on this ideal α-helical coiled coil sequence, several analogues were designed to receive peptides that can react to environmental changes upon alteration of their conformation. The structure of these peptides can be switched from the α-helical coiled coil to an unfolded form (see Fig. 5B) as well as into soluble β-sheets (see Fig. 5C). To maintain the opportunity for a complementary driven α-helical coiled coil folding, modifications of the primary structure have been only carried out at solvent exposed, non-coiled coil dimerization related b, c, and f positions. All the a, d, e, and g positions, which are important for α-helical coiled coil folding, have not been touched, but optimally maintained in terms of favorable hydrophobic and electrostatic interactions. Thus, the generated peptides exhibit two different secondary structure elements that are in direct competition to each other. More importantly, even unfolded and misfolded forms can still be involved in an α-helical coiled coil arrangement. As a result, adding equimolar amounts of the ideal α-helical coiled coil basis sequence (Fig. 5A) forces these peptides to cooperatively fold into a stable heteromeric α-helical structure.
A peptide containing the above-described features of an α-helical coiled coil as well as certain β-sheet inducing elements, has been designed and investigated to study the influence of different environmental parameters on the secondary structure switch from α-helix to β-sheet. The pH was shown to be a strong determinant of the preferred secondary structure and indirectly controls the time-dependent aggregation behavior of the peptide (Fig. 6). Thus, the formation of amyloid fibrils can be perceptibly inhibited or triggered by a change of pH.
 | ||
| Fig. 6 Time-dependent switch of a de novo designed β-sheet forming peptide. (a) CD spectra at different incubation times (0.5 mM peptide, 10 mM acetate buffer, pH 4.0). (b) Transmission electron microscopy (TEM) image of the resulting amyloids after 75 h (0.1 mM peptide, 10 mM acetate buffer, pH 4.0, negatively stained with 1% uranylformate). | ||
Metallochemical reactions are considered to be a common denominator for the development of neurodegenerative diseases as the concentration of heavier metal ions in brain tissue is naturally high.60 The Aβ deposits assembled in the brain of AD patients, as well as the strain variant conformation of PrPsc, have been reported to depend upon Cu2+ and Zn2+ interactions.60,61 The α-helical coiled coil based peptide model could be shown to serve as a simple system for a systematic study of the impact of different metal ions in their different oxidation states on a peptide secondary structure on a molecular level. With this system in hand we can study the impact of metal ion complexation, especially the interplay between the number of His complexation sites, its position within the heptad repeat, the nature of the metal ion as well as its concentration on the secondary structure formation in detail. His mutations were incorporated into the heptad repeat to generate complexation sites for Cu2+ and Zn2+ ions.62
Fig. 7 shows two peptide sequences, both containing several valine residues that decrease the propensity of helical folding. In addition, peptide CCM features four histidines per helix as metal-ion ligation sites. Due to the generally high β-sheet forming propensities of the peptides, trifluoroethanol (TFE) was used to assist a helical starting conformation.
 | ||
| Fig. 7 Helical wheel diagram (a) and sequence drawn as β-sheet (b) of peptide CC and CCM. | ||
Once Zn2+ or Cu2+ are added, the secondary structure with a helical content of 85% converted to 70% β-sheet (Fig. 8). Capturing the metal ions by the scavenger EDTA results in complete reversion of the structural change.
 | ||
| Fig. 8 CD spectra of peptide CCM in 40% TFE at pH 7.4 and at a peptide concentration of 0.1 mM with (a) 0.1 mM CuCl2 and (b) 0.1 mM Zn(OAc)2. | ||
The formation, relative stability, and possible stoichiometries of two (self) complementary peptide sequences (B and E) designed to form either a parallel homodimeric (B + B) or an antiparallel heteromeric (B + E) coiled coil have been investigated by molecular dynamics simulations and CD spectroscopic measurements.63 Peptide B shows a characteristic coiled coil pattern in the CD at pH 7.4, while peptide E has been shown to primarily adopt the unfolded random coil structure under these conditions (Fig. 9). The hydrophobic and charged recognition domains of both peptides are complementary to each other. Peptide E forms an α-helical coiled coil when mixed with peptide B. Thus, the ideally α-helically folded peptide B forces the mainly unfolded peptide E to adopt a helical structure by formation of a heteromeric coiled coil under neutral pH conditions. Molecular dynamics (MD) simulations showed that combinations of B + B and B + E readily form a dimeric coiled coil whereas E + E show a different behavior. However, the simulations strongly suggest the preferred orientation of the helices in the homodimeric α-helical coiled coil is parallel with interactions at the interface, which is quite different from the idealized model. Additionally, the MD simulations suggest an equilibrium between dimers, trimers, and tetramers of α-helices for peptide B.
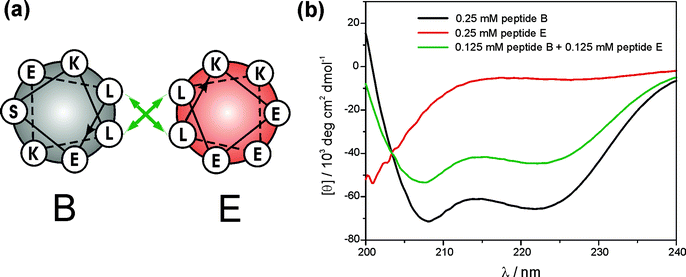 | ||
| Fig. 9 (a) Helical wheel diagram of peptides B and E. (b) CD spectra of peptide B, peptide E and a mixture of both measured in a 10 mM Tris-HCl buffer at pH 7.4 and 250 µM peptide concentration. | ||
Amyloid fibrils are usually formed with β-sheet containing structures as a direct precursor. Thus, the inhibition of the β-sheet formation should prevent amyloid fibrils from depositing. Peptide variants that fold into stable β-sheets (Fig. 5C) can be forced to readopt the helical conformation by forming a heteromeric coiled coil on interaction with the basis α-helical coiled coil peptide (Fig. 5-red/grey). One representative example is the fibril forming peptide presented in Fig. 6. This peptide has been shown to fold into a highly organized fibrillar structure at pH 4.0. The aggregation process can be inhibited by either a change in pH or the addition of a strong helix-forming peptide. This effect is based on the involvement of the β-sheet forming sequence into an α-helical coiled coil arrangement via complementary interactions between the hydrophobic heptad repeat domains (Fig. 10).
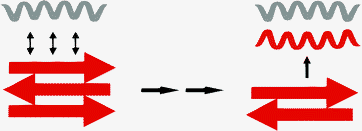 | ||
| Fig. 10 Concept of amyloid breaking by using a corresponding coiled coil peptide. | ||
Conclusion and perspective
A protein folding motif optimized by nature to fold into a stable α-helical coiled coil serves as a tool for the systematic study of the interplay between a primary structure and environmental conditions for the evolution of peptide secondary structure. Those newly designed peptides bear all of the features required for the formation of cooperatively interacting helical structures. Furthermore, they contain domains for cooperative sheet aggregation. In other words, peptides that follow the characteristic heptad repeat of the α-helical coiled coil structural motif are no longer necessarily α-helical. The resulting secondary structure will now strongly depend on the environment. Thus, this system allows us to study the subtle influences that environmental conditions might have on protein folding. Changes can be made stepwise, or in all of the possible combinations that nature applies in vivo. Information gained from these systematic investigations will contribute to the elucidation of complex protein folding processes. The model system introduced exhibits a new opportunity for the selective inhibition of fibril formation.Acknowledgements
We gratefully thank the VW foundation for its financial support and Dr G. Lutsch (Max-Delbrück-Center, Berlin) for the TEM measurements.References
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS.
- C. Soto, Nat. Rev. Neurosci., 2003, 4, 49–60 CrossRef CAS.
- G. Taube, Science, 1996, 271, 1492–1495 CrossRef.
- J. W. Kelly, Curr. Opin. Struct. Biol., 1996, 6, 11–17 CrossRef CAS.
- C. Soto, J. Mol. Med., 1999, 77, 412–418 CrossRef CAS.
- C. Soto, FEBS Lett., 2001, 498, 204–207 CrossRef CAS.
- J. P. Taylor, J. Hardy and K. H. Fischbeck, Science, 2002, 296, 1991–1995 CrossRef CAS.
- A. Schmechel, H. Zentgraf, S. Scheuermann, G. Fritz, R. Pipkorn, J. Reed, K. Beyreuther, T. A. Bayer and G. Multhaupt, J. Biol. Chem., 2003, 278, 35317–35324 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- M. Mutter, R. Gassmann, U. Buttkus and K. H. Altmann, Angew. Chem., 1991, 103, 11, 1504–1506 CrossRef CAS; M. Mutter, R. Gassmann, U. Buttkus and K.-H. Altmann, Angew. Chem. Int. Ed. Engl., 1991, 30, 1514–1516 CrossRef.
- Y. Takahashi, A. Ueno and H. Mihara, Chem. Eur. J., 1998, 4, 2475–2484 CrossRef CAS.
- Y. Takahashi, A. Ueno and H. Mihara, Bioorg. Med. Chem., 1999, 7, 177–185 CrossRef CAS.
- T. Yamashita, Y. Takahashi, T. Takahashi and H. Mihara, Bioorg. Med. Chem. Lett., 2003, 13, 4051–4054 CrossRef CAS.
- R. A. Kaemmerer, D. Kostrewa, J. Zurdo, A. Detken, C. Garcia-Echeverria, J. D. Green, S. A. Müller, B. H. Meier, F. K. Winkler, C. M. Dobson and M. O. Steinmetz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4435–4440 CrossRef CAS.
- H. L. Schenck, G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 12487–12494 CrossRef CAS.
- B. Ciani, E. G. Hutchinson, R. B. Session and D. Woolfson, J. Biol. Chem., 2002, 277, 10150–10155 CrossRef CAS.
- M. Mutter, A. Chandravarkar, C. Boyat, J. Lopez, S. Dos Santos, B. Mandal, R. Mimna, K. Murat, L. Patiny, L. Saucède and G. Tuchscherer, Angew. Chem., Int. Ed., 2004, 43, 4172–4178 CrossRef.
- Y. Peng and U. H. E. Hansmann, Phys. Rev. E, 2003, 68, 041911 CrossRef.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 2, 82–88 CrossRef CAS.
- E. Wolf, P. Kim and B. Berger, Protein Sci., 1997, 6, 1179–1189 CrossRef CAS.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- Y. B. Yu, Adv. Drug Deliv. Rev., 2002, 54, 1113–1129 CrossRef CAS and lit. cited therein.
- J. M. Mason and K. M. Arndt, Chem. Bio. Chem., 2004, 5, 170–176 CrossRef CAS.
- B. Tripet, K. Wagschal, P. Lavigne, C. T. Mant and R. S. Hodges, J. Mol. Biol., 2000, 300, 377–402 CrossRef CAS.
- M. G. Oakley and P. S. Kim, Biochem., 1998, 37, 12603–12610 CrossRef CAS.
- K. Pagel, K. Seeger, B. Seiwert, A. Villa, A. E. Mark, S. Berger and B. Koksch, Org. Biomol. Chem., 2005, 3, 1189–1194 RSC.
- O. D. Monera, C. M. Kay and R. S. Hodges, Biochem., 1994, 33, 3862–3871 CrossRef CAS.
- O. D. Monera, N. E. Zhou, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1993, 268(26), 19218–19227 CAS.
- D. L. McClain, J. P. Binfet and M. G. Oakley, J. Mol. Biol., 2001, 313, 371–383 CrossRef CAS.
- M. G. Oakley and J. J. Hollenbeck, Curr. Opin. Struct. Biol., 2001, 11, 450–457 CrossRef CAS.
- P. Burkhard, S. Ivaninskii and A. Lustig, J. Mol. Biol., 2002, 318, 901–910 CrossRef CAS.
- E. Lacroix, T. Kortemme, M. Lopez de la Paz and L. Serrano, Curr. Opin. Struct. Biol., 1999, 9, 487–493 CrossRef CAS.
- C. Das, S. Raghothama and P. Balaram, Chem. Commun., 1999, 11, 967–968 RSC.
- M. S. Searle, J. Chem. Soc., Perkin Trans. 2, 2001, 1011–1020 RSC.
- M. S. Searle and B. Ciani, Curr. Opin. Struct. Biol., 2004, 14, 458–464 CrossRef CAS.
- T. P. Quinn, N. B. Tweedy, R. W. Williams, J. S. Richardson and D. C. Richardson, Proc. Natl. Acad. Sci. USA, 1994, 91, 8747–8751 CAS.
- Y. Yan and B. W. Erickson, Protein Sci., 1994, 3, 1069–1073 CrossRef.
- T. Kortemme, M. Ramirez-Alvarado and L. Serrano, Science, 1998, 281, 253–256 CrossRef CAS.
- T.-N. Pham, A. Koide and S. A. Koide, Nat. Struct. Biol., 1998, 5, 115–119 CrossRef CAS.
- S. Koide, X. Huang, K. Link, A. Koide, Z. Bu and D. M. Engelmann, Nature, 2000, 403, 456–460 CrossRef CAS.
- P. Y. Chou and G. D. Fasman, Biochemistry, 1974, 13, 222–245 CrossRef CAS.
- C. N. Pace, B. A. Shirley, M. McNutt and K. Gajiwala, FASEB J., 1996, 10, 75–83 Search PubMed.
- G. T. Kilosanidze, A. S. Kutsenko, N. G. Esipova and V. G. Tumanyan, Protein Sci., 2004, 13, 351–357 CrossRef CAS.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef.
- S. Chen, V. Berthelier, J. B. Hamilton, B. O'Nuallain and R. Wetzel, Biochemistry, 2002, 41, 7391–7399 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- Y.-S. Kim, T. W. Randolph, M. C. Manning, F. J. Stevens and J. F. Carpenter, J. Biol. Chem., 2003, 278, 10842–10850 CrossRef CAS.
- F. Yang, G. P. Lim, A. N. Begum, O. J. Ubedal, M. R. Simmons, S. S. Ambegaokar, P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CAS.
- P. Camilleri, N. J. Haskins and D. R. Howlett, FEBS Lett, 1994, 341, 256–258 CrossRef CAS.
- T. Thomas, G. T. Nadackal and K. Thomas, Exp. Clin. Endocrinol. Diabetes, 2003, 111, 8–11 Search PubMed.
- P. Rzepecki, L. Nagel-Steger, S. Feuerstein, U. Linne, O. Molt, R. Zadmard, K. Aschermann, M. Wehner, T. Schrader and D. Riesner, J. Biol. Chem., 2004, 279, 47497–47505 CrossRef CAS.
- A. R. Salomon, K. J. Marcinowski, R. P. Friedland and M. G. Zagorski, Biochemistry, 1996, 35, 13568–13578 CrossRef CAS.
- S. A. Moor, T. N. Huckerby, G. L. Gibson, N. J. Fullwood, S. Turnbull, B. J. Tabner, O. M. A. El-Agnaf and D. Allsop, Biochemistry, 2004, 43, 819–826 CrossRef.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Casaño and B. Frangione, Nat. Med., 1998, 4, 822–826 CrossRef CAS.
- S. Gilead and E. Gazit, Angew. Chem. Int. Ed., 2004, 43, 4041–4044 CrossRef CAS.
- E. Hughes, R. M. Burke and A. J. Doig, J. Biol. Chem., 2000, 275, 25109–25115 CrossRef CAS.
- D. J. Gordon, K. L. Sciarretta and S. C. Meredith, Biochemistry, 2001, 40, 8237–8245 CrossRef CAS.
- K. Pagel, T. Vagt, B. Koksch, A bi-directional peptide switch: α-helical coiled coil interaction versus β-sheet formation in Abstracts of Papers, 3rd International Symposium on Conformational Control of Biolomolecular Functions, sponsored by VW-Foundation, SportSchloss Velen/Germany 2003, Search PubMedunpublished work.
- A. I. Bush, Curr. Opin. Chem. Biol., 2000, 4, 184–191 CrossRef CAS.
- J. D. F. Wadsworth, A. F. Hill, S. Joiner, G. S. Jackson, A. R. Clarke and J. Collinge, Nat. Cell Biol., 1999, 1, 55–59 CrossRef CAS.
- K. Pagel, T. Vagt, T. Kohajda and B. Koksch, Org. Biomol. Chem., 2005, 3, 2500–2502 RSC.
- À. Pineiro, A. Villa, T. Vagt, B. Koksch and A. Mark, Biophys. J., 2005 DOI:10.1529/biophysj.104.055590.
| This journal is © The Royal Society of Chemistry 2005 |