Tetrakis(imidazolium) macrocyclic receptors for anion binding†
Wallace W. H. Wong, Matthew S. Vickers, Andrew R. Cowley, Rowena L. Paul and Paul D. Beer*
Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QR. E-mail: paul.beer@chem.ox.ac.uk
First published on 31st October 2005
Abstract
New (tetrakis)imidazolium macrocyclic receptor systems of variable cavity size have been synthesised by stepwise alkylation reactions of bis(imidazolium) precursor compounds. Proton NMR titration studies reveal the macrocycles to strongly bind halide and benzoate anions, with two receptor systems displaying notable selectivity for fluoride in competitive acetonitrile–water (9 : 1) solvent media.
1. Introduction
Stimulated by the important role negatively charged species play in biological systems, medicine and the environment, interest in the synthesis of new host molecules designed to recognize and sense anions is ever increasing.1 Positively charged ammonium, guanidinium2 and neutral amide, urea and pyrrole groups3 have all been incorporated into various molecular frameworks to produce receptor systems capable of binding anions of diverse sizes and geometries.The imidazolium moiety has been recently shown to be an effective motif for complexing anions via favourable electrostatic interactions and hydrogen bonding.4 Although various acyclic imidazolium receptors have been described,5,6 synthetic routes to macrocyclic analogues are rare, especially those containing more than two imidazolium groups.7 We report here the synthesis of novel tetrakis(imidazolium) and benzimidazolium macrocyclic receptor systems which strongly bind halide and benzoate anions, with two receptors exhibiting notable selectively for fluoride anions in highly competitive aqueous–acetonitrile solvent mixtures.
2. Results and discussion
2.1 Syntheses
In an effort to examine the synergy of multiple imidazolium moieties for anion binding, we designed new tetrakis-imidazolium and benzimidazolium macrocyclic receptor systems, which may be considered as calix[4]imidazolium analogues of calix[4]pyrrole.8 Starting with commercially available imidazole and benzimidazole derivatives, a cascade of simple alkylation reactions was employed to synthesise the macrocycles in good yields.The synthesis of 1,3-bis(N-imidazolyl)propane 1 was achieved by imidazole ring formation starting from commercially avaliable N-(3-aminopropyl)imidazole (Scheme 1).9 The reaction of two equivalents of 1 with 1,3-diiodopropane gave the corresponding imidazole–imidazolium compound 2 in 60% yield. Reaction of 2 with a further equivalent of 1,3-diiodopropane followed by anion exchange with NH4PF6 (aq.) gave the cyclised product 3 in 80% yield (Scheme 1). This remarkably high yield for such a cyclised product suggests the iodide counteranions are playing a significant templating role in macrocycle formation. Indeed, chloride and bromide anions have been shown to exhibit a templation role in the synthesis of bis(imidazolium) macrocycles.10
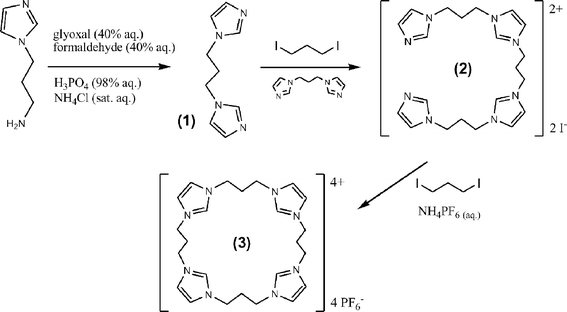 | ||
| Scheme 1 | ||
An analogous synthetic procedure was used to obtain tetrakis(benzimidazolium) receptors of three different macrocyclic sizes: 6, 9 and 11 (Schemes 2–4). Benzimidazole precursors 4 and 7 were obtained from the alkylation of benzimidazole with 1,3-dibromopropane and 1,4-dibromobutane respectively (Schemes 2 and 3). For the synthesis of the four-fold symmetric receptor 6, two equivalents of 4 were reacted with one equivalent of 1,3-diiodopropane to give 5 in 65% yield (Scheme 2). Precursor 5 was then reacted with a further equivalent of 1,3-diiodopropane to give macrocycle 6 in 31% yield (Scheme 2).
 | ||
| Scheme 2 | ||
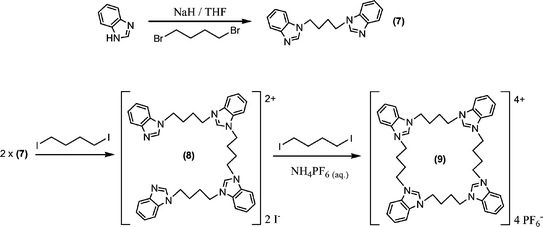 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
Reaction of two equivalents of 7 with 1,4-diiodobutane afforded 8 in 46% yield. The butyl spaced macrocycle 9 was prepared via reaction of 8 with 1,4-diiodobutane in 28% yield (Scheme 3).
The reaction of 7 with 1,3-diiodopropane gave benzimidazole–benzimidazolium compound 10 and subsequent cyclisation of 10 with a further equivalent of 1,3-diiodopropane afforded receptor 11 in 30% yield after anion exchange with aqueous NH4PF6 (Scheme 4).
For comparison purposes a new acyclic tetrapodal imidazolium receptor was also prepared. The synthesis of the tetrapodal resorcinarene cavitand imidazolium receptor 14 involved a simple alkylation reaction of imidazole with tetra(bromomethyl)resorcinarene cavitand1112 followed by reaction with excess 1-bromohexane and anion exchanged to give the pure product in 37% yield (Scheme 5).
 | ||
| Scheme 5 | ||
All four new tetrakis(imidazolium) macrocycles 3, 6, 9, 11 and tetrapodal cavitand imidazolium receptor 14 were characterised by 1H, 13C NMR (Fig. 1) spectroscopy, electrospray mass spectrometry and elemental analysis.†
 | ||
| Fig. 1 13C NMR spectrum or receptor 9 in CD3CN. | ||
It is noteworthy that the high field 1H NMR spectra of 6 and 9 reveal a dominant singlet at around 9 ppm for the respective methine (C–H)+ imidazolium proton superimposed upon a much less intense multiplet (Fig. 2 and 3). No evidence of higher oligomers was seen in the electrospray mass spectrum for any of the four macrocycles, only the one plus charged species corresponding to the tetrameric products plus three counter anions with a difference of one mass unit in the respective isotope envelope pattern was observed. This implies the additional multiplet resonances in this methine imidazolium proton region are due to the presence of multiple conformers in solution. Moreover, the 1H NMR spectrum of 6 also displays more than one set of resonances assigned to the aromatic protons of the benzimidazolium moieties (Fig. 2). Resembling the known conformational isomers of calix[4]arenes, in principle these macrocycles may adopt cone, 1,3-alternate, 1,2-alternate and partial cone conformers in solution.12 Variable temperature 1H NMR spectroscopic experiments up to 100 °C in DMSO-d6 solution however, resulted in no significant changes in the NMR spectra of the macrocycles.
 | ||
| Fig. 2 1H NMR spectrum of receptor 6 in CD3CN. | ||
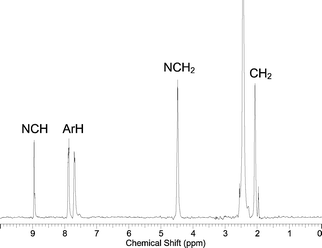 | ||
| Fig. 3 1H NMR spectrum of receptor 9 in CD3CN. | ||
2.2 X-Ray crystal structures
Bis(benzimidazole) precursor compound 4 and receptor 11 were also characterised by single crystal X-ray crystallography. Crystals of 4 suitable for X-ray structural analysis were grown by the slow diffusion of diethyl ether into a chloroform solution of 4. The crystal system is orthorhombic and the molecule lies on a site with no crystallographic symmetry, but closely approximates to local twofold rotational symmetry (Fig. 4). | ||
| Fig. 4 Thermal ellipsoid plot of the bis(benzimidazole) precursor 4. Thermal ellipsoids are drawn at the 40% probability level. | ||
Crystals of 11 suitable for X-ray analysis were grown by the slow evaporation of acetonitrile from a solution of receptor 11 in an acetonitrile–water mixture. The crystal system is triclinic and the macrocyclic tetracation is located on a crystallographic centre of inversion (Fig. 5). Two PF6− ions (related by the inversion centre) lie partly within the macrocyclic ring where the four benzimidazolium moieties are arranged in a cis two up two down 1,2-alternate conformation (Fig. 6).
 | ||
| Fig. 5 Thermal ellipsoid plot of the tetrakis(benzimidazolium) receptor 11. Thermal ellipsoids are drawn at the 50% probability level. The four PF6− counter anions and solvent molecules are omitted for clarity. | ||
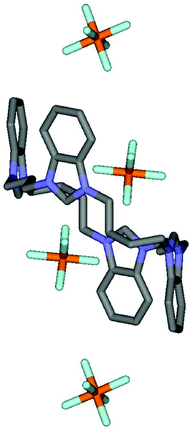 | ||
| Fig. 6 Stick representation of receptor 11 in a 1,2-alternate conformation with two counter PF6− anions lying partially within the macrocyclic cavity. | ||
2.3 Anion binding studies
The complexation of halide and benzoate anions by the macrocyclic receptors 3, 6, 9, 11 and cavitand 14 was investigated by 1H NMR spectroscopic titration experiments. These anionic guests were chosen for their variety in size, shape and charge density in order to probe the differences in the host–guest binding properties of the receptors. Due to the solubility characteristics of the macrocyclic receptors and their receptor–anion complexes, a highly competitive solvent mixture of acetonitrile-d3 and water (9 : 1) was used throughout.In all cases the addition of anions caused significant downfield perturbations of the methine (C–H)+ imidazolium proton resonances of each receptor. Interestingly, with receptor 6 the addition of 1 equivalent of TBA fluoride resulted in the methine proton resonances simplifying to a singlet, which implies that anion complexation results in a single conformer in solution (Fig. 7).
 | ||
| Fig. 7 The aromatic region of the 1H NMR spectra of receptor 6 upon the addition of 1 equivalent of TBAF to a CD3CN solution. | ||
Monitoring the most intense imidazolium methine (C–H)+ proton resonance produced titration curves and Job plots (refer to the electronic supplementary information†) which, after EQNMR,13 a least squares computer fitting program, analysis of the titration binding isotherms (Fig. 8) gave stability constant values displayed in Table 1.
| Receptors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Anion | 3 | 6 | 9 | 11 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Solvent = CD3CN–H2O 9 : 1, temperature = 295 K and errors are ± 10%.b Mixed host–guest binding stoichiometry prevented the calculation of a sensible stability constant value.c Precipitation occurred during titration.d The stoichiometry of receptor–benzoate binding is 1 : 2. K1 and K2 are quoted in the table.e Minimal changes observed in the chemical shift of the imidazolium proton. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| F− | b | >104 | c | >104 | 180 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cl− | b | 1100 | c | 710 | 85 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Br− | b | 1050 | c | 500 | 90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I− | 370 | 560 | 900 | 470 | 125 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BzO−d | K1 1800 | K1 1070 | K1 1700 | K1 2700 | e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| K2 470 | K2 1000 | K2 940 | K2 600 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 | ||
| Fig. 8 1H NMR titration data for the addition of the TBA salt of various halides to a CD3CN–H2O (9 : 1) solution of receptor 6. | ||
Receptors 6 and 11 form extremely strong and selective 1 : 1 stoichiometric complexes with fluoride, with estimated stability constant values of K >104 M−1. Attempts to repeat the titration experiments with increasing amounts of water unfortunately led to solubility problems. Stoichiometric 1 : 1 complexes were also found with chloride, bromide and iodide anions with a halide selectivity trend of F− >> Cl− ≈ Br− > I− for receptor 6 and F− >> Cl− > Br− ≈ I− for the larger macrocycle 11 (Table 1).
Presumably these stable halide complexes result from the four imidazolium moieties of 6 and 11 binding the halide anionic guest in a cooperative manner forming strong imidazolium (C–H)+-X− hydrogen bonds. The equilibrium geometry of macrocycle 6 with a bound fluoride anion was calculated using Merck Molecular Force Field (MMFF) using the SPARTAN 04 modelling program14 and reveals the fluoride anion to be closely bound within the macrocyclic cavity to each of the benzimidazolium moieties (Fig. 9). Similar equilibrium geometries were calculated for both the larger chloride and bromide anions however, indicating that the pronounced fluoride selectivity exhibited by 6 has little to do with receptor cavity–anion size complementarity. This suggests the more basic character of the fluoride anion dictates the observed selectivity trends with these macrocyclic receptors.
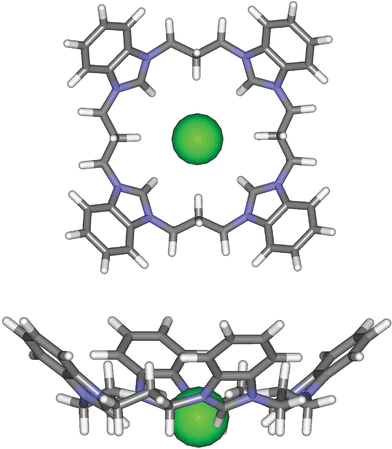 | ||
| Fig. 9 Molecular mechanics model for the 1 : 1 host–guest binding between receptor 6 and fluoride calculated using SPARTAN 04. | ||
It is noteworthy that macrocycle 6 complexes chloride and bromide anions with similar stability (Kca. 1100 M−1). A comparison of the receptor stability constant values with iodide reveals that 9 binds this halide guest anion most strongly, which implies a degree of macrocycle size complementarity.
With receptor 3, a combination of 1 : 1 and 1 : 2 binding stoichiometries were observed with fluoride, chloride and bromide. In a typical titration experiment a steady downfield shift of the imidazolium protons was observed until ca. two equivalents whereupon a large perturbation occurred indicative of a change in binding stoichiometry (Fig. 10). Unfortunately, it was not possible to determine stability constant values from such titration isotherm data.
 | ||
| Fig. 10 Titration curve of receptor 3 and TBA Cl monitoring the methine proton in CD3CN–H2O 9 : 1. | ||
In contrast to macrocycles 6 and 11, the cavitand receptor 14 only weakly binds halide anions (Table 1) which presumably reflects the tetrapodal receptor's unfavourable cavity size and lack of preorganisation. Indeed, molecular mechanics modelling (SPARTAN 04)14 of the cavitand receptor showed the upper rim appended imidazolium moieties to be highly flexible resulting in the absence of a preorganised binding site.
With the benzoate anion, a binding stoichiometry of 1 : 2 was obtained for all four macrocyclic receptors. The relative size of the carboxylate anion may be responsible for this result. Benzoate being an essentially planar molecule and larger than the halide anions, cannot fit inside the macrocyclic cavities of the receptors. Similar to the hexafluorophosphate anions shown in the solid state structure of 11 (Fig. 6), benzoate anions are likely to reside only partially within the macrocyclic cavity of the receptors and hence the observation of 1 : 2 receptor–benzoate binding stoichiometry. The stability constant values (K2) for the binding of the second benzoate are of significant magnitudes (Table 1). This observation suggests the presence of two independent binding sites for benzoate with these macrocyclic systems which also, taking into account the crystal structure of 11, suggests one benzoate anion may be binding to each side of the receptor. Interestingly, there was no evidence of cavitand receptor 14 binding benzoate in this aqueous–acetonitrile solvent mixture.
3. Conclusions
New (tetrakis)imidazolium macrocyclic receptor systems of various sizes have been prepared via sequential alkylation reactions of bis(imidazolium) precursor derivatives. Proton NMR titration investigations revealed the macrocycles to strongly bind halide anions with two macrocycles 6 and 11 exhibiting pronounced selectivity for binding fluoride in a competitive aqueous–acetonitrile (CD3CN–H2O 9 : 1) solvent media. Evidence for a macrocyclic cavity size effect comes from the largest macrocycle 9 forming the strongest complex with iodide.All four macrocycles complex the benzoate anion with a 1 : 2 receptor–anion binding stoichiometry. A new tetrapodal cavitand imidazolium receptor was also synthesized and proved to be an inferior anion complexing agent which modelling studies attribute to the absence of a preorganised binding site. This highlights the importance of the preorganised cyclic (tetrakis)imidazolium receptor design for anion recognition efficacy.
4. Experimental
4.1 General details
NMR spectra were recorded on Varian utility 300 and 500 MHz spectrometers. Mass spectrometry was carried out on a Micromass LCT electrospray mass spectrometer (ESMS, cone voltage = 20–50 V, desolvation temp. = 80 °C, source temp. = 60 °C). Elemental analysis was performed at the Inorganic Chemistry Laboratory, Oxford. Crystal data (Table 2) was obtained using Mo Kα radiation on an Enraf–Nonius KappaCCD diffractometer.‡ Crystals were mounted on a glass fibre and cooled rapidly to 150 K in a stream of cold nitrogen using an Oxford Cryosystems CRYOSTREAM unit. Intensity data were processed using the DENZO-SMN package.15 Structures were solved by direct methods using the SIR92 program.16 Full-matrix least-squares refinement was carried out using the CRYSTALS program suite.17 Hydrogen atoms were positioned geometrically after each cycle of refinement. A Chebychev polynomial weighting scheme was applied.Molecular modelling studies were calculated using Merck Molecular Force Field (MMFF) using the SPARTAN 04 modelling program.14 The molecular models were constructed in three stages using SPARTAN. The geometry of the benzimidazolium moiety was first refined using the basic molecular mechanics refinement in the program. The macrocyclic receptor was constructed by linking the benzimidazolium moieties and then its geometry was refined. The equilibrium geometry of the macrocyclic receptor was calculated using the Merck Molecular Force Field (MMFF). A variety of anions were then inserted into the model of the receptor and, maximising the number of hydrogen bonds, the equilibrium geometry of the receptor plus anion was calculated using MMFF.
MMFF uses a two stage optimisation routine where atoms are allowed to move large distances followed by small distances. The full developmental details of MMFF can be found in reference 18.
| 4 | 11 | |
|---|---|---|
| Chemical formula | C17H16N4 | C46H54F24N10P4 |
| Formula weight | 276.34 | 1326.85 |
| Temperature/K | 150 | 150 |
| Wavelength/Å | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | P b c n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 25.1991(4) | 9.6741(2) |
| b/Å | 8.6736(2) | 12.6292(2) |
| c/Å | 12.6158(2) | 13.1081(2) |
| α (deg) | 90 | 65.7093(10) |
| β (deg) | 90 | 71.7542(9) |
| γ (deg) | 90 | 83.9297(7) |
| Cell volume/Å3 | 2757.40(9) | 1385.83(4) |
| Z | 8 | 1 |
| Calculated density/Mg m−3 | 1.331 | 1.590 |
| Absorption coefficient/mm−1 | 0.082 | 0.262 |
| F000 | 1168 | 676 |
| Crystal size/mm | 0.20 × 0.20 × 0.40 | 0.10 × 0.10 × 0.32 |
| Description of crystal | Colourless prism | Colourless prism |
| Absorption correction | Semi-empirical from equivalent reflections | Semi-empirical from equivalent reflections |
| Transmission coefficients (min, max) | 0.97, 0.98 | 0.92, 0.97 |
| θ range for data collection (deg) | 5.0 ≤ θ ≤ 27.5 | 5.0 ≤ θ ≤ 27.5 |
| Index ranges | 0 ≤ h ≤ 32, 0 ≤ k ≤ 11, 0 ≤ l ≤ 16 | −11 ≤ h ≤ 12, −14 ≤ k ≤ 16, 0 ≤ l ≤ 16 |
| Reflections measured | 18754 | 41262 |
| Unique reflections | 3564 | 6294 |
| Rint | 0.045 | 0.064 |
| Observed reflections (I > 3σ(I)) | 2021 | 3552 |
| Refinement method | Full-matrix least-squares on F | Full-matrix least-squares on F |
| Parameters refined | 190 | 379 |
| Weighting scheme | Chebychev 3-term polynomial | Chebychev 3-term polynomial |
| Goodness of fit | 1.1219 | 1.1089 |
| R | 0.0337 | 0.0526 |
| wR | 0.0381 | 0.0611 |
| Residual electron density (min, max)/e Å−3 | −0.18, 0.19 | −0.49, 0.62 |
4.2 Syntheses
Imidazole, benzimidazole and the alkylating agents used herein were purchased from Sigma Aldrich whilst 3-aminopropyl imidazole was purchased from Acros Organics and were used without further purification. Tetra(bromomethyl)resorcinarene cavitand 12 was synthesised via a literature procedure.111H NMR (CDCl3, 288 K) δ (ppm) = 7.28 (s, 2H, NCHN), 6.88 (s, 2H, NCHCHN), 6.74 (s, 2H, NCHCHN), 3.74 (t, 6.9 Hz, 4H, N–CH2–), 2.10 (quintet, 6.9 Hz, 2H, –CH2–). MS (ESI positive ion, MeOH): m/z 177 (M + H+).
1H NMR (d6-acetone, 288 K) δ (ppm) = 9.05 (s, 2H, NCHN), 8.89 (s, 2H, NCHN), 7.06–7.95 (m, 8H, NCHCHN), 4.33 (m, 12H, N–CH2–), 2.52 (m, 6H, –CH2–). MS (ESI positive ion, MeOH): m/z 521 [M − I−]+.
1H NMR (CD3CN, 288 K) δ (ppm) = 8.51 (s, 4H, imidazole), 7.46 (s, 8H, imidazole), 4.23 (t, 7.2 Hz, 16H, N–CH2–), 2.43 (quintet, 7.2 Hz, 8H, –CH2–). 13C NMR (75 MHz, CD3CN, 288 K) δ (ppm) = 136.26 (NCHN), 123.24 (NCHCHN), 46.84 (N–CH2–), 30.22 (CH2). MS (ESI positive ion, MeOH): m/z 363 [M − 2PF6−. Anal. calcd for C24H36N8P4F24: C, 28.35; H, 3.54; N, 11.02. Found: C, 28.45; H, 3.51; N, 11.47.
1H NMR (CDCl3, 288 K) δ (ppm) = 7.84 (m, 4H, NCHN), 7.29 (m, 6H, ArH), 4.17 (t, 6.6 Hz, 4H, N–CH2–), 2.52 (quintet, 6.6 Hz, 2H, –CH2–). MS (ESI positive ion, MeOH): m/z 277 (M + H+).
1H NMR (d6-DMSO, 288 K) δ (ppm) = 9.99 (m, 2H, NCHN), 9.79 (m, 2H, NCHN), 7.18–8.29 (m, 16H, ArH), 4.30–4.73 (m, 12H, N–CH2–), 2.52 (m, 6H, –CH2–). MS (ESI positive ion, MeOH): m/z 721 [M − I−]+.
1H NMR (CD3CN, 288 K) δ (ppm) = 8.97 (m, 4H, NCHN), 8.01 (m, 2H, ArCH), 7.93 (m, 6H, ArCH), 7.79 (m, 2H, ArCH), 7.74 (m, 6H, ArCH), 4.62 (t, 7.2 Hz, 16H, N–CH2-), 2.65 (quintet, 7.2 Hz, 8H, –CH2–). 13C NMR (75 MHz, CD3CN, 288 K) δ (ppm) = 141.50 (NCHN), 131.97 (ArC), 128.02 (ArCH), 113.92 (ArCH), 44.8 (–NCH2–), 28.8 (CH2). MS (ESI positive ion, MeCN): m/z 1071 [M − PF6−]+. Anal. calcd for C40H44N8P4F24.H2O: C, 38.88; H, 3.73; N, 9.08. Found: C, 38.84; H, 3.84; N, 9.06.
1H NMR (CDCl3, 288 K) δ (ppm) = 7.79 (m, 4H, NCHN), 7.29 (m, 6H, ArH), 4.12 (m, 4H, N–CH2–), 1.88 (m, 4H, –CH2–). MS (ESI positive ion, MeOH): m/z 291 (M + H+).
1H NMR (d6-DMSO, 288 K) δ (ppm) = 9.85 (m, 2H, NCHN), 9.79 (m, 2H, NCHN), 7.18–8.29 (m, 16H, ArH), 4.55 (m, 8H, N–CH2–), 4.31 (m, 4H, N–CH2–), 2.02 (m, 8H, –CH2–), 1.88 (m, 4H, –CH2–). MS (ESI positive ion, MeOH): m/z 763 (M − I−)+, 318 (M2+).
1H NMR (CD3CN, 288 K) δ (ppm) = 8.95 (bs, 4H, NCHN), 7.86 (m, 8H, ArH), 7.69 (m, 8H, ArH), 4.47 (br, 16H, N–CH2–), 2.06 (br, 16H, –CH2–). 13C NMR (75 MHz, CD3CN, 288 K) δ (ppm) = 141.25 (NCHN), 131.95 (ArC), 127.59 (ArCH), 113.94 (ArCH), 47.09 (N–CH2–), 26.15 (–CH2–). MS (ESI positive ion, MeCN): m/z 1127 [M − PF6−]+. Anal. calcd for C44H52N8P4F24: C, 41.52; H, 4.12; N, 8.80. Found: C, 41.60; H, 4.10; N, 8.85.
1H NMR (d6-DMSO, 288 K) δ (ppm) = 8.05 (s, 2H, NCHN), 8.00 (s, 2H, NCHN), 7.14–7.82 (m, 16H, ArH), 4.14–4.60 (m, 12H, N–CH2–), 1.88 (br, 2H, –CH2–), 1.75 (br, 8H, –CH2–). MS (ESI positive ion, MeOH): m/z 749 (M − I−)+.
1H NMR (CD3CN, 288 K) δ (ppm) = 8.86–8.94 (m, 4H, NCHN), 7.90 (m, 8H, ArH), 7.71 (m, 8H, ArH), 4.45–4.62 (br m, 16H, N–CH2–), 2.66 (br m, 4H, propyl CH2), 2.09 (br, 8H, butyl CH2). 13C NMR (75 MHz, CD3CN, 288 K) δ (ppm) = 149.72 (NCHN), 132.00, 131.90 (ArC), 127.73, 127.84 (ArCH), 114.02, 113.80 (ArCH), 47.15 (N–CH2–), 44.70 (N–CH2–), 28.63, 26.07 (–CH2–); MS (ESI positive ion, MeCN): m/z 1099 [M − PF6−]+. Anal. calcd for C42H48N8P4F24: C, 40.91; H, 3.97; N, 9.18. Found: C, 40.53; H, 3.89; N, 9.00.
1H NMR (300 MHz, CDCl3) δ (ppm) = 7.11 (s, 4H, ArH), 6.01 (d, J = 6.6 Hz, 4H, OCH2 [Houter]), 4.75 (t, J = 7.8 Hz, 4H, ArCH), 4.53 (d, J = 6.9 Hz, 4H, OCH2 [Hinner]), 4.40 (s, 8H, CH2Br), 2.17 (m, 8H, ArCHCH2), 1.33 (m, 24H, CH2), 0.89 (t, J = 6.9 Hz, 12H, CH3). Anal. calcd for C56H68Br4O8.½H2O: C, 56.16; H, 5.81; Found: C, 56.06; H, 5.68.
1H NMR (300 MHz, CDCl3, 288 K) δ (ppm) = 7.38 (s, 4H, NCHN), 7.16 (s, 4H, ArH), 6.99 (s, 4H, NCHCHN), 6.87 (s, 4H, NCHCHN), 5.90 (d, J = 6.6 Hz, 4H, OCH2 [Houter]), 4.85 (s, 8H, N–CH2), 4.70 (t, J = 7.8 Hz, 4H, ArCH), 4.08 (d, J = 6.9 Hz, 4H, OCH2 [Hinner]), 2.16 (m, 8H, ArCHCH2), 1.34 (m, 24H, CH2), 0.88 (t, J = 6.9 Hz, 12H, CH3). MS (ESI positive ion, MeCN): m/z 1137 [M + H+].
1H NMR (300 MHz, CD3CN, 288 K) δ(ppm) = 8.53 (s, 4H, NCHN), 7.54 (s, 4H, ArH), 7.38 (s, 8H, NCHCHN), 6.23 (d, J = 6.6 Hz, 4H, OCH2 [Houter]), 5.13 (s, 8H, N–CH2), 4.68 (t, J = 7.8 Hz, 4H, ArCH), 4.55 (d, J = 6.9 Hz, 4H, OCH2 [Hinner]), 4.13 (t, J = 6.9 Hz, 8H, hexyl CH2), 2.16 (m, 8H, ArCHCH2), 1.82 (m, 8H, hexyl CH2), 1.38 (m, 24H, CH2), 3.15 (m, 24H, hexyl CH2), 0.89 (m, 24H, CH3). 13C NMR (75 MHz, CD3CN, 288 K) δ (ppm) = 153.2, 139.0, 135.5, 123.5, 122.6, 122.4, 121.0, 99.7, 49.7, 42.9, 37.5, 31.8, 30.7, 29.4, 27.2, 25.3, 22.3, 22.1, 13.4 and 13.2 ppm. MS (ESI positive ion, MeCN): m/z 883 [M − 2PF6−]2+, 1912 [M − PF6−]+.
4.3 1H NMR titration protocol
1H NMR spectroscopic titration experiments were carried out at ca. 293 K on a 500 MHz Varian Utility Spectrometer. Data were analysed using the computer fitting program EQNMR©. For further details on the workings of the EQNMR computer program please refer to Professor M. J. Hynes.13 To calculate stability constants the binding stoichiometry must be known and the rate of exchange between the complexed and the free receptor must be rapid on the NMR timescale. Therefore the signal is a weighted average of the signals of the two species.The procedure for 1H NMR titrations was as follows: 1.5 × 10−6 moles of host was dissolved in 500 µL of 9 : 1 CD3CN–H2O (so as to avoid deuterium exchange). 3 × 10−5 moles of TBA anion salt was dissolved in 400 µL of the same solvent corresponding to one equivalent having a volume of 20 µL. A proton NMR of the host was recorded and subsequently aliquots of the anion solution were added to the host (11 × 0.2 eq., 2 × 0.4 eq., 2 × 1 eq., 1 × 2 eq. and 1 × 3 eq.). After each addition of anion, another 1H NMR was recorded.
Acknowledgements
We gratefully acknowledge the EPSRC for studentships (W.W.H.W and M.S.V.).References
- (a) R. Martínez-Máňez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef CAS; P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40(3), 486 CrossRef; P. A. Gale, Coord. Chem. Rev., 2003, 240, 191 CAS; F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS; P. D. Beer and D. K. Smith, Prog. Inorg. Chem., 1997, 46, 1 CAS; J. L. Atwood, K. T. Holman and J. W. Steed, Chem. Commun., 1996, 1401 RSC; Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-Espãna, New York, Chichester, Wiley-VCH, 1997 Search PubMed; (b) P. D. Beer, Acc. Chem. Res., 1998, 31, 71 CrossRef CAS; A. P. de Silva, H. Q. N. Guarantee, T. Gunnlaugsson, A. J. Huxley, C. P. McCoy, J. T. Rademacher and T. R. Rice, Chem. Rev., 1997, 97, 1515 CrossRef.
- (a) C. R. Bondy, P. A. Gale and S. J. Loeb, J. Am. Chem. Soc., 2004, 126, 5030 CrossRef CAS; (b) K. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101 CAS; (c) J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17 CAS; (d) F. P. Schmidtchen, Org. Lett., 2002, 4, 431 CrossRef CAS.
- (a) K. Kavallieratos, C. M. Bertao and R. H. Crabtree, J. Org. Chem., 1999, 64, 1675 CrossRef CAS; (b) T. R. Kelly and M. H. Kim, J. Am. Chem. Soc., 1994, 116, 7072 CrossRef CAS; (c) P. A. Gale, J. L. Sessler, V. Kral and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140 CrossRef CAS.
- (a) S. Ramos, E. Alcalde, G. Doddi, P. Mencarelli and L. Perez-Garcia, J. Org. Chem., 2002, 67, 8463 CrossRef CAS; (b) M. S. Vickers, K. Martindale and P. D. Beer, J. Mater. Chem., 2005, 15, 2784 RSC.
- (a) S. K. Kim, N. J. Singh, S. J. Kim, H. G. Kim, J. K. Kim, J. W. Lee, K. S. Kim and J. Yoon, Org. Lett., 2003, 12, 2083 CrossRef; (b) J. Yoon, S. K. Kim, N. J. Singh, J. W. Lee, Y. J. Yang, K. Chellappan and K. S. Kim, J. Org. Chem., 2004, 69, 581 CrossRef CAS; (c) J. Y. Kwon, N. J. Singh, H. N. Kim, S. K. Kim, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2004, 126, 8892 CrossRef CAS.
- (a) S. Yun, H. Ihm, H. G. Kim, C. W. Lee, B. Indrajit, K. S. Oh, Y. J. Gong, J. W. Lee, J. Yoon, H. C. Lee and K. S. Kim, J. Org. Chem., 2003, 68, 2467 CrossRef CAS; (b) H. Ihm, S. Yun, H. G. Kim, J. K. Kim and K. S. Kim, Org. Lett., 2002, 4, 2897 CrossRef CAS; (c) K. Sato, S. Arai and T. Yamagishi, Tetrahedron Lett., 1999, 40, 5219 CrossRef CAS; (d) W. W. H. Wong, D. E. Phipps and P. D. Beer, Polyhedron, 2004, 23, 2821 CrossRef CAS.
- Two examples of tetrakis(imidazolium) macrocycles can be found in the literature: (a) K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899 CrossRef CAS; (b) K. Sato, T. Onitake, S. Arai and T. Yamagishi, Heterocycles, 2003, 60(4), 779 CrossRef CAS.
- P. A. Gale, P. Anzenbacher Jr. and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57–102 CrossRef CAS.
- A. A. Gridnev and I. M. Mihaltseva, Synth. Commun., 1994, 24, 1547 CrossRef CAS.
- E. Alcalde, S. Ramos and L. Perez-Garcia, Org. Lett., 1999, 1, 1035 CrossRef CAS; R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460 CrossRef CAS.
- H. Boerrigter, W. Verboom and D. N. Reinhoudt, J. Org. Chem., 1997, 62, 7148 CrossRef CAS.
- C. D. Gutsche, Calixarenes, 1989 Search PubMed; T. Harada, J. M. Rudzinski, E. Osawa and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1992, 2109 Search PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- Wavefunction Inc., 2004, SPARTAN 04 program for PC Search PubMed.
- Z. Otwinowski, W. Minor, Processing of X-Ray Diffraction Data Collected in Oscillation Mode (Methods Enzymol.), ed C. W. Carter and R. M. Sweet, Academic Press, 1997, p. 276 Search PubMed.
- A. Altomare, G. Cascarano, G. Giacovazzo, A. Guagliardi, M. C. Burla, G. Ploidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- D. J. Watkin, C. K. Prout, J. R. Carruthers, P. W. Betteridge, R. I. Cooper, CRYSTALS issue 11, Chemical Crystallography Laboratory, Oxford, UK, 2001 Search PubMed.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 616 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Titration data, Job plots, ESMS data. See DOI: 10.1039/b510068b |
| ‡ CCDC reference numbers 283001 and 283002. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b510068b |
| This journal is © The Royal Society of Chemistry 2005 |