Glycosylation with in situ separation: carbohydrate chemistry on a TLC plate†
Balaram Mukhopadhyaya, Peter Curab, K. P. Ravindranathan Kartha‡a, Catherine H. Bottingb and Robert A. Field*a
aCentre for Carbohydrate Chemistry, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: r.a.field@uea.ac.uk
bCentre for Biomolecular Sciences, University of St Andrews, St Andrews, UK KY16 9ST
First published on 23rd August 2005
Abstract
Iodine vapour promotes thioglycoside-based glycosylation chemistry on TLC plates, which in turn permits in situ separation by conventional elution with solvent.
Over the past twenty years, the largely neglected biological function of carbohydrates and glycoconjugates has received much attention. However, the precise molecular detail of the role of such biomolecules remains to be established in many, perhaps most, cases.1 There is, therefore, a need for methodologies to provide rapid access to structurally diverse carbohydrate structures. Synthetic procedures have developed substantially of late,2 with orthogonal glycosylation strategies offering scope for minimising manipulations at the anomeric centre during multi-step synthesis,3 as does the application of reactivity tuning4 based on the concept of ‘armed’ and ‘disarmed’ glycosylation reagents.5 Further extension of this strategy has led to ‘programmable’ oligosaccharide synthesis.6 Perhaps the ultimate in oligosaccharide target synthesis has been achieved by Seeberger and co-workers, who adapted technologies from peptide synthesis to develop instrument-based, automated oligosaccharide synthesis.7 Further work from the same group has explored microreactors for optimisation of glycosylation chemistry.8 On a more general front, “random” strategies for preparing carbohydrate libraries have been explored.9
This laboratory has a long-standing interest in the use of iodine in carbohydrate chemistry,10 particularly in relation to glycosyl donor activation.11 Since iodine is readily vapourised and is often used to detect organic compounds on TLC plates, we reasoned that thioglycosides co-spotted with sugar alcohols onto TLC plates might give rise to glycosides on exposure to iodine vapour. We were encouraged to follow this line by literature reports of low-tech chemistries for accessing compound libraries12 and by studies showing silica-supported, solvent-free synthesis of nucleosides,13 the impact of silica on the AgNO3/NCS-mediated cyclisation of an alcohol onto a dithioketal14 and on NBS-promoted glycosylation reactions,15 and the established use of silver silicate as a heterogeneous promoter of SN2-like glycosylation processes.16 Here we report a proof-of-concept for iodine vapour-promoted glycosylation on TLC plates employing armed thioglycosides as donors.17,18
In initial experiments, armed thiogalactoside donor 1 and primary alcohol acceptor methyl 2,3,4-tri-O-benzyl-β-D-galactopyranoside 2 were co-spotted on a standard analytical silica TLC plate and exposed to iodine vapour.19 After 30 min, the plate was removed from the reaction vessel and developed with organic solvent. Subsequent charring of the plate with ethanolic sulfuric acid showed only the conversion of the thioglycoside donor to the corresponding hemiacetal, 3 (Fig. 1);20 no glycoside formation was apparent, as judged by comparison with authentic α/β-disaccharide, 4, produced by conventional solution phase coupling. It was evident that the moisture present in the silica caused the hydrolysis of the donor. When a similar reaction was carried out on pre-dried silica gel plates, traces of disaccharide products, 4, became apparent, but we were unable to produce useful quantities of material for characterisation. The principle product of the reaction still proved to be hemiacetal 3,20 along with smaller amounts of a compound that co-ran with authentic per-O-benzylated 1,1′-linked Gal–Gal disaccharide 5.21 Repetition of the experiment (fifteen 1.5 × 10 cm plates) and extraction of relevant material from the silica (with CHCl3) produced sufficient material for accurate mass characterisation, confirming formation of the 1,1′-linked sugar.22 NMR and mass spectral data for all disaccharides isolated are included in the electronic supplementary information.
 | ||
| Fig. 1 Glycosylation on TLC plates. A–C on a silica plate, D–F on an alumina plate. A – Hemiacetal 3; B – reaction of donor 1 and acceptor 2; C – donor 1 and acceptor 2; D – donor 1 and acceptor 2; E – reaction of donor 1 and acceptor 2 on an alumina plate; F – authentic α/β-disaccharides 4. | ||
Thorough drying of silica TLC plates is not straightforward; when heated at more than 100 °C for a couple of hours, the silica gel on the plate detached from the glass surface. Considering this point, and taking into account that hydrogen iodide generated in situ might lead to acid-catalysed product hydrolysis, we were minded to investigate alumina as an alternative to silica. Glass-backed alumina TLC plates are robust and remain intact after drying at 200 °C for 3 h. The result changed dramatically when pre-dried alumina plates were employed in on-plate glycosylation reactions; reaction between thioglycoside 1 and galactoside primary alcohol 2 led to approximately 40% conversion to the desired disaccharides 4 (Fig. 2). In an attempt to increase the conversion, reaction times were varied. However, for donor 1 the most productive results were obtained from approx. 30–60 min reactions. In the reaction of thioglycoside 1 and alcohol 2, repetition of the on-plate experiment (fifteen 1.5 × 10 cm plates) and extraction of relevant material from the TLC plates19 produced sufficient material for accurate mass measurement as well as for 1H and 13C NMR characterisation. The disaccharide obtained proved to be an approx. 2 : 1 α : β-mixture, as judged by integration of 1H NMR signals for the methyl groups of the reducing terminal sugars (assignment assisted by the relative intensity of anomeric carbon signals in the 13C NMR data).
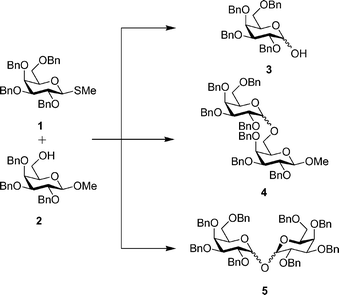 | ||
| Fig. 2 Model glycosylation reaction conducted on TLC plates and promoted by iodine vapour. | ||
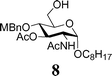
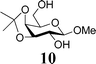
In light of the above success, a range of armed donor thioglycosides and variously protected acceptor sugar alcohols were also investigated. A combination of 3 donors (1, 6, 7) and 4 acceptors (2, 8, 9, 10) was assessed (Table 1); in 6 of the 12 reactions investigated, conversion was sufficient (30–60%) to enable characterisation of the disaccharide products formed. In some of the other reactions investigated, disaccharide formation was evident but tlc separation was poor.23 The relatively mild nature of these on-plate reactions is evident from their compatibility with potentially acid labile protecting groups in all of the acceptor alcohols investigated. Reactions with the ‘armed’ thioglycosides of deoxysugar L-fucose, 7, gave maximum conversion within 15–30 minutes. These observations can be rationalized considering the higher reactivity of the thioglycosides of deoxy-sugars. Of a number of secondary alcohol acceptors investigated (not shown), only the 6-deoxy acceptor 9 gave identifiable disaccharide products, consistent with the greater reactivity of 6-deoxysugar acceptors. In experiments with diol 11, only disaccharide products were identifiable. On the basis of NMR assignment after acetylating the disaccharide obtained (downfield shift of H-2 of galactose from 3.65 to 5.03 ppm upon acetylation), these were identified as the anomeric 1,6-linked sugars, as it to be expected given the greater reactivity of Gal 6-OH over 2-OH.24
| Acceptors | ||||
|---|---|---|---|---|
| Donors |
|
|
|
|
| N.i. - no disaccharide isolable from tlc plate. | ||||
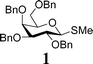 | (4) 2 : 1 | N. i. | (11) 1:1 | (12) 2 : 1 |
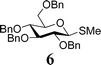 | N. i. | (13) 3 : 1 | N. i. | N. i. |
 | (14) 1 : 1 | N. i. | N. i. | (15) 1 : 1 |

In conclusion, armed thioglycoside donor substrates spotted onto TLC plates can be activated in situ by iodine vapour. Whilst poor results were obtained on silica, alumina plates gave rise to coupling efficiencies of up to ca. 50–60%, enabling the synthesis of a range of disaccharides and their subsequent separation and characterisation on the tens of micrograms scale. This microscale, iodine-mediated glycosylation strategy on a TLC plate minimizes the time and labour of the glycosylation reaction and subsequent purification process. With further optimisation, this approach offers scope for multiple parallel synthesis of glycoside libraries; alternative formats for scale-up are currently being investigated.
Acknowledgements
This work was supported by the EPSRC, the Weston Foundation, the Wellcome Trust and the University of St Andrews. We thank Dr Alan Haines for useful discussions during the course of this work and Colin Macdonald for assistance with acquiring NMR spectra. We gratefully acknowledge the EPSRC Mass Spectrometry Service Centre, Swansea for invaluable support.Notes and references
- A. Varki, Glycobiology, 1993, 3, 97 CrossRef CAS and citations therein.
- (a) Best Synthetic Methods: Carbohydrates, ed. H. M. I. Osborn, Academic Press, London, 2003 Search PubMed; (b) T. Kanemitsu and O. Kanie, Comb. Chem. High Throughput Screening, 2002, 5, 339 CAS; (c) S. N. Baytas and R. J. Linhardt, Mini-Rev. Org. Chem., 2004, 1, 27 Search PubMed.
- O. Kanie, Y. Ito and T. Ogawa, J. Am. Chem. Soc, 1994, 116, 12073 CrossRef CAS.
- D. K. Baeschlin, L. G. Green, M. G. Hahn, B. Hinzen, S. J. Ince and S. V. Ley, Tetrahedron: Asymmetry, 2000, 11, 173 CrossRef CAS.
- (a) B. Fraser-Reid, U. E. Udodong, Z. F. Wu, H. Ottosson, J. R. Merritt, C. S. Rao, C. Roberts and R. Madsen, SYNLETT, 1992, 927 CrossRef CAS; (b) G. H. Veeneman and J. H. van Boom, Tetrahedron Lett., 1990, 31, 275 CrossRef CAS.
- (a) X. S. Ye and C.-H. Wong, J. Org. Chem., 2000, 65, 2410 CrossRef CAS ; reviewed in P. Sears and C.-H. Wong, Science, 2001, 291, 2344 Search PubMed.
- (a) O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523 CrossRef CAS; (b) Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries, ed. P. H. Seeberger, Wiley-Interscience, New York, 2001 Search PubMed.
- D. M. Ratner, E. R. Murphy, M. Jhunjhunwala, D. A. Snyder, K. F. Jensen and P. H. Seeberger, Chem. Commun., 2005, 578 RSC.
- (a) O. Kanie, F. Barresi, Y. L. Ding, J. Labbe, A. Otter, L. S. Forsberg, B. Ernst and O. Hindsgaul, Angew. Chem., Int. Ed., 1996, 34, 2720 CrossRef; (b) Y. L. Ding, J. Labbe, O. Kanie and O. Hindsgaul, Bioorg. Med. Chem., 1996, 4, 683 CrossRef CAS; (c) B. Yu, B. Li, G. W. Xing and Y. Z. Hui, J. Org. Chem., 2001, 3, 404 CAS.
- Iodine, a versatile reagent in carbohydrate chemistry XVIII. For part XVII see: R. M. van Well, K. P. R. Kartha and R. A. Field, J. Carbohydr. Chem., 2005 Search PubMed in press.
- K. P. R. Kartha, T. S. Kärkkäinen, S. J. Marsh and R. A. Field, SYNLETT, 2001, 260 CAS.
- For instance: (a) the exploitation of microwave irradiation in the synthesis of sulfonamides on TLC plates: L. Williams, Chem. Commun., 2000, 435 Search PubMed; (b) the use of cellulose chromatography paper as a novel non-covalent support for synthesis and in situ purification of multi-dimensional arrays: S. E. Shanahan, D. D. Byrne, G. G. A. Inglis, M. Alam and S. J. F. Macdonald, Chem. Commun., 2002, 2554 Search PubMed; (c) solid-phase synthesis of unprotected N-glycopeptide building blocks for SPOT synthesis: L. Jobron and G. Hummel, Angew. Chem., Int. Ed., 2000, 39, 1621 Search PubMed; (d) microwave-accelerated SPOT-synthesis of spatially addressable libraries on cellulose supports: M. D. Bowman, R. C. Jeske and H. E. Blackwell, Org. Lett., 2004, 6, 2019 Search PubMed.
- (a) G. V. Reddy, V. R. Kulkarni and H. B. Mereyala, Tetrahedron Lett., 1989, 30, 4283 CrossRef; (b) H. B. Mereyala and G. V. Reddy, Tetrahedron, 1991, 47, 6435 CrossRef CAS.
- K. C. Nicolaou, M. E. Duggan and C. K. Hwang, J. Am. Chem. Soc., 1986, 108, 2468 CrossRef CAS.
- K. Fukase, A. Hasuoka, I. Kinoshita, Y. Aoki and S. Kusumoto, Tetrahedron, 1995, 51, 4923 CrossRef CAS.
- (a) C. A. A. van Boeckel, T. Beetz and S. F. van Aelst, Tetrahedron, 1984, 40, 4097 CrossRef CAS; (b) C. A. A. van Boeckel, T. Beetz, A. C. Kock-van Dalen and H. van Bekkum, Recl. Trav. Chim. Pays-Bas, 1997, 106, 596 Search PubMed.
- We have previously shown that armed, but not disarmed, thioglycosides can be activated with molecular iodine: K. P. R. Kartha, M. Aloui and R. A. Field, Tetrahedron Lett., 1996, 37, 5175 Search PubMed.
- Other armed glycosyl donors (glycosyl bromides and glycosyltrichloroacetimidates) were investigated in on-plate reactions but glycosylation products were not identifiable; only hemiacetal arising from quenched donor were obtained.
- Typical procedure for glycosylation on a TLC plate: a solution of donor (0.12 µmol) and acceptor (0.10 µmol) in DCM (10 µL) was spotted (1 µL) on a pre-dried TLC plate using a micro-pipette (alumina plates were heated at 200 °C for 2 h and allowed to cool over P2O5 in a desiccator prior to use). The plate was placed in the main chamber of the apparatus (see the electronic supplementary information) and dried over P2O5 under reduced pressure for 1 h. Then iodine vapour was introduced by heating solid iodine kept in the adjacent flask. The TLC plate was kept exposed to iodine vapour for 30–60 min (Table 1). After the reaction, the TLC plate was kept under vacuum for 30 min to remove iodine vapour and it was developed by using a suitable solvent system (e.g.n-hexane–EtOAc 2 : 1). Compounds were identified under UV-light, a relevant band of silica was scratched from the plate, triturated with CH2Cl2–MeOH (1 : 1) and filtered through Celite. The filtrate was evaporated and material obtained was used directly for NMR and mass spectrometry analysis.
- K. Koike, M. Sugimoto, S. Sato, Y. Ito, Y. Nakahara and T. Ogawa, Carbohydr. Res., 1987, 163, 189 CrossRef CAS.
- H. M. Zuurmond, P. A. M. van der Klein, P. H. van der Meer, G. A. van der Marel and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1992, 111, 365 CAS.
- It did not immediately prove possible for this particular experiment to obtain convincing NMR data from this scale of experiment, hence the anomeric stereochemistry of compound 4 was not confirmed. It is well established that the presence of inorganic solids can have a profound impact on the stereochemistry of trehalose formation—see: A. H. Haines, Carbohydr. Res., 2003, 338, 813 Search PubMed and references cited therein.
- A change to use of more lipophilic protecting groups offers scope for substantial changes in chromatographic properties: U. J. Nilsson, E. J. L. Fournier and O. Hindsgaul, Bioorg. Med. Chem., 1998, 6, 1563 Search PubMed.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1982, 21, 155 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental procedures, NMR spectra. See http://dx.doi.org/10.1039/b509417h |
| ‡ Present address: Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, Sector 67, SAS Nagar, Punjab, 160062, India. |
| This journal is © The Royal Society of Chemistry 2005 |