DOI:
10.1039/B508972G
(Paper)
Org. Biomol. Chem., 2005,
3, 3488-3495
A new and efficient method for o-quinone methide intermediate generation: application to the biomimetic synthesis of the benzopyran derived natural products (±)-lucidene and (±)-alboatrin
Received
28th June 2005
, Accepted 25th July 2005
First published on 7th September 2005
Abstract
Lucidene and alboatrin are complex benzopyran derived natural products. A key step in their biogenesis may involve a hetero Diels–Alder cycloaddition between an o-quinone methide intermediate with a simple, or activated tri-substituted olefin. Experimental evidence is provided to support this hypothesis, with the biomimetic synthesis of both (±)-lucidene and (±)-alboatrin successfully achieved using a new and efficient method for o-quinone methide generation.
Introduction
In recent years many structurally novel benzopyran derived natural products have been isolated from organisms as diverse as plants, animals, insects and fungi (Scheme 1). Many of these compounds display interesting biological properties including activity against Plasmodium falciparum,1 phytotoxic properties,2 erythropoietin gene expression,3 and acetylcholine esterase inhibition.4 As part of our continuing efforts directed towards the biomimetic synthesis of natural products, we became interested in studying these class of benzopyranic natural products which include (±)-lucidene 1,5
(+)-alboatrin 2,2
(+)-pughiinin A 3,1 and (+)-epolone A 43
(Scheme 1), and in developing methodology that would allow ready access to the common benzopyran core structure. We have recently reported a biomimetic synthesis of (±)-alboatrin 2, which involved a novel hetero Diels–Alder cycloaddition of an o-quinone methide intermediate and a readily accessible dienophile.6a We now wish to report a complete account of these studies which include a new method for the generation of o-quinone methide intermediates, and their application towards the biomimetic synthesis of complex natural products.
 |
| | Scheme 1 | |
o-Quinone methides are highly reactive transient species, which have been applied as intermediates in the synthesis of several natural products.6,7 Such compounds are known to react with nucleophiles in 1,4-Michael addition type fashion, or with a range of dienophiles to perform [4 + 2] cycloaddition to provide benzopyran type structures. Many strategies have been established in order to generate o-quinone methides in situ. However, problems with such protocols often include undesirable high temperatures,7,8 long reaction times,4,7,8 the need for catalysis,7,9 and/or acidic7 or basic conditions,7,10a which can induce problematic side reactions. In addition, the o-quinone methide precursors necessary for use with existing methodologies are often unstable and relatively inaccessible.7 Thus, we chose to investigate alternative methodologies for the generation of o-quinone methides for the application towards natural product synthesis.
Results and discussion
We have previously reported the generation of o-quinone methide by the thermal driven dehydration of an o-hydroxybenzyl alcohol precursor 5.6c Although this method proved synthetically useful, the reaction temperatures necessary to facilitate dehydration were undesirable and potentially disruptive to delicate structures. However, we were keen to build upon this methodology and uncover a more attractive o-quinone methide precursor. We envisaged a solution to the problem might involve activation of the benzyl alcohol moiety of the o-quinone methide precursor, thus facilitating dehydration through a more attractive acetyl leaving group. To investigate our hypothesis, we prepared the o-quinone methide precursors 6a and 6b
(Scheme 2), since benzopyran sub units are common structural features of several natural products (Scheme 1). Although o-acetoxymethylphenols (including 6b) have been described in the literature, members of this class of compounds have been reported as being labile structures which “can be conserved for several days in dilute solution but polymerise rapidly as soon as they are pure”.10a Their reported synthesis is likewise unattractive; for example Loubinoux has reported the need for a six-step synthesis of o-acetoxymethylphenol (6b) from salicylaldehyde.10b Perhaps as a consequence, reports of potential o-acetoxymethylphenols to serve as o-quinone methide synthons are limited to base promoted chemistry, followed by in situ nucleophilic Michael addition.10a We have been unable to find reports of their use for o-quinone methide generation under purely thermal conditions. In order to avoid the lengthy reported synthesis of 6b,10b we chose to investigate conditions that would allow selective acylation of the primary alcohol moiety of commercially available o-hydroxybenzyl alcohol 5. Subsequently, it was discovered that this selectivity could indeed be achieved by careful control of the reaction temperature and rate of addition of the acylating reagent, leading to the synthesis of 6b in an impressive 89% yield. In order to prepare 6a, we considered that a related selective acylation of the corresponding hydroxymethyl-orcinol derivative 7 could also be used. However, upon reduction of the corresponding aldehydic precursor, compound 7 was found to be unstable and rapidly decomposed under the reaction conditions. To address this problem, it was reasoned that suitable protection of the phenolic position may prevent premature decomposition. To this end, diacetate 811 was prepared, which we reasoned could be reduced to the corresponding alcohol, and that such a compound may facilitate o-quinone methide formation under thermal conditions from an o-acetoxymethylphenol 6a, itself generated via a transesterification mechanism. In practice, the transfer of the phenolic acetate to the adjacent benzyl alcohol group occurred during the reduction of 8 with borane–DMS complex, thus giving the stable compound 6a12
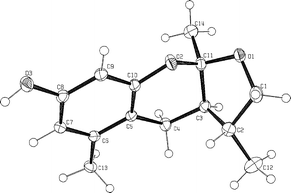
(Fig. 1)13 with a gratifying 84% yield. The proton NMR of 6a displayed a sharp signal at 8.23 ppm, characteristic of a hydrogen bond between the phenolic OH and the benzylic acetate. This was encouraging since it was envisaged that such a hydrogen bond may facilitate the elimination of acetic acid through a six-membered ring transition state, furnishing the desired o-quinone methide under relatively mild conditions.
 |
| | Fig. 1 X-Ray structure of 6a. | |
 |
| | Scheme 2
Reagents and conditions: (a) BH3–MS (1 equiv.), THF, 0 °C, to rt, 1 h, 84%; (b)
(±)-4,5-dihydro-2,4-dimethylfuran 9
(1equiv.), benzene, reflux, 36 h, 93% overall; (c) K2CO3
(3 equiv.), DCM–MeOH–H2O (12 : 7 : 1), rt, 6 h, 97%. | |
To investigate the potential of compounds 6a and 6b as o-quinone methide precursors, several readily available dienophiles were exposed to both compounds under a range of reaction conditions. The results from these studies are summarised in Table 1. Generally, the reaction times, temperatures required and yields obtained compare favourably with those described for other methods,4,7,8,14 and the reaction could be performed on very hindered dienophiles such as α-pinene. To further demonstrate the usefulness of this new method for o-quinone methide generation, we decided to investigate its potential towards the biomimetic synthesis of benzopyran containing natural products. With this in mind, we focused our initial efforts towards the synthesis of alboatrin (±)-2.
Table 1
o-Quinone methide (o-QMS) hetero-Diels–Alder cycloaddition products
|
o-QMS precursor |
Dienophile |
Temperature/°C |
Time/h |
Yield (%)a |
Adduct |
|
Yields were calculated after column chromatography. Reactions were performed in a sealed tube under argon.
This compound was synthesised in four steps starting from propionic acid.4
Dienophile used as solvent at 0.85 M.
The yield quoted refers to a mixture of epimers in a ratio of [12.6 : 1] in favour of (±)-10.
Compound (±)-25 was prepared by deacetylation of compound (±)-24
(see Experimental section).
The reaction has been carried out in toluene using 2.05 equiv. of 6b.
The reaction has been carried out in toluene using 4.10 equiv. of 6b.
Ratios have been determined by 1H NMR.
|
|
6a
|
(±)-4,5-Dihydro-2,4-dimethylfuran 9b |
80 |
36 |
63d |
Acetylalboatrin (±)-10 |
|
6a
|
4,5-Dihydro-2-methylfuran |
100 |
12 |
78c |
|
|
6a
|
3,4-Dihydro-2H-pyran |
100 |
12 |
72c |

|
|
6a
|
(1R)-(+)-α-Pinene |
140 |
12 |
30c |

|
|
6a
|
1-Methylcyclohexene |
140 |
12 |
60c |

|
|
6b
|
Styrene |
100 |
12 |
79c |

|
| (Other o-quinone methide precursors) |
|
(90 |
6.5 |
64)14a |
|
| |
|
(60 |
10.5 |
42)14a |
|
| |
|
(190 |
2 |
68)14a |
|
| |
|
(90–110 |
12 |
56)14b |
|
|
6a
|
α-Humulene 20 |
140 |
12 |
53c |
(±)-24e |
|
6b
|
α-Humulene 20 |
130 |
12 |
52f |
(±)-22 |
| |
|
|
|
38f |
(±)-1/(±)-23
[2.5 : 0.7]h |
|
6b
|
α-Humulene 20 |
130 |
12 |
0g |
(±)-22 |
| |
|
|
|
71g |
(±)-1/(±)-23
[2.5 : 0.7]h |
(+)-Alboatrin22 is a phytotoxic benzopyran natural product reported in 1988 by Ichihara et al., and later structurally corrected by Murphy et al.15 The biosynthesis of (+)-alboatrin 2 may be proposed to proceed through a hetero Diels–Alder cycloaddition of an orcinol-derived o-quinone methide 16, and (R)-4,5-dihydro-2,4 dimethylfuran 9
(Fig. 2). Indeed, Wilson et al. have made a similar connection and demonstrated in a recent study its feasibility as a major pathway to the structurally related natural product (−)-xyloketal A 17 and (−)-xyloketal D 184
(Fig. 3). In order to provide further biosynthetic details regarding the origin of (+)-2, and to further evaluate our methodology towards natural product synthesis, dienophile (±)-4,5-dihydro-2,4-dimethylfuran 9 was prepared according to the procedure of Wilson et al.4 Thereafter, simple heating of 6a in the presence of (±)-9 afforded (±)-acetylalboatrin 10 as the major isolated product (63% yield), (±)-acetyl-epi-alboatrin16
(5% yield), and an inseparable mixture [3 : 2] of diastereoisomers (±)-19
(25% yield). De-acylation of (±)-10 gave target (±)-alboatrin, for which spectral data were identical to the natural material [1H, 13C].2 The structure of (±)-2 was also confirmed by X-ray analysis and was found to be in agreement with Murphy's proposal15
(Fig. 4).13 With the successful application of this methodology towards the synthesis of (±)-2, we next decided to re-investigate the biomimetic synthesis of (±)-lucidene, employing this newly developed approach.
 |
| | Fig. 2 Biomimetic analysis of alboatrin (+)-2. | |
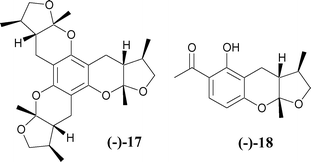 |
| | Fig. 3 | |
 |
| | Fig. 4 X-Ray structure of (±)-2. | |
(±)-Lucidene is a bis(benzopyranyl) sesquiterpene isolated from the root bark of Uvaria lucida ssp. lucida5 in racemic form. The co-isolation of α-humulene 20 from the same species led to speculation that lucidene is the product of two consecutive inverse demand hetero Diels–Alder cycloadditions of 20, with two equivalents of o-quinone methide 21
(Fig. 5).5 This hypothesis is attractive on the grounds that FMO theory supports the regiochemical and syn-stereochemical aspects17 of such inverse electron demand hetero Diels–Alder processes, since electronic factors would favour addition to the more electron rich tri-substituted double bonds at the expense of the more sterically hindered di-substituted double bond. It has also been reported that (E) double bonds in medium sized rings show a higher reactivity than otherwise expected on account of steric strain.18 We have previously provided convincing evidence to support this biosynthetic proposal, where thermolysis of o-hydroxybenzyl alcohol 5 was used to generate the o-quinone methide intermediate (Scheme 3).19 A summary of our previous study is provided in Table 2.6c The experimental results demonstrated that the naturally occurring diastereomer (±)-lucidene 1, is the favoured bis-adduct of α-humulene 20 with unsubstituted o-quinone methide 21 at high temperature. The isolation of monoadduct (±)-22en route to lucidene, offers positive evidence that a similar pathway may occur in the biosynthesis of (±)-lucidene 1. However, it was appreciated that the temperatures employed in the cycloaddition reaction were excessive.
Table 2
| Humulene 20 (equiv.) |
o-Hydroxybenzyl alcohol 5 (equiv.) |
Solvent |
Temp./°C |
Yield of (±)-22 (%) |
Yield of mixture (±)-1/(±)-23 (%)
[ratio] |
| 1 |
2.05 |
Xylene |
170 |
28 |
17 [2.5 : 1] |
| 1 |
2.05 |
Acetonitrile–water 1 : 1 |
170 |
32 |
7 |
| 1 |
2.05 |
1,4-Dioxane |
170 |
23 |
4 |
| 1 |
6 |
Xylene |
170 |
— |
45 (trace of tris-adduct)21 |
 |
| | Fig. 5 Biomimetic analysis of lucidene. | |
 |
| | Scheme 3 | |
To probe for further biosynthetic details, we applied our new method of o-quinone methide generation toward the synthesis of (±)-lucidene 1, as a comparison with our previous method which employed an unactivated o-quinone methide precursor 5.6c In the former case the reaction was carried out in toluene at 130–140 °C, as opposed to heating in xylene at 170 °C. When α-humulene was heated in the presence of 2.05 equivalents of compound 6b, monoadduct (±)-2220 was obtained in 52% yield, along with a mixture of bis-adducts (±)-1/(±)-23 in 38% yield, which favoured the natural product (±)-lucidene 1
[2.5 : 0.7]. The same reaction using 4.10 equivalents of precursor 6b gave an inseparable mixture of compound (±)-1/(±)-23 in the same ratio as above, with an encouraging 71% yield. In this case, no trace of mono-adduct or tris-adduct compounds were detected. In order to investigate the effect of temperature on the diastereoselectivity of the second cycloaddition process, compound (±)-22 was exposed to 2.0 equivalents of 6b in benzene at 90 °C for 36 hours. A mixture of compounds (±)-1/(±)-23 in 32% yield was obtained, with a similar ratio in favour of compound 1
[2.5 : 0.6].
In the same manner, we synthesised compound (±)-25
(Fig. 6)13 by condensation of precursor 6a with α-humulene (20)
(53% yield) to provide adduct (±)-24 and subsequent de-acetylation under basic conditions (82% yield)
(Scheme 4). We believe this building block constitutes a useful starting material for the biomimetic synthesis of pughiinin A 3 and epolone A 4 in racemic form.
 |
| | Fig. 6 X-Ray structure of 25. | |
 |
| | Scheme 4
Reagents and conditions: (a) neat humulene 20
(5 equiv.), 140 °C, 12 h, 53% overall; (c) K2CO3
(1.1 equiv.), DCM–MeOH–H2O (12 : 7 : 1), rt, 6 h, 82%. | |
Conclusion
In conclusion, we have developed a new and efficient method for o-quinone methide generation from o-acetoxymethyl-phenols. The described methodology required no added acid,7 base7,10b or catalyst7,9 for the formation of the Diels–Alder adducts. The elimination of acetic acid was not found to be detrimental to the reaction. The reaction times, temperatures required and yields obtained compare favourably with those described for traditional methods.4,7,8,14 The reaction can be performed on very hindered dienophiles such as α-pinene and on complex non-conjugated polyene such as α-humulene with high regio-, chemo- and stereoselectivity. Our novel method has been applied to a rapid biomimetic synthesis of the complex natural products (±)-alboatrin 2 and (±)-lucidene 1. Application to the biomimetic syntheses of (±)-pughiinin A 3 and (±)-epolone A-4 are objectives.
Experimental
All solvents and reagents were purified by standard techniques,22 or used as supplied from commercial sources as appropriate. Solvents were removed under reduced pressure using a Buchi R110 or R114 Rotavapor fitted with a water condenser. Final traces of solvent were removed from samples using an Edwards E2M5 high vacuum pump with pressures below 2 mmHg. All experiments were carried out under inert atmosphere unless otherwise stated. 1H NMR spectra were recorded at 200, 400 and 500 MHz using, Bruker DPX200, DQX400 and Bruker AMX500 instruments. For 1H spectra recorded in CDCl3, CD3OD, C6D6, chemical shifts are quoted in parts per million (ppm) and are referenced to the residual solvent peak. The following abbreviations are used: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad. Proton assignments and stereochemistry are supported by 1H–1H COSY and NOESY where necessary. Data are reported in the following manner: chemical shift (integration, multiplicity, coupling constant if appropriate). Coupling constants (J) are reported in Hertz to the nearest 0.5 Hz. 13C NMR spectra were recorded at 50.2, 100.6 and 125.8 MHz using Bruker DPX200, Bruker DQX400, and Bruker AMX500 instruments. Carbon spectra assignments are supported by DEPT-135 spectra, 13C–1H (HMQC) correlations where necessary. Chemical shifts are quoted in ppm and are referenced to the appropriate residual solvent peak. Flash column chromatography was carried out using Sorbsil™ C60 (40–63 mm, 230–40 mesh) silica gel. Thin layer chromatography was carried out on glass plates pre-coated with Merck silica gel 60 F254 which were visualised by quenching of UV fluorescence or by staining with 10% w/v ammonium molybdate in 2 M sulfuric acid or 1% w/v potassium permanganate in aqueous alkaline solution followed by heat, as appropriate. Melting points were recorded using a Cambridge Instruments Gallen™ III Kofler Block melting apparatus or a Buchi 510 capillary apparatus and are uncorrected. Infrared spectra were recorded either as a thin film between NaCl plates on a Perkin-Elmer Paragon 1000 Fourier Transform spectrometer with internal referencing. Absorption maxima are reported in wavenumbers (cm−1) and the following abbreviations are used: w, weak; m, medium; s, strong; br, broad. Low resolution mass spectra were recorded on V. G. Micromass ZAB 1 F and V. G. Masslab instruments as appropriate with modes of ionisation being indicated as CI, EI, ES or APCI with only molecular ions. High resolution mass spectrometry was measured on a Waters 2790-Micromass LCT electrospray ionisation mass spectrometer and on a VG autospec chemical ionisation mass spectrometer.
To a stirred solution of 2-hydroxybenzyl alcohol 5
(5.0 g, 40.2 mmol) in dry DCM (80 ml) was added slowly at 0 °C pyridine (3.3 ml, 40.2 mmol) and acetyl chloride (2.9 ml, 40.2 mmol) under nitrogen. The reaction mixture was then warmed to room temperature and stirred one hour. The reaction mixture was quenched with a saturated solution of ammonium chloride (100 ml) and washed three times with a saturated solution of copper sulfate (3 × 100 ml). The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to afford 6b as a colourless oil (6.0 g, 89%) which was used without further purification. RF 0.3 [80 : 20 30–40 petroleum ether (PE) : EtOAc]; νmax/cm−1
(film) 3369 (s), 1711 (s), 1490 (m), 1458 (m), 1383 (m), 1281 (s), 1244 (s), 909 (s), 734 (s); δH
(200 MHz, CDCl3) 2.12 (3H, s), 5.13 (2H, s), 6.88–7.00 (2H, m), 7.22–7.35 (2H, m), 7.81 (1H, s); δC
(100 MHz, CDCl3) 21.0, 63.4, 117.9, 120.6, 121.7, 131.3, 132.3, 155.6, 173.9; m/z
(ES−) 165.14 ([M − H]−). 6b was stable for over three months at 0 °C.
In a sealed tube was stirred 2-acetoxymethylphenol 6b
(200 mg, 1.2 mmol) in styrene (1.4 ml, 12.2 mmol) at 100 °C, under argon for 12 hours. After evaporation of excess of styrene under reduced pressure, the yellow oil was purified by flash silica gel chromatography (98 : 2 30–40 PE : EtOAc) to give 15 as a white solid (200 mg, 79%). Mp = 40–41 °C; RF 0.6 (95 : 5 30–40 PE : EtOAc); νmax/cm−1
(film) 3063 (m), 3029 (m), 2927 (m), 2846 (m), 1582 (s), 1488 (s), 1455 (s), 1235 (s), 754 (s); δH
(200 MHz, CDCl3) 2.02–2.32 (2H, m), 2.77–2.89 (1H, ddd, J 16.5, 5.0, 3.5 Hz), 2.96–3.13 (1H, m), 5.10 (1H, dd, J 9.5, 3.0 Hz), 6.89–6.99 (2H, m), 7.12–7.25 (2H, m), 7.32–7.51 (5H, m); δC
(50 MHz, CDCl3) 25.1, 30.0, 77.8, 117.0, 120.4, 121.9, 126.0 (2C), 127.4, 127.9, 128.6 (2C), 129.6, 141.8, 155.2.
2,4-Dihydroxy-6-methylbenzaldehyde11
Phosphorous oxychloride (5.6 ml, 60.5 mmol) was added dropwise over 10 minutes to stirring DMF (20 ml) at −10 °C under nitrogen, and the mixture stirred for a further 20 minutes. Orcinol (7.5 g, 60.5 mmol) in DMF (25 ml) was then added to the solution at −10 °C and the mixture allowed to warm to room temperature over 2 hours. To the reaction was then added ice and 10% aqueous NaOH until pH 9–10 was achieved, and a precipitate formed. The mixture was then heated to boiling for 10 minutes then cooled to room temperature. The acidity was then adjusted to pH 3, and the precipitate then formed was filtered and washed with water until neutral, then dried in a vacuum desiccator to give the 2,4-dihydroxy-6-methylbenzaldehyde as a yellow solid (3.5 g, 38%) which was used without further purification. Mp = 178–180 °C; RF 0.2 (75 : 25 30–40 PE : EtOAc); νmax/cm−1
(KBr) 3080 (m), 2926 (m), 1628 (s), 1482 (s), 1303 (s), 1233 (s), 1170 (s); δH
(400 MHz, MeOD) 2.50 (3H, s), 6.11 (1H, d, J 2.0 Hz), 6.22 (1H, d, J 2.0 Hz), 10.02 (1H, s); δC
(100 MHz, MeOD) 17.3, 100.5, 110.8, 113.0, 145.3, 166.2, 166.5, 193.2; HRMS (APCI) Calculated for C8H9O3
([M + H]+): 153.0552, Found: 153.0551.
To a stirred solution of 2,4 dihydroxy-6-methylbenzaldehyde (4.0 g, 26.3 mmol) in dry DCM (100 ml) was slowly added at 0 °C pyridine (4.6 ml, 58.0 mmol) and acetyl chloride (2.0 ml, 29.0 mmol) under nitrogen. The reaction mixture was warmed to room temperature, stirred two hours and then cooled to 0 °C when another quantity of acetyl chloride was slowly added (2.0 ml, 29.0 mmol). The reaction was stirred for a further two hours then quenched with a saturated solution of ammonium chloride (100 ml) and washed three times with a saturated solution of copper sulfate (3 × 150 ml). The combined organic layers were dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to give a yellow oil which was purified by flash silica gel chromatography (80 : 20 30–40 PE : EtOAc) to afford the desired product 8 as a colourless oil (4.4 g, 71%). RF 0.5 (70 : 30 30–40 PE : EtOAc); νmax/cm−1
(film) 2930 (w), 1774 (s), 1692 (s), 1610 (s), 1369 (m), 1193 (s), 1126 (s); δH
(200 MHz, CDCl3) 2.28 (3H, s), 2.34 (3H, s), 2.61 (3H, s), 6.85 (1H, d, J 2.0 Hz), 6.91 (1H, d, J 2.0 Hz), 10.30 (1H, s); δC
(50 MHz, CDCl3) 20.6, 20.9, 21.2, 114.8, 122.5, 123.9, 144.0, 153.7, 154.3, 168.4, 169.1, 188.8; m/z
(ES+) 254.19 ([M + NH4]+).
To a stirred solution of 2,4-diacetoxy-6-methylbenzaldehyde 8
(3.8 g, 16.1 mmol) in dry THF (90 ml) was slowly added a 2 M THF solution of borane–DMS complex (8.0 ml, 16.0 mmol) at 0 °C under nitrogen. The reaction was stirred for one hour at room temperature and was then quenched at 0 °C with water (2 ml). The mixture was evaporated to dryness under reduced pressure to give a crude product which was purified by flash silica gel chromatography (70 : 30 30–40 PE : EtOAc). The title compound was obtained as a white solid which was crystallised from ether to afford 6a as white crystals (3.2 g, 84%). Mp = 95–96 °C; RF 0.3 (70 : 30 30–40 PE : EtOAc); νmax/cm−1
(film) 3413 (br), 1779 (s), 1735 (s), 1708 (s), 1599 (m), 1370 (m), 1209 (s), 1133 (s); δH
(400 MHz, CDCl3) 2.10 (3H, s), 2.27 (3H, s), 2.38 (3H, s), 5.12 (2H, s), 6.53 (1H, s), 6.54 (1H, s) 8.23 (1H, s); δC
(100 MHz, CDCl3) 19.6, 21.0, 21.2, 59.6, 109.3, 115.8, 118.8, 141.2, 152.1, 157.3, 169.5, 174.3; HRMS (ES−) Calculated for C12H13O5
([M − H]−): 237.0763, Found: 237.0763. Crystal data for6a: C12H14O5, M
= 238.24, monoclinic, a
= 29.1890(14), b
= 4.9244(3), c
= 19.8157(11)
Å, U
= 2363.5(2)
Å3, T
= 150 K, space group C2/c, Z
= 8, µ(Mo-Kα)
= 0.105 mm−1, 11482 reflections measured, 2983 unique (Rint
= 0.045) which were used in calculations. The final wR was 0.0599.13
(±)-5a(R*),14a(S*),(E)-(E)-5a,6,9,10,13,14,14a,15-Octahydro-5a,9,9,12-tetramethylcycloundeca[1,2-b]benzopyran (22)
Procedure A.
In a sealed tube was stirred 2-acetoxymethylphenol 6b
(750 mg, 4.5 mmol) in dry toluene (6 ml) with α-humulene (0.52 ml, 2.2 mmol) at 130 °C, under argon for 12 hours. After evaporation of toluene under reduced pressure, the yellow oil was purified by flash silica gel chromatography (99 : 1 30–40 PE : EtOAc) to give the monoadduct (±)-22 as a white solid (353 mg, 52%) and an inseparable mixture (346 mg, 38%), [2.5 : 0.7] of diadduct (±)-lucidene 1 and (±)-isolucidene 23 as a white solid.
Procedure B.
In a sealed tube was stirred 2-acetoxymethylphenol 6b
(1.5 g, 9.0 mmol) in dry toluene (6 ml) with α-humulene (0.52 ml, 2.2 mmol) at 130 °C, under argon for 12 hours. After evaporation of toluene under reduced pressure, the yellow oil was purified by flash silica gel chromatography (99 : 1 30–40 PE : EtOAc) to give a white solid (641 mg, 71%) which was an inseparable mixture [2.5 : 0.7] of diadduct (±)-lucidene 1 and (±)-isolucidene 23. The compounds (±)-1 and (±)-23 were separated, for the purpose of characterisation by preparative HPLC. A reverse phase Hypersil C18 (25 cm × 0.25 inch) column was found to achieve the required separation with an aqueous acetonitrile solvent system (4 : 1 MeCN : H2O).
Compound (±)-22.
Mp = 118 °C; RF 0.7 (98 : 2 30–40 PE : EtOAc); νmax/cm−1
(KBr) 3056 (w), 2986 (m), 2928 (s), 2852 (m), 1584 (m), 1492 (s), 1456 (s), 1250 (s), 1136 (m), 1040 (m), 982 (s), 757 (s); δH
(500 MHz, CDCl3) 1.06 (3H, s), 1.10 (3H, s), 1.15 (3H, s), 1.17 (1H, m), 1.38 (1H, dd, J 13.0, 11.0 Hz), 1.68, (3H, s), 1.80 (1H, dd, J 12.5, 4.0 Hz), 1.88 (1H, dd, J 13.0, 11.0 Hz), 1.94 (1H, ddd, J 12.5, 9.0, 5.5 Hz), 2.16 (1H, dd, J 13.0, 7.5 Hz), 2.22 (1H, t, J 12.5 Hz), 2.35 (1H, dd, J 14.5, 10.0 Hz), 2.50 (1H, dd, J 16.5, 12.5 Hz), 2.58 (1H, dt, J 14.5, 2.0 Hz), 2.96 (1H, dd, J 16.5, 5.5 Hz), 5.06 (1H, br dd, J 12.5, 4.0 Hz), 5.16 (1H, dd, J 16.0, 2.0 Hz), 5.24 (1H, ddd, J 16.0, 10.0, 2.0 Hz), 6.86 (1H, d, J 7.5 Hz), 6.87 (1H, t, J 7.5 Hz), 7.10 (1H, d, J 7.5 Hz), 7.15 (1H, t, J 7.5 Hz); δC
(125 MHz, CDCl3) 17.2, 20.2, 24.3, 29.5, 30.3, 30.4, 35.6, 37.8, 38.2, 41.4, 43.1, 80.2, 117.0, 119.2, 121.0, 121.8, 123.1, 127.2, 128.8, 136.5, 142.0, 154.0; HRMS (CI+) Calculated for C22H31O ([M + H]+): 311.2375, Found: 311.2378.
(±)-5a(R*),10a(S*),16a(R*),18a(S*)-(E)-5a,6,9,10,10a,16a,17,18,18a,19-Decahydro-5a,9,9,16a-tetramethyl-11H-cycloundeca[1,2-b:5,6-b′]bisbenzopyran (lucidene)
(1)5
Mp = 208–211 °C; RF 0.6 (98 : 2 30–40 PE : EtOAc); νmax/cm−1
(film) 2921 (s), 1610 (w), 1585 (m), 1487 (s), 1454 (s), 1378 (m), 1304 (m), 1259 (s), 1220 (m), 753 (m); δH
(500 MHz, CDCl3) 1.09 (1H, dd, J 14.0, 5.5 Hz), 1.09 (3H, s), 1.13 (3H, s), 1.27 (6H, s), 1.55 (1H, m), 1.71 (1H, m), 1.83 (2H, m), 1.97 (1H, m) 2.06 (1H, br m), 2.22 (1H, m), 2.58 (1H, m,), 2.59 (2H, d, J 7.0 Hz), 2.67 (1H, br d, J 16.0 Hz), 2.81 (1H, dd, J 17.0, 5.5 Hz), 2.82 (1H, br m), 5.65 (1H, dt, J 16.0, 8.0 Hz), 5.79 (1H, d, J 16.0 Hz), 6.76 (1H, d, J 8.0 Hz), 6.77 (1H, d, J 8.0 Hz), 6.79 (1H, td, J 7.0, 1.0 Hz), 6.82 (1H, td, J 7.0, 1.0 Hz), 7.00 (1H, d, J 7.0 Hz), 7.06 (1H, d, J 7.0 Hz), 7.07 (2H, t, J 7.0 Hz); δC
(125 MHz, CDCl3) 19.6, 21.3, 23.7, 26.7, 29.1, 29.6, 30.0, 31.8, 32.6, 35.7, 40.8, 46.0, 49.0, 79.4, 79.7, 116.7, 117.2, 119.3, 119.6, 121.7, 122.8, 124.5, 127.1, 127.3, 128.7, 129.1, 143.9 (2C), 153.5; HRMS (CI+) Calculated for C29H37O2
([M + H]+): 417.2794, Found: 417.2782.
(±)-5a(R*),10a(R*),16a(S*),18a(S*)-(E)-5a,6,9,10,10a,16a,17,18,18a,19-Decahydro-5a,9,9,16a-tetramethyl-11H-cycloundeca[1,2-b:5,6-b′]bisbenzopyran (isolucidene)
(23)
Mp = 121 °C; RF 0.6 (98 : 2 30–40 PE : EtOAc); νmax/cm−1
(film) 2919 (s), 1610 (w), 1586 (s), 1499 (s), 1456 (s), 1377 (m), 1310 (m), 1254 (s), 1141 (m), 1102 (m), 1032 (m), 940 (w), 754 (s); δH
(500 MHz, CDCl3) 1.05 (1H, m), 1.05 (3H, s), 1.09 (3H, s), 1.15 (1H, br m), 1.20 (3H, s), 1.26 (3H, s), 1.55–1.68 (3H, m), 1.87–2.04 (3H, m), 2.41 (1H, dd, J 13.0, 7.5 Hz), 2.50–2.62 (3H, m), 2.78 (1H, dd, J 16.5, 5.5 Hz), 2.83 (1H, dd, J 16.5, 5.5 Hz), 5.48 (1H, ddd, J 15.5, 7.0, 6.0 Hz), 5.55 (1H, d, J 15.5 Hz), 6.76 (1H, d, J 7.5 Hz), 6.78 (1H, d, J 7.5 Hz), 6.82 (2H, t, J 7.5 Hz), 7.03 (1H, d, J 7.5 Hz), 7.05 (1H, d, J 7.5 Hz), 7.09 (1H, t, J 7.5 Hz), 7.10 (1H, t, J 7.5 Hz); δC
(125 MHz, CDCl3) 19.1, 20.9, 24.4, 29.6, 30.7, 30.8, 30.9, 32.6, 36.3, 38.3, 41.3, 47.6, 48.4, 79.6, 80.6, 116.7, 116.7, 119.3, 119.5, 121.6, 121.6, 122.5, 127.2, 127.3, 128.4, 128.8, 143.5, 153.4, 153.6; HRMS (CI+) Calculated for C29H37O2
([M + H]+): 417.2794, Found: 417.2784.
Procedure C.
In a sealed tube was stirred compound (±)-22
(130 mg, 0.42 mmol) in dry benzene (0.5 ml) with 2-acetoxymethylphenol 6b
(140 mg, 0.84 mmol) at 90 °C, under argon during 36 hours. After evaporation of benzene under reduced pressure, the yellow oil was purified by flash silica gel chromatography (99 : 1 30–40 PE : EtOAc) to give a white solid (56 mg, 32%) which was an inseparable mixture [2.5 : 0.6] of (±)-lucidene 1 and (±)-isolucidene 23. The mixture of two compounds was crystallized from hexane to afford selectively white crystals of (±)-1.
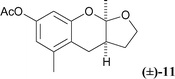
(±)-3a(R*),9a(R*)-2,3,3a,9a-Tetrahydro-5,9a-dimethyl-7-acetoxy-4H-furo[2,3-b]chroman (3-nor-methyl-acetyl-alboatrin)
(11)6a
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(203 mg, 0.85 mmol) in 4,5-dihydro-2-methylfuran (1.0 ml, 10.9 mmol) at 100 °C, under argon for 12 hours. After evaporation under reduced pressure, the yellow oil obtained was purified by flash silica gel chromatography (95 : 5 30–40 PE : EtOAc) to give (±)-11 as a white solid (175 mg, 78%). Mp = 115–117 °C; RF 0.2 (90 : 10 30–40 PE : EtOAc); νmax/cm−1
(film) 3056 (w), 2982 (w), 1757 (s), 1597 (m), 1482 (w), 1368 (w), 1265 (w), 1213 (w), 1109 (w), 736 (s); δH
(400 MHz, CDCl3) 1.51 (3H, s), 1.75–1.85 (1H, m), 2.01–2.09 (1H, m), 2.21 (3H, s), 2.26 (3H, s), 2.43–2.49 (1H, m), 2.78 (1H, s), 2.79 (1H, s), 3.95 (1H, dd, J 16.5, 8.5 Hz), 4.03 (1H, td, J 8.5, 3.0 Hz), 6.43 (1H, br s), 6.50 (1H, br s); δC
(100 MHz, CDCl3) 19.4, 21.2, 22.4, 23.5, 29.0, 40.5, 66.7, 106.3, 108.2, 115.0, 115.3, 138.1, 149.5, 153.8, 169.7; HRMS (ES+) Calculated for C15H19O4
([M + H]+): 263.1283, Found: 263.1289.
(±)-4a(R*),10a(S*)-3,4,4a,10a-Tetrahydro-6-methyl-8-acetoxy-2H,5H-pyrano[2,3-b]chroman (12)6a
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(203 mg, 0.85 mmol) in 3,4-dihydro-2H-pyran (1.0 ml, 10.9 mmol) at 100 °C, under argon for 12 hours. After evaporation under reduced pressure, the yellow oil obtained was purified by flash silica gel chromatography (95 : 5 30–40 PE : EtOAc) to give (±)-12 as a colourless oil (161 mg, 72%). RF 0.3 (90 : 10 30–40 PE : EtOAc); νmax/cm−1
(film) 3055 (m), 2934 (w), 1759 (s), 1598 (m), 1422 (m), 1265 (s), 1217 (s), 1093 (s), 897 (m), 739 (s); δH
(400 MHz, CDCl3) 1.62–1.70 (5H, m), 2.18 (3H, s), 2.26 (3H, s), 2.50 (1H, dd, J 16.5, 4.5 Hz), 2.72 (1H, dd, J 16.5, 6.5 Hz), 3.69–3.73 (1H, m), 3.98–4.04 (1H, m), 5.28 (1H, d, J 2.5 Hz), 6.49 (1H, s), 6.50 (1H, s); δC
(100 MHz, CDCl3) 19.4, 21.2, 23.6, 24.4, 26.5, 31.6, 62.7, 96.3, 107.8, 115.6, 116.4, 138.3, 149.4, 153.3, 169.7; HRMS (CI+) Calculated for C15H19O4
([M + H]+): 263.1283, Found: 263.1288.
(−)-1(S),3(R),4a(S),10a(R)-6,10a,11,11-Tetramethyl-8-acetoxybicyclo[3.1.1]heptan[2,3-b]chroman (13)6a
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(203 mg, 0.85 mmol) in (1R)-(+)-α-pinene (1.0 ml, 6.3 mmol) at 140 °C, under argon for 12 hours. After evaporation under reduced pressure, the yellow oil obtained was purified by flash silica gel chromatography (99 : 1 30–40 PE : EtOAc) to give (±)-13 as a viscous colourless oil (81 mg, 30%). [α]25D
=
− 2.9 (c
= 1, CHCl3); RF 0.6 (98 : 2 30–40 PE : EtOAc); νmax/cm−1
(film) 2922 (s), 1758 (s), 1559 (s), 1483 (m), 1369 (m), 1256 (s), 1216 (s), 1127 (s), 1015 (m), 739 (s); δH
(500 MHz, CDCl3) 1.04 (3H, s), 1.16 (1H, d, J 10.5 Hz), 1.28 (3H, s), 1.31 (3H, s), 1.32–1.35 (1H, m), 1.81–1.84 (1H, m), 2.04–2.15 (2H, m), 2.17 (1H, t, J 5.5 Hz), 2.24 (3H, s), 2.25 (3H, s), 2.43 (1H, dd, J 15.0, 5.0 Hz), 2.47–2.53 (1H, m), 2.73 (1H, dd, J 15.0, 6.0 Hz), 6.40 (1H, d, J 2.5 Hz), 6.49 (1H, d, J 2.5 Hz); δC
(125 MHz, CDCl3) 19.2, 21.2, 23.2, 26.8, 27.5, 28.0, 29.3, 34.2, 34.5, 40.2, 40.4, 54.9, 84.0, 108.3, 114.9, 121.3, 136.8, 149.3, 157.3, 169.7; HRMS (ES+) Calculated for C20H27O3
([M + H]+): 315.1960, Found: 315.1963.
(±)-4a(S*),9a(S*)-2,3,4,4a,9,9a-Hexahydro-4a,8-dimethyl-6-acetoxy-1H-xanthene (14)6a
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(203 mg, 0.85 mmol) in 1-methylcyclohexene (1.0 ml, 8.5 mmol) at 140 °C, under argon for 12 hours. After evaporation under reduced pressure, the yellow oil was purified by flash silica gel chromatography (99 : 1 30–40 PE : EtOAc) to give (±)-14 as a white solid (141 mg, 60%). Mp = 100–101 °C; RF 0.6 (98 : 2 30–40 PE : EtOAc); νmax/cm−1
(film) 2933 (s), 1758 (s), 1596 (s), 1370 (m), 1265 (s), 1218 (s), 1125 (s), 737 (s); δH
(400 MHz, CDCl3) 1.17 (3H, s), 1.24–1.31 (2H, m), 1.36–1.44 (2H, m), 1.48–1.52 (1H, m), 1.57–1.75 (3H, m), 1.93 (1H, br d, J 13.5 Hz), 2.18 (3H, s), 2.24 (1H, d, J 17.0 Hz), 2.25 (3H, s), 2.81 (1H, dd, J 17.0, 6.5 Hz), 6.40 (1H, d, J 2.0 Hz), 6.44 (1H, d, J 2.0 Hz); δC
(100 MHz, CDCl3) 19.4, 21.2, 21.7, 25.4, 25.6, 27.1, 28.8, 36.8, 38.3, 74.7, 108.0, 114.3, 116.4, 138.4, 149.1, 153.8, 169.8; HRMS (CI+) Calculated for C17H23O3
([M + H]+): 275.1647, Found: 275.1644.
Bp = 100 °C at atmospheric pressure; δH
(400 MHz, C6D6) 0.86 (3H, d, J 7.0 Hz), 1.67 (3H, d, J 2.0 Hz), 2.72–2.82 (1H, m), 3.70 (1H, dd, J 8.5, 6.5 Hz), 4.21 (1H, dd, J 9.5, 8.5 Hz), 4.43–4.44 (1H, m); δC
(100 MHz, C6D6) 13.6, 20.9, 37.8, 77.1, 101.4, 155.0.
(±)-3(R*),3a(R*),9a(R*)-2,3,3a,9a-Tetrahydro-3,5,9a-trimethyl-7-acetoxy-4H-furo[2,3-b]chroman (acetylalboatrin)
(10)6a
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(200 mg, 0.84 mmol) with (±)-4,5-dihydro-2,4-dimethylfuran 9
(82 mg, 0.84 mmol) in benzene (1 ml) at 80 °C under argon for 36 hours. After evaporation under reduced pressure, the colourless oil obtained was purified by flash silica gel chromatography (98 : 2 30–40 PE : EtOAc) to give the spiroacetal (±)-19 as a viscous colourless oil (57 mg, 25%, as an inseparable mixture [3 : 2 from 1H NMR] of two diastereoisomers, RF 0.5 (90 : 10 30–40 PE : EtOAc)), and (±)-acetylalboatrin 10 and (±)-acetyl-epi-alboatrin as a white solid (158 mg, 68%, as a mixture [12.6 : 1 from 1H NMR], RF 0.3 (90 : 10 30–40 PE : EtOAc)).
Data for (±)-10.
ν
max/cm−1
(film) 3054 (s), 2987 (s), 1760 (m), 1603 (m), 1421 (s), 1262 (s), 1215 (m), 896 (s), 752 (s); δH
(200 MHz, CDCl3) 1.04 (3H, d, J 6.0 Hz), 1.50 (3H, s), 1.93 (1H, ddd, J 11.0, 4.5, 3.0 Hz), 2.12–2.17 (1H, m), 2.22 (3H, s), 2.26 (3H, s), 2.71–2.73 (2H, m), 3.52 (1H, t, J 8.0 Hz), 4.17 (1H, t, J 8.0 Hz), 6.42 (1H, d, J 2.0 Hz), 6.49 (1H, d, J 2.0 Hz); δC
(100 MHz, CDCl3) 15.9, 19.5, 21.2, 22.0, 22.8, 35.4, 47.9, 74.1, 107.1, 108.3, 114.9, 115.3, 138.1, 149.5, 153.6, 169.7; HRMS (ES+) Calculated for C16H21O4
([M + H]+): 277.1440, Found: 277.1431. The 1H NMR spectra of the mixture of acetylalboatrin and acetyl-epi-alboatrin displayed signals which compare favourably with the structure of epi-alboatrin described by Murphy et al.
(ref. 15). e.g. for acetyl-epi-alboatrin δH
(200 MHz, CDCl3) 0.85 (3H, d, J 7.0 Hz); for epi-alboatrin δH
(200 MHz, CDCl3) 0.88–0.90 (3H, d, J 7.0 Hz). The two epimers could be separated and characterised by GCMS, LRMS (CI+) 277 for each. The Dept 135 experiment of the mixture of two spiroacetals (±)-19 displays two new CH2's for each compound and the disappearance of one CH and one CH3. The 1H NMR spectra also confirms the disappearance of one CH3 for each compound and the HRMS of the mixture of compounds is in agreement with both proposed structures calculated for C6H21O4
([M + H]+): 277.1440, Found: 277.1431. Likewise the structure of the spiroacetal compounds (±)-19 was in agreement with quinone methide derived products described by Wilson et al.4 in an analogous study.
(±)-3(R*),3a(R*),9a(R*)-2,3,3a,9a-Tetrahydro-3,5,9a-trimethyl-7-hydroxy-4H-furo[2,3-b]chroman (alboatrin)
(2)2
To a stirred solution of (±)-acetylalboatrin 10 and (±)-acetyl-epi-alboatrin [12.6 : 1]
(480 mg, 1.74 mmol) in 9 ml of DCM–MeOH–H2O (12 : 7 : 1) was added potassium carbonate (721 mg, 5.21 mmol) at room temperature under nitrogen. The mixture was stirred for 6 hours and was then extracted with ethyl acetate (3 × 50 ml). The combined organic layers were washed with a saturated solution of ammonium chloride (3 × 50 ml), dried over magnesium sulfate and concentrated under reduced pressure. The crude obtained was purified by flash silica gel chromatography (98 : 2 30–40 PE : EtOAc) to give a white solid. The solid was crystallised from ether to afford (±)-alboatrin 2
(368 mg, 97%) as white crystals.
Mp = 148–149 °C; RF 0.1 (90 : 10 30–40 PE : EtOAc); νmax/cm−1
(film) 3400 (s), 2958 (m), 1620 (s), 1599 (s), 1494 (m), 1462 (m), 1335 (m), 1204 (m), 1148 (s), 1117 (s), 986 (m), 845 (m); δH
(200 MHz, CDCl3) 1.04 (3H, d, J 6.5 Hz), 1.51 (3H, s), 1.93 (1H, ddd, J 11.0, 4.5, 3.5 Hz), 2.01–2.14 (1H, m), 2.19 (3H, s), 2.68 (2H, br s), 3.51 (1H, t, J 8.5 Hz), 4.17 (1H, t, J 8.5 Hz), 4.80 (1H, br s), 6.26 (1H, s), 6.29 (1H, s); δC
(100 MHz, CDCl3) 16.0, 19.4, 21.5, 23.2, 35.5, 48.4, 74.0, 101.9, 107.5, 109.6, 110.0, 138.2, 153.7, 155.1; HRMS (ES−) Calculated for C14H17O3
([M − H]−): 233.1178, Found: 233.1180. Crystal data for2: C14H18O3, M
= 234.30, monoclinic, a
= 8.8435(3), b
= 13.4040(5), c
= 10.6982(3)
Å, U
= 1221.60(7)
Å3, T
= 150 K, space group P21/n, Z
= 4, µ(Mo-Kα)
= 0.088 mm−1, 1990 reflections measured, 2890 unique (Rint
= 0.024) which were used in calculations. The final wR was 0.0498.13
(±)-5a(R*),14a(S*),(E)-(E)-5a,6,9,10,13,14,14a,15-Octahydro-5a,9,9,12,16-pentamethyl-18-acetoxycycloundeca[1,2-b]benzopyran (24)
In a sealed tube was stirred 2-acetoxymethyl-3-methyl-5-acetoxyphenol 6a
(203 mg, 0.85 mmol) with α-humulene (1 ml, 4.35 mmol) at 140 °C, under argon for 12 hours. The yellow mixture obtained was purified by flash silica gel chromatography (pure 30–40 PE, then 99 : 1 30–40 PE : EtOAc) to give (±)-24 as a white solid (173 mg, 53%). Mp = 130–132 °C; RF 0.4 (90 : 10 30–40 PE : EtOAc); νmax/cm−1
(film) 2925 (s), 1761 (s), 1598 (s), 1452 (m), 1367 (m), 1211 (s), 1126 (s), 1046 (m), 911 (m); δH
(400 MHz, CDCl3) 1.02 (3H, s), 1.04 (3H, s), 1.05 (3H, s), 1.14 (1H, td, J 13.0, 8.0 Hz), 1.34 (1H, dd, J 13.0, 11.0 Hz), 1.62 (3H, s), 1.75 (1H, dd, J 12.5, 4.0 Hz), 1.79–1.88 (2H, m), 2.07–2.18 (3H, m), 2.21–2.30 (1H, m), 2.22 (3H, s), 2.26 (3H, s), 2.50 (1H, br d, J 14.5 Hz), 2.82 (1H, dd, J 16.5, 5.5 Hz), 5.00 (1H, dd, J 11.5, 4.0 Hz), 5.09 (1H, dd, J 16.0, 1.5 Hz), 5.16 (1H, ddd, J 16.0, 10.0, 2.0 Hz), 6.42 (1H, d, J 2.5 Hz), 6.44 (1H, d, J 2.5 Hz); δC
(100 MHz, CDCl3) 16.1, 18.1, 18.8, 19.9, 23.3, 26.1, 29.3, 29.5, 34.4, 36.8, 37.1, 40.3, 41.8, 78.7, 106.8, 112.9, 117.0, 119.7, 122.0, 135.2, 136.3, 140.9, 148.1, 153.3, 168.5; HRMS (ES+) Calculated for C25H35O3
([M + H]+): 383.2586, Found: 383.2581.
A repeated reaction using the same conditions with only one equivalent of humulene in xylene gave the same product (±)-24 in 48% yield.
(±)-5a(R*),14a(S*),(E)-(E)-5a,6,9,10,13,14,14a,15-Octahydro-5a,9,9,12,16-pentamethyl-18-hydroxycycloundeca[1,2-b]benzopyran (25)
To a stirred solution of (±)-24
(820 mg, 2.14 mmol) in 20 ml of a mixture (12 : 7 : 1) of DCM–MeOH–H2O was added potassium carbonate (330 mg, 2.38 mmol) at room temperature under nitrogen. The mixture was stirred for 6 hours and then extracted with ethyl acetate (3 × 100 ml). The combined organic layers were washed with a saturated solution of ammonium chloride (3 × 100 ml), dried over magnesium sulfate and concentrated under reduced pressure. The solid obtained was purified by flash silica gel chromatography (98 : 2 30–40 PE : EtOAc) to give a white solid which was crystallised from ether to afford (±)-25
(601 mg, 82%) as white crystals. Mp = 155–157 °C; RF 0.4 (90 : 10 30–40 PE : EtOAc); νmax/cm−1
(film) 3400 (s), 2925 (s), 2859 (m), 1616 (s), 1600 (s), 1461 (s), 1381 (m), 1324 (m), 1265 (m), 1145 (s), 1046 (m), 991 (m), 839 (m), 738 (s); δH
(400 MHz, CDCl3) 1.03 (3H, s), 1.05 (3H, s), 1.06 (3H, s), 1.14 (1H, td, J 13.5, 8.5 Hz), 1.34 (1H, dd, J 13.5, 10.5 Hz), 1.63 (3H, s), 1.75 (1H, dd, J 13.0, 4.5 Hz), 1.81–1.87 (2H, m), 2.04–2.17 (3H, m), 2.19 (3H, s), 2.28 (1H, dd, J 14.5, 10.0 Hz), 2.50 (1H, br d, J 14.5 Hz), 2.80 (1H, dd, J 16.5, 5.5 Hz), 4.72 (1H, br s), 5.02 (1H, d, J 12.0, 4.5 Hz), 5.11 (1H, dd, J 16.0, 1.5 Hz), 5.18 (1H, ddd, J 16.0, 10.0, 2.5 Hz), 6.20 (1H, d, J 2.5 Hz), 6.26 (1H, d, J 2.5 Hz); δC
(100 MHz, CDCl3) 17.3, 19.3, 19.9, 24.4, 27.0, 30.4, 30.7, 35.8, 38.0, 38.3, 41.5, 43.0, 79.9, 101.4, 108.6, 113.1, 121.0, 123.1, 136.7, 137.8, 142.0, 154.3, 154.7; HRMS (ES−) Calculated for C23H31O2
([M − H]−): 339.2324, Found: 339.2331.Crystal data for25: C23H32O2, M
= 240.51, monoclinic, a
= 19.4765(3), b
= 9.6966(2), c
= 21.0587(4)
Å, U
= 3964.72(13)
Å3, T
= 150 K, space group P21/c, Z
= 8, µ(Mo-Kα)
= 0.071 mm−1, 32823 reflections measured, 9525 unique (Rint
= 0.051) which were used in calculations. The final wR was 0.0524.13
Acknowledgements
We thank Dr Andrew Cowley for crystallographic analysis, and Dr Barbara ODell for NMR assistance. We thank the EPSRC for funding JEM.
References
- P. Pittayakhajonwut, M. Theerasilp, P. Kongsaeree, A. Rungrod, M. Tanticharoen and Y. Thebtaranonth, Planta Med., 2002, 68, 1017 CrossRef CAS.
- A. Ichihara, M. Nonaka, S. Sakamura, R. Sato and A. Tajimi, Chem. Lett., 1988, 1, 27.
- P. Cai, D. Smith, B. Cunningham, S. Brown-Shimer, B. Katz, C. Pearce, D. Venables and D. Houck, J. Nat. Prod., 1998, 61, 791 CrossRef CAS.
- J. D. Pettigrew, J. A. Bexrud, R. P. Freeman and P. D. Wilson, Heterocycles., 2004, 62, 445 CrossRef CAS.
- H. Weenen and M. H. H. Nkunya, J. Org. Chem., 1990, 55, 5107 CrossRef CAS.
- For example, see:
(a) R. Rodriguez, R. M. Adlington, J. E. Moses, A. Cowley and J. E. Baldwin, Org. Lett., 2004, 6, 3617 CrossRef CAS;
(b) J. E. Baldwin, A. V. W. Mayweg, K. Neumann and G. J. Pritchard, Org. Lett., 1999, 1, 1933 CrossRef CAS;
(c) R. M. Adlington, J. E. Baldwin, G. J. Pritchard, A. J. Williams and D. J. Watkin, Org. Lett., 1999, 1, 1937 CrossRef CAS;
(d) R. M. Adlington, J. E. Baldwin, A. V. W. Mayweg and G. J. Pritchard, Org. Lett., 2002, 4, 3009 CrossRef CAS.
- For a recent review see: R. W. Van De Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 Search PubMed.
- T. Katada, S. Eguchi, T. Esaki and T. Sasaki, J. Chem. Soc., Perkin Trans. 1, 1984, 2649 RSC.
- K. Chiba, T. Hirano, Y. Kitano and M. Tada, Chem. Commun., 1999, 8, 691 RSC.
-
(a) B. Loubinoux, J. Miazimbakana and P. Gerardin, Tetrahedron Lett., 1989, 30, 1939 CrossRef CAS;
(b) B. Loubinoux, S. Tabbache, P. Gerardin and J. Miazimbakana, Tetrahedron, 1988, 44, 6055 CrossRef CAS.
- E. M. Gruneiro and E. G. Gros, An. Asoc. Quim. Argent., 1971, 59, 259 CAS.
- Stable at room temperature for over four months.
- CCDC reference numbers 2 239364, 6a 237660, 25 239363. See http://dx.doi.org/10.1039/b508972g for crystallographic data in CIF or other electronic format.
-
(a) M. Yato, T. Ohwada and K. Shudo, J. Am. Chem. Soc., 1990, 112, 5341 CrossRef CAS;
(b) S. Nakamura, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 5282 CrossRef CAS.
- S. R. Graham, J. A. Murphy and A. R. Kennedy, J. Chem. Soc., Perkin Trans. 1, 1999, 3071 RSC.
- Deacetylated to epi-alboatrin15.
- A regiochemically controlled reaction of o-quinone methide 21 to (−)-alloaromadendrene to provide (−)-tanzanene has been reported, see: S. K. Paknikar, K. P. Fondekar and R. Mayer, Nat. Prod. Lett., 1996, 8, 253 Search PubMed . For related Diels–Alder approaches employing functionalized o-quinone methides, see for example: L. O. Chapman, M. R. Engel, J. P. Springer and J. C. Clardy, J. Am. Chem. Soc., 1971, 93, 6696 CrossRef CAS; J. P. Marino and S. L. Dax, J. Org. Chem., 1984, 49, 3671 Search PubMed; Y. Genisson, P. C. Tyler and R. N. Young, J. Am. Chem. Soc., 1994, 116, 759 CrossRef CAS.
-
K. Nakanishi, T. Goto, S. Ito, S. Natori and S. Nazoe, Natural Product Chemistry, Academic Press, New York, 1974, vol. 1, p. 87 Search PubMed.
- This dehydration route to o-quinone methide 21 proved superior to others, see: J. I. Cunneen, E. H. Farmer and H. P. Koch, J. Chem. Soc., 1943, 472 Search PubMed.
- Structure was confirmed by single-crystal X-ray diffraction. See reference 6c.
- Mass spectrum: ([M + H]+): 523.
-
D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.