Efficient synthesis of methyl lycotetraoside, the tetrasaccharide constituent of the tomato defence glycoalkaloid α-tomatine
Nigel A.
Jones
a,
Sergey A.
Nepogodiev
*a and
Robert A.
Field
*ab
aCentre for Carbohydrate Chemistry, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: s.nepogodiev@uea.ac.uk; r.a.field@uea.ac.uk
bDepartment of Biological Chemistry, John Innes Centre, Colney Lane, Norwich, NR4 7UH, UK
First published on 27th July 2005
Abstract
Branched oligosaccharide lycotetraose, β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-glucopyranosyl-(1→4)-β-D-galactopyranose, is a key constituent of many steroidal saponins, including glycoalkaloid α-tomatine, which is involved in protection of plants from invading pathogens. A new synthesis of the methyl β-lycotetraoside (2) is described. Key steps of the synthesis include two successive glycosylation reactions of disaccharide acceptor methyl (4,6-O-benzylidene-3-O-p-methoxybenzyl-β-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranoside with readily available benzoylated trichloroacetimidates of α-D-glucopyranose and α,β-D-xylopyranose. This scheme allows sequential glycosylation in one-pot on account of the convenient in situ removal of a p-methoxybenzyl protecting group under the acid conditions of the first glycosylation step. Following deprotection, tetrasaccharide 2 was obtained in 19% yield over eight steps.
Introduction
Saponins are a structurally diverse family of glycosylated triterpenes and steroids constitutively present in a variety of plant species1 and marine organisms.2 They possess a wide range of pharmacological activities; some such compounds are known to be the active principles in traditional herbal medicines.3 The promising pharmaceutical properties of saponins have prompted the search for new compounds of this type from natural sources, followed by investigation of their biological and pharmacological activities.1 Since isolation of saponins from plants is a formidable task, considerable efforts have also been devoted to their chemical synthesis.4 In the plant kingdom, saponins often play a role as antimicrobials toxic to various pathogens, including fungi, and thus contribute to plant resistance to infection.5 The proposed mechanism of saponin antimicrobial activity is believed to be based on complexation of sterols in the pathogen membrane, causing a loss of the membrane integrity and hence cell lysis.6 The main saponin of tomato plants, the steroidal glycoalkaloid α-tomatine,7 has a potent fungicidal activity in vivo.8 However, this activity can be completely suppressed by some infectious fungi, which produce α-tomatine-detoxifying enzymes known as tomatinases.9 These enzymes are glycosyl hydrolases which can remove one or more sugar residues from α-tomatine leading to a modified saponin or aglycone (tomatidine), which are substantially less toxic to fungi. Furthermore, it has recently been shown that degradation products resulting from tomatinase action suppress other mechanisms of plant resistance to fungal attack.10 To gain insight into the mechanism of action of α-tomatine-detoxifying enzymes, the complete oligosaccharide structure of α-tomatine as well as fragments are needed. α-Tomatine 1 consists of a branched tetrasaccharide, β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-glucopyranosyl-(1→4)-D-galactose, attached to O-3 of the steroidal aglycone, tomatidine (Fig. 1). This tetrasaccharide, known as lycotetraose,11 is a recurrent glycone motif of glycoalkaloids found in a very wide variety of plant and marine species,12 including members of the Solanaceae family13 such as tomatoes, potatoes and other plants important in the human diet. Preparative enzymatic synthesis of lycotetraose from readily available α-tomatine was recently reported by our laboratory.14 In this synthesis, the target tetrasaccharide was liberated from the glycoalkaloid by cleavage of the β-galactoside linkage between lycotetraose and tomatidine by a recombinant endo-glycosidase from the plant pathogen Fusarium oxysporum f. sp. lycopersici.15 Continuing our efforts16 on synthesis of oligosaccharide components of saponins important for plant defence mechanisms, we describe herein the chemical synthesis of branched methyl lycotetraoside 2. The flexibility of the route described offers potential for construction of lycotetraose analogues. | ||
| Fig. 1 Structure of α-tomatine 1 and methyl lycotetraoside 2. | ||
Syntheses of lycotetraose17 and lycotetraose-containing saponin desgalactotigonin18 have been reported in the literature. Takeo and co-workers17 synthesised lycotetraose as a reducing sugar in 12 steps in an overall 13% yield. The key building block in their synthesis was disaccharide derivative 3 (Scheme 1), which was 2-O-glycosylated, then 3-O-deprotected and 3-O-glycosylated to give protected lycotetraoside 4. Danishefsky and Randolph18 commenced the synthesis of desgalactotigonin 9 with the preparation of a steroid glycoside in the form of stannyl ether derivative 6, which served as a glycosyl acceptor in the next step (Scheme 2). Coupling 6 with disaccharide epoxide 5 produced trisaccharide 7 having a conveniently deprotected 2-OH group in the second glucopyranosyl residue. However, glycosylation of this position in compound 7 proved to be difficult; the authors only succeeded with the use of glucopyranosyl fluoride 8, which was “armed” by the presence of three benzyl protecting groups. Using this reaction scheme, the synthesis of tetrasaccharide saponin 9 was achieved in 13% overall yield, though this yield is calculated for the linear reaction sequence, which does not consider the lengthy syntheses of glycosyl donors 5 and 8.
 | ||
| Scheme 1 Key glycosylation steps in the synthesis of the lycotetraose by Takeo et al.17 | ||
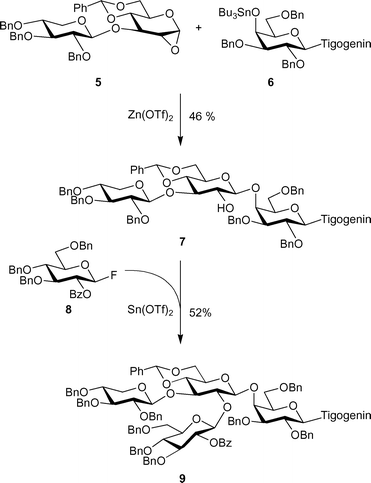 | ||
| Scheme 2 Key glycosylation steps in the synthesis of desgalactotigonin by Danishefsky et al.18 Tigogenin denotes a steroidal aglycone. | ||
Results and discussion
In our hands, synthesis of methyl lycotetraoside 2 (Fig. 1) began with the construction of β-Glc1-(1→4)-β-Gal disaccharide unit. Glucosylation of the known galactose derivative 1019 with trichloroacetimidate 1120 in the presence of TMSOTf in CH2Cl2 gave β-linked disaccharide 12 in 80% yield (Scheme 3). De-O-benzoylation of 12 with NaOMe in MeOH followed by 4,6-O-benzylidenation gave diol 13 in 75% yield over two steps.21 The relative reactivity of the 2-OH and 3-OH groups in glycosylation of 4,6-O-benzylidene derivatives of glucopyranosides is known to be dependent on many factors,22 amongst which are the nature and anomeric orientation of an aglycone, the method of glycosylation and the structure of the glycosyl donor. With the expectation of higher reactivity of the 3-OH in 13, which was based on steric considerations, diol 13 was treated with 1.1 equiv. of xylopyranosyl donor 1423 in the presence of TMSOTf, producing a single regioisomer, 15, in 81% yield. The regioselectivity was established by comparison of the 1H NMR spectra of trisaccharide 15 and its 2′-O-acetylated derivative. The resonance of H-2′ in the spectra of 15 appeared ∼1 ppm upfield of the corresponding signal in the spectra of the 2′-acetate of 15, thus confirming the presence of a (1→3)-linkage in the trisaccharide. Coupling constants observed for the xylopyranosyl residue (J1,2 = 3.3 Hz, J2,3 = J3,4 = 5.1 Hz) indicated that the protected trisaccharide 15 has the desired β-configuration, but its pyranose ring adopts an unusual 1C4 conformation instead of the more common 4C1 form. Similar conformational behaviour for benzoylated xylopyranosyl residues has been observed before.24 | ||
| Scheme 3 Reagents and conditions: (i) TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 1 h; (ii) NaOMe, MeOH, 20 °C, 30 h; (iii) PhCH(OMe)2, TsOH, MeCN, 20 °C, 3 h; (iv) TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 1 h. | ||
To complete the construction of the target tetrasaccharide, the remaining 2′-OH group in trisaccharide 15 needed to be β-glucosylated. In an attempt to complete this task, a number of readily available acylated glucopyranosyl donors were investigated. Reactions of 15 were carried out with glycosyl bromide 16, trichloroacetimidates 14 and 17, thioglycoside 18 and selenoglycoside 19 in the presence of typical promoters (Table 1). However, these attempts failed to produce the desired tetrasaccharide, giving only hydrolysed donor and the unchanged trisaccharide. Apparently, steric hindrance of the 2′-position makes trisaccharide 15 a poor glycosyl acceptor. The reactivity limitations which were observed previously in the glycosylation of trisaccharide acceptor 7 (similar to 15) were overcome by Danishefsky et al. by application of more potent, partly benzylated glucosyl fluoride 8.18 However, in our hands, this partly “armed” donor, which required seven steps for its own preparation, did not react satisfactorily with alcohol 15 using conditions described in the literature. Further attempts to glycosylate the 2′-position in trisaccharide 15 were made using readily available fully benzylated thioglucoside 20,25 known to be a very reactive “armed” glycosyl donor.26 This glycosylation was carried out in the presence of MeCN, which is known to provide β-stereoselctivity through the stereoselective kinetic formation of a reactive α-nitrilium ion intermediate.27 Once again, the route was unsuccessful, in this case because of the complex mixture of products formed in this reaction which made separation impractical.

![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H)CCl3
H)CCl3Because of the lack of reactivity of the 2′-OH group in trisaccharide 15 for glycosylation, an alternative route to the construction of branched-chain tetrasaccharide 2 was investigated. In this route the 2′-OH of the β-Glc1-(1→4)-β-Gal disaccharide needed to be glycosylated first to produce β-Glc2-(1→2)-β-Glc1-(1→4)-β-Gal trisaccharide. Attachment of xylopyranose residue to this trisaccharide would then furnish target branched tetrasaccharide β-Glc2-(1→2)-[β-Xyl-(1→3)]-β-Glc1-(1→4)-β-Gal. This approach required a temporary protecting group to be installed in the 3′-position of the β-Glc1-(1→4)-β-Gal disaccharide, which can be achieved using 3-O-p-methoxybenzylated β-glucopyranosyl donor 2120b as a precursor of the β-Glc1 unit (Scheme 4). Furthermore, it was demonstrated by Nilsson et al.28 that an O-p-methoxybenzyl (PMB) group is removable in situ, which is useful for “one-pot multistep” assemblies of complex oligosaccharides. Coupling of glycosyl donor 21 with acceptor 10 promoted by NIS/TfOH afforded disaccharide 22 in 62% yield. Disaccharide glycosyl acceptor 23, prepared in 87% yield by debenzoylation of 22, was subsequently glycosylated with glucopyranosyl trichloroacetimidate 11 in the presence of TMSOTf. Under these conditions, formation of the trisaccharide was accompanied by a significant loss of the PMB protecting group in the product, giving a partially deprotected trisaccharide 24. Treatment of the reaction mixture with an excess of TMSOTf led to 24 as the sole trisaccharide product, which was isolated in 64% yield. Finally, glycosylation of 24 with xylopyranosyl donor 14 afforded protected tetrasaccharide 25 in 91% yield.
 | ||
| Scheme 4 Reagents and conditions: (i) NIS, TfOH, CH2Cl2–Et2O (1 : 1), 4 Å MS, 0 °C, 1 h; (ii) NaOMe, MeOH, 20 °C, 30 h; (iii) 1. TMSOTf (0.07 equiv.), 4 Å MS, CH2Cl2, 0 °C, 1 h, 2. TMSOTf (0.3 equiv.); (iv) TMSOTf, 4 Å MS, CH2Cl2, 0 °C; (v) 1. TMSOTf (0.14 equiv.), 4 Å MS, CH2Cl2, 0 °C, 30 min, 2. TMSOTf (0.5 equiv.), (vi) TMSOTf, 4 Å MS, CH2Cl2, 20 °C, 20 min; (vii) Pd–C, H2, EtOH, EtOAc, 24 h; (viii) NaOMe, MeOH, 20 °C, 17 h. | ||
The possibility of one-pot synthesis of tetrasaccharide 25 starting from disaccharide 23 was also explored. In this approach, glycosylation of 23 with 10 (1.5 equiv.) was carried out in the presence of NIS/TMSOTf (0.2 equiv.) until complete consumption of the glycosyl acceptor, as judged by TLC. Addition of more TMSOTf (0.8 equiv.) allowed complete conversion of the initially formed trisaccharide into 24. Xylopyranosyl trichloroacetimidate 11 (3.0 equiv.) was finally added to the reaction mixture, affording tetrasaccharide 25, which was easy to isolate from the mixture, in an overall 40% yield. Deprotection of 25 by catalytic hydrogenation followed by debenzoylation gave the target, methyl lycotetraoside 2, in an 87% yield.
The structure of tetrasaccharide 2 was confirmed by the presence of four anomeric signals both in 1H and 13C NMR spectra. On the basis of JH-1,H-2 coupling constants (7–8 Hz) it can be concluded that in deprotected tetrasaccharide 2 the β-D-xylopyranosyl residue has the expected 4C1 conformation. Overall, NMR data obtained for the synthetic methyl glycoside are consistent with those for authentic lycotetraose.14,29
Conclusion
In summary, we have developed a convenient route to the construction of lycotetraose, the saccharide component of plant defence glycoalkaloid α-tomatine, in the form of methyl glycoside. This oligosaccharide, as well its smaller trisaccharide fragments which were prepared en route to the complete structure, will be useful probes for studying the mechanism of α-tomatine action in protecting plants against pathogenic fungi. The reaction scheme applied for the synthesis of lycotetroside 2 was based on readily available monosaccharide building blocks and led to the target product in 19% yield over eight steps. It was demonstrated that the branched structure of lycotetraose β-Glc2-(1→2)-[β-Xyl-(1→3)]-β-Glc1-(1→4)-Gal can only be assembled efficiently by first attaching a β-Glc2 unit to the 2′-position of β-Glc1-(1→4)-Gal disaccharide, followed by attaching β-Xyl to the 3′-position of trisaccharide β-Glc2-(1→2)-β-Glc1-(1→4)-Gal. The reverse order of glycosylation, involving attachment of β-Glc residue to the 2′-position of readily available trisaccharide β-Xyl-(1→3)-β-Glc1-(1→4)-Gal, failed because of the very low reactivity of the 2′-position in the glycosyl acceptor based on this trisaccharide. The reaction sequence used for the preparation of lycotetroside 2 is applicable to syntheses of related oligosaccharides, whilst the one-pot double glycosylation approach described offers scope for compound library synthesis.Experimental
General
Reactions were carried out in dry solvents using septa and syringes for addition of reagents. Dry CH2Cl2 and MeCN were prepared by distillation from CaH2, MeOH was distilled from Mg(OMe)2 and stored over 4 Å molecular sieves. Cation-exchange resins were washed with water and dry MeOH before use. TLC was performed on precoated aluminium plates (Silica Gel 60 F254, Merck). Spots were visualized by exposure to UV light or by immersion in 5% ethanolic H2SO4 followed by heating to 150 °C. Solutions of reaction products were dried over MgSO4 and solvents were evaporated under reduced pressure at 25–40 °C. Column chromatography was performed on silica gel (40–70 µm, BDH-Merck). Optical rotations were measured at 25 °C using a Perkin–Elmer 141 polarimeter. 1H and 13C NMR spectra were recorded at 24 °C with a Varian Unity Plus spectrometer at 400 and 100 MHz, respectively, using TMS (for solution in CDCl3) or Me2CO (δ 49.9, for solutions in D2O) as internal standards. Resonance assignments were made with the aid of gCOSY and gHSQC experiments. In cases where spectral dispersion was poor, only selected diagnostic NMR data are given; other spectral features were in accord with the proposed structures. Accurate electrospray ionisation mass spectra (HR ESI-MS) were obtained using positive ionization mode on a Finnigan MAT 900 XLT mass spectrometer. For compounds with molecular masses over 1000, low resolution ESI-MS were obtained on the same instrument and experimental data were matched to theoretical isotope patterns.Method A. Trisaccharide 24 (67 mg, 0.052 mmol), imidate 14 (62 mg, 0.104 mmol) and 4 Å MS (0.12 g) were stirred in CH2Cl2 (4 ml) at room temperature for 1 h. The reaction mixture was cooled to 0 °C, TMSOTf (2 µl, 0.01 mmol) was added and the reaction was stirred for 1 h at this temperature. Et3N (0.2 ml) was added, the mixture was diluted with CH2Cl2 (15 ml), filtered through Celite, washed with water (3 × 10 ml), dried (MgSO4) and concentrated in vacuo. Column chromatography (toluene–EtOAc 17 : 1) gave tetrasaccharide 25 as a colourless oil (82 mg, 91%); [α]D −3 (c 1.00, CHCl3); δH (400 MHz, CDCl3): 2.67 (1 H, dd, J4,5a = 7.5 Hz, J5a,5b = 12.1 Hz, H-5a Xyl), 2.89–3.01 (1 H, m, H-5 Glc2), 3.21–3.28 (1 H, m, H-5 Glc1), 3.44–3.69 (9 H, m, H-3 Gal, H-5 Gal, H-6a Gal, H-6b Gal, H-4 Glc1, H-6a Glc1, OMe), 3.87 (1 H, dd, J1,2 = 7.3 Hz, J2,3 = 9.2 Hz, H-2 Glc1), 3.99 (1 H, dd, J2,3 = J3,4 = 9.2 Hz, H-3 Glc1), 4.09 (1 H, s, H-4 Gal), 4.12–4.23 (3 H, m, H-2 Gal, H-6b Glc1, H-5b Xyl), 4.23 (2 H, s, OCH2Ph), 4.33 (1 H, d, J1,2 = 7.1 Hz, H-1 Gal), 4.41 (1 H, dd, J5,6b = 3.7 Hz, J6a,6b = 11.7 Hz, H-6a Glc2), 4.58–4.66 (2 H, m, H-6b Glc2, OCH2Ph), 4.92–4.98 (2 H, m, H-1 Glc2, H-1 Xyl), 4.99 (1 H, d, Jgem = 12.1 Hz, OCH2Ph), 5.13 (1 H, d, J1,2 = 7.3 Hz, H-1 Glc1), 5.16–5.24 (3 H, m, H-4 Xyl, OCH2Ph), 5.48–5.60 (2 H, m, H-2 Xyl, CHPh), 5.58 (1 H, dd, J3,4 = J4,5 = 9.3 Hz, H-4 Glc2), 5.72–5.81 (3 H, m, H-2 Glc2, H-3 Glc2, H-3 Xyl), 7.01–8.21 (55 H, m, Ph); δC (100 MHz, CDCl3): 57.4 (OMe), 61.2 (C-5 Xyl), 64.5 (C-6 Glc2), 66.1 (C-5 Glc1), 69.1 (C-6 Glc1), 69.6 (C-6 Gal), 69.8 (C-4 Xyl), 71.0, 71.2, 71.5, 71.6, 71.8, 73.2 (× 2), 73.3, 73.5 (×2 OCH2Ph), 76.1 (OCH2Ph), 77.5, 79.1, 79.3, 80.4, 82.3, 99.5 and 99.7 (C-1 Xyl and C-1 Glc2), 100.6 (C-1 Glc), 101.3 (CHPh), 105.2 (C-1 Gal), 126.2–130.3, 133.2–133.9, 137.4, 138.6, 139.1, 139.3 (Ph), 163.8–166.0 (7 × CO); ESI MS: found m/z 1760.6 [M + Na]+ calcd. for C101H92O27Na 1760.6.
Method B. Disaccharide 23 (40 mg, 0.048 mmol), trichloroacetimidate 11 (53 mg, 0.072 mmol) and 4 Å MS (0.10 g) were stirred in CH2Cl2 (4 ml) at room temperature for 1 h. TMSOTf (2 µl, 0.01 mmol) was added at 0 °C, the reaction was stirred for 30 min, a second portion of TMSOTf (8 µl, 0.04 mmol) added, and the reaction stirred until TLC (EtOAc–hexane 1 : 1) indicated complete removal of the p-methoxybenzyl group, as described in the synthesis of 24. A solution of xylopyranosyl donor 14 (58 mg, 0.096 mmol) in CH2Cl2 (1 ml) was added, the reaction was stirred at room temperature for 20 min, treated with Et3N (0.5 ml), diluted with CH2Cl2 (15 ml) and filtered through Celite. The filtrate was washed with water (3 × 10 ml), dried (MgSO4) and concentrated in vacuo. Purification of the residue by chromatography (toluene–EtOAc 17 : 1) gave the tetrasaccharide 25 as a colourless oil (33 mg, 40%).
Acknowledgements
This research was supported by the UK Engineering and Physical Sciences Research Council and the Weston Foundation. We are indebted to the EPSRC Mass Spectrometry Service Centre, Swansea, for invaluable support.References and notes
- K. Hostettmann and A. Martson, Saponins, Cambridge University Press, Cambridge, UK, 1995 Search PubMed.
- M. Iorizzi, S. D. Marino and F. Zollo, Curr. Org. Chem., 2001, 5, 951–973 CrossRef CAS.
- Saponins used in traditional and modern medicine, ed. G. R. Waller and K. Yamasaki, Plenum Press, New York, 1996 Search PubMed.
- H. Pellissier, Tetrahedron, 2004, 60, 5123–5162 CrossRef CAS.
- G. R. Fenwick, K. R. Price, C. Tsukamoto and K. Okubo, ‘Saponins’, in: Toxic substances in crop plants, ed. J. P. F. D'Mello, C. M. Duffus and J. H. Duffus, Royal Society of Chemistry, Cambridge, UK, 1991, pp. 285–327 Search PubMed.
- (a) E. A. J. Keukens, T. de Vrije, C. van den Boom, P. de Waard, H. H. Plasman, F. Thiel, V. Chupin, W. M. F. Jongen and B. de Kruijff, Biochim. Biophys. Acta, 1995, 1240, 216–228 CAS; (b) E. A. J. Keukens, T. de Vrije, L. A. M. Jansen, H. de Boer, M. Janssen, A. I. P. M. de Kroon, W. M. F. Jongen and B. de Kruijff, Biochim. Biophys. Acta, 1996, 1279, 243–250 CAS.
- T. D. Fontaine, G. W. Irving, R. Ma, J. B. Poole and S. P. Doolittle, Arch. Biochem., 1948, 18, 467–475 Search PubMed.
- J. Roddick, Phytochemistry, 1974, 13, 9–25 CrossRef CAS.
- (a) J. P. Morrissey and A. E. Osbourn, Microbiol. Mol. Biol. Rev., 1999, 63, 708–724 CAS; (b) A. Osbourn, Trends Plant Sci., 1996, 1, 4–9 CrossRef; (c) M. Ruiz-Rubio, A. Pérez-Espinosa, K. Lairini, T. Roldán-Arjona, A. Dipietro and N. Anaya, in: Studies in natural products chemistry, ed. Atta-ur-Rahman, Elsevier, Oxford, UK, 2001, vol. 25, pp. 293–326 Search PubMed.
- (a) K. Bouarab, R. Melton, J. Peart, D. Baulcombe and A. Osbourn, Nature, 2002, 418, 889–892 CrossRef CAS; (b) S. Ito, T. Eto, S. Tanaka, N. Yamauchi, H. Takahara and T. Ikeda, FEBS Lett., 2004, 571, 31–34 CrossRef CAS.
- R. Kuhn, I. Low and H. Trischmann, Chem. Ber., 1957, 90, 203–218 CrossRef CAS.
- 110 saponins incorporating lycotetraose attached to O-3 of a steroidal aglycon are registered in the Chem. Abstr. database (May 2005).
- (a) M. Friedman and G. M. McDonald, Crit. Rev. Plant Sci., 1997, 16, 55–132 CrossRef CAS; (b) M. Friedman, J. Agric. Food Chem., 2002, 50, 5751–5780 CrossRef CAS.
- K. Woods, C. J. Hamilton and R. A. Field, Carbohydr. Res., 2004, 339, 2325–2328 CrossRef CAS.
- T. Roldán-Arjona, A. Pérez-Espinosa and M. Ruiz-Rubio, Mol. Plant-Microbe Interact., 1999, 12, 852–861 Search PubMed.
- B. Mukhopadhyay and R. A. Field, Carbohydr. Res., 2004, 339, 1285–1291 CrossRef CAS.
- K. Takeo, T. Nakaji and K. Shinmitsu, Carbohydr. Res., 1984, 133, 275–287 CrossRef CAS.
- (a) J. T. Randolph and S. J. Danishefsky, J. Am. Chem. Soc., 1993, 115, 8473–8474 CrossRef CAS; (b) J. T. Randolph and S. J. Danishefsky, J. Am. Chem. Soc., 1995, 117, 5693–5700 CrossRef CAS.
- M. Sakagami and H. Hamana, Tetrahedron Lett., 2000, 41, 5547–5551 CrossRef CAS.
- (a) R. R. Schmidt, J. Michel and M. Roos, Liebigs Ann. Chem., 1984, 1343–1357 CrossRef CAS; (b) R. Verduyn, M. Douwes, P. A. M. van der Klein, E. M. Mosinger, G. A. van der Marel and J. H. van Boom, Tetrahedron, 1993, 49, 7301–7316 CrossRef CAS.
- Removal of O-benzoyl groups in tetrabenzoate 12 by Zemplen deacylation was unusually slow, requiring 30 h for completion. This slow de-O-benzoylation may be a consequence of the increased stability of a 2-O-benzoyl group on a galactopyranosyl residue. Similar behaviour of 2-O-acyl protecting groups was noticed previously,17 in some cases removal of 2-benzoates being impossible, see: S. Deng, B. Yu, Y. Hui, H. Yu and X. Han, Carbohydr. Res., 1999, 317, 53–62 Search PubMed.
- (a) H. B. Mereyala, S. Bhavani and A. P. Rudradas, Synth. Commun., 2004, 34, 1057–1063 CrossRef CAS; (b) B. Fraser-Reid, J. C. Lopez, K. V. Radhakrishnan, M. Mach, U. Schlueter, A. M. Gomez and C. Uriel, J. Am. Chem. Soc., 2002, 124, 3198–3199 CrossRef CAS; (c) Y. Zeng and F. Z. Kong, Carbohydr. Res., 2003, 338, 843–849 CrossRef CAS; (d) P. G. Evans, H. M. I. Osborn and W. G. Suthers, Tetrahedron Lett., 2002, 43, 7855–7857 CrossRef CAS; (e) Y. Zeng, J. Ning and F. Kong, Tetrahedron Lett., 2002, 43, 3729–3733 CrossRef CAS.
- Xylopyranose trichloroacetimidate 14 was prepared as an anomeric mixture, the α-anomer adopting the 1C4 conformation and the β-anomer the 4C1 conformation: L. Q. Chen and F. Z. Kong, Carbohydr. Res., 2002, 337, 2335–2341 Search PubMed.
- (a) G. Gu, Y. Du and R. J. Linhardt, J. Org. Chem., 2004, 69, 5497–5500 CrossRef CAS; (b) K. Ple, M. Chwalek and L. Voutquenne-Nazabadioko, Tetrahedron, 2005, 61, 4347–4362 CrossRef CAS.
- G. H. Veeneman and J. H. van Boom, Tetrahedron Lett., 1990, 31, 275–278 CrossRef CAS.
- B. Fraser-Reid, Z. F. Wu, U. E. Udodong and H. Ottosson, J. Org. Chem., 1990, 55, 6068–6070 CrossRef CAS.
- (a) I. Braccini, C. Derouet, J. Esnault, C. Hervé du Penhoat, J.-M. Mallet, V. Michon and P. Sinaÿ, Carbohydr. Res., 1993, 246, 23–41 CrossRef CAS; (b) K. Fukase, A. Hasuoka, I. Kinoshita, Y. Aoki and S. Kusumoto, Tetrahedron, 1995, 51, 4923–4932 CrossRef CAS.
- S. Bhattacharyya, B. G. Magnusson, U. Wellmar and U. J. Nilsson, J. Chem. Soc., Perkin Trans. 1, 2001, 886–890 RSC.
- W. Willker and D. Leibfritz, Magn. Reson. Chem., 1992, 30, 645–650 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |