One-pot multi-step synthesis: a challenge spawning innovation
Steven J.
Broadwater
,
Shoshannah L.
Roth
,
Kristin E.
Price
,
Muris
Kobašlija
and
D. Tyler
McQuade
*
Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: DTM25@Cornell.edu
First published on 20th July 2005
Abstract
Creating one-pot synthetic routes is a challenge that is already spawning new chemistry, enzymes, materials, and mechanistic insight. Through one-pot reactions, the chemical products that add value to our lives can be produced with less waste and greater economic benefits. Within this Emerging Area, we describe models for designing one-pot reactions as well as advanced catalysts created to facilitate their realization.
 Steven Broadwater | Steven Broadwater received his B.A. in chemistry from Western Maryland College in 2001. He is currently pursuing his Ph.D. at Cornell University in the McQuade group. His research is focused on the development of encapsulated metal catalysts. |
 Shoshannah Roth | Shoshannah L. Roth received a B.S. in chemistry and a B.A. is psychology from the University of Iowa in 2002. Currently, she is a Ford Foundation predoctoral fellow working towards her Ph.D. with D. Tyler McQuade. Her work focuses on the directed evolution of bond-forming enzymes. |
 Kristin Price | Kristin Price received her B.A. in chemistry from Franklin and Marshall College in 2001. Since 2001, she has been pursuing her doctoral studies at Cornell University in the McQuade group. Her research is focused on mechanism-based design of encapsulated organocatalysts. |
 Muris Kobašlija | Muris Kobašlija, born in Bosnia and Herzegovina in 1979, studied chemistry at Canisius College in Buffalo, NY. After completing his B.S. degree in 2002 he joined McQuade group at Cornell University. He is currently involved in encapsulation of enzymes as a means to achieve multi-step, one-pot syntheses. |
 Tyler McQuade | D. Tyler McQuade, Assistant Professor of Chemistry and Chemical Biology-Cornell University, is currently a Dreyfus, 3M, Rohm and Haas, Beckman, and NYSTAR Young Investigator and a 2004 MIT Tech Review 100. He received a B.S. in Chemistry and Biology from UC-Irvine and a Ph.D. in Chemistry from UW-Madison with Professor Samuel Gellman. His interests include performing highly efficient one-pot syntheses using site-isolated catalysts. Early McQuade Group innovations nucleated an enterprise that won the 2005 BRV Business Idea Competition. |
Introduction
Chemistry has made an impact on almost every aspect of daily life from toothpaste to life-saving medicines. The essential feature of this central science is synthesis. In particular, organic synthetic methods have progressed dramatically since the initial synthesis of urea by Wöhler.1 The top-selling pharmaceutical agent Lipitor™, for example, is an optically pure, entirely synthetic product.2 This progress has prompted some to declare synthetic chemistry a mature field. What is neglected in this myopic analysis, however, is the resource-intensive nature of the synthetic enterprise. In order to continue meeting the world's demands, new approaches, methods, and tools are needed to make synthetic chemistry a more sustainable process.3Iterative organic synthesis: an infinite resources model
Effective organic synthesis is predicated on site-isolation, the physical separation of reagents or catalysts from each other. Synthetic organic chemists typically achieve site-isolation by using separate flasks or reactors. Separate vessels prevent incompatible catalysts or reagents from fouling or yielding intractable mixtures. This reliance on ‘multiple pots’ is both a triumph and a curse. Iterative transformation and purification has been an enormously successful model, but it is plagued by waste, mostly manifested in the form of solvents. Solvents are often incinerated and if the precursors to solvents are a finite resource, current chemical synthesis will only be possible for a limited amount of time. Beyond solvent, high-yielding reactions often produce salts and other impurities that must be removed to avoid deleterious effects on downstream transformations. Serial reactions and purifications require massive amounts of solvents and materials. The average pharmaceutical synthesis yields 25–100 kg (including solvent) of waste per kilogram of product, according to Sheldon.4,5 Inputs used by the pharmaceutical industry are highly purified, processed, and refined fine/bulk chemicals that are also wasteful to produce. Currently, inputs for pharmaceuticals and fine chemical synthesis are plentiful, allowing synthesis to proceed at a reasonable price. As resources become more scarce and expensive, however, synthesis will become increasingly cost-prohibitive unless made sustainable.Biosynthesis offers an alternative to the organic chemist's current model of synthesis. In a cell, myriad incompatible reactions occur in each organelle and the cytosol. By using enzymes, all of the catalysts in a given compartment are site-isolated and substrate-selective, preventing fouling and cross-reactivity. Cells use both reversible and irreversible reactions along with recyclable reagents to create millions of tons of complex materials in ‘one-pot’ systems each year. Polyketide antibiotics such as Zithromax® are produced in ton quantities by fermentation.6 Metabolic engineering, the field dedicated to understanding and applying fermentation, maintains a venerable place in both industrial and academic research.7
Metabolic engineering is limited, however, to natural substrates and structural motifs because it relies on cellular machinery. A brief survey of the top 25 highest grossing drugs reveals that only 30% are natural products or their derivatives while the remaining 70% are made using synthetic chemistry. In rare cases where non-natural elements are incorporated into biosynthesized products, the non-natural elements must be synthetically charged onto proteins or cofactors, thus limiting the scale on which these abiotic products can be produced.8 Biosynthesis can be far more efficient and environmentally benign than traditional organic synthesis, which begs the question, ‘How can we merge the best of organic synthesis and metabolic engineering to create synthetic routes with excellent atom efficiency9 or E-factor?’10–16 One way is to increase the number of reactions performed per pot in the same way that metabolic pathways run many reactions in the same environment.
Examining two models for one-pot synthesis17,18
The value of one-pot reactions was recognized early in the history of organic synthesis.19 Since inception, one-pot reactions have grown in two directions.17,18 In one case, multiple orthogonal, irreversible steps are combined. In the other, multiple reversible steps are coupled to one irreversible step using an enzymatic catalyst. Scheme 1 depicts minimal versions of these two models for one-pot reactions. | ||
| Scheme 1 Two potential approaches to complex one-pot reactions: (a) coupled irreversible reactions, and (b) cascade of equilibria coupled to an irreversible step. | ||
Coupled irreversible reactions
Scheme 1a depicts the coupling of irreversible reactions. A single product is realized from orthogonal reactions by controlling the order of addition and tuning the relative rates. Leckta's ‘Sequentially-Linked Columns’ represents one example of coupled irreversible reactions.20Scheme 2 illustrates Leckta's β-lactam synthesis where reagents are flowed over a series of solid phase reagents and catalysts. The use of flow reactors is gaining ground, and issues of reagent recycling and reaction linking are areas of active research.21 | ||
| Scheme 2 Leckta's flow through approach. | ||
Cascade of equilibria coupled to an irreversible step
Scheme 1b illustrates an equilibria cascade coupled to one irreversible step. If each of the pre-equilibria is fast and the by-products do not react with starting materials, intermediates will be guided by the substrate-selectivity of the irreversible step. The Baylis–Hillman (BH) reaction is a reaction that illustrates this cascade of equilibria concept, Scheme 3. The BH reaction is a series of three reversible equilibria coupled to an irreversible elimination step.22 The BH reaction suggests that with appropriate design, many equilibrating reactions can be coupled in the same way. Multicomponent condensations (e.g. Ugi, Passerini, etc.) are a class of reactions that use a single catalyst and transition through a series of complex equilibria.19,23 | ||
| Scheme 3 The Baylis–Hillman mechanism. | ||
A more complex example of multiple equilibria coupled to an irreversible reaction is illustrated by Zimmermann's one-pot synthesis of D-xylulose 5-phosphate from hydroxypyruvate and D-fructose 1,6-biphosphate.24 The route begins with a D-fructose 1,6-bisphosphate aldolase (FruA, EC 4.1.2.13) catalyzed retro-aldolization of D-fructose 1,6-bisphosphate followed by a triosephosphate isomerase (TPI, EC 5.3.1.1) catalyzed isomerization (Scheme 4). Both of these initial steps are reversible and these equilibria are coupled to an irreversible transketolation catalyzed by transketolase A (TK, EC 2.2.1.1).25 The specificity of the TK step, which prevents decarboxylation of the starting material, allows this equilibria cascade to function and be driven to a single product.
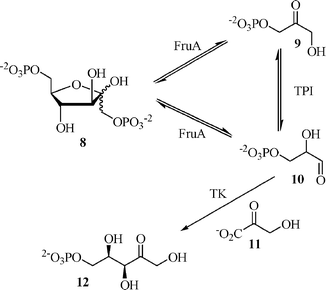 | ||
| Scheme 4 A coupled equilibria reaction: Zimmermann's D-xylulose 5-phosphate synthesis. | ||
A handful of non-enzymatic coupled equilibria have been reported, but many of these reactions are either simple proof of concept experiments or a series of reactions catalyzed by the same catalyst.17 To move to the next level, organic chemists must develop tools allowing more reactions to be linked in both the irreversible and equilibrium-coupled models.
Creating tools to facilitate a broader range of one-pot syntheses
Nature has shown that reactions can be coupled to synthesize complex products. Many natural product chemists model their key steps within a total synthesis after analogous biosynthetic reactions. In the same way, organic chemists can borrow ideas from coupled reactions in nature. Nature has two tools that are essential for coupled reactions: site-isolation and substrate-selectivity. The remainder of this Emerging Area discussion will focus on these two tools.
Site-isolation is the first tool that we will discuss. As mentioned earlier, site-isolation is a crucial element that enables biosynthesis. Enzymes effectively shield their active sites allowing for vastly different polarities, pHs, metals, oxidation states, and a variety of other properties.26 Site-isolation of non-natural catalysts is a relatively new area that is growing out of the august field of catalyst immobilization, where homogeneous catalysts are attached to organic or inorganic solid supports.27,28 Immobilization does not necessarily site-isolate. Many immobilized metal catalysts, for example, serve as reservoirs that release small amounts of highly active homogeneous catalysts.29,30 For a one-pot multi-catalyst system to function well, catalysts must remain completely site-isolated to prevent catalyst–catalyst fouling and reagent–catalyst fouling. Before moving forward, methods to determine site-isolation should be examined.
A variety of techniques are available to determine if a catalyst is site-isolated. The three-phase test is one of the most thorough methods.31–37 This technique involves anchoring one reaction partner to a solid support while the other remains homogeneous. The resulting ‘macromolecular reagent’ cannot diffuse and can no longer interact with a site-isolated catalyst. The catalyst could be bound to a solid support, encapsulated within a polymer or encased in inorganic material. If the reaction proceeds with the macromolecular reagent, the catalyst has become homogeneous during the reaction and is therefore not site-isolated.
Kinetic evaluation is also a powerful tool in the identification of site-isolated catalysts.29,30,38–43 Observation of a sigmoidal kinetic profile is a strong indication of in situ catalyst formation. An example of this phenomenon was recently reported independently by Bergbreiter et al.44 and Jones, Weck and co-workers,45,46 where both groups concluded that immobilized Pd–SCS pincer complexes decomposed during the course of the reaction to form soluble, catalytically active species. In addition to these two techniques, a host of other tools exist, including catalyst poisoning,47–52 TEM38,53 and the Maitlis filtration test.54
Only a few groups have articulated the value of catalyst site-isolation in the context of one-pot synthesis.24,55 Avnir and Blum have reported several seminal examples of exploiting sol–gel-based catalyst isolation for one-pot multi-step reactions.56–59 Frechet and co-workers have demonstrated the use of site-isolation to improve the photophysical properties of dye mixtures.60,61 The most prominent use of site-isolated catalysts and reagents has been demonstrated in an increasing body of work from the Ley laboratory. Ley and co-workers use site-isolation not for one-pot synthesis but for purification, and have used site-isolated catalysts and reagents sequentially, in many pots, to yield complex natural products.62,63 Since no one material or site-isolation approach has emerged as ideal for one-pot synthesis, we will discuss a few broad classes including soluble and bead-based polymeric supports, polymer incarceration, dendrimers, polymer encapsulation, and inorganic supported catalysts.
Soluble and bead-based polymeric supports
A common method of catalyst immobilization involves the use of soluble or insoluble polymeric supports.64–75 The simplest polymeric organic catalysts are linear polymers whose side chains or end groups are modified to contain catalytic groups, Scheme 5a. Since these polymers are not cross-linked, they are soluble in many organic solvents. Soluble polymer catalysts have been built off of a number of backbones including poly(ethylene glycol) and polystyrene.76,77 Soluble catalysts tend to be more active than their heterogenized counterparts, but often suffer from the need for a precipitation step to separate them from product. Thermomorphic and thermoregulated catalysts are systems that are soluble at one temperature and not at another. These interesting advances allow easy recovery of soluble, polymer-supported catalysts, taking advantage of their temperature-dependent solubility properties.71,72 Linear polymer catalysts show high reaction rates and turnover numbers in many reactions, including the Staudinger reaction,78 the Mitsonobu reaction,79 and the aza-Baylis–Hillman reaction.80 Soluble polymer catalysts can also be made by modifying the backbone to be a ligand for a transition metal, creating an organometallic catalyst.76 Although soluble polymers can be removed completely from the reaction mixture, they cannot protect their catalyst cargo from other catalysts in solution. | ||
| Scheme 5 (a) Coupling a catalyst to a soluble support; (b) functionalization of a macroporous bead with a catalyst. | ||
The most popular polymer-based heterogenization approach is covalent attachment of a ligand or catalyst to polystyrene beads, Scheme 5b. Polymer bead-supported catalysts have received attention due to ease of recovery and recycling. Catalyst heterogenization, however, often results in low catalyst activity. Cross-linked polystyrene beads can be modified with a variety of catalytic units, including phosphines, chiral boranes, chicona alkaloids and a variety of small molecules that serve as ligands for organometallic catalysts.81 Beyond polystyrene and poly(ethylene glycol), techniques that rely on gels formed by ring opening metathesis polymerizations have also recently been introduced, allowing for more porous polymer beads and higher catalyst loading to mitigate the decrease in rate due to the heterogenization.82 Although catalyst sites on a single bead interact,83,84 catalysts on separate beads have been shown to be site-isolated.82
Polymer encapsulation and polymer incarceration
Kobayashi and co-workers have used coacervation, another polymer-based heterogenization method, to create insoluble precipitates. This approach was used to encapsulate active Os, Sc, Pd and Ru catalysts.85–87 Kobayashi and co-workers expanded on this technology by performing the coacervation step with polymers containing pendant epoxide and alcohol functionalities. These coacervates were then thermally cross-linked to produce polymer incarcerated (PI) catalysts, Scheme 6. This method was used to prepare active Pd and Pt catalysts.88–90 Many of the catalytic systems described are easily recovered and reused without substantial loss in activity. Evidence exists, however, that these catalysts are not actually site-isolated. Use of coordinating solvents results in a substantial increase in leaching.89 In addition, many of the Pd-catalyzed transformations require excess phosphine ligands in the reaction mixture in order to achieve good reactivity.91 These observations, along with the high catalyst loading required in PI-mediated reactions, suggest that PI catalysts are serving as reservoirs for homogeneous, catalytically active species. These catalysts have a great deal of promise, but no experiments have been reported that directly establish site-isolation.92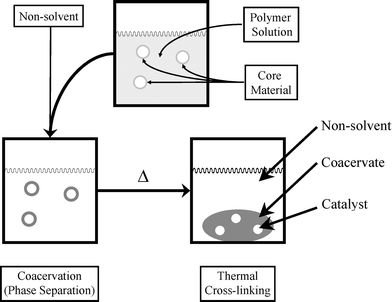 | ||
| Scheme 6 Polymer incarceration procedure: a powdered core material is added to a polymer solution with stirring. Non-solvent addition affords coacervated capsules that are cured to cross-link. | ||
Dendrimers
Dendrimers and star polymers represent a middle ground between the soluble and insoluble polymer supports.93,94 Dendrimers are soluble polymers consisting of a small molecule core connected to repetitively branching arms. Star polymers, in contrast, consist of a small molecule core connected to linear polymer arms. By modifying the core units, the macromolecule can be converted into a site-isolated catalyst, Fig. 1a. Dendrimeric catalysts can also be created by modifying the branches to include catalytic species, though this limits the site-isolation because the periphery of a dendrimer is solvent-exposed, Fig. 1b,c. The site-isolation provided by the branches of dendrimers and star polymers has been established and compared to linear analogues using photophysical studies.95,96 Catalyst site-isolation, however, has not been established. | ||
| Fig. 1 Metal species located (a) in the core, (b) the internal dendritic structure, and (c) at the periphery of dendritic catalysts. | ||
There have been examples of organocatalysts attached to dendrimers including phosphines that catalyze various condensation reactions and amine bases for substrate-selective alkylations.97,98 Recent applications of catalytically active dendrimer-supported metals include Rh-catalyzed hydroformylation99–101 and asymmetric hydrogenation,102 Pd-catalyzed cabonylation,103,104 oxidation,105 Sonogashira coupling,106,107 Heck coupling,108 Suzuki coupling,109 ethylene polymerization110 and allylic amination,111,112 Cu-mediated addition of Et2Zn to aldehydes,113 Os-catalyzed dihydroxylation,114 Ni-catalyzed ethylene oligomerization,115 Mn-catalyzed epoxidation116,117 and Ti-catalyzed Diels–Alder reactions.118 A major drawback of using these catalysts is the lengthy synthesis required to build the dendrimer, although this can be somewhat lessened by substituting a star polymer.94
Catalyst encapsulation
An alternative approach to site-isolation is the encapsulation of a catalyst in liquid-filled capsules via emulsion polymerization, Scheme 7. Immobilization of the catalyst can be accomplished by attachment within the shell, attachment on the interior surface of the shell, or by entrapping a macromolecular catalyst (polymer-bound or enzymatic) in the core, Fig. 2.119 Our group has reported the use of nanocapsules120,121 and Ley has reported the use of micron size capsules.62,63 Using microemulsion-templated polymerization, Ley and co-workers have created polyurea matrices containing OsO4122 for use in olefin dihydroxylation and Pd(OAc)273,123–126 for use in carbonylations, Heck couplings, Suzuki couplings, Stille couplings and olefin and imine hydrogenation. In addition, formic acid reduction of PdEnCat™ produced encapsulated Pd(0) (Pd0EnCat™) capable of performing transfer hydrogenation of aryl ketones127 and hydrogenolysis of epoxides.128 These heterogeneous materials are easily recovered from reaction mixtures by filtration or centrifugation, allowing for repeated use. The materials are reported to be low leaching and highly recyclable, making them attractive catalysts for one-pot, multi-step reactions. Despite the promise of these capsule-based methods, no demonstration of site-isolation has been reported. | ||
| Scheme 7 Catalyst encapsulation procedure: an emulsion containing a catalyst and a monomer in the dispersed phase is established. A second monomer is then added to the continuous phase to create capsules. | ||
 | ||
| Fig. 2 Various catalyst attachment points in a microcapsule: (a) within the shell, (b) attached to the shell, and (c) confined within the shell. | ||
Sol–gel encapsulation
Inorganic supports are widely used to heterogenize catalysts. The most popular supports are composed of silicates and aluminates. The work of Avnir and Blum is the most developed with respect to site-isolation. Avnir, Blum and co-workers have applied sol–gel technology to create a series of encapsulated catalysts.129,130 Of particular interest are the examples where sol–gels have been shown to effectively site-isolate incompatible catalysts, allowing them to be used in the same reaction mixture. Examples that have been used in one-pot reactions include an enzyme and a transition metal,57 an oxidant and a reductant,56 and an acid and a base.58 These ‘mutually destructive’ materials were able to function in the same reaction without deactivation of either species. The use of inorganic supports has a number of advantages including stability towards harsh conditions, low swelling, and consistent binding sites for the catalyst.131 Despite these advantages, inorganic supports, as a rule, lack the synthetic flexibility of organic shells. This deficiency may limit their broad use.
Although a number of viable site-isolation methods exist, currently the sol–gel supports are the only materials studied in context of one-pot synthesis. To realize the full potential of one-pot multiple catalyst reactions, new synthetic methods using the site-isolation methods need to be developed and examined in detail. Given the variety of methods for site-isolation, why do so few examples of one-pot multi-catalyst reactions exist? The problem is substrate-selectivity. Many of the catalysts mentioned can perform reversible or irreversible reactions but few can discriminate between two molecules with similar functionality. Consider a tandem BH–Heck reaction, as shown in Scheme 8, where the BH reaction would be catalyzed by an isolated Lewis base and the Heck reaction by an isolated palladium catalyst. The relative reaction rates would dictate the product distribution and since the BH reaction is slow, the expected product would be the Heck coupling of 13 and 16 and not the Heck coupling between 15 and 16. The reaction outcome, therefore, would be a mixture of both products. This lack of substrate-selectivity is a major obstacle to realizing efficient reactions and to realizing reactions with new product outcomes. The next section will discuss methods of including biosynthetic substrate-selectivity within a synthetic one-pot reaction.132
 | ||
| Scheme 8 Competition between a Baylis–Hillman–Heck reaction and a Heck reaction. | ||
Many commercial products are currently produced in biocatalytic processes, including fructose, acrylamide, aspartame and L-DOPA.134 The vast majority of the enzymes used in industry are mutated to improve properties over wild-type enzymes.132 Directed evolution has been used recently to make enzymes more suitable for use in industrial processes, including the synthesis of fine chemicals.134–136 Substantial progress has been made in the directed evolution of bond-breaking enzymes, and many mutants have found their way into industrial processes.132Bond-forming enzymes also have a wide variety of applications, but are difficult to evolve.132,144 Several main classes of bond-forming enzymes have found success as industrial biocatalysts, including lipases/esterases,137–139 aldolases,140,141 and glycosynthases142,143 using both natural and non-natural substrates.132
Directed evolution is a process in which the properties of an enzyme can be manipulated.145 Directed evolution can change substrate specificity, stability, enantioselectivity,146–148 and reaction rate.149 The first step in directed evolution is to identify a starting protein. The gene for the initial protein is then randomized. Several mutagenesis methods have been developed, falling into three general categories: mutator strains, polymerase chain reaction (PCR)-based, and oligonucleotide-based.150 After mutagenesis has been used to create a library of genes, it can be expressed as a library of proteins. The expression method must connect each genotype (DNA) and its phenotype (protein), allowing the experimenter to access the nucleic acid sequence of a selected protein. This linkage can be formed in several ways for both in vivo and in vitro protein expression systems.132 After the protein has been expressed, a screening or selection step must be performed. This step allows separation of improved enzymes from other members of the library.151
For evolved enzymes to be integrated into one-pot syntheses, they must be rendered more robust and more compatible with the one-pot conditions. Strategies for evolving enzymes that are more thermally and solvent-stable have been developed and a number of successes are reported.152,153 In general, enzyme stability must be reinforced by some alternate means like chemical cross-linking. Directed evolution and cross-linking are means to render enzymes stable towards organic solvent, but activities are typically sacrificed.154,155 A method is needed to allow enzymes to be trapped in an ideal environment while still functioning within an organic medium. Enzymes in emulsions and encapsulated enzymes have come close, but neither has been perfected.
The future of one-pot multi-step synthesis
We have emphasized the valuable role that one-pot synthesis plays in creating more sustainable synthetic routes to small molecules. Two elements that can enable one-pot reactions are catalyst site-isolation and substrate-selective catalysis. Current site-isolation procedures have been presented but none are ideally suited for one-pot syntheses. The field of substrate-selective catalysts, in terms of practical reactions, is limited to enzymes. The field of enzyme engineering is relatively new and has a great deal of growth potential.Challenges for the future with respect to site-isolation are the realization of a general method that immobilizes a catalyst in a protective, optimized microenvironment that allows indefinite recycling. On the selectivity side, highly active bond-forming enzymes that can withstand a variety of solvents and reaction temperatures are needed. Beyond site-isolation and substrate-selective catalysts, other missing elements that have received little attention are new reactor designs and recyclable reagents specifically designed for one-pot reactions.
The justification for complex natural product synthesis has long been that new reaction methods and mechanisms would be discovered en route. In the same vein, the challenge of one-pot synthesis will spawn new chemistry, enzymes, materials, and mechanistic insight. Thanks to the efforts of synthetic chemists before us, we can begin to create methods that mimic the efficient processes that nature uses. In the end, to throw away finite natural resources needlessly by not creating new efficient synthetic methods is hubris.
References and notes
- F. Wohler, Ann. Phys. Chem., 1828, 88 Search PubMed.
- W. A. Greenberg, A. Varvak, S. R. Hanson, K. Wong, H. J. Huang, P. Chen and M. J. Burk, Proc. Natl. Acad. Sci. USA, 2004, 101, 5788–5793 CrossRef CAS.
- P. T. Anastas and T. C. Williamson, Green chemistry: frontiers in benign chemical synthesis and processes, Oxford University Press, New York, 1998 Search PubMed.
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 68, 381–388 CrossRef CAS.
- R. A. Sheldon, Chemtech, 1994, 24, 38–47 CAS.
- N. Bahal and M. C. Nahata, Ann. Pharmacother., 1992, 26, 46–55 Search PubMed.
- W. R. Strohl, Metab. Eng., 2001, 3, 4–14 CrossRef CAS.
- C. T. Walsh, Science, 2004, 303, 1805–1810 CrossRef CAS.
- B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS.
- J. Andraos, Org. Proc. Res. Dev., 2005, 9, 149–163 Search PubMed.
- C. Jimenez-Gonzalez, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess., 2004, 9, 114–121 Search PubMed.
- A. E. Marteel, J. A. Davies, W. W. Olson and M. A. Abraham, Annu. Rev. Environ. Resour., 2003, 28, 401–428 Search PubMed.
- P. J. Dunn, S. Galvin and K. Hettenbach, Green Chem., 2004, 6, 43–48 RSC.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- M. Eissen and J. O. Metzger, Chem.-Eur. J., 2002, 8, 3580–3585 CrossRef CAS.
- D. J. C. Constable, A. D. Curzons, L. M. F. dos Santos, G. R. Geen, R. E. Hannah, J. D. Hayler, J. Kitteringham, M. A. McGuire, J. E. Richardson, P. Smith, R. L. Webb and M. Yu, Green Chem., 2001, 3, 7–9 RSC.
- J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–312 RSC.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Proc. Res. Dev., 2003, 7, 622–640 Search PubMed.
- C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51–80 CAS.
- A. M. Hafez, A. E. Taggi, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2001, 123, 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 853–10859 CrossRef CAS.
853–10859 CrossRef CAS. - G. Jas and A. Kirschning, Chem.-Eur. J., 2003, 9, 5708–5723 CrossRef CAS.
- K. E. Price, S. J. Broadwater, H. M. Jung and D. T. McQuade, Org. Lett., 2005, 7, 147–150 CrossRef CAS.
- D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602–1634 CrossRef CAS.
- F. T. Zimmermann, A. Schneider, U. Schorken, G. A. Sprenger and W. D. Fessner, Tetrahedron: Asymmetry, 1999, 10, 1643–1646 CrossRef CAS.
- N. J. Turner, Curr. Opin. Biotechnol., 2000, 11, 527–531 CrossRef CAS.
- C. T. Walsh, Enzymatic Reaction Mechanisms, WH Freeman and Company, New York, 1979 Search PubMed.
- L. E. Manzer, in Benign by Design, P. T. Anastas and C. A. Farris, eds., ACS Symp. Ser. vol. 577, American Chemical Society, Washington, 1994, pp. 144–154 Search PubMed.
- A. Deratani, T. Maraldo, G. D. Darling and J. M. J. Frechet, React. Polym., 1988, 9, 19–28 CAS.
- J. A. Widegren, M. A. Bennett and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10301–10310 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- J. Rebek and F. Gavina, J. Am. Chem. Soc., 1974, 96, 7112–7114 CrossRef CAS.
- J. Rebek, D. Brown and S. Zimmerman, J. Am. Chem. Soc., 1975, 97, 454–455 CrossRef CAS.
- J. Rebek and F. Gavina, J. Am. Chem. Soc., 1975, 97, 1591–1592 CrossRef CAS.
- J. Rebek, Tetrahedron, 1979, 35, 723–731 CrossRef CAS.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139–10140 CrossRef CAS.
- R. Grubbs, C. P. Lau, R. Cukier and C. Brubaker, J. Am. Chem. Soc., 1977, 99, 4517–4518 CrossRef CAS.
- P. Jayalekshmy and S. Mazur, J. Am. Chem. Soc., 1976, 98, 6710–6711 CrossRef CAS.
- Y. Lin and R. G. Finke, Inorg. Chem., 1994, 33, 4891–4910 CrossRef CAS.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- J. D. Aiken and R. G. Finke, J. Am. Chem. Soc., 1998, 120, 9545–9554 CrossRef.
- K. S. Weddle, J. D. Aiken and R. G. Finke, J. Am. Chem. Soc., 1998, 120, 5653–5666 CrossRef CAS.
- J. A. Widegren, J. D. Aiken, S. Ozkar and R. G. Finke, Chem. Mater., 2001, 13, 312–324 CrossRef CAS.
- C. M. Hagen, J. A. Widegren, P. M. Maitlis and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 4423–4432 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, Adv. Synth. Catal., 2005, 347, 172–184 CrossRef CAS.
- K. Q. Yu, W. Sommer, M. Weck and C. W. Jones, J. Catal., 2004, 226, 101–110 CrossRef CAS.
- K. Q. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161–171 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819–1830 CrossRef CAS.
- M. N. Vargaftik, V. P. Zagorodnikov, I. P. Stolarov, II Moiseev, D. I. Kochubey, V. A. Likholobov, A. L. Chuvilin and K. I. Zamaraev, J. Mol. Cat., 1989, 53, 315–348 CrossRef CAS.
- R. Vanasselt and C. J. Elsevier, J. Mol. Cat., 1991, 65, L13–L19 CrossRef CAS.
- S. Klingelhofer, W. Heitz, A. Greiner, S. Oestreich, S. Forster and M. Antonietti, J. Am. Chem. Soc., 1997, 119, 10116–10120 CrossRef.
- B. J. Hornstein, J. D. Aiken and R. G. Finke, Inorg. Chem., 2002, 41, 1625–1638 CrossRef CAS.
- L. N. Lewis and N. Lewis, J. Am. Chem. Soc., 1986, 108, 7228–7231 CrossRef CAS.
- J. E. Hamlin, K. Hirai, A. Millan and P. M. Maitlis, J. Mol. Catal., 1980, 7, 543–544 CAS.
- S. Hecht and J. M. J. Frechet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- F. Gelman, J. Blum and D. Avnir, New J. Chem., 2003, 27, 205–207 RSC.
- F. Gelman, J. Blum and D. J. Avnir, J. Am. Chem. Soc., 2002, 124, 14460–14463 CrossRef CAS.
- F. Gelman, J. Blum, H. Schumann and D. J. Avnir, Sol–Gel Sci. Technol., 2003, 26, 43–46 Search PubMed.
- F. Gelman, J. Blum and D. Avnir, Angew. Chem., Int. Ed., 2001, 40, 3647–3649 CrossRef CAS.
- P. Furuta, J. Brooks, M. E. Thompson and J. M. Frechet, J. Am. Chem. Soc., 2003, 125, 13165–13172 CrossRef CAS.
- P. Furuta and J. M. J. Frechet, J. Am. Chem. Soc., 2003, 125, 13173–13181 CrossRef CAS.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815–4195 RSC.
- S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discov., 2002, 1, 573–586 CrossRef CAS.
- P. H. Toy and K. D. Tanda, Acc. Chem. Res., 2000, 33, 546–554 CrossRef CAS.
- S. Bhattacharyya, Comb. Chem. High Throughput Screen., 2000, 3, 65–92 Search PubMed.
- B. M. Bhanage and M. Arai, Catal. Rev. Sci. Eng., 2001, 43, 315–344 CrossRef CAS.
- U. Kragl and T. Dwars, Trends Biotechnol., 2001, 19, 442–449 CrossRef CAS.
- B. Clapham, T. S. Reger and K. D. Janda, Tetrahedron, 2001, 57, 4637–4662 CrossRef CAS.
- Q. H. Fan, Y. M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3465 CrossRef.
- Y. Uozumi, in Immobilized Catalysts, A. Kirschning, ed., Springer-Verlag, Berlin, 2004, vol. 242, pp. 77–112 Search PubMed.
- D. E. Bergbreiter, in Immobilized Catalysts, A. Kirschning, ed., Springer-Verlag, Berlin, 2004, vol. 242, pp. 113–176 Search PubMed.
- Y. Hao, M. Andersson, C. Virto, I. Y. Galaev, B. Mattiasson and R. Hatti-Kaul, Biocatal. Biotransform., 2001, 19, 341–359 CrossRef CAS.
- PdEnCats are highlighted in: N. End, K. U. Schoning, in Immobilized Catalysts, A. Kirschning, ed., Springer-Verlag, Berlin, 2004, vol. 242, pp. 241–271 Search PubMed.
- V. Farina, Adv. Synth. Catal., 2004, 346, 1553–1582 CrossRef CAS.
- S. Bhattacharyya, Curr. Opin. Drug Discov. Dev., 2004, 7, 752–764 Search PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3343 CrossRef CAS.
- D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489–509 CrossRef CAS.
- T. Holletz and D. Cech, Synthesis, 1994, 789–791 CrossRef CAS.
- F. Sieber, P. Wentworth, J. D. Toker, A. D. Wentworth, W. A. Metz, N. N. Reed and K. D. Janda, J. Org. Chem., 1999, 64, 8058 CrossRef CAS.
- J. W. Huang and M. Shi, Adv. Synth. Catal., 2003, 345, 953–958 CrossRef CAS.
- C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3299 CrossRef CAS.
- A. G. M. Barrett, B. T. Hopkins and J. Kobberling, Chem. Rev., 2002, 102, 3301–3323 CrossRef CAS.
- E. S. Chandrasekaran, R. H. Grubbs and C. H. Brubaker, J. Organomet. Chem., 1976, 120, 49–63 CrossRef.
- S. L. Regen and D. P. Lee, Macromolecules, 1977, 10, 1418–1419 CrossRef CAS.
- S. Nagayama, M. Endo and S. Kobayashi, J. Org. Chem., 1998, 63, 6094–6095 CrossRef CAS.
- T. Ishida, R. Akiyama and S. Kobayashi, Adv. Synth. Catal., 2003, 345, 576–579 CrossRef CAS.
- S. Kobayashi and R. Akiyama, Chem. Commun., 2003, 449–460 RSC.
- R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3412–3413 CrossRef CAS.
- H. Hagio, M. Sugiura and S. Kobayashi, Synlett, 2005, 813–816 CAS.
- K. Okamoto, R. Akiyama and S. Kobayashi, Org. Lett., 2004, 6, 1987–1990 CAS.
- K. Okamoto, R. Akiyama, H. Yoshida, T. Yoshida and S. Kobayashi, J. Am. Chem. Soc, 2005, 127, 2125–2135 CrossRef CAS.
- The authors of ref. 90 investigated the nature of the PI–Pd catalyst using the three-phase test and conclude that the catalysts are truly heterogeneous. However, the method described by Davies et al. (ref. 35) demonstrates that a solution phase reaction must be performed simultaneously with the heterogeneous reaction. Unfortunately, the authors of ref. 90 did not do this and therefore no conclusion can be made about the nature of the catalytically active species.
- R. van Heerbeek, P. C. J. Kamer, P. van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717–3756 CrossRef.
- S. Hecht, J. Polym. Sci. Polym. Chem., 2003, 41, 1047–1058 Search PubMed.
- S. Hecht, N. Vladimirov and J. M. J. Frechet, J. Am. Chem. Soc., 2001, 123, 18–25 CrossRef CAS.
- E. M. Harth, S. Hecht, B. Helms, E. E. Malmstrom, J. M. J. Frechet and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 3926–3938 CrossRef CAS.
- A. Sarkar, P. Ilankumaran, P. Kisanga and J. G. Verkade, Adv. Synth. Catal., 2004, 346, 1093–1096 CrossRef CAS.
- T. Marquardt and U. Luning, Chem. Commun., 1997, 1681–1682 RSC.
- J. N. H. Reek, D. de Groot, G. E. Oosterom, P. C. J. Kamer and P. van Leeuwen, C. R. Chim., 2003, 6, 1061–1077 Search PubMed.
- Y. Y. Huang, H. L. Zhang, G. J. Deng, W. J. Tang, X. Y. Wang, Y. M. He and Q. H. Fan, J. Mol. Catal. A: Chem., 2005, 227, 91–96 CrossRef CAS.
- T. Mizugaki, Y. Miyauchi, M. Murata, K. Ebitani and K. Kaneda, Chem. Lett., 2005, 34, 286–287 CrossRef CAS.
- G. D. Engel and L. H. Gade, Chem.-Eur. J., 2002, 8, 4319–4329 CrossRef CAS.
- S. Antebi, P. Arya, L. E. Manzer and H. Alper, J. Org. Chem., 2002, 67, 6623–6631 CrossRef CAS.
- R. Touzani and H. Alper, J. Mol. Catal. A: Chem., 2005, 227, 197–207 CrossRef CAS.
- P. P. Zweni and H. Alper, Adv. Synth. Catal., 2004, 346, 849–854 CrossRef CAS.
- K. Heuze, D. Mery, D. Gauss, J. C. Blais and D. Astruc, Chem.-Eur. J., 2004, 10, 3936–3944 CrossRef CAS.
- D. Astruc, K. Heuze, S. Gatard, D. Mery, S. Nlate and L. Plault, Adv. Synth. Catal., 2005, 347, 329–338 CrossRef CAS.
- L. K. Yeung and R. M. Crooks, Nano Lett., 2001, 1, 14–17 CrossRef CAS.
- M. Pittelkow, K. Moth-Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682–7684 CrossRef CAS.
- G. Smith, R. Chen and S. Mapolie, J. Organomet. Chem., 2003, 673, 111–115 CrossRef CAS.
- M. Ooe, M. Murata, A. Takahama, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32, 692–693 CrossRef CAS.
- Y. Ribourdouille, G. D. Engel, M. Richard-Plouet and L. H. Gade, Chem. Commun., 2003, 1228–1229 RSC.
- A. M. Arink, R. van de Coevering, B. Wieczorek, J. Firet, J. Jastrzebski, R. Gebbink and G. van Koten, J. Organomet. Chem., 2004, 689, 3813–3819 CrossRef CAS.
- W. J. Tang, N. F. Yang, B. Yi, G. J. Deng, Y. Y. Huang and Q. H. Fan, Chem. Commun., 2004, 1378–1379 RSC.
- C. Muller, L. J. Ackerman, J. N. H. Reek, P. C. J. Kamer and P. van Leeuwen, J. Am. Chem. Soc., 2004, 126, 14960–14963 CrossRef.
- J. Bu, Z. M. A. Judeh, C. B. Ching and S. Kawi, Catal. Lett., 2003, 85, 183–187 CrossRef CAS.
- Z. W. Yang, Q. X. Kang, H. C. Ma, C. L. Li and Z. Q. Lei, J. Mol. Catal. A: Chem., 2004, 213, 169–176 CrossRef CAS.
- B. M. Ji, Y. Yuan, K. L. Ding and A. B. Meng, Chem.-Eur. J., 2003, 9, 5989–5996 CrossRef CAS.
- T. M. S. Chang, Eur. J. Pharm. Biopharm., 1998, 45, 3–8 CrossRef CAS.
- H. M. Jung, K. E. Price and D. T. McQuade, J. Am. Chem. Soc., 2003, 125, 5351–5355 CrossRef CAS.
- K. E. Price and D. T. McQuade, Chem. Commun., 2005, 1714–1716 RSC.
- S. V. Ley, C. Ramarao, A. L. Lee, N. Ostergaard, S. C. Smith and I. M. Shirley, Org. Lett., 2003, 5, 185–187 CrossRef CAS.
- C. Ramarao, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAlmeida, Chem. Commun., 2002, 1132–1133 RSC.
- S. V. Ley, C. Ramarao, R. S. Gordon, A. B. Holmes, A. J. Morrison, I. F. McConvey, I. M. Shirley, S. C. Smith and M. D. Smith, Chem. Commun., 2002, 1134–1135 RSC.
- N. Bremeyer, S. V. Ley, C. Ramarao, I. M. Shirley and S. C. Smith, Synlett, 2002, 1843–1844 CAS.
- PdEnCats are highlighted in: D. A. Pears and S. C. Smith, Aldrichimica Acta, 2005, 38, 23–33 Search PubMed.
- J. Q. Yu, H. C. Wu, C. Ramarao, J. B. Spencer and S. V. Ley, Chem. Commun., 2003, 678–679 RSC.
- S. V. Ley, C. Mitchell, D. Pears, C. Ramarao, J. Q. Yu and W. Z. Zhou, Org. Lett., 2003, 5, 4665–4668 CrossRef CAS.
- L. Sarussi, J. Blum and D. Avnir, J. Sol–Gel Sci. Technol., 2000, 19, 17–22 CrossRef CAS.
- J. Blum, F. Gelman, R. Abu-Reziq, I. Miloslavski, H. Schumann and D. Avnir, Polyhedron, 2000, 19, 509–512 CrossRef CAS.
- Z. L. Lu, E. Lindner and H. A. Mayer, Chem. Rev., 2002, 102, 3543–3577 CrossRef CAS.
- A. S. Bommarius and B. R. Riebel, Biocatalysis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- B. Hauer and S. M. Roberts, Curr. Opin. Chem. Biol., 2004, 8, 103–105 CrossRef CAS.
- K. A. Powell, S. W. Ramer, S. B. del Cardayre, W. P. C. Stemmer, M. B. Tobin, P. F. Longchamp and G. W. Huisman, Angew. Chem., Int. Ed., 2001, 40, 3948–3959 CrossRef CAS.
- J. R. Cherry and A. L. Fidantsef, Curr. Opin. Biotechnol., 2003, 14, 438–443 CrossRef CAS.
- M. Adamczak and S. H. Krishna, Food Technol. Biotechnol., 2004, 42, 251–264 Search PubMed.
- S. Lutz, Tetrahedron: Asymmetry, 2004, 15, 2743–2748 CrossRef CAS.
- A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS.
- N. J. Turner, Curr. Opin. Chem. Biol., 2004, 8, 114–119 CrossRef CAS.
- J. J. Hao and A. Berry, Protein Eng. Des. Sel., 2004, 17, 689–697 Search PubMed.
- H. J. Lamble, M. J. Danson, D. W. Hough and S. D. Bull, Chem. Commun., 2005, 124–126 RSC.
- A. M. Daines, B. A. Maltman and S. L. Flitsch, Curr. Opin. Chem. Biol., 2004, 8, 106–113 CrossRef CAS.
- Y. W. Kim, S. S. Lee, R. A. J. Warren and S. G. Withers, J. Biol. Chem., 2004, 279, 42787–42793 CrossRef CAS.
- M. Lafferty and M. J. Dycaico, Methods in Enzymology, 2004, 388, 119–134 CAS.
- J. L. Jestin and P. A. Kaminski, J. Biotechnol., 2004, 113, 85–103 CrossRef CAS.
- M. T. Reetz, Proc. Natl. Acad. Sci. USA, 2004, 101, 5716–5722 CrossRef CAS.
- P. A. Dalby, Curr. Opin. Struct. Biol., 2003, 13, 500–505 CrossRef CAS.
- M. Alexeeva, R. Carr and N. Turner, J. Org. Biomol. Chem., 2003, 1, 4133–4137 Search PubMed.
- K. Hult and P. Berglund, Curr. Opin. Biotechnol., 2003, 14, 395–400 CrossRef CAS.
- S. Lutz and W. M. Patrick, Curr. Opin. Biotechnol., 2004, 15, 291–297 CrossRef CAS.
- M. Olsen, B. Iverson and G. Georgiou, Curr. Opin. Biotechnol., 2000, 11, 331–337 CrossRef CAS.
- F. H. Arnold, Trends Biotechnol., 1990, 8, 244–249 CrossRef CAS.
- F. H. Arnold, K. Q. Chen, C. Economou, W. Chen, P. Martinez, K. P. Yoon and M. Vandam, in Biocatalyst Design for Stability and Specificity, M. E. Himmel and B. Georgiou, eds., ACS Symp. Ser. vol. 516, American Chemical Society, Washington, 1993, pp. 109–113 Search PubMed.
- F. H. Arnold, Acc. Chem. Res., 1998, 31, 125–131 CrossRef CAS.
- M. T. Reetz and K. E. Jaeger, in Biocatalysis: from Discovery to Application, W. D. Fessner, ed., Springer-Verlag, Berlin, 1999, vol. 200, pp. 31–57 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |