Design and synthesis of aromatic inhibitors of anthranilate synthase
Richard J. Paynea, Miguel D. Toscanoa, Esther M. M. Bullocha, Andrew D. Abellb and Chris Abell*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 336405
bDepartment of Chemistry, University of Canterbury, Christchurch, New Zealand
First published on 6th May 2005
Abstract
Aromatic analogues of chorismate were synthesised as potential inhibitors of anthranilate synthase. Molecular modelling using GOLD2.1 showed that these analogues docked into the active site of Serratia marcescens anthranilate synthase in the same conformation as chorismate. Most compounds were found to be micromolar inhibitors of S. marcescens anthranilate synthase. The most potent analogue, 3-(1-carboxy-ethoxy)-4-hydroxybenzoate (KI 3 µM), included a lactyl ether side chain. This appears to be a good replacement for the enol-pyruvyl side chain of chorismate.
Introduction
The product of the shikimate pathway, chorismate (1), is a branchpoint for five biosynthetic pathways leading to the formation of the major aromatic metabolites, including the aromatic amino acids phenylalanine, tyrosine and tryptophan. The folate coenzymes, benzoid and naphthenoid quinones and siderophores are also produced through these pathways (Fig. 1).1 Three of the branchpoint enzymes; anthranilate synthase, 4-amino-4-deoxychorismate synthase and isochorismate synthase, appear to share many mechanistic features and are believed to have diverged from a common ancestor.2–6 Chorismate-utilising enzymes only occur in plants, bacteria, fungi and apicomplexan parasites. Inhibitors of these enzymes may therefore have herbicidal, antibiotic, fungicidal or antiparasitic activity.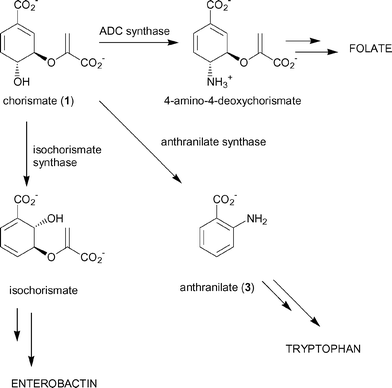 | ||
| Fig. 1 Chorismate-utilising enzymes. | ||
Anthranilate synthase catalyses the first committed step in the biosynthesis of tryptophan, in which chorismate is first aminated and then the enol-pyruvyl side chain cleaved to form the aromatic product anthranilate (3).7 Anthranilate synthase is a multifunctional enzyme composed of a small TrpG and large TrpE subunit encoded by the trpG and trpE genes, respectively. In most bacteria the enzyme consists of either a TrpG–TrpE heterodimer or TrpG2–TrpE2 heterotetramer. TrpG belongs to the family of “triad” glutamine amidotransferases that hydrolyse the amido side chain of glutamine. In the case of anthranilate synthase, the nascent ammonia is believed to be transferred through an intramolecular channel to the synthase active site of TrpE.8 The TrpE subunit catalyses the production of anthranilate in two steps (Scheme 1).
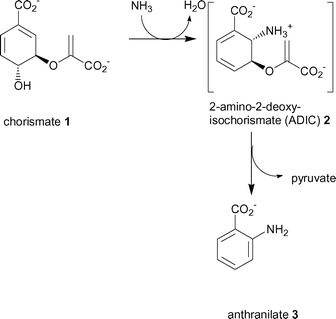 | ||
| Scheme 1 The two reactions catalysed by the TrpE subunit of anthranilate synthase. | ||
The first reaction catalysed by TrpE is the nucleophilic attack at C-2 of chorismate (1) with ammonia (produced by the TrpG subunit) to give an intermediate, 2-amino-2-deoxyisochorismate (ADIC) (2).9 The second reaction is the elimination of pyruvate from ADIC to produce anthranilate (3). It has been shown that the TrpE subunit alone is capable of producing anthranilate from chorismate when ammonia is supplied.10
Previous inhibition studies on anthranilate synthase have focused on substrate analogues based on cyclohexadiene (as observed in chorismate, 1) or cyclohexene rings with varied functionality.11,12 The inhibition constants against the Serratia marcescens enzyme ranged from sub-micromolar to millimolar.12 The most potent of these inhibitors are shown in Fig. 2. We have designed and synthesised a series of substrate analogues built around an aromatic core. The use of this simplified core allowed us to explore the functionality around the ring. The flat aromatic ring is a reasonable mimic of the almost planar cyclohexadiene core of chorismate.
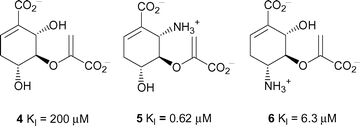 | ||
| Fig. 2 Inhibition constants for inhibitors 4–6 against anthranilate synthase from S. marcescens.12 | ||
Results and discussion
Inhibitor design and modelling
Crystal structures of anthranilate synthase from Sulfolobus solfataricus (1QDL),9Salmonella typhimurium (1I1Q)13 and Serratia marcescens (1I7Q and 1I7S)8 have been reported. Of the two structures reported for S. marcescens, one contains a tryptophan bound to an allosteric binding site, whilst the other has benzoate and pyruvate ligands bound in the TrpE active site. The latter structure was used in this study.8,14 The TrpE active site of S. marcescens anthranilate synthase was prepared for modelling studies using SYBYL6.5.15 This involved first removing the benzoate and pyruvate ligands. Chorismate was then docked into the vacant active site using GOLD2.1 (Genetic-Optimisation Ligand Docking).16 Chorismate docks with the C-1 carboxylate forming an electrostatic interaction with an enzyme bound magnesium ion, while the C-4 hydroxyl is in the vicinity of a glutamate residue (Glu309) (Fig. 3A). The enol-pyruvyl side chain at C-3 sits in a binding pocket where it can form hydrogen bonds to Arg469 and Tyr449. The RMSD of the common heavy atoms in the chorismate docking compared with the benzoate ligand in the original crystal structure is 0.62 Å.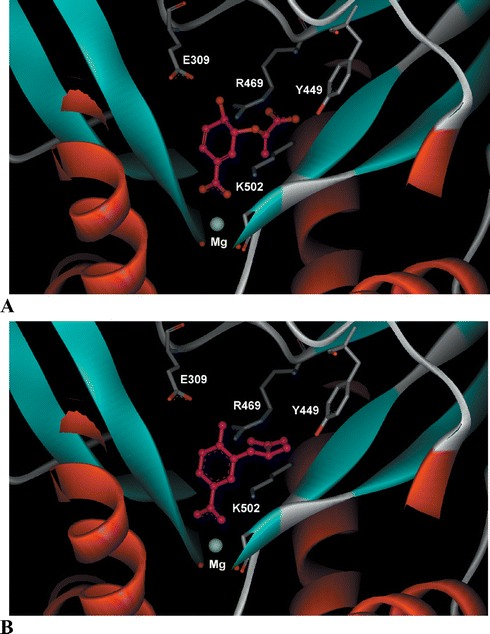 | ||
| Fig. 3 (A) Chorismate (1) and (B) aromatic analogue (R-21) docked into the active site of S. marcescens anthranilate synthase (1I7Q).8 | ||
In considering the inhibitor design the proposal was to use a flat aromatic scaffold with a C-1 carboxylate, which was assumed to bind to the magnesium ion in the active site of the enzyme. A variety of substituents were incorporated at C-3 and C-4. The substituents at C-4 included OMe (7–9), NH2 (10–12) and OH (13–21) (Fig. 4). These were intended to probe the role of the C-4 hydroxyl in chorismate, which is central to the enzyme reaction and was therefore expected to be relatively intolerant to change. The different side chains at C-3 were designed to explore the enol-pyruvyl binding pocket of anthranilate synthase and to identify groups that could be used to replace the enol-pyruvyl side chain of chorismate, which is difficult to incorporate synthetically.
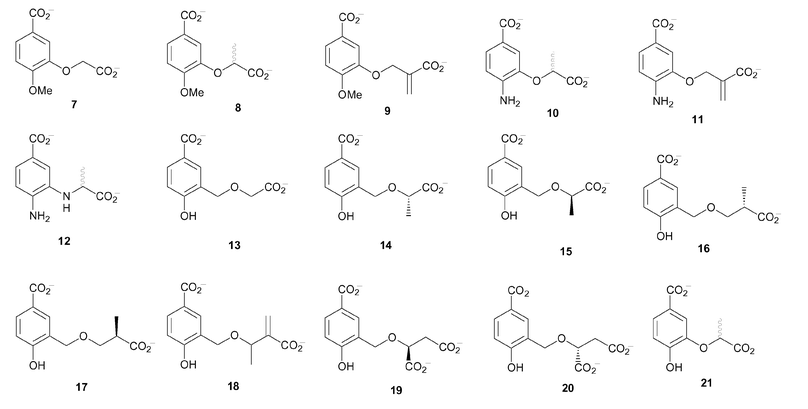 | ||
| Fig. 4 | ||
The first series of inhibitors were the 4-methoxybenzoate analogues (7–9), a simple replacement for hydroxyl that introduces possible steric interactions. The second series were the 4-aminobenzoate analogues (10–12). In this series, two different C-3 substituents were used; one retaining the ether linkage (10, 11) of chorismate and the other with a secondary amine linkage (12).
The final series of compounds proposed as targets were the 4-hydroxybenzoate analogues (13–21). These analogues most closely mimic chorismate, but with the secondary hydroxyl at C-4 replaced by a more acidic phenolic hydroxyl. Again, two types of side chains were used at C-3. One contained an ether linkage as found in chorismate (21). The other series of analogues incorporated an additional methylene unit before the ether linkage on the C-3 side chain (13–20). Inspection of the active site of anthranilate synthase indicated that there was unoccupied space where the enol-pyruvyl side chain of chorismate binds, which could be explored by extending the side chain. The second step of the reaction catalysed by anthranilate synthase involves elimination of the enol-pyruvyl side chain. It was envisaged that extension of the side chain may mimic this departure.
Molecular docking was used to predict the binding of the target compounds in the active site of S. marcescens anthranilate synthase. The ligands were first built in SYBYL6.5 and energy minimised using the Tripos force field.15 The aromatic analogues (7–21) were docked into the active site using GOLD2.1.16 The docking results suggested that these ligands would bind in a similar orientation to chorismate (the docking result of the R enantiomer of 21 is shown in Fig. 3B). A number of general observations were made about the dockings. The C-1 carboxylate of the analogues docked in such a way that it would interact with the metal ion, while the para-substituents were situated close to Glu309, suggesting a hydrogen-bonding interaction. The para-methoxy group in ligands (7–9) cannot donate a hydrogen bond to Glu309, however this does not affect the orientation of the ring in the docking. The C-3 side chains docked in the same binding pocket as the enol-pyruvyl side chain of chorismate (1), where they could potentially form hydrogen bonding interactions with Arg469 and Tyr449. The docking of the ligands with larger C-3 side chains (13–20) resulted in conformations where the aromatic ring was displaced slightly. This movement of the ring would affect both the interaction of the C-4 substituent with Glu309 and the C-1 carboxylate with the magnesium ion.
Synthesis
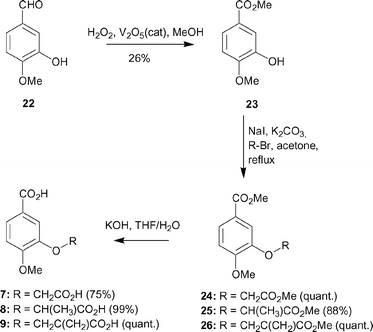 | ||
| Scheme 2 | ||
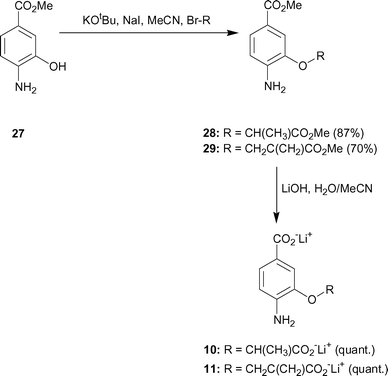 | ||
| Scheme 3 | ||
Synthesis of the 4-amino analogue 12 began from methyl 3,4-diaminobenzoate. The meta-amine of 30 is the most nucleophilic, so a weak base was used to alkylate at this position. Reaction of methyl-2-bromo-propionate with 30 in the presence sodium iodide and anhydrous potassium carbonate gave the desired diester 31 in moderate yield (Scheme 4). Finally, deprotection with aqueous lithium hydroxide yielded the dicarboxylate 12.
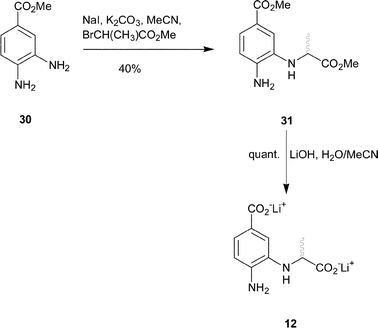 | ||
| Scheme 4 | ||
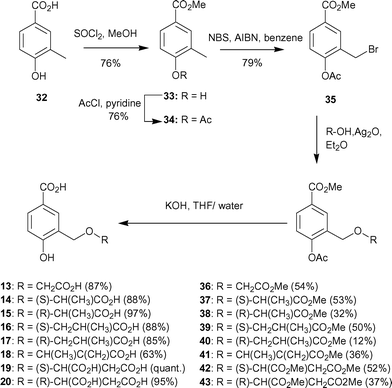 | ||
| Scheme 5 | ||
Ethers were formed by treatment of 35 with silver oxide in diethyl ether using a variety of alcohols. The reactions proceeding in moderate yields (35–50%). A number of commercially available alcohols were selected to produce the small library of triesters 36–43 with side chains of differing length and functionality. Methyl ester and acetate hydrolysis was achieved in one step in generally high yields by treatment with aqueous potassium hydroxide to afford the diacids 13–20.
Synthesis of compound 21 was achieved from methyl-3,4-dihydroxybenzoate (44). Selective alkylation of the meta-hydroxyl of methyl-3,4-dihydroxybenzoate proved to be more problematic than for previous analogues. Treatment of methyl-3,4-dihydroxybenzoate (44) with methyl-2-bromopropionate and sodium hydride in DMF at 25 °C for five days, gave the dialkylated product (16%) and an inseparable mixture of both mono-alkylated products (27%). The mixture of mono-alkylated products was further treated with benzoyl chloride in pyridine in order to protect the free alcohol (Scheme 6). Column chromatography of the crude product allowed separation of a small amount of the desired C-3 alkylated product. The regiochemistry of 45 was confirmed by NOE analysis. Irradiation of the doublet signal arising from the methyl group of the lactyl side chain caused enhancement of the doublet for the C-2 ring proton and vice versa. Hydrolysis of the methyl and benzoyl esters with aqueous potassium hydroxide and acidic work-up gave the desired diacid 21 as well as benzoic acid, which could be separated by HPLC.
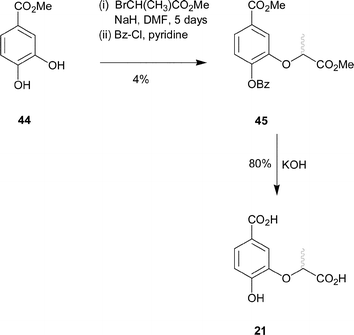 | ||
| Scheme 6 | ||
Assay Results
The aromatic chorismate analogues synthesised (7–21) were tested against S. marcescens anthranilate synthase using a previously reported fluorescence assay.18 All the compounds were found to be competitive reversible inhibitors. The inhibition results are shown in Table 1.Several general conclusions can be made from the inhibition data. Extension of the C-3 side chain (13–20) had a detrimental effect on binding of these analogues to the enzyme. Analogues containing an extra methylene linkage before the ether side chain at C-3 were relatively weak inhibitors of the enzyme, with inhibition constants ranging from 160–1600 µM. The introduction of methyl substituents in the side chain further reduced the affinity of these analogues and, in general, the longer the side chain the lower the potency of the inhibitor. The inclusion of a double bond in the side chain of analogue 18 recovered some binding affinity, possibly due to its similarity to the enol-pyruvyl side chain of chorismate.
Compounds 8, 10, 12 and 21, which all contain a lactyl side chain, were the most potent inhibitors of anthranilate synthase with KI's of 25, 43, 50 and 3 µM, respectively. It is unclear if this is because the lactyl side chain is a good mimic of the enol-pyruvyl side chain of chorismate. Alternatively, there may be additional bonding interactions associated with the stabilisation of the transition state that leads to the loss of the side chain and aromatisation of ADIC to form anthranilate.
Somewhat surprisingly, anthranilate synthase appears to be relatively insensitive to the functionality at C-4. Analogues 9 and 11, both containing an acrylate side chain and a methoxy and amino moiety at C-4, respectively, exhibited identical inhibition constants (120 µM).
The most potent of the aromatic chorismate analogues was 21 (Fig. 5), which exhibited a KI of 3 µM. This compound is five times less potent than 5. Of all the compounds tested it is the closest structural analogue of chorismate, with the lactyl group replacing the enol-pyruvyl side chain and an aromatic ring replacing the cyclohexadiene ring in chorismate.
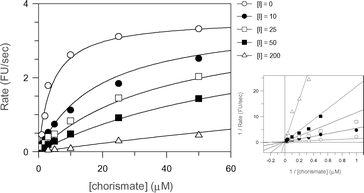 | ||
| Fig. 5 Least squares fitting and Lineweaver–Burk plots for the reversible competitive inhibition of anthranilate synthase by 21. | ||
These studies indicate that the use of an aromatic template in place of a more saturated ring system does not lead to a marked decrease in inhibition against anthranilate synthase when compared to those synthesised previously.12 Compound 21 is the second most potent inhibitor of anthranilate synthase reported to date and serves as a lead for future studies in this area.
Conclusions
New aromatic inhibitors of anthranilate synthase were designed and synthesised. Molecular modelling suggests that these inhibitors bind in a similar manner to the substrate. The inhibition results suggest that the lactyl side chain at C-3 is a good replacement for the enol-pyruvyl side chain of chorismate. Interestingly, altering the substitution at C-4 does not appear to affect the inhibition of anthranilate synthase substantially. This will be explored further and will be the focus of future work in this area.Experimental
General
All organic solvents were freshly distilled prior to use. Milli-Q deionised water was used for all biochemical work. Analytical thin layer chromatography was carried out on commercial silica gel 60 0.25 mm plates using either UV absorption or potassium permanganate stain for visualisation (3 g potassium permanganate, 20 g potassium carbonate, 5 ml of 5% sodium hydroxide, 300 ml water). RF values are quoted with respect to the solvent system used to develop the plate. Column chromatography was carried out using 230–400 mesh silica gel 60. Petroleum ether refers to the fraction distilled between 40–60 °C. Unless specified in the experimental, 1H NMR spectra were recorded on a Bruker AM-400 spectrometer in deuterated solvents, as indicated. Otherwise, 1H NMR spectra were recorded on a Bruker Avance 500 spectrometer. Unless specified in the experimental, all 13C NMR spectra were recorded on a Bruker AM-400 spectrometer operating at 100 MHz. Otherwise, 13C NMR spectra were carried out on a Bruker Avance 500 spectrometer operating at 125 MHz. All chemical shifts are quoted in parts per million (ppm) δ. Coupling constants for 1H NMR spectroscopy are assigned where possible and are given in Hz. Infrared spectra were recorded on an ATI Mattson Genesis FTIR infrared spectrometer using NaCl plates or on a Perkin Elmer Spectrum One FTIR spectrometer using attenuated transmittance reflectance (ATR). High resolution mass spectrometry was carried out using a Micromass Quadrapole-Time of Flight (Q–Tof) spectrometer. HPLC purification was carried out on a Gilson HPLC system fitted with a Gilson 118 UV/Vis detector, with detection at 254 nm. Where noted, compounds were analysed using a Waters Xterra MS C18 5 µm 4.6 × 50 mm column and purified using a Waters Xterra MS C18 5 µm 19 × 50 mm column. Columns were eluted with a gradient of between 5 : 95 and 95 : 5 acetonitrile : water, both containing 0.1% TFA. Liquid-chromatography mass-spectrometry (LCMS) was carried out using an Alliance HT Waters 2795 Separations Module coupled to a Waters Micromass ZQ Quadrapole Mass Analyzer. Samples were detected using a photomultiplier detection system. Samples were run on a gradient from 10 mM ammonium acetate containing 0.1% formic acid to 95% acetonitrile over a period of 8 min.Cloning and purification of anthranilate synthase
The vector ptttSmEDC containing S. marcescens trpEGDC was kindly supplied by Glen Spraggon (Genomics Institute of the Novartis Research Foundation, San Diego, California) and Charles Yanofsky (Stanford University, Stanford, California). The trpEG gene fragment was amplified with a BamHI restriction enzyme site at the 5′-end and a NcoI site at the 3′-end by PCR using the pttSmEDC vector as a template. The primers were 5′-CGC GGA TCC ATG AAC ACC AAA CCA CAA TTG ACA C-3′ (forward) and 5′-GCA TGC CAT GGT TAC TTC GCC AGC GCC CAG-3′ (reverse). The trpEG fragment was inserted between the BamHI and NcoI sites of the expression vector mini-pRSETA. Anthranilate synthase is over-expressed from the resulting trpEG/mini-pRSETA vector with a hexahistidine tag (MRGSHHHHHHGLVPRGS) attached to the N-terminus of the TrpE subunit.Cells of the Escherichia coli strain C41(DE3) were transformed with trpEG/mini-pRSETA and used to inoculate a 10 ml LB (50 µg ml−1 ampicillin) preculture. Precultures were grown for 16 h at 30 °C with 200 rpm shaking. A 1 L 2xYT (50 µg ml−1 ampicillin) culture was inoculated with the preculture and grown at 37 °C with 250 rpm shaking. Once an optical density (600 nm) of 0.6–0.8 was reached, induction of over-expression of the His6-tagged anthranilate synthase was carried out by the addition of IPTG (1 mM). Growth was continued for approximately 4 h. Cells were harvested by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 15 min and cell pellets were stored at −80 °C.
000 × g for 15 min and cell pellets were stored at −80 °C.
The following steps were all carried out on ice or at 4 °C. Cell pellets were defrosted and resuspended in Buffer N1 (50 mM sodium phosphate buffer pH 8.0, 300 mM NaCl, 10 mM imidazole, 1 mM β-mercaptoethanol). Cells were lysed by sonication and cell debris was removed by centrifugation at 39000 × g for 30 min. The cell-free lysate was applied to a 5 ml Ni–NTA (nitriloacetic acid) column that had been pre-equilibriated with Buffer N1. The column was washed with 24 column volumes of Buffer N2 (Buffer N1 with 50 mM imidazole). His6-tagged anthranilate synthase was eluted from the column with 4–5 column volumes of Buffer N3 (Buffer N1 with 250 mM imidazole). The eluted protein was dialysed into storage buffer (50 mM Tris HCl pH 7.5, 1 mM DTT and 0.1 mM EDTA), separated into aliquots (100–500 µl) and flash frozen in liquid nitrogen. Aliquots were stored at −80 °C.
Anthranilate synthase assay
Kinetic parameters for S. marcescens anthranilate synthase were determined using the kinetic fluorescence assay developed by Bauerle et al.18 Fluorescence was detected at 390 nm after initial excitation at 313 nm at 25 °C. Reactions were initiated by the addition of anthranilate synthase (20 µl of a 0.01 mg ml−1 solution) to the following assay mixture (total volume 200 µl): 100 mM potassium phosphate buffer (pH = 7.0); 10 mM magnesium chloride solution; 20 mM glutamine solution. For the inhibitor studies, various concentrations of inhibitor (10 µM–1 mM) were added to the assay mixture using chorismate concentrations varying from 1–50 µM. Initial rates were determined by measuring the increase in fluorescence over the first minute.Enzyme Kinetics Pro19 or Grafit20 software were used to construct Michaelis–Menton plots of the kinetic data and carry out a least squares fitting for the inhibition of anthranilate synthase. The software was also utilised to calculate the inhibition constants KI and associated standard errors, assuming reversible competitive inhibition.
Gold docking
All ligands and the receptor were prepared using SYBYL6.5 and used as MOL2 files.15 The ligands were prepared as carboxylate anions and their structures were energy minimised using the Tripos force-field. Gasteiger–Huckel charges were calculated prior to docking. Each ligand was docked using GOLD2.1 in 25 independent genetic algorithm (GA) runs. For each of these, a maximum number of 100000 GA operations was performed on a single population of 50 individuals.16 Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10 respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of the active site was introduced and the radius was set to 10 Å, with the automatic active-site detection selected.3-Hydroxy-4-methoxy-benzoic acid methyl ester 2317
Vanadium pentoxide (0.24 g, 1.32 mmol) was added to 30% H2O2 (14.9 ml, 0.13 mol) and the solution stirred at 0 °C until all of the vanadium pentoxide dissolved and the solution became reddy–brown. This solution was added dropwise over a period of 1 h to an ice-cold solution of iso-vanillin (5 g, 0.03 mol) in methanol (165 ml) containing 70% HClO4 (1.6 ml, 0.02 mol). The reaction was stirred for 6 h. The solvent was removed in vacuo and the resulting residue redissolved in ethyl acetate (50 ml) and applied to a silica column (eluent 4 : 1 v/v petroleum ether–ethyl acetate) to afford 23 as an orange solid (1.56 g, 26% isolated).RF
[4 : 1 petroleum ether : ethyl acetate]
= 0.18; νmax.
(ATR): 3411 (br, OH stretch) 3016, 2956 (Ar C–H stretch), 1697 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1612, 1589 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 3.85 (3H, s, CH3), 3.90 (3H, s, CH3), 5.88 (1H, br s, OH), 6.83 (1H, d, J 8.2 Hz, ArH, H-5), 7.57 (1H, dd, J 8.2, 2.1 Hz, ArH, H-6), 7.59 (1H, d, J 2.1 Hz ArH, H-2); 13C NMR (CDCl3)
δ 52.3, 56.4, 110.3, 116.0, 123.2, 123.7, 145.7, 150.9, 167.3; LCMS (MH+)
= 183.2 (ret. time = 3.3 min); HRMS calcd for C9H10O4Na: MNa+, 205.0477. Found: MNa+, 205.0473.
O), 1612, 1589 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 3.85 (3H, s, CH3), 3.90 (3H, s, CH3), 5.88 (1H, br s, OH), 6.83 (1H, d, J 8.2 Hz, ArH, H-5), 7.57 (1H, dd, J 8.2, 2.1 Hz, ArH, H-6), 7.59 (1H, d, J 2.1 Hz ArH, H-2); 13C NMR (CDCl3)
δ 52.3, 56.4, 110.3, 116.0, 123.2, 123.7, 145.7, 150.9, 167.3; LCMS (MH+)
= 183.2 (ret. time = 3.3 min); HRMS calcd for C9H10O4Na: MNa+, 205.0477. Found: MNa+, 205.0473.
4-Methoxy-3-methoxycarbonylmethoxy-benzoic acid methyl ester 24
Methylbromoacetate (0.20 ml, 2.14 mmol) was added dropwise to a stirred solution of 23 (300 mg, 1.65 mmol), sodium iodide (0.04 g, 0.30 mmol) and anhydrous potassium carbonate (0.46 g, 3.29 mmol) in acetone (18 ml). The reaction was heated at reflux for 18 h. The reaction was allowed to cool and the solvent was removed in vacuo. The residue was redissolved in DCM (10 ml) and washed with water (10 ml). The aqueous fraction was back extracted with DCM (2 × 10 ml). The combined organic fractions were dried (MgSO4), and the solvent was removed in vacuo to afford the crude product as a brown solid. Purification by column chromatography (eluent 4 : 1 v/v petroleum ether–ethyl acetate) gave 24 as a white solid (0.42 g, quant).RF [4 : 1 petroleum ether : ethyl acetate] = 0.16; 1H NMR (CDCl3) δ 3.78 (3H, s, CH3,), 3.85 (3H, s, CH3), 3.92 (3H, s, CH3), 4.72 (2H, s, CH2) 6.90 (1H, d, J 8.5 Hz, ArH, H-5), 7.46 (1H, d, J 1.9 Hz, ArH, H-2), 7.70 (1H, dd, J 8.5, 1.9 Hz, ArH, H-6); 13C NMR (CDCl3) δ 53.8, 54.1, 57.9, 67.9, 112.8, 116.3, 124.4, 126.8, 148.6, 155.3, 168.3, 170.8; HRMS calcd for C12H14O6Na: MNa+, 277.0688. Found: MNa+, 277.0698.
3-Carboxymethoxy-4-methoxy-benzoic acid 7
Diester 24 (0.15 g, 0.59 mmol) was dissolved in THF–water (1 : 1 v/v,8 ml). Potassium hydroxide (0.03 g, 2.35 mmol) was added and the reaction stirred at 40 °C for 2 h. The solvent was removed in vacuo and the resulting residue dissolved in water. The aqueous fraction was washed with ethyl acetate (10 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 20 ml), dried (MgSO4) and solvent removed in vacuo to afford the desired diacid 7 as a white solid (0.10 g, 75%).νmax.
(ATR): 3432, 3090, (br, acid OH stretch) 2931, 2570 (Ar C–H stretch), 1732, 1690 (CO), 1606, 1581 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 3.90 (3H, s, OCH3), 4.77 (2H, s, CH2), 7.07 (1H, d, J 8.5 Hz, ArH, H-5), 7.53 (1H, d, J 2.0 Hz, ArH, H-2), 7.68 (1H, dd, J 8.5, 2.0 Hz, ArH, H-6); 13C NMR (d6-acetone)
δ 57.9, 68.0, 114.0, 117.6, 125.3, 127.1, 149.8, 156.3, 168.9, 171.8; HRMS calcd for C10H10O6Na: MNa+, 249.0375. Found: MNa+, 249.0364.
4-Methoxy-3-(1-methoxycarbonylethoxy)-benzoic acid methyl ester 25
Methyl-2-bromopropionate (0.24 ml, 2.14 mmol) was added dropwise to a stirred solution of 23 (300 mg, 1.65 mmol), sodium iodide (0.04 g, 0.30 mmol) and anhydrous potassium carbonate (0.46 g, 3.29 mmol) in acetone (18 ml). The reaction was heated at reflux for 17 h. Work up was carried out as described for 24 and the product purified by column chromatography (eluent 4 : 1 v/v petroleum ether–ethyl acetate) to afford 25 as a white solid (0.38 g, 88%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.29; νmax.
(ATR): 2957, 2946 (Ar C–H stretch), 1731, 1711 (CO), 1599, 1587 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.63 (3H, d, J 6.8 Hz, CH3), 3.73 (3H, s, CH3,), 3.84 (3H, s, CH3), 3.88 (3H, s, CH3), 4.81 (1H, q, J 6.8 Hz, CH), 6.87 (1H, d, J 8.5 Hz, ArH, H-5), 7.48 (1H, d, J 1.9 Hz, ArH, H-2), 7.68 (1H, dd, J 8.5, 1.9 Hz, ArH, H-6); 13C NMR (CDCl3)
δ 16.8,50.3, 50.6, 54.4, 72.2, 109.6, 114.9, 121.0, 123.4, 144.8, 152.4, 164.9, 170.5; HRMS calcd for C13H16O6Na: MNa+, 291.0845. Found: MNa+, 291.0828
3-(1-Carboxyethoxy)-4-methoxy-benzoic acid 8
Methyl ester 25 (0.18 g, 0.67 mmol) was dissolved in THF–water (1 : 1 v/v, 10 ml). Potassium hydroxide (0.15 g, 2.68 mmol) was added and the reaction stirred at 40 °C for 2 h. Acidification and extraction as described for 7, gave 8 as a white solid (0.16 g, 99%).νmax.
(ATR): 2989 (br, acid OH stretch) 2596 (Ar C–H stretch), 1718, 1676 (CO), 1603, 1583 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.60 (3H, d, J 6.8 Hz, CH3), 3.91 (3H, s, OCH3), 4.85 (1H, q, J 6.8 Hz, CH), 7.07 (1H, d, J 8.4 Hz, ArH, H-5), 7.55 (1H, d, J 2.0 Hz, ArH, H-2), 7.69 (1H, dd, J 8.4, 2.0 Hz, ArH, H-6); 13C NMR (d6-acetone)
δ 19.2, 56.7, 74.6, 112.8, 117.8, 124.0, 126.1, 148.2, 155.5, 167.7, 173.5; HRMS calcd for C11H12O6Na: MNa+, 263.0532. Found: MNa+, 263.0529.
4-Methoxy-3-(2-methoxycarbonyl-allyloxy)-benzoic acid methyl ester 26
Methyl-2-(bromomethyl)acrylate (0.26 ml, 2.14 mmol) was added dropwise to a stirred solution of 23 (300 mg, 1.65 mmol), sodium iodide (0.04 g, 0.30 mmol) and anhydrous potassium carbonate (0.46 g, 3.29 mmol) in acetone (18 ml). The reaction was heated at reflux for 14 h. Work up was carried out as described for 24 and the product purified by column chromatography (eluent 4 : 1 v/v petroleum ether–ethyl acetate) to afford 26 as a white solid (0.48 g, quant.).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.45; νmax.
(NaCl plate): 2952 (br, acid OH stretch), 1719, 1655 (CO), 1438 (CC) cm−1; 1H NMR (CDCl3)
δ 3.79 (3H, s, CH3), 3.87 (3H, s, CH3), 3.91 (3H, s, CH3), 4.83 (2H, s, CH2), 6.20 (1H, s, CC–H), 6.40 (1H, s, CC–H), 6.88 (1H, d, J 8.5 Hz, ArH, H-5), 7.54 (1H, d, J 2.0 Hz, ArH, H-2), 7.68 (1H, dd, J 8.5, 2.0 Hz, ArH, H-6); 13C NMR (CDCl3)
δ 52.4, 56.4, 67.6, 111.2, 114.9, 123.1, 124.7, 127.2, 135.8, 147.7, 154.0, 166.2, 167.1; HRMS calcd for C14H16O6Na: MNa+, 303.0845. Found: MNa+, 303.0846.
3-(2-Carboxy-allyloxy)-4-methoxy-benzoic acid 9
Methyl ester 26 (0.15 g, 0.53 mmol) was dissolved in THF–water (1 : 1 v/v, 8 ml). Potassium hydroxide (0.12 g, 2.13 mmol) was added and the reaction stirred at 40 °C for 2 h. Acidification and extraction as described for 7, gave the desired product 9 as a white solid (0.17 g, quant.).νmax.
(ATR): 2845 (br, acid OH stretch) 2588 (Ar C–H stretch), 1679 (CO), 1635, 1601, 1586 (CC, ar), 1436 (CC) cm−1; 1H NMR (CD3OD)
δ 3.89 (3H, s, OMe), 4.77 (2H, s, CH2), 5.99 (1H, s, CC–H), 6.35 (1H, s, CC–H), 7.02 (1H, d, J 8.5 Hz, ArH, H-5), 7.56 (1H, d, J 2.0 Hz, ArH, H-2), 7.67 (1H, dd, J 8.5, 2.0 Hz, ArH, H-6); 13C NMR (CD3OD)
δ 56.9, 69.1, 112.7, 116.7, 124.5, 126.2, 127.7, 138.5, 149.1, 155.5, 169.1, 170.0; HRMS calcd for C12H12O6Na: MNa+, 275.0532. Found: MNa+, 275.0522.
Methyl 4-amino-3-(1-methoxycarbonyl-ethoxy)-benzoate 28
Methyl-2-bromopropionate (200 µl, 1.80 mmol) was added to a solution of methyl 3-hydroxy-4-aminobenzoate (300 mg, 1.80 mmol), potassium tert-butoxide (202 mg, 1.80 mmol) and sodium iodide (54 mg, 0.36 mmol) in acetonitrile (2 ml) and stirred at 22 °C, under argon for 6 h. The mixture was diluted with ethyl acetate (50 ml) and washed with saturated aqueous sodium bicarbonate solution (3 × 50 ml). The organic layer was dried over Na2SO4, filtered and the solvent removed in vacuo. Purification by column chromatography (eluent: 1 : 1 v/v ethyl acetate–hexane) gave 28 as a white solid (400 mg, 87%).RF
[1 : 1 hexane : ethyl acetate]
= 0.62; νmax
(ATR) 3065 (NH), 1721, 1692 (CO), 1609 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.57 (3 H, d, J 6.9 Hz, CH3), 3.74 (3 H, s, CH3), 3.88 (3 H, s, CH3), 4.68 (1 H, q, J 6.9 Hz, CH), 6.88 (1 H, d, J 8.1 Hz, H-5), 7.63 (1 H, d, J 1.7 Hz, H-2), 7.66 (1 H, dd, J 8.1, 1.7 Hz, H-6); 13C NMR (DEPT, CDCl3)
δ 15.1 (CH3), 50.9 (OCH3), 52.2 (CH3), 72.1 (C), 114.2 (CH), 117.2 (CH), 123.3 (CH), 124.8 (C), 129.4 (C), 141.5 (C), 165.0 (C),167.5 (C); LC/MS (ret. time = 3.5 min) (ESI+)
m/z 254 (MH+).
Lithium 4-amino-3-(1-carboxy-ethoxy)-benzoate 10
Methyl ester 28 (100 mg, 0.39 mmol) and lithium hydroxide (41 mg, 0.98 mmol) in H2O–MeCN (1 : 1 v/v, 2 ml) was stirred at 22 °C for 8 h. The solvent was removed in vacuo to give the dicarboxylate 10 as a white solid, (88 mg, quant).νmax
(ATR) 3067 (NH), 1682 (CO), 1611 (CC, ar) cm−1; 1H NMR (D2O)
δ 1.27 (3 H, d, J 6.9 Hz, CH3), 4.52 (1 H, q, J 6.9 Hz, CH), 6.81 (1 H, d, J 8.1 Hz, H-5), 7.28 (1 H, d, J 1.9 Hz, H-2), 7.38 (1 H, dd, J 8.1, 1.9 Hz, H-6); 13C NMR (DEPT, D2O)
δ 16.5 (CH3), 72.8 (CH), 116.9 (CH), 119.9 (CH), 124.2 (CH), 130.8 (C), 137.0 (C), 142.7 (C), 175.4 (C), 174.3 (C); LC/MS (ret. time = 3.1 min) (ESI+)
m/z 226 (MH+).
Methyl 4-amino-3-[(2-methoxycarbonylallyl)-oxy]-benzoate 29
Potassium tert-butoxide (136 mg, 1.20 mmol) was added to a solution of methyl 3-hydroxy-4-aminobenzoate (200 mg, 1.20 mmol) and sodium iodide (36 mg, 0.24 mmol) in acetonitrile (10 ml) and the reaction stirred at 22 °C, under argon. After 20 min, methyl 2-(bromomethyl)acrylate (148 µl, 1.20 mmol) was added dropwise and the reaction was stirred for a further 1 h at 22 °C. The solution was diluted with ethyl acetate (50 ml) and washed with saturated aqueous sodium bicarbonate solution (3 × 30 ml). The organic layer was dried (Na2SO4) and the solvent removed in vacuo. Purification by column chromatography (eluent: 1 : 1 v/v ethyl acetate–hexane) gave 29 as a white solid (220 mg, 70%).RF
[1 : 1 hexane : ethyl acetate]
= 0.69; νmax 3340 (NH), 1709, 1680 (CO), 1624 (CC), 1593 (CC, ar) cm−1;
1H NMR (CDCl3)
δ 3.81 (3 H, s, OCH3), 3.85 (3 H, s, OCH3), 4.13 (2H, bs, NH2), 4.81 (2H, s, CH2), 5.95 (1 H, s, CHH), 6.41 (1 H, s, CHH), 6.67 (1 H, d, J 8.2 Hz, H-5), 7.49 (1 H, d, J 1.7 Hz, H-2), 7.57 (1 H, dd, J 8.2, 1.7 Hz, H-6); 13C NMR (100 MHz, DEPT, CDCl3), 52.1 (CH3), 52.5 (CH3), 67.6 (CH2), 114.0 (CH), 114.1 (CH), 120.0 (C), 125.2 (CH), 127.6 (CH2), 136.1 (C), 142.0 (C), 145.0 (C), 166.3 (C), 167.5 (C); LC/MS (ret. time = 3.7 min) (ESI+)
m/z 266 [MH+].
Lithium 4-amino-3-[(2-carboxyallyl)-oxy]-benzoate 11
A solution of 29 (200 mg, 0.75 mmol) and lithium hydroxide (94 mg, 2.25 mmol) in H2O–MeCN (1 : 1 v/v, 2 ml) was stirred for 10 h at 22 °C. The solution was diluted with water (10 ml), washed with ethyl acetate (2 × 10 ml) and lyophilised to give the dicarboxylate 11 as a yellow solid (165 mg, 93%).νmax
(ATR) 3042 (NH), 1591 (CO + CC, ar str) cm−1; 1H NMR (500 MHz, D2O)
δ 4.57 (2 H, s, CH2), 5.47 (1 H, s, CHH), 5.73 (1 H, s, CHH), 6.63 (1 H, d, J 8.1 Hz, H-5), 7.18 (1 H, d, J 8.1 Hz, H-6), 7.22 (1 H, s, H-2); 13C NMR (100 MHz, DEPT, CDCl3)
δ 69.8 (CH2), 115.0 (CH), 115.7 (CH), 123.2 (CH2), 124.2 (CH), 127.1 (C), 140.3 (C), 142.0 (C),145.8 (C), 174.9 (C), 175.7 (C); LC/MS (ret. time = 3.3 min) (ESO+)
m/z 238 (MH+); HRMS calcd for C11H10NO5: (M–H)−, 236.0564. Found: (M–H)−, 236.0565.
Methyl 4-amino-3-(1-methoxycarbonyl-ethylamino)-benzoate 31
Methyl 2-bromopropionate (335 µl, 3.00 mmol) was added dropwise to a solution of methyl 3,4-diaminobenzoate (500 mg, 3.00 mmol), anhydrous potassium carbonate (620 mg, 4.50 mmol) and potassium iodide (90 mg, 0.60 mmol) in acetonitrile (3 ml) and the reaction was stirred under nitrogen at 22 °C, for 24 h. The reaction was diluted with ethyl acetate (50 ml) and washed with saturated aqueous sodium bicarbonate solution (3 × 50 ml). The organic layer was dried (Na2SO4) and the solvent removed in vacuo. Purification by column chromatography (eluent: 1 : 1 v/v ethyl acetate–hexane) gave 31 as a yellow solid (300 mg, 40%).RF
[1 : 1 hexane : ethyl acetate]
= 0.31; νmax
(ATR) 3352 (NH), 1676 (CO), 1601 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.35 (3 H, d, J 6.7 Hz, CH3), 3.74 (3 H, s, CH3), 3.84 (3 H, s, CH3), 3.90 (1 H, q, J 6.7 Hz, CH), 6.80 (1 H, d, J 8.6 Hz, H-5), 7.34 (2 H, m, H-2 and H-6); 13C NMR (DEPT, MeOD)
δ 17.9 (CH3), 51.9 (CH), 52.4 (OCH3), 52.6 (OCH3), 115.7 (CH), 115.9 (CH), 122.5 (CH), 126.3 (C), 131.8 (C), 135.6 (C), 168.6 (C), 171.6 (C); LC/MS (ret. time = 3.1 min) (ESI+)
m/z 221 ([M–MeOH]H+).
Lithium 4-amino-3-(1-carboxy-ethylamino)-benzoate 12
A solution of 31 (100 mg, 0.40 mmol) and lithium hydroxide (41 mg, 0.84 mmol) in H2O–MeCN (1 : 1 v/v, 2 ml) was stirred at 22 °C for 2 h. The acetonitrile was removed in vacuo and the remaining solution lyophilised to afford the dicarboxylate 12 as a white solid. (94 mg, quant.).νmax
(ATR) 3340 (NH), 1668 (CO), 1610 (CC, ar) cm−1; 1H NMR (D2O)
δ 1.16 (3 H, d, J 6.8 Hz, CH3), 3.74 (1 H, q, J 6.8 Hz, CH), 6.75 (1 H, d, J 8.1 Hz, H-5), 7.19 (1 H, d, J 1.8 Hz, H-2), 7.25 (1 H, dd, J 8.1, 1.8 Hz, H-6); 13C NMR (DEPT, D2O)
δ 17.1 (CH3), 51.4 (CH), 116.0 (CH), 118.1 (CH), 122.1 (CH), 131.2 (C), 133.7 (C), 134.4 (C), 175.0 (C), 175.7 (C).
Methyl 4-hydroxy-3-methylbenzoate 33
To a suspension of 4-hydroxy-3-methyl benzoic acid (2.5 g, 0.02 mol) in freshly distilled methanol (40 ml) was added 20 drops of thionyl chloride. The reaction mixture was heated at reflux under nitrogen for 16 h. The solution was allowed to cool to 22 °C before the solvent was removed in vacuo to give the crude product as a white solid. Purification by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) gave 33 as a white solid. (2.08 g, 76%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.41; νmax.
(ATR): 3260 (br OH stretch), 2961 (Ar C–H stretch), 1683 (CO), 1598, 1509 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 2.27 (3H, s, CH3), 3.87 (3H, s, CH3), 5.66 (1H, br s, OH), 6.80 (1H, d, J 8.5 Hz, ArH, H-5), 7.78 (1H, dd, J 8.5, 1.7 Hz, ArH, H-6), 7.83 (1H, d, J 1.7 Hz, ArH); 13C NMR (CDCl3)
δ 13.9, 50.2, 113.0, 120.7, 122.2, 127.7, 131.1, 156.5, 165.5; HRMS calcd for C9H10O3Na: MNa+, 189.0528. Found: MNa+, 189.0533.
4-Acetoxy-3-methyl-benzoic acid methyl ester 34
Methyl ester 33 (2.03 g, 0.012 mol) in dry pyridine (10 ml) was cooled to 0 °C. Acetyl chloride (1.30 ml, 0.018 mol) was added dropwise at 0 °C and the reaction stirred at 22 °C for 6 h. The reaction was diluted with ethyl acetate (30 ml), washed with saturated ammonium chloride solution (2 × 20 ml), brine (20 ml), dried (MgSO4), and solvent removed in vacuo to afford the crude product as an orange oil. Purification by column chromatography (eluent 5 : 1 v/v petroleum ether–ethyl acetate) gave the acetate 34 as a white solid (1.95 g, 76%).RF
[5 : 1 petroleum ether : ethyl acetate]
= 0.50; νmax.
(ATR) 2954 (Ar C–H stretch), 1686 (CO), 1610, 1591 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 2.27 (3H, s, CH3) 3.87 (3H, s, CH3), 6.79 (1H, d, J 8.4 Hz, ArH, H-5), 7.77 (1H, dd, J 8.4, 1.5 Hz, ArH, H-6), 7.83 (1H, d, J 1.5 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 17.6, 20.5, 51.9, 123.0, 127.0, 129.1, 130.9, 135.6, 149.0, 166.0, 168.7; HRMS calcd for C11H12O4Na: MNa+, 231.0633. Found: MNa+, 231.0623.
4-Acetoxy-3-bromomethyl-benzoic acid methyl ester 35
To a solution of 34 (1.90 g, 9.08 mmol) in benzene (70 ml) under nitrogen was added N-bromosuccinimide (1.62 g, 9.08 mmol) and AIBN (0.08 g, 0.45 mmol). The reaction was heated at reflux (85 °C) for 6 h. The reaction was allowed to cool to 22 °C before dilution with DCM (50 ml). The organic fraction was washed with aqueous saturated sodium carbonate solution (70 ml), water (70 ml), dried (MgSO4) and the solvent removed in vacuo. The crude product was recrystallised from petroleum ether–ethyl acetate to afford bromide 35 as colourless needles. (2.05 g, 79%).νmax.
(NaCl plate): 2954 (Ar C–H stretch), 1767, 1724 (CO), 1613, 1595 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 2.38 (3H, s, CH3), 3.90 (3H, s, CH3), 4.43 (2H, s, CH2) 7.22 (1H, d, J 8.5 Hz, ArH, H-5), 8.01 (1H, dd, J 2.1, 8.5 Hz, ArH, H-6), 8.10 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 22.7, 28.5, 54.0, 125.0, 129.8, 131.6, 132.9, 134.0, 154.2, 167.5, 170.0; HRMS calcd for C11H11O4BrNa: MNa+, 308.9758. Found: MNa+, 308.9741.
4-Acetoxy-3-methoxycarbonyl-methoxymethyl)-benzoic acid methyl ester 36
Methyl glycolate (20 µl, 0.25 mmol) was added dropwise to a solution of 35 (80 mg, 0.28 mmol) in freshly distilled diethyl ether (1.5 ml). Silver oxide (129 mg, 0.56 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring, additional silver oxide (50 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 18 h. The reaction was filtered through a small plug of silica and the silica washed with diethyl ether (30 ml). The combined organic fractions were dried (MgSO4), and the solvent removed in vacuo to afford the crude product as a colourless oil. Purification by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) gave 36 as a colourless oil. (40 mg, 54%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.48; νmax.
(NaCl plate): 2953 (Ar C–H stretch), 1759, 1722 (CO), 1612, 1592 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 2.33 (3H, s, CH3), 3.75 (3H, s, CH3), 3.90 (3H, s, CH3), 4.06 (2H, s, CH2), 4.62 (2H, s, CH2), 7.15 (1H, d, 1H, d, J 8.4 Hz, ArH, H-5), 8.02 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.13 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 20.8, 51.8, 52.2, 67.0, 68.1, 122.8, 128.0, 129.5, 130.8, 131.5, 152.7, 166.1, 168.8, 170.4; HRMS calcd for C11H12O6Na: MNa+, 319.0794. Found: MNa+, 319.0803.
3-Carboxymethoxymethyl-4-hydroxy benzoic acid 13
Potassium hydroxide (91 mg, 1.62 mmol) was added to a solution of 36 (20 mg, 0.07 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction was heated to 50 °C for 12 h. The reaction was allowed to cool to 22 °C before dilution with water (2 ml). The aqueous layer was washed with ethyl acetate (4 ml) before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 5 ml). The organic fraction was dried (MgSO4) and the solvent removed in vacuo to afford the desired diacid 13 as a white solid. (13 mg, 85%).νmax.
(ATR): 3289, (br, acid OH stretch and ar OH), 2897, 2567 (Ar C–H stretch), 1676 (CO str), 1617, 1586 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 4.26 (2H, s, CH2), 4.69 (2H, s, CH2), 6.92 (1H, d, J 8.4 Hz, ArH, H-5), 7.89 (1H, dd, J 2.1, 8.5 Hz, ArH, H-6), 7.97 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 65.6, 68.2, 114.6, 120.1, 123.3, 130.5, 130.6, 159.2, 166.0, 172.4; HRMS calcd for C10H10O6Na: MNa+, 249.0375. Found: MNa+, 249.0367.
4-Acetoxy-3-(1-methoxycarbonyl-ethoxy-(S)-methyl)-benzoic acid methyl ester 37
Methyl-(S)-lactate (30 µl, 0.31 mmol) was added dropwise to a solution of 35 (100 mg, 0.35 mmol) in freshly distilled diethyl ether (1.5 ml). Silver oxide (161 mg, 0.67 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (30 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 29 h. Work up was carried out as described for 36. The product was purified by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) to afford 37 as a colourless oil (50 mg, 53%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.33; νmax.
(NaCl plate): 2953, 2921 (Ar C–H stretch), 1747, 1722 (CO, ar), 1614, 1592 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.41 (3H, d, J 6.9 Hz, CH3), 2.33 (3H, s, CH3), 3.75 (3H, s, CH3), 3.90 (3H, s, CH3), 4.00 (1H, q, J 6.9 Hz, CH), 4.38 (1H, d, J 12.2 Hz, CHH), 4.70 (1H, d, J 12.2 Hz, CHH), 7.15 (1H, d, J 8.4 Hz, ArH, H-5), 8.01 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.13 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 19.0, 21.3, 52.4, 52.6, 67.2, 74.4, 123.2, 128.4, 130.4, 131.0, 131.9, 153.0, 166.6, 169.2, 173.7; HRMS calcd for C15H18O7: MH+, 310.1053. Found: MH+, 310.1051.
3-(1-Carboxy-ethoxy-(S)-methyl)-4-hydroxy benzoic acid 14
Potassium hydroxide (108 mg, 1.92 mmol) was added to a solution of 37 (25 mg, 0.08 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction was heated to 50 °C for 12 h. Acidification and extraction were carried out as described for 13 to afford the desired diacid 14 as a white solid. (17 mg, 88%).νmax.
(ATR): 3290 (br, acid OH stretch and ar OH), 2926, 2547 (Ar C–H stretch), 1731 (CO), 1673, 1616, 1587 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.43 (3H, d, J 7.0 Hz, CH3), 4.24 (1H, q, J 7.0 Hz, CH), 4.59 (1H, d, J 11.2 Hz, CHH), 4.72 (1H, d, J 11.2 Hz, CHH), 6.91 (1H, d, J 8.5 Hz, ArH, H-5), 7.87 (1H, dd, J 2.2, 8.5 Hz, ArH, H-6), 7.94 (1H, d, J 2.2 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 20.3, 70.5, 75.9, 118.0, 123.9, 126.0, 133.8, 162.8, 168.9, 178.1; HRMS calcd for C11H12O6Na: MNa+, 263.0532. Found: MNa+, 263.0539.
4-Acetoxy-3-(1-methoxycarbonyl-ethoxy-(R)-methyl)-benzoic acid methyl ester 38
Compound 38 was synthesised in an identical manner to 37, above, using methyl-(R)-lactate in place of methyl-(S)-lactate. Purification by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) gave 38 as a colourless oil (30 mg, 32%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.33; νmax.
(NaCl plate): 2953, 2921 (Ar C–H stretch), 1747, 1722 (CO), 1614, 1592 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.40 (3H, d, J 6.9 Hz, CH3), 2.33 (3H, s, CH3), 3.75 (3H, s, CH3), 3.90 (3H, s, CH3), 4.00 (1H, q, J 6.9 Hz, CH), 4.39 (1H, d, J 12.2 Hz, CHH), 4.70 (1H, d, J 12.2 Hz, CHH), 7.15 (1H, d, J 8.1 Hz ArH, H-5), 8.00 (1H, dd, J 2.1, 8.1 Hz, ArH, H-6), 8.13 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 19.0, 21.3, 52.4, 52.6, 67.2, 74.4, 123.2, 128.4, 130.4, 131.0, 131.9, 153.0, 166.6, 169.2, 173.7; LCMS (MH+
= 311.2) (ret. time = 3.73 min); HRMS calcd for C15H18O7Na: MNa+, 333.0950. Found: MNa+, 333.0942.
3-(1-Carboxy-ethoxy-(R)-methyl)-4-hydroxy benzoic acid 15
Potassium hydroxide (91 mg, 1.62 mmol) was added to a solution of 38 (20 mg, 0.06 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction heated at 50 °C for 12 h. Acidification and extraction were carried out as described for 13 to afford the desired diacid 15 as a white solid. (15 mg, 97%).νmax.
(ATR): 3290, 2987 (br, acid OH stretch and ar OH), 2930, 2547 (Ar C–H stretch), 1731 (CO), 1674, 1615, 1587 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.43 (3H, d, J 7.0 Hz, CH3), 4.24 (1H, q, J 7.0 Hz, CH), 4.59 (1H, d, J 11.2 Hz, CHH), 4.72 (1H, d, J 11.2 Hz, CHH), 6.91 (1H, d, J 8.5 Hz, ArH, H-5), 7.87 (1H, dd, J 2.2, 8.5 Hz, ArH, H-6), 7.94 (1H, d, J 2.2 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 20.3, 70.5, 75.9, 118.0, 123.9, 126.0, 133.8 (× 2), 162.8, 168.9, 178.1;HRMS calcd for C11H12O6Na: MNa+, 263.0532. Found: MNa+, 263.0540.
4-Acetoxy-3-(2-methoxycarbonyl-propoxy-(S)-methyl)-benzoic acid methyl ester 39
Methyl (S)-3-hydroxy-2-methylpropionate (52 µl, 0.47 mmol) was added dropwise to a solution of 35 (150 mg, 0.52 mmol) in freshly distilled diethyl ether (2 ml). Silver oxide (242 mg, 1.05 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (75 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 20 h. Work up was carried out as described for 36 and the product was purified by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) to afford 39 as a colourless oil (76 mg, 50%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.40; νmax.
(NaCl plate): 2980, 2953, 2878 (Ar C–H stretch), 1766, 1730 (CO), 1614, 1593 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.15 (3H, d, J 7.1 Hz, CH3), 2.30 (3H, s, CH3), 2.74 (1H, ddd, J 5.8, 7.1, 7.3 Hz, CH), 3.45 (1H, dd, J 5.8, 9.0 Hz, CHH), 3.61 (1H, dd, J 7.3, 9.0 Hz, CHH), 3.68 (3H, s, CH3), 3.89 (3H, s, CH3), 4.42 (1H, d, J 12.6 Hz, CHH), 4.48 (1H, d, J 12.6 Hz, CHH), 7.12 (1H, d, J 8.4 Hz, ArH, H-5), 7.98 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.08 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 13.8, 20.7, 39.9, 51.6, 52.0, 67.9, 72.2, 122.4, 127.8, 130.2, 130.4, 130.9, 152.2, 166.1, 168.5, 175.0; HRMS calcd for C16H20O7Na:MNa+, 347.1107. Found: MNa+, 347.1109.
3-(2-Carboxy-propoxy-(S)-methyl)-4-hydroxy-benzoic acid 16
Potassium hydroxide (198 mg, 3.54 mmol) was added to a solution of 39 (72 mg, 0.22 mmol) in THF–water (1 : 1 v/v, 4 ml) and the reaction was heated at 50 °C for 12 h. Acidification and extraction was carried out as described for 13 to afford the desired diacid 16 as a white solid (50 mg, 88%).νmax.
(ATR): 3191, 2987 (br, acid OH stretch and ar OH), 2947, 2886 (Ar C–H stretch), 1680 (CO str), 1610 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.18 (3H, d, J 7.1 Hz, CH3), 2.79 (1H, ddd, J 5.7, 7.0, 7.1 Hz, CH), 3.63 (1H, dd, J 5.7, 9.2 Hz, CHH), 3.75 (1H, dd, J 7.0, 9.2 Hz, CHH), 4.62 (2H, s, CH2), 6.91 (1H, d, J 8.5 Hz, ArH, H-5), 7.83 (1H, dd, J 2.2, 8.5 Hz, ArH, H-6), 7.99 (1H, d, J 2.2 Hz, ArH, H-2), 9.05 (1H, brs, OH); 13C NMR (d6-acetone)
δ 12.4, 38.6, 67.2, 71.4, 114.0, 125.2, 123.7, 129.8 (× 2), 158.3, 170.9, 179.1; HRMS calcd for C12H14O6Na: MNa+, 277.0688. Found: MNa+, 277.0677.
4-Acetoxy-3-(2-methoxycarbonyl-propoxy-(R)-methyl)-benzoic acid methyl ester 40
Methyl (R)-3-hydroxy-2-methylpropionate (52 µl, 0.47 mmol) was added dropwise to a solution of 35 (150 mg, 0.52 mmol) in freshly distilled diethyl ether (2 ml). Silver oxide (242 mg, 1.05 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (75 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 20 h. Work up was carried out as described for 36 and the product was purified by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) to afford 40 as a colourless oil (20 mg, 12%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.39; νmax.
(NaCl plate): 2953, 2878 (Ar C–H stretch), 1765, 1725 (CO), 1610, 1591 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 1.15 (3H, d, J 7.1 Hz, CH3), 2.30 (3H, s, CH3), 2.74 (1H, ddd, J 5.8, 7.1, 7.3 Hz, CH), 3.45 (1H, dd, J 5.8, 9.0 Hz, CHH), 3.61 (1H, dd, J 7.3, 9.0 Hz, CHH), 3.68 (3H, s, CH3), 3.89 (3H, s, CH3), 4.42 (1H, d, J 12.6 Hz, CHH), 4.48 (1H, d, J 12.6 Hz, CHH), 7.12 (1H, d, J 8.4 Hz, ArH, H-5), 7.98 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.08 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 13.8, 20.7, 39.9, 51.6, 52.0, 67.9, 72.2, 122.4, 127.8, 130.2, 130.4, 130.9, 152.2, 166.1, 168.5, 175.0; HRMS calcd for C16H20O7Na: MNa+, 347.1107. Found: MNa+, 347.1097.
3-(2-Carboxy-propoxy-(R)-methyl)-4-hydroxy-benzoic acid 17
Potassium hydroxide (58 mg, 1.03 mmol) was added to a solution of 40 (21 mg, 0.01 mmol) in THF–water (1 : 1 v/v, 2 ml). The reaction was heated at 50 °C for 12 h. Acidification and extraction was carried out as described for 13 to afford the desired diacid 17 as a white solid. (14 mg, 85%).νmax.
(ATR): 3185 (br, acid OH stretch and ar OH), 2948, 2879 (Ar C–H stretch), 1682 (CO, str), 1610 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 1.18 (3H, d, J 7.1 Hz, CH3), 2.79 (1H, ddd, J 5.7, 7.0, 7.1 Hz, CH), 3.63 (1H, dd, J 5.7, 9.2 Hz, CHH), 3.75 (1H, dd, J 7.0, 9.2 Hz, CHH), 4.62 (2H, s, CH2), 6.91 (1H, d, J 8.5 Hz, ArH, H-5), 7.83 (1H, dd, J 2.2, 8.5 Hz, ArH, H-6), 7.99 (1H, d, J 2.2 Hz, ArH, H-2), 9.05 (1H, brs, OH); 13C NMR (d6-acetone)
δ 12.4, 38.6, 67.2, 71.4, 114.0, 125.2, 123.7, 129.8 (× 2), 158.3, 170.9, 179.1; HRMS calcd for C12H14O6Na: MNa+, 277.0688. Found: MNa+, 277.0678.
4-Acetoxy-3-(2-methoxycarbonyl-1-methyl-allyloxymethyl)-benzoic acid methyl ester 41
Methyl-3-hydroxy-2-methylenebutyrate (57 µl, 0.47 mmol) was added to a solution of 35 (150 mg, 0.52 mmol) in freshly distilled diethyl ether (2 ml). Silver oxide (242 mg, 1.05 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (75 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 20 h. Work up was carried out as described for 36 and the product was purified by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) to afford 41 as a colourless oil (60 mg, 36%).RF
[3 : 1 petroleum ether:ethyl acetate]
= 0.43; νmax.
(NaCl plate): 2980, 2952 (Ar C–H stretch), 1765, 1720 (CO), 1630, 1599 (CC, ar), 1438 (CC) cm−1; 1H NMR (CDCl3)
δ 1.33 (3H, d, J 6.4 Hz, CH3), 2.29 (3H, s, CH3), 3.76 (3H, s, CH3), 3.89 (3H, s, CH3), 4.39 (1H, q, J 6.4 Hz, CH), 4.35 (1H, d, J 12.3 Hz, CHH), 4.47 (1H, d, J 12.3 Hz, CHH), 5.91 (1H, d, J 1.2 Hz, CCHH), 6.29 (1H, d, J 1.2 Hz, CCHH), 7.13 (1H, d, J 8.4 Hz, ArH, H-5), 7.98 (1H, dd, J 2.2, 8.4 Hz, ArH, H-6), 8.14 (1H, d, J 2.2 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 21.2, 22.2, 52.2, 52.6, 66.0, 74.1, 122.9, 125.0, 128.4, 130.6, 131.2, 131.5, 142.5, 152.6, 166.7, 167.0, 169.0; HRMS calcd for C17H20O7Na: MNa+, 359.1107. Found: MNa+, 347.1104.
3-[1-(1-Carboxy-ethyl)-vinyloxymethyl]-4-hydroxy-benzoic acid 18
Potassium hydroxide (107 mg, 1.90 mmol) was added to a solution of 41 (40 mg, 0.19 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction heated to 50 °C for 12 h. Acidification and extraction was carried out as described for 13 to afford the desired diacid 18 as a white solid (20 mg, 63%).νmax.
(ATR): 3183 (br, acid OH stretch and ar OH), 2948 (Ar C–H stretch), 1650 (CO, str), 1586 (CC, ar), 1427 (CC) cm−1; 1H NMR (d6-acetone)
δ 1.36 (3H, d, J 6.4 Hz, CH3), 4.48 (1H, q, J 6.4 Hz, CH), 4.53 (1H, d, J 12.5 Hz, CHH), 4.62 (1H, d, J 12.5 Hz, CHH), 5.98 (1H, d, J 1.4 Hz, CCHH), 6.30 (1H, d, J 1.4 Hz, CCHH), 6.91 (1H, d, J 8.4 Hz, ArH, H-5), 7.83 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.05 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 22.3, 67.2, 75.1, 116.2, 123.0, 125.1, 126.3, 132.0 (× 2), 144.0, 160.5, 168.0, 168.1; HRMS calcd for C13H14O6Na: MNa+, 289.0688. Found: MNa+, 289.0677.
2-(2-Acetoxy-5-methoxycarbonyl-(S)benzyloxy)-succinic acid dimethyl ester 42
Dimethyl-(S)-malate (62 µl, 0.47 mmol) was added dropwise to a solution of 35 (150 mg, 0.52 mmol) in freshly distilled diethyl ether (2 ml). Silver oxide (242 mg, 1.05 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (75 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 42 h. Work up was carried out as described for 36 and the product was purified by column chromatography (eluent 2 : 1 v/v petroleum ether–ethyl acetate) to afford 42 as a colourless oil (90 mg, 52%).RF
[3 : 1 petroleum ether : ethyl acetate]
= 0.17; νmax.
(NaCl plate): 2955, 2855 (Ar C–H stretch), 1756, 1731 (CO), 1612 (CC, ar) cm−1. 1H NMR (CDCl3)
δ 2.32 (3H, s, CH3), 2.77 (2H, dd, J 5.1, 7.6 Hz, CH2), 3.66 (3H, s, CH3), 3.75 (3H, s, CH3), 3.89 (3H, s, CH3), 4.34 (1H, dd, J 5.1, 7.6 Hz, CH), 4.48 (1H, d, J 11.9 Hz, CHH), 4.75 (1H, d, J 11.9 Hz, CHH), 7.14 (1H, d, J 8.4 Hz, ArH, H-5), 7.99 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.12 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 23.0, 39.8, 54.1, 54.4, 54.4, 69.8, 76.7, 124.8, 130.1, 131.8, 132.9, 133.9, 154.7, 168.3, 170.9, 172.5, 173.6; HRMS calcd for C17H20O9Na: MNa+, 391.1005. Found: MNa+, 391.1006.
2-(2-Acetoxy-5-carboxy-(S)-benzyloxy)-succinic acid 19
Potassium hydroxide (141 mg, 2.51 mmol) was added to a solution of 42 (58 mg, 0.16 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction heated to 50 °C for 12 h. Acidification and extraction was carried out as described for 13 to afford the desired triacid 19 as a white solid (49 mg, quant.).νmax.
(ATR): 3254, 2900 (br, acid OH stretch and ar OH), 2948, 2607 (Ar C–H stretch), 1729, 1693 (CO, str), 1668, 1618 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 2.78 (1H, dd, J 8.5, 16.3 Hz, CH), 2.93 (1H, dd, J 3.8, 16.3 Hz, CH), 4.54 (1H, dd, J 3.8, 8.5 Hz, CH), 4.66 (1H, d, J 11.4 Hz, CHH), 4.84 (1H, d, J 11.4 Hz, CHH), 6.91 (1H, d, J 8.4 Hz, ArH, H-5), 7.98 (1H, dd, J 1.8, 8.4 Hz, ArH, H-6), 7.95 (1H, d, J 1.8 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 36.0, 68.2, 73.3, 114.7, 120.4, 122.4, 130.6, 130.6, 159.4, 166.0, 169.8, 172.6; HRMS calcd for C12H12O8Na: MNa+, 307.0430. Found: MNa+, 307.0438.
2-(2-Acetoxy-5-methoxycarbonyl-(R)-benzyloxy)-succinic acid dimethyl ester 43
Dimethyl-(R)-malate (62 µl 0.47 mmol) was added dropwise to a solution of 35 (150 mg, 0.52 mmol) in freshly distilled diethyl ether (2 ml). Silver oxide (242 mg, 1.05 mmol) was added and the reaction heated at reflux (42 °C) under nitrogen. After 1 h of stirring additional silver oxide (75 mg) was added and the reaction stirred at reflux for a further 2.5 h before stirring at 22 °C for 42 h. Work up was carried out as described for 36 and the product was purified by column chromatography (eluent 2 : 1 v/v petroleum ether–ethyl acetate) to afford 43 as a colourless oil (64 mg, 37%).RF
[2 : 1 petroleum ether : ethyl acetate]
= 0.37; νmax.
(NaCl plate): 2956, 2918 (Ar C–H stretch), 1758, 1739 (CO), 1613 (CC, ar) cm−1; 1H NMR (CDCl3)
δ 2.32 (3H, s, CH3), 2.77 (2H, dd, J 5.1, 7.6 Hz, CH2), 3.66 (3H, s, CH3), 3.75 (3H, s, CH3), 3.89 (3H, s, CH3) 4.34 (1H, dd, J 5.1, 7.6 Hz, CH), 4.48 (1H, d, J 11.9 Hz, CHH), 4.75 (1H, d, J 11.9 Hz, CHH), 7.14 (1H, d, J 8.4 Hz, ArH, H-5), 7.99 (1H, dd, J 2.1, 8.4 Hz, ArH, H-6), 8.12 (1H, d, J 2.1 Hz, ArH, H-2); 13C NMR (CDCl3)
δ 23.0, 39.8, 54.1, 54.4, 54.4, 69.8, 76.7, 124.8, 130.1, 131.8, 132.9, 133.9, 154.7, 168.3, 170.9, 172.5, 173.6; HRMS calcd for C17H20O9Na: MNa+, 391.1005. Found: MNa+, 391.1019.
2-(2-Acetoxy-5-carboxy-(R)-benzyloxy)-succinic acid 20
Potassium hydroxide (160 mg, 2.82 mmol) was added to a solution of 43 (65 mg, 0.18 mmol) in THF–water (1 : 1 v/v, 2 ml) and the reaction heated to 50 °C for 12 h. Acidification and extraction was carried out as described for 13 to afford the desired triacid 20 as a white solid (42 mg, 85%).νmax.
(ATR): 3251, 2900 (br, acid OH stretch and ar OH), 2948, 2869 (Ar C–H stretch), 1728, 1691 (CO, str), 1670, 1618 (CC, ar) cm−1; 1H NMR (d6-acetone)
δ 2.78 (1H, dd, J 8.5, 16.3 Hz, CH), 2.93 (1H, dd, J 3.8, 16.3 Hz, CH), 4.54 (1H, dd, J 3.8, 8.5 Hz, CH), 4.66 (1H, d, J 11.4 Hz, CHH), 4.84 (1H, d, J 11.4 Hz, CHH), 6.91 (1H, d, J 8.4 Hz, ArH, H-5), 7.98 (1H, dd, J 1.8, 8.4 Hz, ArH, H-6), 7.95 (1H, d, J 1.8 Hz, ArH, H-2); 13C NMR (d6-acetone)
δ 36.0, 68.2, 73.3, 114.7, 120.4, 122.4, 130.6, 130.6, 159.4, 166.0, 169.8, 172.6; HRMS calcd for C12H12O8Na: MNa+, 307.0430. Found: MNa+, 307.0420.
4-Benzoyl-3-(1-methoxycarbonyl-ethoxy)-benzoic acid methyl ester 45
To a solution of methyl-3,4-dihydroxybenzoate (1.11 g, 6.60 mmol) in dry DMF (7 ml) was added sodium hydride (264 mg, 6.60 mmol, 60% dispersion in oil), followed by the dropwise addition of methyl-2-bromopropionate (0.74 ml, 6.6 mmol). The reaction was stirred at 22 °C for 5 days at which point the solvent was removed in vacuo. The crude residue was purified by column chromatography (eluent: 3 : 1 v/v petroleum ether–ethyl acetate) to give a mixture of the two mono-alkylated products (0.28 g, 17%), which was used in the next step.The mixture of monoalkylated products (0.28 g, 1.12 mmol) were dissolved in pyridine (7 ml) and cooled to 0 °C. Benzoyl chloride (0.195 ml, 1.68 mmol) was added dropwise and the reaction was stirred at 22 °C for 24 h. The reaction was diluted with ethyl acetate (15 ml) and washed sequentially with saturated aqueous NH4Cl solution (20 ml), brine (20 ml), dried (MgSO4) and the solvent removed in vacuo. Purification by column chromatography (eluent 3 : 1 v/v petroleum ether–ethyl acetate) gave the desired mono-benzoylate 50 as a white solid (0.08 g, 4% over the two steps).
1H NMR (CDCl3) δ 1.51 (3H, d, J 6.8 Hz, CH3), 3.90 (3H, s, CH3), 4.81 (1H, q, J 6.7 Hz CH), 7.26 (1H, d, J 8.5 Hz, ArH, H-5,), 7.50 (2H, m, ArH), 7.59 (1H, d, J 1.7 Hz ArH, H-2), 7.62 (1H, m, ArH), 7.74 (1H, dd, J 8.5, 1.7 Hz, ArH, H-6, ArH), 8.20 (2H, m, ArH); NOE: irridiation at δ 4.81 ppm caused enlargement of the signal at δ 7.59 ppm and vice versa. 13C NMR (CDCl3) δ 20.3, 54.2 (× 2), 76.0, 118.0, 125.3, 125.8, 130.5, 130.7, 131.0, 132.2, 135.6, 146.8, 151.4, 166.2, 168.0, 173.5.
3-(1-Carboxy-ethoxy)-4-hydroxy-benzoic acid 21
Potassium hydroxide (38 mg, 0.67 mmol) was added to a solution of 45 (60 mg, 0.17 mmol) in THF–water (1 : 1 v/v, 2.6 ml). The reaction was stirred at 22 °C for 4 h before dilution with water (5 ml). The aqueous fraction was washed with ethyl acetate (10 ml), before acidifying to pH 1 with 1 M HCl. The aqueous fraction was extracted with ethyl acetate (2 × 10 ml), before the combined organic fractions were dried (MgSO4) and the solvent removed in vacuo to afford the desired product and benzoic acid. HPLC purification and lyophilisation gave the desired diacid 23 as a white solid (30 mg, 80%).1H NMR (d6-acetone) δ 1.64 (3H, d, J 6.9 Hz, CH3), 4.90 (1H, q, J 6.9 Hz, CH), 6.92 (1H, d, J 8.3 Hz, ArH, H-5), 7.60 (1H, d, J 1.9 Hz, ArH, H-2), 7.62 (1H, dd, J 8.3, 1.9 Hz, ArH, H-6); 13C NMR (d6-acetone) δ 17.0, 73.7, 114.4, 116.2, 120.9, 124.4, 144.5, 151.3, 165.2, 172.2; HRMS calcd for C10H10O6: M+, 226.0477. Found: M+, 226.0484.
Acknowledgements
We would like to thank the Gates Cambridge Trust for PhD funding (R. J. P. and E. M. M. B.) and the BBSRC.References
- C. T. Walsh, J. Liu, F. Rusnak and M. Sakaitani, Chem. Rev., 1990, 90(7), 1105–1129 CrossRef CAS.
- P. Goncharoff and B. P. Nichols, J. Bacteriol., 1984, 159, 57–62 CAS.
- B. A. Ozenberger, T. J. Brickman and M. A. McIntosh, J. Bacteriol., 1989, 171, 775–783 CAS.
- M. F. Elkins and C. F. Earhart, FEMS Microbiol. Lett., 1988, 56, 35–40 CrossRef CAS.
- Z. He, K. D. Stigers Lavoie, P. A. Bartlett and M. D. Toney, J. Am. Chem. Soc., 2004, 126, 2378–2385 CrossRef CAS.
- E. M. M. Bulloch, M. A. Jones, E. J. Parker, A. P. Osbourne, E. Stephens, G. M. Davies, J. R. Coggins and C. Abell, J. Am. Chem. Soc., 2004, 126(32), 9912–9913 CrossRef CAS.
- F. Dosselaere and J. Vanderleyden, Crit. Rev. Microbiol., 2001, 27(2), 75–131 CAS.
- G. Spraggon, C. Kim, X. Nguyen-Huu, M. Yee, C. Yanofsky and S. Mills, Proc. Natl. Acad. Sci. USA, 2001, 98(11), 6021–6026 CrossRef CAS.
- T. Knoechel, A. Ivens, G. Hester, A. Gonzalez, R. Bauerle, M. Wilmanns, K. Kirschner and J. N. Jansonius, Proc. Natl. Acad. Sci. USA, 1999, 96, 9479–9484 CrossRef CAS.
- H. Zalkin and J. L. Smith, Adv. Enzymol. Relat. Areas Mol. Biol., 1998, 72, 87 Search PubMed.
- C. T. Walsh, M. D. Erion, A. E. Walts, J. J. Delany and G. A. Berchtold, Biochemistry, 1987, 26, 4734–4745 CrossRef CAS.
- M. C. Kozlowski, N. J. Tom, C. T. Seto, A. M. Sefler and P. A. Bartlett, J. Am. Chem. Soc., 1995, 117, 2128–2140 CrossRef CAS.
- A. A. Morollo and M. J. Eck, Nat. Struct. Biol., 2001, 8, 243–247 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- Tripos SYBYL version 6.5; Tripos, Inc., 1699, South Hanley Road, St Louis, Missouri, 63144, USA Search PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- R. Gopinath and B. K. Patel, Org. Lett., 2000, 2(5), 577–579 CrossRef CAS.
- (a) Y. Gozo, H. Zalkin, P. S. Kein and R. L. Heinrisburg, J. Biol. Chem., 1976, 251, 941–944; (b) R. Bauerle, J. Hess and S. French, Methods Enzymol., 1987, 142, 366–386 CAS.
- Enzyme Kinetics! Pro (version: 2.33), 1999–2000, SynexChem, LLC, USA Search PubMed.
- GraFit (version 5.0.6), 1989–2003, Erithacus Software Limited, UK Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |