Inhibition studies on salicylate synthase†
Richard J.
Payne
a,
Olivier
Kerbarh
a,
Ricardo Nunez
Miguel
b,
Andrew D.
Abell
c and
Chris
Abell
*a
aUniversity of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 336405
bUniversity of Cambridge, Department of Biochemistry, 80 Tennis Court Road, Cambridge, UK CB2 1GA
cDepartment of Chemistry, University of Canterbury, Christchurch, New Zealand
First published on 5th May 2005
Abstract
Analogues of chorismate and isochorismate were designed and tested as potential inhibitors in the first inhibition study against a salicylate synthase.
Iron is essential for growth of pathogenic bacteria.1–3 One of several mechanisms by which bacteria acquire iron is using siderophores, which are low molecular weight organic chelators that sequester ferric iron.4–6 Salicylate 1 serves as a building block in the biosynthesis of some siderophores. In Pseudomonas aeruginosa and P. fluorescens it has been shown that salicylate 1 is derived from chorismate 2via isochorismate 3 (Scheme 1), the two reactions being catalysed by isochorismate synthase and isochorismate pyruvate lyase, respectively.7–9
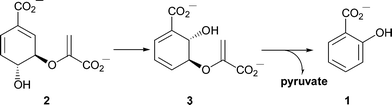 | ||
| Scheme 1 Formation of salicylate from chorismate. In Pseudomonas the two reactions are catalysed by separate enzymes. Irp9 is bifunctional and catalyses both steps. | ||
However, there is also genetic evidence for the existence of a salicylate synthase.10 It has been postulated that Irp9, which is involved in the biosynthesis of the siderophore yersiniabactin 4 in Yersinia enterocolitica (Fig. 1), converts chorismate 2 into salicylate 1.10 We have now characterised Irp9 biochemically and demonstrated that it does indeed form salicylate from chorismate 2via the intermediate isochorismate 3 (Scheme 1).11
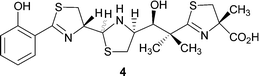 | ||
| Fig. 1 Structure of yersiniabactin. | ||
In order to investigate the mechanism of the Y. enterocolitica salicylate synthase, a variety of chorismate and isochorismate analogues were prepared (Fig. 2) and tested as inhibitors. The first series, 5–13, are analogues of chorismate, where the cyclohexadiene ring has been replaced by a synthetically more tractable aromatic ring and the enol pyruvyl side chain replaced by a lactyl group. The substituent at C-4 was varied to probe the site of departure of the C-4 hydroxyl in the conversion of chorismate 1 to isochorismate 2. Analogues 14–16 were designed to mimic the isochorismate intermediate in the Irp9-catalysed reaction, probing the sensitivity to substitution at C-2.
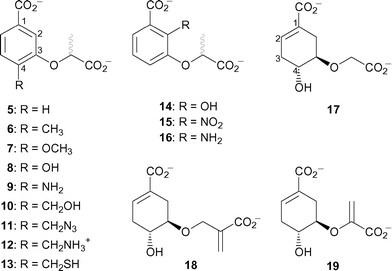 | ||
| Fig. 2 Chorismate and isochorismate analogues tested as inhibitors of Irp9. | ||
There have been no previous inhibition studies on a salicylate synthase, however Koslowski et al. reported several potent inhibitors of E. coli isochorismate synthase,12 which catalyses the first part of the salicylate synthase reaction. These inhibitors (20–22) were reduced chorismate analogues, with substituents at C-4 and C-6. Inhibition constants ranged from 0.053 µM for 21 to 0.45 µM for 22 (Fig. 3). Simplified analogues 17–19 of these compounds were therefore designed to examine the dependence of the C-6 substituent for binding.
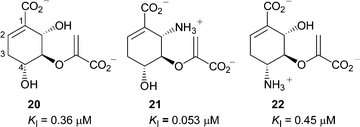 | ||
| Fig. 3 Inhibitors of E. coli isochorismate synthase.12 | ||
In order to provide a structural rationale for inhibitor design, a model of salicylate synthase was constructed.13 The model was built using the primary amino acid sequence of Y. enterocolitica Irp9 and four templates of closely related enzymes for which crystal structures are known on the protein databank.14 Anthranilate synthase from Salmonella typhimurium (1I1Q),15Serratia marcescens (1I7Q),16Sulfolobus solfataricus (1QDL)17 and E. coli PabB (1K0E)18 were the template enzymes chosen. The alignment between the templates and the target primary amino acid sequence were initially obtained using FUGUE.19 The software MODELLER was then used to obtain the model structure of Y. enterocolitica Irp9.20 PROCHECK21 and Verify3D22 software programs were used for validation of the model. Several iterations of the above processes were carried out in order to produce the best possible model.13 The proposed analogues were each modelled into this structure to identify potential interactions with the enzyme. The model of the active site of Irp9 is shown in Fig. 4.
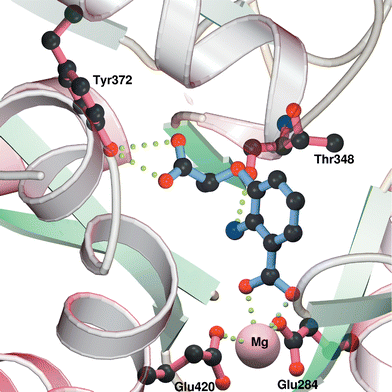 | ||
| Fig. 4 Model of the active site of Y. enterocolitica Irp9 showing possible interactions with inhibitor 16. | ||
The figure shows the possible binding interactions of inhibitor 16 (R enantiomer shown). The C-1 carboxylate interacts with the magnesium ion, whilst the side chain carboxylate is hydrogen bonded with the phenol of Tyr372. The C-2 amino group is also shown hydrogen-bonded to the backbone carbonyl of Thr348.
In order to test the inhibitors, Y. enterocolitica Irp9 was over-expressed as a (His)6-tagged protein in E. coli. Compounds 5–19 were assayed by following the formation of salicylate from chorismate by fluorescence (ex. 305 nm, abs. 440 nm).7,23 The compounds all exhibited competitive inhibition with respect to chorismate, with the inhibition constants (KI) summarized in Table 1.
| Compounds | K I/µM |
|---|---|
| 5 | 200 ± 20 |
| 6 | 270 ± 30 |
| 7 | >2000 |
| 8 | 1110 ± 170 |
| 9 | 43 ± 6 |
| 10 | 460 ± 90 |
| 11 | 330 ± 60 |
| 12 | 1100 ± 140 |
| 13 | 100 ± 20 |
| 14 | 19 ± 5 |
| 15 | 270 ± 40 |
| 16 | 24 ± 2 |
| 17 | >2000 |
| 18 | 440 ± 70 |
| 19 | 160 ± 9 |
Analysis of these inhibition data provide insight into aspects of substrate recognition. Analogues 5–13 only differ by substitution at the C-4 position of the aromatic ring. Their inhibition constants range between 43 µM–2 mM, highlighting the sensitivity to functionality at this part of the molecule. The most potent compound 9 has a C-4 amino group, which would not be protonated under the conditions of the assay. Somewhat surprisingly, replacement of this group with a hydroxyl group (the C-4 functionality in chorismate) increased the inhibition constant twenty-fold (8, KI = 1110 µM). The C-4 methoxy compound was even less potent, possibly due to steric interactions. In contrast, the compounds substituted at C-4 with a methyl group 6 or a hydrogen 5 were moderately potent (KI = 270 and 200 µM, respectively).
Compounds 10–13 were designed to extend the hydrogen bonding group further from the carbocyclic core, mimicking the departure of the C-4 hydroxyl in the proposed enzyme mechanism. All showed intermediate levels of potency, with 12 (R = CH2NH3+, KI = 1100 µM), being the least potent and 13 (R = CH2SH) being the most potent (KI = 100 µM). Taken together, the inhibition results of 5–13 suggest that the part of the active site around C-4 of chorismate is more hydrophobic than might have been expected.
The series 14–16 are simplified mimics of the isochorismate intermediate. These compounds were generally more potent than the C-4 series, with 14 (R = OH, KI = 19 µM) and 16 (R = NH2, KI = 24 µM) being the two most potent compounds tested. Significantly, both these compounds have hydrogen bond donating groups at C-2. Modelling of 16 into the model of the enzyme active site (Fig. 4) identified a potential hydrogen bonding interaction with the backbone carbonyl of Thr348. Compound 15 (R = NO2) which cannot form this hydrogen bond was much less potent (KI = 270 µM). Compounds 5–16 all contained racemic lactyl side chains. Modelling of individual enantiomers of these compounds into the model of the enzyme active site did not show any significant discrimination between them.
The final series of analogues 17–19 maintain the stereochemistry at C-3 and C-4 found in chorismate, but vary in C-5 substitution. These compounds are less potent than most of the aromatic analogues. The most potent being 19 (KI = 160 µM), which contains the enol-pyruvyl side chain found in chorismate. Compound 19 is similar to the potent inhibitors of isochorismate synthase 20 and 21,12 but lacks the substituent at C-6. On the reasonable assumption that the inhibition studies on isochorismate synthase are relevant to inhibition of salicylate synthase, this finding reinforces the importance of the substituent at C-6 (C-2 in the aromatic series) seen in comparing the first two series of inhibitors above.
This initial inhibition study on the newly discovered salicylate synthase has identified key aspects of substrate and intermediate recognition and may serve as a starting point for the inhibition of yersiniabactin biosynthesis in Y. enterocolitica. The trends seen in the inhibition of Irp9 may be relevant to homologous proteins, notably MbtI, which is involved in the biosynthesis of the siderophore mycobactin produced by Mycobacterium tuberculosis, and YbtS, involved in yersiniabactin biosynthesis in Y. pestis.
Acknowledgements
R. J. P. would like to thank the Gates Cambridge Trust for PhD funding. This work was also supported by the Biotechnology and Biological Sciences Research Council, UK.References
- U. E. Schaible and S. H. E. Kaufmann, Nat. Rev. Microbiol., 2004, 2, 946–953 Search PubMed.
- C. Ratledge, Tuberculosis, 2004, 84, 110–130 CrossRef.
- C. Ratledge and L. G. Dover, Annu. Rev. Microbiol., 2000, 54, 881–941 CrossRef CAS.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 CrossRef CAS.
- V. Braun and H. Killmann, Trends Biochem. Sci., 1999, 24, 104–109 CrossRef CAS.
- J. B. Neilands, J. Biol. Chem., 1995, 270, 26723–26726 CAS.
- C. Gaille, C. Reimmann and D. Haas, J. Biol. Chem., 2003, 278, 16893–16898 CrossRef CAS.
- C. Gaille, P. Kast and D. Haas, J. Biol. Chem., 2002, 277, 21768–21775 CrossRef CAS.
- J. Mercado-Blanco, K. M. G. M. van der Drift, P. E. Olsson, J. E. Thomas-Oates, L. C. van Loon and P. Bakker, J. Bacteriol., 2001, 183, 1909–1920 CrossRef CAS.
- C. Pelludat, D. Brem and E. Heesemann, J. Bacteriol., 2003, 185, 5648–5653 CrossRef CAS.
- O. Kerbarh, A. Ciulli, N. I. Howard and C. Abell, submitted for publication.
- M. C. Kozlowski, N. J. Tom, C. T. Seto, A. M. Sefler and P. A. Bartlett, J. Am. Chem. Soc., 1995, 117, 2128–2140 CrossRef CAS.
- O. Kerbarh, R. J. Payne, R. Nunez Miguel and C. Abell, unpublished results.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weisseg, N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- A. A. Morollo and M. J. Eck, Nat. Struct. Biol., 2001, 8, 243–247 CrossRef CAS.
- G. Spraggon, C. Kim, X. Nguyen-Huu, M. Yee, C. Yanofsky and S. Mills, Proc. Natl. Acad. Sci. USA, 2001, 98, 6021–6026 CrossRef CAS.
- T. Knoechel, A. Ivens, G. Hester, A. Gonzalez, R. Bauerle, M. Wilmanns, K. Kirschner and J. N. Jansonius, Proc. Natl. Acad. Sci. USA, 1999, 96, 9479–9484 CrossRef CAS.
- J. F. Parsons, P. Y. Jensen, A. S. Pachikara, A. J. Howard, E. Eisenstein and J. E. Ladner, Biochemistry, 2002, 41, 2198–2208 CrossRef CAS.
- J. Shi, T. L. Blundell and K. Mizuguchi, J. Mol. Biol., 2001, 310, 243–257 CrossRef CAS.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef.
- R. Luthy, J. U. Bowie and D. Eisenberg, Nature, 1992, 356, 83–85 CrossRef CAS.
- See supplementary material for assay conditions†.
Footnote |
| † Electronic supplementary information (ESI) available: Irp9 enzyme assay conditions and analytical data for 5–19. See http://www.rsc.org/suppdata/ob/b5/b503800f/ |
| This journal is © The Royal Society of Chemistry 2005 |