New directions in supramolecular transition metal catalysis
Matthew J. Wilkinson, Piet W. N. M. van Leeuwen and Joost N. H. Reek*
van't Hoff Institute for Molecular Sciences, University of Amsterdam, Nieuwe Achtergracht 166, 1018 WV Amsterdam, The Netherlands. E-mail: reek@science.uva.nl; Fax: (+31)20-525-6422; Tel: (+31)20-525-6437
First published on 17th May 2005
Abstract
Supramolecular chemistry has grown into a major scientific field over the last thirty years and has fueled numerous developments at the interfaces with biology and physics, clearly demonstrating its potential at a multidisciplinary level. Simultaneously, organometallic chemistry and transition metal catalysis have matured in an incredible manner, broadening the pallet of tools available for chemical conversions. The interface between supramolecular chemistry and transition metal catalysis has received surprisingly little attention. It provides, however, novel and elegant strategies that could lead to new tools in the search for effective catalysts, as well as the possiblity of novel conversions induced by metal centres that are in unusual environments. This perspective describes new approaches to transition metal catalyst development that evolve from a combination of supramolecular strategies and rational ligand design, which may offer transition metal catalysts for future applications.
 Matthew J. Wilkinson Matthew J. Wilkinson | Matthew J. Wilkinson was born in Cuckfield, England in 1977. He received his MSci in Chemistry from the University of Bristol in 1999 and stayed there to study for his PhD, entitled ‘Bulky Arylphosphines and Arylarsines for Catalysis of C–C Bond-Forming Reactions’, under the supervision of Professor Paul G. Pringle. He received his doctorate in 2003 before moving to the Universiteit van Amsterdam to work for J. N. H. Reek as a postdoctoral researcher on the encapsulation and immobilisation of transition metal catalysts within vesicles for use in aqueous-phase catalysis. |
 Piet W. N. M. van Leeuwen Piet W. N. M. van Leeuwen | Piet W. N. M. van Leeuwen is Professor of homogeneous catalysis at the University of Amsterdam since 1989 and group leader at the Institute of Chemical Research of Catalonia in Tarragona since 2004. For many years he worked with Shell Research in Amsterdam where he was head of the research group in the area of homogeneous catalysis and organometallic chemistry. His present research aims at the development of novel homogeneous catalysts, using the full range of available tools and techniques. |
 Joost N. H. Reek Joost N. H. Reek | Joost Reek finished his masters at the University of Nijmegen in 1991 and did his PhD with Prof. R. J. M. Nolte at the same university in supramolecular chemistry. He attended the group of Prof. M. J. Crossley in Sydney as a postdoctoral fellow in 1996, where he gained experience in porphyrin chemistry. Among other things, he developed guest templated molecular capsules using porphyrins and dendrimers. Since January 1998 he is lecturer (senior lecturer since 2003) in the group of Prof. Van Leeuwen with research activities focusing on transition metal catalysis, supramolecular chemistry and supramolecular catalysis, catalyst immobilization and dendritic transition metal catalysis. He is currently especially interested in developing new concepts in supramolecular catalysis. |
Introduction
Nature provides excellent examples of selective and efficient catalytic conversions executed by enzymes, metabolons, organelles and complete cells. In all these examples supramolecular organisation plays a crucial role. It is therefore not surprising that nature has served as an immense source of inspiration for scientists involved in supramolecular catalysis and supramolecular chemistry in general.With respect to catalysis, we can distinguish different levels of organization that affect the rate and selectivity of the transformation. When we consider systems that have a transition metal in the active site, the first level of control starts with the ligation of the metal centre determining its electronic properties. It is well-known that small changes in the conformation of a protein backbone, caused by the binding of a substrate molecule, for example, can change the ligation by the protein and thereby (de-)activate the metal centre.
A second level of organisation comprises the binding of the substrate(s) in proximity to the active centre in the enzyme pocket, which is generally based on several noncovalent interactions, shape complementarities and solvent effects. The effect on catalysis has been discussed for more then fifty years1 and can be divided in two main contributions:2 1) the transition state is bound more strongly than the ground states, thereby lowering the energy barrier for reaction; and 2) pre-organization of the substrate results in a gain in entropy, helping to drive the reaction.
Side effects that can play a role but are scarcely documented are the desolvation of the substrate upon complexation in the cavity and the local environment (polarity) where the reaction takes place, which can differ significantly from the bulk solvent. An interesting example where the desolvation of the substrate plays a crucial role is the hydration of carbon dioxide carried out in the cavity of carbonic anhydrase. Theoretical work shows that the reaction barrier in water is associated with the displacement of the solvent shell from the hydroxy anion.3
Organisation on a macroscopic level is also of utmost importance for biological systems. For example, the anabolism and catabolism (biosynthesis and degradation processes respectively) occur simultaneously in the cell.4 The conflicting demands are managed by proper separate regulation of the metabolic needs and physical separation of the competing pathways in different cellular compartments. Macroscopic organisation of catalyst systems has been utilised by this group5 and others6 by using amphiphilic compounds that form assemblies to increase the catalyst activity. In addition, it may be possible to use this type of approach to protect fragile catalyst systems from harmful reagents and to organise catalysts for cascade-type reactions.
Without doubt, most supramolecular catalysts reported so far can be classified as host–guest catalysts since they combine a binding site for substrates with a reactive centre aiming at enzyme-like behaviour. In fact, most scientists do not differentiate between supramolecular catalysis and host–guest catalysis, underscoring the dominance of this approach. There are several reviews on this topic and therefore in this perspective we will not discuss this traditional approach to supramolecular catalysis.7 Although this approach is very interesting from a fundamental point of view and research in this area contributed significantly to the understanding of enzymes, so far there are no examples of such catalytic systems that have evolved to commercial applicability. A notable type of host–guest catalysis, pioneered by Kumada et al.8 in 1982 and reviewed in 1992,7i involves transition metal complexes of ligands that bear pendant substrate-directing groups. The key-point behind this approach is that the substrate is bound by two interactions during the transformation, the metal–substrate and the metal–ligand interaction, providing highly selective catalysts. The success of this approach has led to considerable interest in these metalloenzyme-mimics and future developments are expected to offer additional exciting examples.7j,9
There are two new interesting approaches in the area of host–guest catalysis that might lead to a revival, which we would like to mention. One relatively new approach is based on the selection and amplification of active catalysts from dynamic combinatorial libraries (DCL).10 In this strategy a mixture of potential supramolecular catalysts is formed by the reversible assembly of small building blocks. The reversibility ensures that the DCL is in thermodynamic equilibrium and responsive to external influences, such as the addition of a transition state analogue or substrate. Stabilisation of the more active species will occur, leading to an equilibrium shift amplifying the strong binders at the expense of the weaker binders in the library.11 The second new direction in this area is the construction of bio–chemo-hybrid catalysts, whereby a protein cavity can function as a supramolecular biocavity, inducing substrate recognition and enantiodiscrimination, while the chemocatalyst delivers the desired reactivity. Inspired by the seminal work of Kaiser and Lawrence12 and Wilson and Whitesides,13 Reetz14 and Ward et al.15 have modified enzymes with transition metal catalyst moieties to yield artificial metalloenzymes. While Reetz has approached this problem by utilising covalent interactions between the catalyst and cysteine moieties in the protein, Wilson and Whitesides and Ward and cowrokers have utilised supramolecular interactions. The latter approach makes use of the high affinity (Ka = 1014 M−1) of biotin for both avidin and streptavidin proteins. The streptavidin protein has a deeper binding pocket, possibly making it more suitable for enantiodiscrimination. Functionalisation of achiral bidentate ligands with the biotin moiety, allows anchoring into the protein and unique catalysis is observed. For instance, in the asymmetric hydrogenation of acetamidoacrylic acid, when no host protein is used racemic products are formed, whereas when the catalyst is located within a protein cavity products with ee's of up to 96% are obtained.
In this perspective we will discuss new strategies in supramolecular transition metal catalysis that arise from the implementation of supramolecular strategies in traditional catalyst development. In the approaches that arise noncovalent interactions are used to construct the catalyst rather then assist the reaction to proceed. The interesting new opportunities that emerge will be discussed, but before doing so we will first briefly elaborate on traditional catalyst development to give the reader the background information required to appreciate the new directions in supramolecular transition metal catalysis described in this perspective.
Traditional homogeneous catalysis
The most powerful tool in transition metal catalysis is ligand variation. The delicate interplay of steric and electronic effects between a catalytically active metal and the auxiliary ligands determines the properties of the catalyst. Traditionally, the catalytic performance of an organometallic complex is rationalised in terms of ligand parameters. The electronic properties of phosphine ligands were first quantified by Tolmann, who introduced the X-parameter.16 The X-value was determined from the CO-frequency of [Ni(L)(CO)3] complexes for a large set of ligands to quantify the electronic donor properties of the ligands. Tolmann also introduced the cone angle (Θ) as a means of describing the steric properties of monodenate ligands (see Fig. 1). | ||
| Fig. 1 The cone angle (Θ) and the bite angle (βn) are important ligand parameters that aid rational catalyst design. | ||
Much later, the importance of the geometry of bidentate ligands was recognised and the “natural bite angle” was introduced as a way of describing the properties of bidentate chelating ligands.17 The natural bite angle is defined as the angle at which two donor atoms of a chelating bidentate ligand ‘bite’ into a transition metal, βn (see Fig. 1) and can be calculated using simple molecular modelling of the backbone. A strongly related parameter is the flexibility range, which is defined as the accessible range of bite angles within less than 3 kcal mol−1 excess strain energy from the calculated bite angle. For example, the rigid Xantphos backbone has a narrower flexibility range (97–135°) than that of BISBI (101–148°)18 and this has a dramatic effect on the ability of the ligand to stabilise the metal in different geometries.
These ligand parameters have a great influence on catalyst performance and in many examples the effects can be easily rationalised. Several catalytic cycles, especially those of cross coupling reactions, include oxidative addition and reductive elimination steps. Electron rich ligands promote the oxidative addition step relative to electron poor ligands, whereas for reductive elimination the opposite is true. In a similar fashion, it can be rationalised that wide bite angle ligands stablize tetrahedral complexes relative to square planar complexes and will, therefore, promote reductive elimination from square planar nickel(II) and palladium(II) complexes.
The effect of the steric bulk of a ligand is very hard to generalise, but often the effect of steric bulk determines the number of ligands that can be coordinated to the metal centre, which can have a large impact on the catalyst properties. Van Leeuwen et al.19 have shown that the use of bulky monodentate phosphite ligands in the rhodium-catalysed hydroformylation allows otherwise unreactive olefins (2-methyl-1-hexene, limonene, cyclohexene and methylcyclohexene) to be hydroformylated under mild conditions. This high activity was ascribed to the fast coordination of the olefin to the mono-ligand species, which due to steric hindrance is favoured over the bis-ligand. Another example is given by van Strijdonck et al.,20 who systematically varied the electronic and steric properties of monodentate phosphorus ligands in the palladium-catalysed Heck reaction. If the steric bulk is too small more than one ligand can coordinate to the palladium, leading to a less reactive catalyst. If, however, the steric bulk is too large a reactive species is formed, but due to the steric bulk of the ligand the approach of the substrate is hindered and the reaction is slow. Clearly, these ligand parameters can be used to aid rational ligand design. Indeed, recent work by Fu et al.,21 Buchwald et al.22 and Hartwig and coworkers23 has emphasised the high activity of low ligated complexes in various coupling reactions.
The design of catalysts for asymmetric conversions is more difficult and is to a large extent still based on trial and error and sophisticated guesses. Several attempts have been made to rationalise asymmetric catalysis by various computational techniques, but none of these have reached a level of general acceptance.24 More recently, combinatorial approaches and high throughput experimentation have been introduced to speed up the processes of lead finding and catalyst optimisation.25
Besides the development of new catalysts by rational ligand design and combinatorial approaches, a lot of research is devoted to the complete system approach including catalyst recycling. Various elegant concepts for catalyst separation and recycling have been developed.26 Two-phase catalysis is a very general method and involves the immobilisation of the catalyst in a solvent that is immiscible with the product phase. The introduction of water-soluble groups to the ligands of a transition metal allow the catalyst to be immobilised in an aqueous phase.27 The feasibility of this approach has been shown by the commercialisation of many processes, such as that of the Rhone–Poulenc–Ruhrchemie two-phase hydroformylation of propene using TPPTS–rhodium complexes.28 Other strategies include supported aqueous phase catalysis,29 fluorous phase catalysis,30 the use of ionic liquids31 and supercritical fluids.32 A widely studied approach to facilitate catalyst-product separation is the attachment of homogeneous catalysts to dendritic,33 polymeric organic, inorganic or hybrid supports.34,35 Here the ligand is functionalised with a group that enables anchoring to such a support.
Unfortunately, there is no “holy grail” that provides a general solution for catalyst-separation and for every new catalytic process the strategy for catalyst separation and recycling needs to be studied. General properties such as solubility of the substrates, catalyst instability and metal leaching during the recycling procedure strongly limit some of the concepts. For example, aqueous-phase catalysis is limited to substrates that are soluble in water. Immobilisation to inorganic materials such as silica has advantages due to physical strength and chemical inertness, but the activity is generally lower than the homogeneous system. The decrease in activity is usually not a problem for catalysts on soluble dendrimeric or hyperbranched polymer supports, but here the availability of suitable membrane materials complicates practical use.
New strategies in supramolecular catalysis
Considering the short introduction on rational ligand design and catalyst immobilisation, the catalyst given in Fig. 2 is an illustrative example because it includes all the variables. The Xantphos series of diphosphine ligands36 with wide bite angles was designed to give high selectivity in the hydroformylation reaction, the catalytic performance was optimised by fine-tuning the electronic properties of the ligand by changing substitutents on the phenyl rings and on the backbone and the ligand has been immobilised on silica support to obtain a stable, easily recyclable catalyst.37 Inspection of Fig. 2 with the idea of introducing these design elements or functionalities via supramolecular interactions, leads to the suggestion that this is possible at least at three positions:1) The ligand can be anchored to a support using noncovalent interactions.
2) Two monodenate ligands can be assembled via noncovalent interactions to form a chelating bidentate ligand by assembly.
3) The electronic and steric properties of the ligand as well as the chiral environment can be modified by the assembly of functional groups.
 | ||
| Fig. 2 An example of a bidentate ligand designed for the selective hydroformylation, anchored onto silica support. The green arrows show the locations where supramolecular strategies can be implemented in the development of novel supramolecular catalysts. | ||
While the above approaches to rational ligand design have led to active and selective catalysts, the new strategies described in this perspective, which are based on the implementation of supramolecular interactions, open a new area with novel opportunities. We will discuss illustrative examples, with no intention to be comprehensive, that clearly explain these novel strategies in supramolecular transition metal catalysis.
1. Noncovalent anchoring of catalysts to supports
The essence of homogeneous catalysts is, by definition, the fact that the substrate, products and catalyst are all dissolved in the same phase. This can hamper catalyst separation and subsequent recycling and therefore many methodologies for separating catalysts from the products have been developed. These methodologies usually involve tedious synthetic procedures to create a complex supported catalyst that is optimised for only one process. We will focus on the reversible noncovalent anchoring of discrete catalyst species to soluble and insoluble supports that provide simple and efficient recycling methods, while also allowing re-functionalisation of the support and variation of the catalyst loading even during catalysis (Scheme 1). | ||
| Scheme 1 The concept of reversible anchoring of catalysts to a (dendritic) support that is functionalised with a binding motif complementary to that of the catalyst. | ||
Schweb and Mecking38 utilised electrostatic interactions to bind phosphine ligands with multiple sulfonate groups, such as NaTPPTS (tris(sodium-m-sulfonatophenyl)phosphine), to soluble polyelectrolytes which can be recovered and recycled by ultrafiltration. The electrostatically polymer-bound complex was employed in the hydroformylation of 1-hexene, yielding turn-over-frequencies of up to 160 TO h−1 at 80 °C and 30 bar. The system exhibits typical selectivity (l : b = 2.5–3.1) for a bis-triphenylphosphine bound rhodium catalyst.
In contrast to the above example, our approach involved the noncovalent anchoring of the catalyst to a dendrimer support using well-defined binding sites in order to keep control over the exact location of the catalyst on the support (Scheme 2). To this end, we have utilised the fifth generation poly(propylene imine) dendrimer functionalised with urea adamantyl units at the periphery (1)39 that allows the directional noncovalent anchoring of 32 guest molecules. For our purpose we used phosphine ligands with the complementary-binding motif (2).40 The binding of guest molecules into the periphery of 1 was studied in detail and appeared to be sufficiently high to warrant further study. The dendrimer complex (1[(2)2Pd(allyl)Cl]16) remained intact after size exclusion chromatography (SEC). The dendritic host containing 32 phosphine ligands 2 assembled to the periphery of 1 was used as a multidentate ligand in the Pd-catalysed allylic amination of crotyl acetate by piperidine. High reaction rates were observed, similar to the unbound analogue, indicating that every active site on the dendrimer acts as an independent catalyst. The supramolecular anchoring of the catalysts does not decrease the activity or the selectivity, in contrast to the general observation for catalysts immobilised on insoluble supports. The noncovalently functionalised dendritic catalysts were used in these batchwise reactions was recycled using SEC, but the catalyst was partly decomposed during such a procedure.
 | ||
| Fig. 3 Polyoxometallate clusters anchored to soluble support via ionic interactions. | ||
 | ||
| Scheme 2 The urea–adamantyl functionalised dendrimers. | ||
The noncovalently functionalised dendritic catalysts were also applied in a continuous-flow membrane reactor. The retention measured for the supramolecular acid–dendrimer complex [(2)Pd(crotyl)Cl]32–dendrimer was 99.4%, implying that 99.4% of the catalyst remains inside the reactor after each “cycle”. Indeed, the dendritic supramolecular catalyst was successfully applied in the allylic amination reaction in the continuous-flow membrane reactor. The conversion remained fairly constant during the first 10 h of the experiment, after which a small decrease was observed due to catalyst deactivation (not leaching!). These experiments clearly demonstrate that noncovalently supported catalysts can be used as recyclable catalysts. These supramolecular supports can be conveniently reloaded with new catalysts, in contrast to their covalent analogues.
A similar approach has been taken by van de Coevering et al.,41 who utilised ion-pairing interactions to tether transition metal complexes with sulfonated anionic tails to cationic dendrimeric supports. These assemblies were successfully applied as Lewis acid catalysts in the aldol condensation of benzaldehyde and methyl isocyanate.
Kaneda et al.42 have recently utilised acid–base interactions to noncovalently attach diphenylphosphine-4-benzoic acid palladium complexes to the exterior of poly(propyleneimine) dendrimers and to cavities within these dendrimers via the interaction of the benzoic acid with the carboxyl groups of amino acids positioned within the dendrimer. Interestingly, the supramolecular dendritic catalyst gave rise to much more active catalysts for the Heck reaction. In addition, selectivity for mono-substitution in the Heck reaction of p-diiodobenzene with n-butyl acrylate was observed for the dendritic encapsulated system, whereas the parent complex gave rise to a mixture of products. In the allylic amination of cinnamyl methyl carbonate, using morpholine as the nucleophile, the supramolecular catalysts also gave rise to higher selectivities compared to the non supported analogue. Functionalisation of the exterior of the dendritic scaffold with triethoxybenzoyl chloride instead of decanoyl chloride makes the dendrimer insoluble in aliphatic solvents, but soluble in polar solvents such as DMF. These properties enabled a simple, recyclable thermomorphic biphasic system for the allylic amination to be devised.
The structural perfection of dendritic support is not required for every application in catalysis, and hyperbranched polymers provide interesting and cheap alternatives as catalyst supports.43 These hyperbranched polymers are obtained from a simple one-pot syntheses, yielding globular polymeric structures with broad weight distributions when compared to their dendritic analogues. Optically active hyperbranched polymers have been synthesised and used to immobilise platinum NCN–pincer complexes both covalently and noncovalently.44,45 These chiral supports exhibit slight circular dichroic activity, but no enantioselectivity is observed upon using these systems to catalyse Michael additions between methyl vinyl ketone and ethyl-α-cyanopropionate.
Polyoxometallate clusters (POM's), a class of fascinating well-defined structures that are rich in redox chemistry and therefore highly suitable for oxidation catalysis, have been covalently46 attached to dendritic support to aid separation, recycling and stability. Very recently Astruc et al.47 noncovalently linked POM's to a dendritic support by utilising ion-pairing interactions between the anionic POM fragments and a cationic dendrimer. The polyoxometallate fragment [PO4{WO(O2)2}4]3− (Fig. 3) was noncovalently linked to tri-cationic tripod fragments of the dendrimer, yielding an epoxidation catalyst that was as active as its unbound analogue. The catalysts were recovered by precipitation and filtration with a recovery rate of up to 96%. These catalysts could then be re-used with no loss of activity.
 | ||
| Fig. 4 Two examples of supported hydrogen-bonded catalysts introduced by Bianchini et al.: the catalyst is anchored to the silica support via hydrogen bonds between the sulfonate of the ligand and the hydroxyl groups of the silica surface. | ||
Perez et al.50 have also used hydrogen bonding as a tool to succesfully immobilise catalyst complexes. They anchored polypyrazolylborate copper(I) complexes directly to silica supports via a hydrogen bonding interaction between the silanol groups and B–H and/or pyrazolyl nitrogen atoms of the ligand. The activity of the supported olefin cyclopropanation catalyst was reported to be similar to that of the homogeneous analogue and, interestingly, the catalyst was recycled ten times with retention of this activity.
Recently, we introduced a new approach in which well-defined binding sites based on different binding motifs are immobilised on silica that can be noncovalently functionalised with catalysts that have ancillary ligands with the complementary motif (Scheme 3). This offers a high level of control and flexibility since in this approach the noncovalent anchoring is independent of the type of support and the transition metal.51
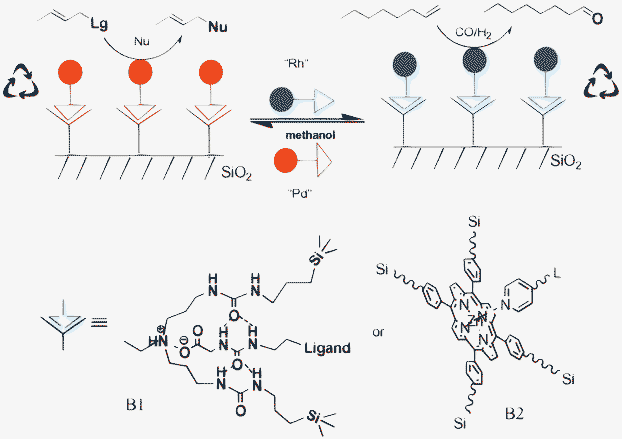 | ||
| Scheme 3 The concept of supramolecular anchoring of catalysts to binding sites immobilised on silica (A) and the binding motifs based on hydrogen bonding (HB) and metal–ligand interactions (ML). | ||
Two binding motifs, either a complementary hydrogen bonding unit (HB) or metal–ligand interactions (ML) have been utilised to immobilise the catalyst. The supramolecular interaction between the transition metal catalyst and the binding site is sufficiently strong to enable efficient catalyst recycling. In addition, the support can be readily re-functionalised with different catalyst systems by washing with methanol to remove the first catalyst system and then attaching the new catalyst system by simply stirring in apolar solvent such as toluene (Scheme 3).
The resulting noncovalently immobilised complexes have been used as ligand systems for both the Pd-catalysed allylic amination reaction and Rh-catalysed hydroformylation. An immobilised glycine–urea functionalised PPh3 ligand, 3(S), attached to the HB support attains similar yields and product distributions as the homogeneous analogue for the Pd-catalysed allylic amination of crotyl acetate by piperidine, while exhibiting a reduced rate as is commonly observed for heterogenised systems (90% conversion achieved after 30 min compared with 5 min for the homogeneous system). Interestingly, the catalyst could be recycled three times via a simple filtration step. Subsequently, the catalyst was separated from the support and the support was uploaded with hydroformylation catalysts.

Ligand 3 was first studied in the rhodium-catalysed hydroformylation of 1-octene and gave chemo- and regio-selectivities typical of bis-triphenylphosphine based rhodium complexes. Reactions proceeded to 80–90% conversion and, as usual for supported catalysts, the activity was less than the analogous homogeneous systems. The catalyst can be recycled up to eight times with only a slight drop in activity, which is due to metal-leaching. A rhodium catalyst based on the glycine–urea functionalised Xantphos ligand (4) was subsequently used in combination with the same support material, as a catalyst for the hydroformylation of 1-octene. In 11 consecutive reactions the catalyst did not show any deterioration or metal-leaching. Similar to previously observed for covalently anchored systems (5),37 a decrease in activity and selectivity is observed compared to the homogeneous system. Interestingly, higher activity and selectivity for the linear aldehyde are observed for the noncovalently anchored ligand compared to covalently anchored Nixantphos (5), while in the homogeneous phase these Nixantphos and Xantphos ligands show similar activity and selectivity.52
When the binding motif ML was used to immobilise ligand 6 to the silica support similar results to those obtained for ligand 4 bound to HB were obtained for the hydroformylation of 1-octene, although the recyclability of the system was diminished. This was ascribed to reaction of the aldehyde, formed during the reaction, with the amine of the ligand, required for the binding process. The same system was used in the allylic alkylation of crotyl acetate by diethyl 2-methylmalonate, where similar chemo- and regio-selectivies are observed to the homogeneous system, with the activity similar to covalently supported systems. These noncovalently anchored catalysts in general exhibit a behavior similar to their covalently bound analogues, but can now be separated from the support after the reactions.
One of the questions that arises when catalysts are noncovalently bound to supports is the required binding strength of the complex to the support. This depends on too many aspects to give a clearcut answer, but association constants should be greater than 103 M−1. The binding of the catalyst to the support must be much stronger in continuous-flow systems than in batch-wise systems as any unbound catalyst will be continuously washed away. In a continuous-flow system, the retention of the non-supported (free) complex also plays a crucial role. If the free complex shows a high retention, as is the case for 2 applied in a membrane reactor,40 then small amounts of free complex will not cause a dramatic depletion of catalyst. In contrast, if the free catalyst is not retained at all, which will be the case for solid supports that can be separated by large-pore filters, then small amounts of free catalyst will lead to substantial leaching. In all these experiments the absolute catalyst concentration plays an important role, since it determines, together with the association constant, the amount of free catalyst. A trick to prevent the presence of unbound catalyst is the use of excess binding sites.
2. Chelating ligands formed by assembly
The spatial orientation of donor atoms coordinating to the catalytically active transition metal is of crucial importance for the catalytic properties of complexes.53 Traditionally, these donor atoms have been attached covalently to a ligand backbone thereby, depending on the rigidity of the backbone, forcing a certain coordination geometry around the metal. In this section we will elaborate on the potential of self-assembled bidentate ligands. We can distinguish homobidentate ligands from heterobidentate ligands, the latter consisting of two different donor atoms. Although for many reactions bidentate ligands with two different donor atoms offer a higher level of control with respect to the selectivity of the reaction, this class of ligands is generally harder to synthesise. It is this type of ligand that would especially benefit from a supramolecular approach that involves merely mixing of the proper monodentate ligands functionalised with complementary binding motifs.There are two principle strategies that can be followed (Scheme 4):
1) a template can be used that contains binding sites for the selective assembly of two different ligands, or
2) the ligands can be functionalised with complemetary binding motifs.
An important difference between these two strategies is the number of components involved. Approach 1 leads to a three-component-assembly and, as a consequence, the system is more complicated, but it does easily produce large ligand libraries (10 × 10 × 10 = 1000 members based on 30 compounds). Approach 2 is simpler, but more building blocks are required to arrive at large ligand libraries (30 × 30 = 900 members based on 60 compounds).
 | ||
| Scheme 4 Schematic representation of two methods to form chelating bidentate ligands by assembly: (1) via a template and (2) via complementary binding motifs directly attached to the ligand. | ||
The supramolecular approaches will lead to dynamic ligand systems, in which exchange-processes between associated and dissociated states can occur. The exchange rates can be modified by changing the interaction that is involved in the association process. So far it is unclear how this may affect the selectivity of reactions, but we believe that, as a first approach, the dynamics should be treated similarly to flexibility of ligands. Indeed ligand flexibility is required for some reactions, whereas in other reactions higher selectivities and activities are obtained with more rigid ligand systems.

![Transition metal catalyst [HRh(7(9)2)(CO)2] formed by self-assembly of 4-pyridyldiphenylphosphine 9 on dimeric zinc(ii) porphyrin 7 and in the presence of a rhodium precursor.](/image/article/2005/OB/b503407h/b503407h-s5.gif) | ||
| Scheme 5 Transition metal catalyst [HRh(7(9)2)(CO)2] formed by self-assembly of 4-pyridyldiphenylphosphine 9 on dimeric zinc(II) porphyrin 7 and in the presence of a rhodium precursor. | ||
The assemblies based on 7 and pyridine phosphorus ligands 8–11(R) were used as supramolecular ligands in the rhodium-catalysed hydroformylation and typical bidentate behaviour has been observed. The chelating bidentate assembly exhibited lower activities (a factor of three) compared to the monodentate analogue. Only a slightly higher selectivity for the linear aldehyde was observed. The chiral ligand assemblies based on 10(S) and 11(R) were studied in the asymmetric rhodium-catalysed hydroformylation of styrene.55,56 The rhodium complexes based on monodentates 10(S) and 11(R), as well as their zinc(II) porphyrin supramolecular complexes, resulted in low enantiomeric excess (approximately 7%), which is in line with previous results for monodentate ligands.57 Interestingly, the templated ligand assemblies 7(10(S))2 and 7(11(R))2 resulted in significantly higher enantioselectivity (33%), along with an increase in activity. So far, only moderate enantioselectivities and activities in the rhodium-catalysed hydroformylation of styrene have been observed. However, these results are very promising considering the challenge that is involved.
In the previous example bis-porphyrins were used as templates to form bidentate ligands. Alternatively, ditopic nitrogen ligands such as DABCO (diaza-[2.2.2]-bicyclooctane) can be used as templates to form 2 : 1 assemblies with zinc(II) porphyrins. If these porphyrins are functionalised with ligands, i.e. phosphines or phosphites, this self-assembly process can also lead to chelating bidentate ligands (see Fig. 5).
 | ||
| Fig. 5 Schematic representation of the formation of bidentate ligands using ditopic nitrogen ligands as templates for the association of functionalised zinc(II) porphyrins. | ||
Novel phosphite–porphyrins were prepared to investigate the properties of this type of assembly (Scheme 6). The phosphites were based on chiral binapthol and can, in principle, be used as ligands for asymmetric catalysis. UV–vis spectroscopic titrations of zinc(II) porphyrin phosphite 12(S) and DABCO 13 showed that 13 binds two monomeric zinc(II) porphyrin phosphite 7 units with sufficiently high binding constants, K1 = 5.9 × 104 M−1 and K2 = 1.6 × 104 M−1, respectively. UV–vis spectroscopic titrations of an insitu formed rhodium complex,58 using [HRh(CO)(12(S))2(PPh3)], showed that the binding constant of the ditopic template DABCO 13, on forming the 1 : 1 complex [HRh(CO)(13(12(S))2)(PPh3)], increased considerably, K = 4.4 × 105 M−1. This shows that the pre-organisation of the two porphyrins via coordination to the rhodium metal does indeed lead to stronger binding of the ditopic template, indicating a positive cooperative binding process.
 | ||
| Scheme 6 The assembly of chiral phosphite ligands (12(S)) on a template (13) to form chelating complexes that can be applied in asymmetric catalysis. | ||
Initial studies using complex [HRh(CO)(13(12)2)(PPh3)] in the hydroformylation of 1-octene, indicated that the assembled bidentate gave only slightly different results. The activity was still 80% of that of the catalyst without template, an effect that was ascribed to the bidentate character, and the selectivity was only slightly changed.
A more rigid assembly was anticipated to provide a catalyst with a more typical bidentate behaviour and, to this end, trisporphyrin–phosphite ligand 14 was prepared.59 Upon addition of a metal prescursor and 1.5 eq. of DABCO with respect to the trisporphyrin phosphite ligand 14, a multicomponent supramolecular assembly was formed in which two ligands are firmly fixed by three bridging DABCO ligands (Scheme 7). The stoichiometry of the supramolecular complex formed (13 : [Rh(acac)(14)2] = 3 : 1) was determined by UV–vis titrations and high-pressure NMR spectroscopy, revealing an average binding constant calculated to be 1.9 × 107 M−1, typical of ditopic complexation. This increase in the binding constant was also reflected in the performance of the assembly in the rhodium-catalysed hydroformylation of 1-octene.55 The monomeric tris zinc(II) porphyrin phosphite 14 yields a rhodium catalyst with an activity and regioselectivity that is typical of rhodium–bis-phosphite complexes (TOF = 2.0 × 103 and l : b = 2.5).55 The rhodium catalyst based on assembly 15 shows a high linear to branched ratio (l : b = 15.1) and a lower activity, which is characteristic for bidentate chelating ligand systems.55 This shows that the multi-component ligand assembly based on noncovalent interactions acts as a chelating disphosphite! Surprisingly, lowering the temperature to 30 °C resulted in an even more selective catalyst and the linear to branched ratio increased to 22.8,60 suggesting that the supramolecularly-assembled catalyst system 15 exhibits less dynamic behaviour at lower temperatures. The selectivity is very sensitive to the 13 : 14 ratio used and it was shown that three bridging DABCO ligands are required to obtain high selectivity.
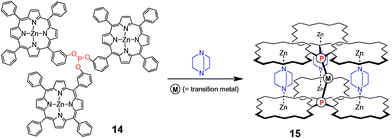 | ||
| Scheme 7 Schematic representation of a multicomponent assembly fixing two phosphites in space that form a chelating bidentate ligand. | ||
In this section we have demonstrated the formation of chelating ligand systems by using metal–ligand interactions for the assembly process. In the examples given, zinc(II) porphyrins have been explored in combination with selective nitrogen coordination. Of course, the concept is by no means limited to this well-studied binding motif and many other metals can, in principle, be used as templates for the assembly process. For example, the class of diamines (phenanthroline, bipyridine, BIAN etc.) can be functionalised with phosphorus ligands,61 to provide an interesting set of molecules that can form bidentates by self-assembly around metal-templates.
Indeed, in 1981, Rauchfuss et al.62 utilised the (o-(diphenylphosphino)benzoyl)pinacolone ligand to prepare various heterobimetallic complexes featuring both hard and soft metals. Iron–catacholate complexes are another interesting motif that could be used in the study of the current concepts. Many heterobimetallic species have been studied in catalysis, but here the focus has been mainly on dinuclear catalysis63 or on the variation of catalyst properties due to allosteric64 or other65 effects, rather using one of the metals as an assembly motif to create bidentate ligands.
Secondary phosphine oxides (SPOs) form an unusual class of ligands,66 that are stable and inert to water. This is due to the tautomerisation equilibrium that is in favour of the stable oxide, but in 1968 it was found that the equilibrium can be shifted by the addition of a metal precursor (Scheme 8)67 and in 1975 the formation of platinum complexes based on SPOs were reported.68 Although they were not specifically designed as supramolecular ligands, this class of compounds represent the first example of ligands that form a bidentate via a hydrogen bond, in this particular case between the phosphine oxide and the alcohol, as was later confirmed by X-ray analysis.69 In 1986 it was reported that these platinum complexes are active in the hydroformylation and hydrogenation reaction and that the ligands were operating as bidentate ligands.70 Much later it was found that the class of ligands can be used for a variety of reactions such palladium-catalysed cross coupling reactions,71 platinum catalysed nitrile hydrolysis and iridium catalysed hydrogenation. In most examples, however, the ligands were employed as monodentate ligands rather than supramolecular bidentate ligands. In some of the reactions, the ligands might rearrange into the supramolecular bidentate, but in many reports where the ligands were added in situ and the ligation was not investigated in detail. Various chiral SPOs have been prepared by the groups of Feringa and de Vries, which provide fascinating new opportunities in this field.72
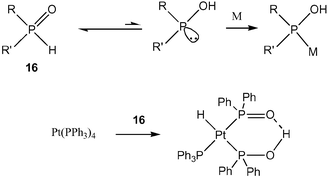 | ||
| Scheme 8 The tautomerization equilibrium of SPOs. | ||
Nakamura et al. developed the tridentate ligand Pyphos 17 (6-(diphenylphosphino)-2-pyridone) that was used to form tetranuclear metal complexes.73 They also prepared dinuclear palladium and platinum complexes using these ligands, in which case a hydrogen bond between two ligands was formed in the solid-state structure.74 Breit and Seiche75 have reported the use of this interesting phosphine ligand as a self-assembled bidentate ligand. It is known that the 2-pyridone and its 2-hydroxypyridine tautomer in apolar solvents give rise mainly to the self-assembled 2-pyridone homodimer pair. Therefore, it was anticipated that pre-organisation of the two monodentate phosphines by coordination to a transition metal would lead to the assembly of a chelating diphospine held together by hydrogen bonds. Indeed, the X-ray structure of a PtL2Cl2 complex revealed the formation of the expected hydrogen bond motif. In addition, the C–N and C–O bond distances observed in the structure indicated that one of the ligands was in the 2-hydroxypyridine tautomeric form, whereas the other was in the 2-pyridone form. The structure also suggests significant contribution from a π–π stacking interaction between a phenyl of one ligand with hydroxypyridine of the other ligand (see Scheme 9).
 | ||
| Scheme 9 Schematic representation of the metal-mediated self-assembly process of the Pyphos ligand and a part of the solid state structure of the PtCl2(Pyphos)2 complex clearly showing the hydrogen bonding interactions between the ligands and a π–π interaction. | ||
The bidentate character of rhodium complexes of the supramolecular ligand was investigated by using the complexes in the rhodium catalysed hydroformylation of 1-octene. The ligand performed very well and gave selectivities similar to that of the well-established bidentate ligand Xantphos (l : b = 32 compared to 49 observed for Xantphos) and higher activities. Interestingly, catalysis performed at temperatures between 50 °C and 140 °C revealed that the high selectivity typical for bidentate ligands was retained up to 110 °C, indicating the surprising thermal stability of the supramolecular ligand assembly. The chelate was functional group tolerant, in that alkenes with amides, alcohols and esters were also hydroformylated with high selectivity for the linear aldehyde. Hydroformylation experiments in methanol as the solvent showed that in this solvent the Pyphos ligand gave similar selectivities as triphenylphosphine, indicating that the assembly was not stable in protic solvents.
We have reported a supramolecular strategy to make bidentate ligands based on metal–ligand interactions that involves simply mixing monomeric ligand building blocks (Fig. 6).76 We had previously54,59,77 shown that zinc(II) porphyrins and nitrogen donor ligands form a complimentary motif that is suited for this purpose since the binding is sufficiently strong and selective.78 We prepared phosphite functionalised porphyrins that could combine with nitrogen-containing phosphorus ligands for the self-assembly of bidentate ligands. Importantly, this approach leads to the formation of bidentate ligands with two different donor atoms (Scheme 10 and Fig. 7).
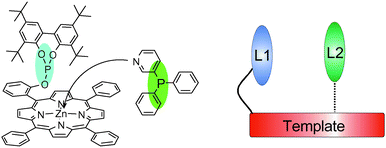 | ||
| Fig. 6 Schematic presentation of the formation of a chelating bidentate ligand formed by assembly of a pyridyl appended phosphine ligand. | ||
 | ||
| Fig. 7 Building blocks that have been used to construct a library of 48 bidentate ligands by self-assembly. | ||
 | ||
| Scheme 10 According to high pressure NMR experiments, the supramolecular complex can be prepared via two routes. The route via the triphenylphosphine complex clearly demonstrates the chelating behavior of the bidentate ligand assembly. | ||
The coordination behaviour of the supramolecular ligands was investigated, using bidentate ligand 18·b as a typical example. UV–vis titrations and NMR spectroscopy experiments show that the pyridyl moiety of b selectively coordinates to the zinc(II) porphyrin 18, with a binding constant in the expected range (K(1·b) = 3.8 × 103 M−1). An increase in the association constant of 18·b is observed in the presence of [HRh(a)3(CO)] (K = 64.5 × 103 M−1), proving the formation of a bidentate chelating system with a corresponding chelate energy of 7 kJ mol−1. Bidentate ligand assembly 19·b gave similar results.
The chelating behaviour can also be observed by NMR techniques (Scheme 10), which indicate that the ligand assembly 18·b coordinates in an equatorial–equatorial fashion to the rhodium metal centre. Importantly, the presence of triphenylphosphine a did not influence the formation of this complex and the addition of b to a solution of HRh(CO)2(18)PPh3 resulted in the formation of the same complex [HRh(CO)2(18·b)], proving the chelating behavior of the ligand assembly.
The chelating behaviour of the assembly is also reflected in the rhodium-catalysed hydroformylation of styrene.55 An increase in the selectivity for the branched product (b : l = 10) and a decrease in activity (TOF = 398) is observed when using supramolecular catalyst 18·b compared to 18 (TOF = 2900, b : l = 2.6). These experiments show that via selective pyridine–zinc interactions two monodentate phosphorus ligands form a chelating bidentate ligand-assembly.
After proving that assembly of bidentate ligands using phosphite porphyrins and pyridyl phosphines gave interesting supramolecular bidentate ligands, we prepared six phosphite functionalised porphyrins (18–23) that in combination with eight monodentate phosphorus ligands (b–i) provided a library of 48 chelating ligands formed by assembly. The supramolecular ligand library based on monodentate phosphorus ligands a–i and 18–23 was tested in the palladium-catalysed asymmetric allylic alkylation79 of rac-1,3-diphenyl-2-propenyl acetate with dimethyl malonate at rt. The matrix of the supramolecular bidentate ligands gave rise to a catalyst library of 60 members by just mixing stock-solutions of the 16 monodentate ligands. For the tested catalysts the enantiomeric excess ranged from 85% (S) to 86% (R). Interestingly, the ee of the products depends strongly on the ligand assembly used, both of the components being important. As expected, the optically inactive building blocks 18 and 19 only afforded chiral products (up to 40% ee) when combined with chiral building blocks g–i. It is important to note that all reactions in the presence of triphenylphosphine (a) as the second donor ligand gave products with no ee, indicating that the catalysis is dominated by palladium triphenylphosphine species and highlighting the importance of the chelate effect induced by the binding motif.
The best catalyst systems were subsequently studied at −20 °C (Table 1). The catalyst formed from monomeric ligand 20 gave products in up to 97% ee (S), a higher enantioselectivity than observed using phosphoramidites as ligands in this reaction.80 Similar high enantioselectivity has been observed when using the chiral MeO-MOP (2-diphenylphosphino-2′-methoxy-1,1′-binapthyl) ligands developed by Hayashi and Kawatsura.81
| Ligand | Conversion (%) | Ee (%) |
|---|---|---|
| a [[Pd(allyl)Cl]2] = 0.1 mmol L−1, [ligand] = 0.6 mmol L−1, the reaction was stopped after 24 h, T = −20 °C. | ||
| 20 | 56 | 97 (S) |
| 20·b | 100 | 60 (R) |
| 20·c | 100 | 0 |
| 20·d | 100 | 44 (S) |
| 21 (R) | 54 | 96 (R) |
| 21·b (R) | 100 | 60 (S) |
| 22 (S) | 73 | 42 (S) |
| 22·b (S) | 42 | 70 (S) |
The catalyst based on ligand assembly 20·b gave products with ee's of 60% (R), which did not change in the presence of excess b, again demonstrating the chelate effect under catalytic conditions. The assembly 20·b proved to be more active than the catalyst based on 20, since the yield after 24 h was 100% compared to 56%. The catalyst based on the bidentate assembly 20·d resulted in the formation of the S-product with an enantiomeric excess of 44%. As shown previously for covalently linked phosphine–phosphite ligands,76 a small difference in the length of the bridge between the phosphine and phosphite resulted in a large difference in enantioselectivity (20·b 60% (R) and 20·d 44% (S)). At −20 °C the catalyst derived from 22 yielded products with 42% ee (S), whereas the bidentate assembly 22·b formed products with up to 70% ee (S). In this case the assembly 22·b gave a slightly slower catalyst (40% yield compared to 73% for 22).
Importantly, small changes to the phosphite zinc(II) porphyrin and the phosphorus ligands a–i have a large influence on the enantioselectivity, as well as the activity of the assembled catalyst system. The diversity of the relatively small supramolecular catalyst library is already sufficient to give catalysts with selectivities ranging from 70% (S) to 60% (R).
Recently, Takacs et al.82 reported a strategy to prepare chiral bidentate bis-phosphite ligands using a modular assembly around a structural metal to form a heteroleptic complex (Scheme 11). The bifunctional subunits have a second set of ligating groups (TADDOL-based phosphites) that are suitably disposed for binding a second metal that acts as the catalytically active centre. A series of subunits were synthesised from which a ligand library of 50 assembled ligands was constructed. Enantioselectivites between 20% and 97% were observed in the palladium-catalysed asymmetric allylic amination, compared to 48% ee for the bis-monodentate TADDOL phenylphosphine analogues. Nine ligand combinations gave products with ee's above 90%, indicating that the bidentate ligands clearly outperformed the monodentates.
 | ||
| Scheme 11 The concept used by Takacs et al.82 to construct bidentate phosphite ligands by assembly via a heteroleptic complex. | ||
Although in the current examples in the literature the number of catalysts generated by these supramolecular ligands is not too large, the strategies are well-suited for combinatorial approaches83 since catalyst libraries become accessible that overshadow those based on traditional methods. A library based on two-component assemblies generates 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bidentate ligands from 200 building blocks. The three-component assembly approach (the template strategy for example) generates almost 300000 ligands from 200 building blocks. It is clear that if such large numbers of catalysts are produced, smart screening of the catalyst libraries will be of the utmost importance. Several methods have already been developed in the past decade.84 An ingenious method utilising ESI-MS techniques to screen the activity of cationic metal polymerisation catalysts has been developed recently by Chen.85 Due to the mild ionisation method, a growing polymer chain will stay attached to the catalyst during analysis. As the most active catalysts will carry the longest polymer chains, the polymer chain length, measured by ESI-MS, becomes a measure of the catalyst activity. Since the signals of various catalyst intermediates appear at different m/z ratios, the screening of catalyst mixtures is possible. Makert and Pfaltz86 have extended this approach to measure directly the inherent enantioselectivity of chiral catalysts. They found it was possible to obtain reliable selectivity data from the kinetic resolution of allylic esters by palladium catalyst mixtures in an allylic substitution reaction. The selectivity of the catalysts for mass-labelled enantiomers (also known as pseudoenantiomers) is determined by measuring the ratio between the two resting state intermediates formed. In principle, there is no limit to the number of catalysts that can be screened, as long as the m/z do not overlap. It is important, however, that the reactivities of the individual catalysts are of the same order of magnitude.
000 bidentate ligands from 200 building blocks. The three-component assembly approach (the template strategy for example) generates almost 300000 ligands from 200 building blocks. It is clear that if such large numbers of catalysts are produced, smart screening of the catalyst libraries will be of the utmost importance. Several methods have already been developed in the past decade.84 An ingenious method utilising ESI-MS techniques to screen the activity of cationic metal polymerisation catalysts has been developed recently by Chen.85 Due to the mild ionisation method, a growing polymer chain will stay attached to the catalyst during analysis. As the most active catalysts will carry the longest polymer chains, the polymer chain length, measured by ESI-MS, becomes a measure of the catalyst activity. Since the signals of various catalyst intermediates appear at different m/z ratios, the screening of catalyst mixtures is possible. Makert and Pfaltz86 have extended this approach to measure directly the inherent enantioselectivity of chiral catalysts. They found it was possible to obtain reliable selectivity data from the kinetic resolution of allylic esters by palladium catalyst mixtures in an allylic substitution reaction. The selectivity of the catalysts for mass-labelled enantiomers (also known as pseudoenantiomers) is determined by measuring the ratio between the two resting state intermediates formed. In principle, there is no limit to the number of catalysts that can be screened, as long as the m/z do not overlap. It is important, however, that the reactivities of the individual catalysts are of the same order of magnitude.
3. Supramolecular variation of the ligand substituents
The third approach that can be derived from Fig. 2 comprises the modification of catalyst environment by the association of functional groups. So far, there are not many examples reported that exactly followed this approach. We have explored if it was possible at all to steer catalyst performance via supramolecular assembly of building blocks.87 Pyridyl functionalised phosphines and phosphites have been modified by the assembly of porphyrins to the pyridine (see Fig. 8 for typical examples). These first experiments have shown that variation of the substituents on the porphyrin system influence the electronic properties of the phosphine, but only if 4-pyridyl-phosphine ligands were used. The electronic effect, that was measured using IR spectroscopy monitoring the CO stretching frequency of the series of [Rh(L)2(CO)Cl] complexes, was not observed for 3-pyridyl-phosphines. The change in electronic properties of the phosphine ligands on association to various porphyrins via the 4-pyridyl moiety was reflected in their catalysis, by a small but distinct increase in activity in the hydroformylation of 1-octene.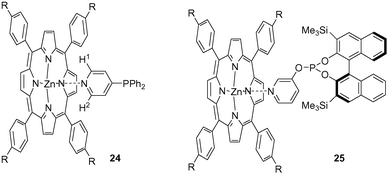 | ||
| Fig. 8 Assembly of 4-pyridyl-diphenylphosphine and (S)-(1,1′-binaphthyl-2,2′-diyl)-(3-pyridyl) phosphite i a on zinc(II)TPP. | ||
The change in steric properties upon assembly formation is obvious and can be fine-tuned by using more sterically demanding porphyrins for the assembly process. Complexes based on (S)-(1,1′-binaphthyl-2,2′-diyl)-(3-pyridyl) and various porphyrins were studied in the rhodium-catalysed hydrogenation of dimethyl itaconate. The enantioselectivity of the catalyst strongly depends on the porphyrin building block and products with ee's varying from 2–50% were afforded (no porphyrin, 16% ee). In a related approach, chiral zinc(II) porphyrins were assembled as co-factors to non-chiral ligands (Fig. 9). In these novel assemblies, the chiral information completely originates from the supramolecularly associated components! While the products of the palladium-catalysed allylic alkylation of 1,3-diphenyl-allylacetate were obtained with only modest enantiomeric excess (18.5%), the proof of concept has been clearly demonstrated.
 | ||
| Fig. 9 Crystal structure of zinc(II)-complex87 of meso-octahydro-1,4–5,8-dimethylanthracene porphyrin (left) and modelled structure of the assembly of this porphyrin (white) and 4-pyridyldiphenylphosphine c (dark grey) (right). | ||
It may be possible to combine this supramolecular approach with the quest for substrate recognition7,8,9 by using suitably functionalised building blocks to hydrogen bond substrate molecules, with the aim of improving selectivity and activity. Variation of the hydrogen bonding moieties should also be simpler on a supramolecular building block than a complicated ligand system. Indeed, by linking substrate recognition sites onto chiral building blocks we may impart catalysts with enzyme-like efficiency.
Conclusions
By combining the principles derived from traditional homogeneous catalysis and supramolecular chemistry new concepts for the development of transition metal catalysts can be envisaged. These new strategies comprise the supramolecular anchoring of catalysts to supports and the formation of new ligands by the selective assembly of rationally designed building blocks. The inherent dynamic character of the noncovalent bonds provides new opportunities, such as the easy separation of the catalyst from its support and the formation of ligand libraries by simply mixing building blocks.It is important to note our trepidation at trying to control such complex systems. Chemists have struggled for years to decipher catalyst mechanisms and while the major pathways and side reactions may be known, many phenomena are still not fully understood. When we are not always sure of the form of the active catalyst using traditional ligand systems, how can we dream of controlling this extra layer of complexity?
The use of well-defined, complentary hydrogen-bonding motifs to anchor rationally designed ligands to dendrimers can result in recyclable catalysts with no appreciable loss of rate compared to the homogeneous analogues, while also being suitable for use in continuous-flow membrane reactors. Using a similar supramolecular strategy, silica suppports can be covalently functionalised with tethers bearing similar well-defined binding motifs to allow the specific and reversible binding of catalyst complexes that retain their high selectivity even if a reduction in activity is observed. These supports can be defunctionalised by washing with methanol and simply refunctionalised with new catalysts, an advantage that clearly originates from the supramolecular approach.
Noncovalent interactions can also be used to form bidentate ligands from monodentate ligand building blocks by assembly. This can be achieved via a direct interaction between the building blocks, or via the assembly on a template. In the latter case more noncovalent interactions are involved in the formation of the assembly. For both approaches, large ligand libraries become easily accessible: 100 building blocks can generate a library of 2,500 bidentate ligands, a number that dwarfs the current largest phosphorus-based bidentate library. In addition to its obvious application in combinatorial catalysis with the aim of discovering more selective and active catalysts, the reversibility of the bond between the ligand building blocks can give rise to new properties, such as adaptive ligands.
By developing various catalyst assembly building blocks with well-defined, specific and benign binding motifs, we may, one day, be able to create libraries of catalysts based on numerous supramolecular interactions, allowing the variation of every conceivable catalyst parameter in a fast and efficient manner. Since catalyst recycling strategies are based on similar supramolecular interactions, implementation of separation processes should become straight forward. Clearly, this field of scientific research is still in its infancy, but already it promises a bright future for catalysis.
Acknowledgements
We would like to thank all co-workers that have participated in and contributed to our research efforts. NWO and the University of Amsterdam are kindly acknowledged for financial support for J. N. H. R. via a VICI-grant.References
- (a) L. Pauling, Nature, 1946, 161, 707; (b) A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707 CrossRef CAS.
- More recently it was shown that the protein dynamic motions also contribute to the increase of reaction rate: (a) E. Z. Eisenmesser, D. A. Bosco, M Akke and D. Kern, Science, 2002, 295, 1520 CrossRef CAS; (b) T. C. Bruice and S. J. Benkovic, Biochemistry, 2000, 39, 6267 CrossRef CAS.
- A. V. Nemukhin, I. A. Topol, B. L. Grigorenko and S. K. Burt, J. Phys. Chem. B, 2002, 106, 1734 CrossRef CAS.
- R. H. Garrett and C. M. Grisham, Biochemistry, Saunders Collge Publishing, New York, 2nd edn., 1999 Search PubMed.
- (a) M. S. Goedheijt, B. E. Hanson, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2000, 122, 1650 CrossRef; (b) K. Ramkisoensing, P. W. N. M. van Leeuwen and J. N. H. Reek, unpublished results; (c) M. J. Wilkinson, P. W. N. M. van Leeuwen and J. N. H. Reek, unpublished results.
- (a) M. T. Zarka, M. Bortenshlager, K. Wurst, O. Nuyken and R. Weberskirch, Organometallics, 2004, 23, 4817 CrossRef CAS; (b) J. E. Klijn and J. B. F. N. Engberts, Org. Biomol. Chem., 2004, 2, 1789 RSC; (c) Y. M. A. Yamada, K. Takeda, H. Takahashi and S. Ikegami, J. Org. Chem., 2003, 68, 7733 CrossRef CAS; (d) E. Paetzold, I. Jovel and G. Oehme, J. Mol. Catal. A: Chem., 2004, 214, 241 CrossRef CAS; (e) Y. M. A. Yamada, K. Takeda, H. Takahashi and S. Ikegami, Tetrahedron, 2004, 60, 4097 CrossRef CAS; (f) M. T. Zarka, O. Nuyken and R. Weberskirch, Macromol. Rapid Commun., 2004, 25, 858 CrossRef CAS; (g) G. Oehme, I. Grassert, E. Paetzold, H. Fuhrmann, T. Dwars, U. Schmidt and I. Iovel, Kinet. Catal., 2003, 44, 766 CAS; (h) A. Solsona, J. Suades and R. Mathiue, J. Organomet. Chem., 2003, 669, 172 CrossRef CAS; (i) S. Gaemers, K. Keune, A. M. Kluwer and C. J. Elsevier, Eur. J. Inorg. Chem., 2000, 1139 CrossRef CAS; (j) K. Nozaki, M. Yoshida and H. Takaya, J. Organomet. Chem., 1994, 473, 253 CrossRef CAS.
- (a) J.-M. Lehn, Science, 1985, 227, 849 CrossRef CAS; (b) A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707 CrossRef CAS; (c) Y. Murakami, J. Kikuchi, Y. Hisaeda and O. Hayashida, Chem. Rev., 1996, 96, 721 CrossRef CAS; (d) J. K. M. Sanders, Chem. Eur. J., 1998, 4, 1378 CrossRef; (e) W. B. Motherwell, M. J. Bingham and Y. Six, Tetrahedron, 2001, 57, 4663 CrossRef CAS; (f) R. Pascal, Eur. J. Org. Chem., 2003, 1813 CrossRef CAS; (g) M. C. Feiters, Supramolecular catalysis, in Comprehensive Supramolecular Chemistry, ed. D. N. Reinhoudt, Elsevier, Oxford, UK, 1996, pp. 267––360 Search PubMed; (h) M. C. Feiters, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2000, 29, 375 RSC; (i) M. Sawamura and Y. Ito, Chem. Rev., 1992, 857 CrossRef CAS; (j) G. J. Rowlands, Tetrahedron, 2001, 57, 1865 CrossRef CAS.
- T. Hayashi, K. Kanehira, H. Tsuchiya and M. Kumada, J. Chem. Soc., Chem. Commun., 1982, 1162 RSC.
- (a) D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232 CrossRef CAS; (b) D. B. Grotjahn, S. Van, D. Combs, D. A. Lev, C. Schneider, C. D. Incarvito, K.-C. Lam, G. Rossi, A. L. Rheingold, M. Rideout, C. Meyer, G. Hernandez and L. Mejorado, Inorg. Chem., 2003, 42, 3347 CrossRef CAS; (c) D. B. Grotjahn, C. D. Incarvito and A. L. Rheingold, Angew. Chem., Int. Ed., 2001, 40, 3884 CrossRef CAS; (d) R. H. Crabtree, Dalton Trans., 2003, 3985 RSC; (e) D.-H. Lee, R. H. Crabtree and S.-K. Park, Bull. Korean Chem. Soc., 2002, 23, 1157 CAS; (f) E. Clot, O. Eisenstein and R. H. Crabtree, New J. Chem., 2001, 25, 66 RSC; (g) R. H. Crabtree, J. A. Loch, K. Gruet, D.-H. Lee and C. Borgmann, J. Organomet. Chem., 2000, 600, 7 CrossRef CAS; (h) K. Gruet, R. H. Crabtree, D.-H. Lee, L. Liable-Sands and A. L. Rheingold, Organometallics, 2000, 19, 2228 CrossRef CAS.
- (a) P. A. Brady and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1997, 3237 RSC; (b) J.-M. Lehn and A. V. Eliseev, Science, 2001, 291, 2331 CrossRef CAS; (c) J.-M. Lehn, Chem. Eur. J., 1999, 9, 2455 CrossRef; (d) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 899 CAS; (e) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590 CrossRef CAS.
- (a) B. Brisig, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2003, 45, 1270 CrossRef CAS and references therein; (b) P. T. Corbett, S. Otto and J. K. M. Sanders, Chem. Eur. J., 2004, 10, 3139 CrossRef CAS.
- E. T. Kaiser and D. S. Lawrence, Science, 1984, 226, 505 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- (a) M. T. Reetz, Tetrahedron, 2002, 58, 6595 CrossRef CAS and references therein; (b) M. T. Reetz, (Studiegesellschaft Kohle MBH) WO 02/103009 A2, 2002.
- (a) J. Collot, J. Gradinaru, N. Humbert, M. Skander, A. Zocchi and T. R. Ward, J. Am. Chem. Soc., 2003, 125, 9030 CrossRef CAS; (b) C. Letondor, N. Humbert and T. R. Ward, Proc. Natl. Acad. Sci. USA, 2005, 102, 4683 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- (a) C. P. Casey and G. T. Whiteker, Isr. J. Chem., 1990, 30, 299 CAS; (b) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes;, Chem. Rev., 2000, 100, 2741 CrossRef CAS.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen and C. F. Roobeek, J. Organomet. Chem., 1983, 258, 343 CrossRef CAS; (b) A. van Rooy, E. N. Orij, P. C. J. Kamer, F. van de Aardweg and P. W. N. M. van Leeuwen, J. Chem. Soc., Chem. Commun., 1991, 1096 RSC.
- G. P. F. van Strijdonck, M. D. K. Boele, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 1999, 1073 CrossRef CAS.
- A. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- (a) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722 CrossRef CAS; (b) J. P. Wolfe and S. L. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413 CrossRef CAS; (c) J. P. Wolfe, R. A. Singerm, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550 CrossRef CAS.
- J. P. Stambuli, M. Buhl and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9346 CrossRef CAS.
- (a) D. Gleig and W. A. Herrmann, Organometallics, 1999, 18, 4354 CrossRef CAS; (b) P. O. Norrby, T. Rasmussen, J. Haller, T. Strassner and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 10186 CrossRef CAS; (c) G. Ujaque, F. Maseras and A. Lledos, J. Am. Chem. Soc., 1999, 121, 1317 CrossRef CAS.
- Combinatorial approaches:S. R. Wilson and A. W. Czarnik, Combinatorial Chemistry, Wiley, New York, 1997 Search PubMed.
- (a) D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS; (b) Chem. Rev., 2002, Search PubMed102, issue 10 thematic issue.
- B. Cornils and W. A. Herrmann, Applied homogeneous catalysis with organometallic compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinhein, 1996, ch. 3 Search PubMed.
- (a) E. Kuntz, (Rhone-Poulenc Industrie) French Pat. 2349562, 1976; (b) E. Kuntz, (Rhone-Poulenc Industrie) French Pat. 2314910, 1975.
- (a) J. P. Arhanchet, M. E. Davis, J. S. Merola and B. E. Hanson, Nature, 1989, 339, 454 CrossRef CAS; (b) M. E. Davis, Chem. Tech., 1992, 498 CAS; (c) A. J. Sandee, V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 1999, 17, 1633 RSC.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 CrossRef CAS; (b) I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef CAS; (c) L. P. Barthel-Rosa and J. A. Gladysz, Coord. Chem. Rev., 1999, 190–192, 587 CrossRef CAS; (d) C. Rocaboy, D. Rutherford, B. L. Bennett and J. A. Gladysz, J. Phys. Org. Chem., 2000, 13, 596 CrossRef CAS; (e) B. Richter, A. L. Spek, G. van Koten and B.-J. Deelman, J. Am. Chem. Soc., 2000, 122, 3945 CrossRef CAS; (f) E. de Wolf, G. van Koten and B.-J. Deelman, Chem. Soc. Rev., 1999, 28, 37 RSC; (g) R. H. Fish, Chem. Eur. J., 1999, 5, 1677 CrossRef CAS.
- (a) P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH Verlag, Weinheim, Germany, 2003 Search PubMed; (b) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS; (c) P. Wasserscheid, H. Waffenschmidt, P. Machnitzki, K. W. Kottsieper and O. Stelzer, Organometallics, 2000, 19, 3818 CrossRef CAS; (d) R. P. J. Bronger, S. M. Silva, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 2002, 3044 RSC; (e) W Keim, D. Vogt, H. Waffenschmidt and P. J. Wasserscheid, J. Catal., 1999, 186, 481 CrossRef CAS; (f) P. Wasserscheid, H. Waffenschmidt, P. Machnitzki, K. W. Kottsieper and O. Stelzer, Chem. Commun., 2001, 451 RSC.
- (a) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475 CrossRef CAS; (b) J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495 CrossRef CAS.
- (a) R. van Heerbeek, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717 CrossRef; (b) R. Kreiter, A. W. Kleij, R. J. M. Klein Gebbink and G. van Koten, in Dendrimers IV: Metal Coordination, Self Assembly, Catalysis, ed. F. Vögtle and C. A. Schalley, Springer-Verlag, Berlin, 2001. vol. 217, p. 163 Search PubMed; (c) G. E. Oosterom, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 1828 CrossRef CAS; (d) R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yueng, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS; (e) J. N. H. Reek, D. de Groot, G. E. Oosterom, P. C. J. Kamer and P. W. N. M. van Leeuwen, Rev. Mol. Biotechnol., 2002, 90, 159 CrossRef CAS; (f) D. Astruc and F. Chardac, Chem. Rev., 2001, 101, 2991 CrossRef CAS.
- Extensive reviews on polymer immobilised catalysts include: (a) F. R. Hartley and P. N. Vezey, Adv. Organomet. Chem., 1977, 15, 189 CAS; (b) F. R. Hartley, Supported Metal Complexes. A New Generation of Catalysts, ed. R. Ugo and B. R. James, Reidel, Dordrecht, 1985 Search PubMed; (c) Y. Iwasawa, Tailored Metal Catalysts, ed. R. Ugo and B. R. James, Reidel, Dordrecht, 1986 Search PubMed; (d) W. Keim and B. Driessen-Hölscher, Handbook of heterogeneous catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, Weinhein, 1997, vol. 1 Search PubMed; (e) E. Lindner, T. Schneller, F. Auer and H. A. Mayer, Angew. Chem., Int. Ed., 1999, 38, 2154 CrossRef; (f) J. M. Thomas, Angew. Chem., Int. Ed., 1999, 38, 3588 CrossRef.
- Other interesting contributions: (a) A. Nailt Ajjou and H. Alper, J. Am. Chem. Soc., 1998, 120, 1466 CrossRef CAS; (b) M. R. Buchmeiser and K. Wurst, J. Am. Chem. Soc., 1999, 121, 11101 CrossRef CAS; (c) D. E. Berbreiter, Y.-S. Liu and P. L. Osburn, J. Am. Chem. Soc., 1998, 120, 4250 CrossRef CAS; (d) M. K. Richmond, S. L. Scott and H. Alper, J. Am. Chem. Soc., 2001, 123, 10521 CrossRef CAS; (e) D. E. Bergbreiter, Catal. Today, 1998, 42, 389 CrossRef CAS; (f) D. E. Berbreiter, P. L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531 CrossRef CAS; (g) K. Nozaki, Y. Itoi, F. Shibahara, E. Shirakawa, T. Ohta, H. Takaya and T. Hiyama, J. Am. Chem. Soc., 1998, 120, 4051 CrossRef CAS.
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, CATTECH, 2002, 6, 116 CrossRef CAS.
- (a) A. J. Sandee, L. A. van der Veen, J. N. H. Reek, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 3231 CrossRef CAS; (b) A. J. Sandee, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468 CrossRef CAS; (c) A. J. Sandee, D. G. I. Petra, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Eur. J., 2001, 7, 1202 CrossRef CAS; (d) A. J. Sandee, R. S. Ubale, M. Makkee, J. N. H. Reek, P. C. J. Kamer, J. A. Moulijn and P. W. N. M. van Leeuwen, Adv. Synth. Catal., 2001, 1, 201 CrossRef.
- E. Schweb and S. Mecking, Organometallics, 2001, 20, 5504 CrossRef CAS.
- M. W. P. L. Baars, A. J. Karlsson, V. Sorokin and B. F. M. Meijer, Angew. Chem., Int. Ed., 2000, 66, 2136.
- D. de Groot, B. F. M. de Waal, J. N. H. Reek, A. P. H. J. Schenning, P. C. J. Kamer, E. W. Meijer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8453 CrossRef CAS.
- R. van de Coevering, M. Kuil, R. J. M. Klein Gebbink and G. van Koten, Chem. Commun., 2002, 1636 RSC.
- (a) M. Ooe, M. Murata, A. Takahama, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32(8), 692 CrossRef CAS; (b) M. Ooe, M. Murata, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 1604 CrossRef CAS.
- A. Sunder, J. Heinemann and H. Frey, Chem. Eur. J., 2000, 6, 2499 CrossRef CAS.
- E.-E. Stiriba, M. Q. Slagt, H. Kautz, R. J. M. Klein Gebbink, R. Thomann, H. Frey and G. van Koten, Chem. Eur. J., 2004, 10, 1267 CrossRef.
- M. Q. Slagt, S.-E. Stiriba, H. Kautz, R. J. M. Klein Gebbink, H. Frey and G. van Koten, Organometallics, 2004, 23, 1525 CrossRef CAS.
- H. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., Int. Ed., 2000, 39, 1772 CAS.
- L. Plault, A. Hauseler, S. Nlate, D. Astruc, J. Ruiz, S. Gatard and R. Neumann, Angew. Chem., Int. Ed., 2004, 43, 2924 CrossRef CAS.
- (a) C. Bianchini, D. G. Burnaby, J. Evans, P. Frediani, A. Meli, W. Oberhauser, R. Psaro, L. Sordelli and F. Vizza, J. Am. Chem. Soc., 1999, 121, 5961 CrossRef CAS; (b) C. Bianchini, V. D. Santo, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Organometallics, 2000, 19, 2433 CrossRef CAS; (c) C. Bianchini, P. Barbaro, V. D. Santo, R. Gobetto, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Adv. Synth. Catal., 2001, 343(1), 41 CrossRef CAS.
- W. P. Mul, K. Ramkisoensing, P. C. J. Kamer, J. N. H. Reek, A. J. van der Linden, A. Marson and P. W. N. M. Van Leeuwen, Adv. Synth. Catal., 2002, 344, 293 CrossRef CAS.
- M. M. Diaz-Requejo, T. R. Belderrain, M. C. Nicasio and P. J. Perez, Organometallics, 2000, 19, 285 CrossRef CAS.
- R. Chen, R. P. J. Bronger, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126(44), 14557 CrossRef CAS.
- L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872–883 CrossRef CAS.
- Z. Frexia and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC.
- V. F. Slagt, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2003, 2474 RSC.
- Rhodium-Catalysed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publishers, Dordecht, 2000 Search PubMed.
- (a) F. Agbossou, J.-F. Carpentier and A. Morteux, Chem. Rev., 1995, 95, 2485 CrossRef CAS; (b) S. Gladiali, J. C. Bayon and C. Claver, Tetrahedron: Asymmetry, 1995, 6, 1453 CrossRef CAS.
- S. Gladiali, A. Dore, D. Fabbri, O. De Lucchi and M. Manassero, Tetrahedron: Asymmetry., 1995, 6, 1453 CrossRef CAS.
- [Rh(acac)(CO)2] and [HRh(CO)(PPh3)3] were mixed with zinc(II) porphyrin phosphite 1 in a 1 : 2 ratio, forming [Rh(acac)(1)2] and [HRh(CO)(1)2(PPh3)], respectively.
- V. F. Slagt, P. W. N. M. van Leeuwen and J. N. H. Reek, Angew. Chem., Int. Ed., 2003, 42, 5619 CrossRef CAS.
- Usually the l : b ratio decreases at lower temperatures: (a) R. Lazzaroni, G. Uccello-Barretta, S. Scamuzzi, R. Settambolo and A. Caiazzo, Organometallics, 1996, 15, 4657 CrossRef CAS; (b) C. Botteghi, L. Cazzolato, M. Marchetti and S. Paganelli, J. Org. Chem., 1995, 60, 6612 CrossRef CAS.
- (a) G. R. Newkome, Chem. Rev., 1993, 93, 2067 CrossRef CAS; (b) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499 CrossRef CAS.
- (a) T. B. Rauchfuss, S. R. Wilson and D. A. Wrobleski, J. Am. Chem. Soc., 1981, 103, 6769 CrossRef CAS; (b) D. A. Wrobleski and T. B. Rauchfuss, J. Am. Chem. Soc., 1982, 104, 2314 CrossRef CAS; (c) D. A. Wrobleski, C. S. Day, B. A. Goodman and T. B. Rauchfuss, J. Am. Chem. Soc., 1984, 106, 5464 CrossRef CAS.
- (a) M. P. Garcia, A. M. Lopez, M. A. Estruelas, F. J. Lahoz and L. A. Oro, J. Chem. Soc., Chem. Commun., 1988, 793 RSC; (b) E. K. van den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985 CrossRef CAS; (c) P. Braunstein, J. Durand, X. Morise, A. Tripicchio and F. Ugozzoli, Organometallics, 2000, 19, 444 CrossRef CAS; (d) P. Braunstein, G. Clerc, X. Morise, R. Welter and G. Mantovani, Dalton Trans., 2003, 1601 RSC.
- (a) L. Kovbasyuk, H. Pritzkow, R. Kramer and I. O. Fritsky, Chem. Commun., 2004, 880 RSC; (b) N. C. Gianneschi, S.-H. Cho, S. T. Nguyen and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 5503 CrossRef CAS.
- Other papers of interest: (a) M. Quirmbach, A. Kless, J. Holz, V. Tararov and A. Borner, Tetrahedron: Asymmetry, 1999, 10, 1803 CrossRef CAS; (b) Y. Sun, M. Ahmed, R. Jackstall, M. Beller and W. R. Thiel, Organometallics, 2004, 23, 5260 CrossRef CAS.
- For a recent highlight see: N. V. Dubrovina and A. Börner, Angew. Chem., Int. Ed., 2004, 43, 5883 Search PubMed.
- J. Chatt and B. T. Heaton, J. Chem. Soc. A, 1968, 2745 RSC.
- W. B. Beaulieu, T. B. Rauchfuss and D. M. Roundhill, Inorg. Chem., 1975, 14, 1732 CrossRef CAS.
- P. W. N. M. van Leeuwen, C. F. Roobeek, J. H. G. Frijns and A. G. Orpen, Organometallics, 1990, 9, 1211 CrossRef.
- (a) P. W. N. M. van Leeuwen, C. F. Roobeek, R. L. Wife and J. H. G. Frijns, J. Chem. Soc., Chem. Commun., 1986, 31 RSC; (b) P. W. N. M. van Leeuwen and C. F. Roobeek, New. J. Chem., 1990, 14, 487 Search PubMed.
- G. Y. Li, J. Org. Chem., 2002, 67, 3643 CrossRef CAS.
- (a) X.-B. Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. F. Boogers and J. G. de Vries, Org. Lett., 2003, 5, 1503 CrossRef CAS; (b) X.-B. Jiang, M. van den Berg, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Tetrahedron: Asymmetry, 2004, 15, 2223 CrossRef CAS; (c) X.-B. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327 CrossRef CAS.
- K. Mashima, H. Nakano and A. Nakamura, J. Am. Chem. Soc., 1993, 115, 11632 CrossRef CAS.
- K. Mashima, M. Tanaka, K. Tani, H. Nakano and A. Nakamura, Inorg. Chem., 1996, 35, 5244 CrossRef CAS.
- B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608 CrossRef CAS.
- (a) V. F. Slagt, M. Röder, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 4056 CrossRef CAS; (b) J. N. H. Reek, M. Röder, P. E. Goudriaan, P. C. J. Kamer, P. W. N. M. van Leeuwen and V. Slagt, J. Organomet. Chem., DOI: 10.1016/j.jorganchem.2005.02.026 Search PubMed.
- V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271 CrossRef CAS.
- Phosphorus donor atoms do not coordinate to the zinc(II) porphyrin.
- (a) J. Tsuji, Tetrahedron, 1986, 42, 4361 CrossRef CAS; (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (c) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS; (d) A. Pfaltz, Acc. Chem. Res., 1993, 26, 339 CrossRef CAS; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS.
- M. D. K. Boele, P. C. J. Kamer, M. Lutz, A. L. Spek, J. G. de Vries, P. W. N. M. van Leeuwen and G. P. F. van Strijdonck, Chem. Eur. J., 2004, 10, 6232 CrossRef CAS.
- T. Hayashi and M. Kawatsura, Chem. Commun., 1997, 561 RSC.
- J. M. Takacs, D. S. Reddy, S. A. Moteki, D. Wu and H. Palencia, J. Am. Chem. Soc., 2004, 126, 4494 CrossRef CAS.
- (a) M. T. Reetz, T. Sell, A. Meiswinkel and G. Mehler, Angew. Chem., Int. Ed., 2003, 42, 790 CrossRef CAS; (b) M. T. Reetz, Angew. Chem., Int. Ed., 2001, 40, 284 CrossRef CAS; (c) M. T. Reetz, Angew. Chem., Int. Ed., 2002, 41, 1335 CrossRef CAS; (d) S. J. Taylor and J. P. Morken, Science, 1998, 280, 267 CrossRef CAS; (e) M. B. Francis and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 937 CrossRef CAS; (f) R. H. Crabtree, Chem. Commun., 1999, 1611 RSC; (g) M. B. Francis, T. F. Jamison and E. N. Jacobsen, Curr. Opin. Chem. Biol., 1998, 2, 422 CrossRef CAS.
- (a) J. P. Stambuli, S. R. Stauffer, K. H. Shaughnessy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 2677 CrossRef CAS; (b) R. F. Harris, A. A. J. Natio, G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 2000, 122, 11270 CrossRef CAS; (c) K. H. Shaughnessy, P. Kim and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 2123 CrossRef CAS; (d) T.-Y. Yue and W. A. Nugent, J. Am. Chem. Soc., 2002, 124, 13692 CrossRef CAS; (e) M. T. Reetz, K. M. Kuhling, A. Deege, H. Hinrichs and D. Belder, Angew. Chem., Int. Ed., 2000, 39, 3891 CrossRef CAS; (f) M. T. Reetz, Angew. Chem., Int. Ed., 2002, 41, 1335 CrossRef CAS; (g) J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 799 CrossRef CAS; (h) S. R. Stauffer and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 6977 CrossRef CAS.
- P. Chen, Angew. Chem., Int. Ed., 2003, 42, 2832 CrossRef CAS.
- C. Makert and A. Pfaltz, Angew. Chem., Int. Ed., 2004, 43, 2498 CrossRef.
- V. F. Slagt, PhD thesis, University of Amsterdam, The Netherlands, 2003. This work was partly done in collaboration with Prof. A. Berkessel, manuscript in preparation.
| This journal is © The Royal Society of Chemistry 2005 |