Deprotonation–electrophile trapping of terminal epoxides
David M.
Hodgson
*a,
Eirene H. M.
Kirton
a,
Steven M.
Miles
a,
Stéphanie L. M.
Norsikian
a,
Nigel J.
Reynolds
a and
Steven J.
Coote
b
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, UK OX1 3TA
bGlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, UK SG1 2NY
First published on 13th April 2005
Abstract
Organolithium-induced deprotonation of terminal epoxides in the presence of appropriate diamine ligands allows trapping with a range of electrophiles, yielding functionalised di- and tri-substituted epoxides in good yields and with control of stereochemistry at the epoxide.
Introduction
In view of the many roles that epoxides play in organic synthesis, efficient (asymmetric) methods for the direct introduction of this important functional group would be highly valued.1 At the outset of our studies the coupling of oxiranyl organometallics with electrophiles was one method for incorporation of epoxides that had yet to be fully developed, especially as an asymmetric transformation.2 The first systematic studies of stabilised oxiranyl anions were reported in 1976 by Eisch and Galle.3 Their pioneering research demonstrated the apparent necessity of an anion-stabilising group attached to the epoxide ring to enhance the facility of metalation of epoxides and to increase the thermal stability of the oxiranyl anions produced. Metalation of cis-disubstituted trialkylsilyl epoxides and reaction of the resultant oxiranyl anions with simple electrophiles, such as Me3SiCl, Me3SnCl, CO2, MeI, amides, and D2O, led to high yields of the desired products, with complete configurational stability being observed. This early work has subsequently been extended in many directions, expanding the range of electrophiles and anion-stabilising groups, and even generating and trapping destabilised (alkyl-substituted) oxiranyl anions by ligand exchange (desulfinylation) of sulfinyloxiranes.4 Until the last few years it was still generally believed that, as stated in a 1996 review on the synthesis and utility of oxiranyl anions, the “metal–hydrogen exchange reaction is obviously not useful for generation of nonstabilised (H substituent) oxiranyl anions”.2aArising from our studies concerning the lithiation–rearrangement of epoxides,5 we envisioned that an appropriate ligand might be able to serve the dual role of accelerating deprotonation and reducing the rate of oxiranyl anion decomposition, enabling the direct deprotonation–electrophile trapping of unfunctionalised terminal epoxides. Compared to lithiation–electrophile trapping using epoxides of medium-sized cycloalkenes, terminal epoxides (e.g.1a) represent a greater challenge due to comparatively reduced epoxide C–H acidity,6 combined with a higher propensity of both the starting and α-lithiated epoxides to undergo ring-opening reactions. In this paper we report a full account of our studies in this area.7
Results and discussion
We started our investigations with a study of various conditions for the deprotonation–deuteration of the simple terminal epoxide 1,2-epoxydodecane 1a (Scheme 1). The reaction was first carried out in Et2O in the absence of a ligand. Whilst no reaction occurred at −90 °C, we performed the addition of a solution of 1a in Et2O to iPrLi (2.5 eq.) in Et2O at −78 °C over a period of 10 min followed after 1.5 min by D2O quench. This run led after chromatography to recovered 1a (35% yield, 0% D incorporation), trans-alkene 3a (R = iPr, 34%) arising from reductive alkylation,8 and alcohol 4a (R = iPr, 22%). The same reaction, but carried out in the presence of TMEDA (2.5 eq.) was found to accelerate the deprotonation step. In this case, in Et2O at −78 °C or −90 °C the alkene dominated the reaction mixture (1a (0% D) : 3a = 20 : 80 and 50 : 50, respectively) with no 4a (R = iPr) detected. On switching from Et2O to THF, no reaction of 1a was observed at −90 °C with iPrLi in the presence of TMEDA. | ||
| Scheme 1 Deprotonation–deuteration studies of 1,2-epoxydodecane 1a. | ||
Significantly, replacing TMEDA with commercially available (−)-sparteine 5 (Fig. 1) as the ligand, we found that D incorporation was possible. When 1a was added to a mixture of 5 (2.5 eq.) and either nBuLi, tBuLi or iPrLi (2.5 eq.) in Et2O at −90 °C, followed after 15 min by D2O quench, 1a was recovered with 0% D, 10% D and 22% D incorporation, respectively. Increasing the reaction time to 1 h with iPrLi in Et2O before D2O quench led to 1a in 50% yield with 45% D incorporation, along with 3a (R = iPr, 18%) and 4a (R = iPr, 11%). The reaction performed under the same conditions (iPrLi–5 at −90 °C for 1 h) but with hexane as solvent allowed us to recover 1a in 60% yield with the same level of D incorporation but with significantly less 3a (R = iPr, 6%) (Table 1, entry 1). Better results were obtained using sBuLi in hexane, which after only 15 min at −90 °C gave 1a in 70% yield with 75% D incorporation (Table 1, entry 3) with 9% of 3a (R = sBu) isolated. Longer reaction times resulted in increased formation of by-products and diminished recovery of 1a (Table 1, entries 2 and 4). Incorporation of deuterium occurred exclusively trans to the alkyl substituent on the oxirane ring.
| Entrya | RLi | Ligand | Time/min | Amount of Db in 1a (%) | Yieldc of 1a (%) |
|---|---|---|---|---|---|
| a Reactions were carried out by the addition over 10 min of a solution of 1a (1 eq.) in hexane to a mixture of RLi–ligand (2.5 eq. each) in hexane at −90 °C, followed by the addition of CD3OD after the time indicated. b Determined by 1H NMR spectroscopy. c Yield following chromatography. d 4a (R = sBu) was only observed (5%) in this experiment. | |||||
| 1 | i PrLi | 5 | 60 | 46 | 60 |
| 2 | i PrLi | 5 | 180 | 50 | 50 |
| 3 | s BuLi | 5 | 15 | 75 | 70 |
| 4d | s BuLi | 5 | 60 | 90 | 40 |
| 5 | s BuLi | 6 | 15 | 45 | 82 |
| 6 | s BuLi | 7 | 15 | 50 | 85 |
| 7 | s BuLi | 7 | 60 | 52 | 80 |
| 8 | s BuLi | 8 | 15 | 63 | 91 |
| 9 | s BuLi | 8 | 60 | 70 | 75 |

The above encouraging results with 5 as ligand prompted us to examine ligands such as 6–89 that all possess the 3,7-diazabicyclo[3.3.1]nonane structural feature of 5 (Table 1, entries 5–8). For these ligands, it was found that the deprotonation step was slower than with 5. However, in these experiments little or no alkene was formed, which demonstrates the ability of these ligands to stabilise the intermediate oxiranyl anion 2a. The best results were obtained using sBuLi–8 in hexane at −90 °C (Table 1, entries 8 and 9). D2O quenching after 1 h afforded 1a in 75% yield with 70% D incorporation along with 3a (R = sBu, 8%). Using sBuLi–8 in Et2O was less effective and provided after 1 h a mixture of 1a and 3a (96 : 4 respectively), with only 48% D incorporation in 1a.
Attempts to trap the intermediate oxiranyl anion 2a with other external electrophiles (Me3SiCl, ClCO2Me, PhCHO, EtCHO) were found to be initially unsuccessful under the above conditions. However, reactions carried out with Me3SiCl present during the generation of the oxiranyl anion led to α,β-epoxysilanes 9 in good yields (Scheme 2, Table 2). α,β-Epoxysilanes are interesting synthons for organic chemistry since, for example, they can be hydrolysed to give carbonyl compounds, undergo regio- and stereospecific α-opening by a range of nucleophiles to give diastereomerically pure β-hydroxysilanes and can serve as vinyl cation equivalents for the preparation of olefins of known stereochemistry.10
| Entrya | 1 | Time/h | Yield of 9 (%)b | ||
|---|---|---|---|---|---|
| a Reactions were carried out at −90 °C (−83 °C for entry 3) for the time indicated, followed by warming to −80 °C over 5 min (to −50 °C over 30 min for entries 1 and 7–10). b Yield following chromatography. c Ligand 5 used in place of ligand 8. | |||||
| 1 |

|
1a | 2 |

|
9a |
| 2 |

|
1b | 2.5 |

|
9b |
| 3 |

|
1c | 4.5 |

|
9c |
| 4 |

|
1d | 2 |

|
9d |
| 5 |

|
1e | 3 |

|
9e |
| 6 |

|
1f | 4.5 |

|
9f |
| 7 |

|
1g | 3 |

|
9g |
| 8c |

|
1a | 0.25 |

|
(1S,2S)-9a |
| 9c |

|
(S)-1a | 0.25 |
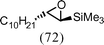
|
(1S,2S)-9a |
| 10c |

|
(R)-1a | 0.25 |

|
(1R,2R)-9a |
 | ||
| Scheme 2 Silylation of terminal epoxides 1. | ||
As shown in Table 2, the process is compatible with a variety of functionalised epoxides11 leading to stereochemically pure trans-α,β-epoxysilanes (entries 2–5).12 The reaction can also be performed with 2,2-disubstituted epoxides affording trisubstituted epoxides (entries 6 and 7).13 For the unsymmetrical epoxide 1g (entry 7), silylation was found to proceed with a high degree of selectivity (97 : 3) trans to the phenyl substituent.13b
Chiral non-racemic ligand 5 could in theory effect kinetic resolution on a racemic epoxide, and this was found to be the case to an extent (entry 8, ca. 30% ee for the enantiomer shown based on the specific rotation of the product). When employing 0.6 eq. each of sBuLi, 5 and Me3SiCl, the product (1S,2S)-9a was returned in 18% yield and ca. 46% ee (based on its specific rotation), along with starting material 1a (63%) and alkene 3a (R = sBu) from reductive alkylation (5%). Importantly, it was demonstrated that sBuLi–5 could be used to silylate either enantiomer of epoxide 1a in high yield and with complete trans selectivity (entries 9 and 10). This is useful given the commercial availability of ligand 5.
A minor side-reaction of bis-silylation was observed in the silylation chemistry with sparteine 5 as the ligand. This led to the formation of an epoxydisilane 10, presumably via lithiation–silylation of 9 (Scheme 3, ca. 10% bis-silylation observed for R = C10H21).3,10,13a,14
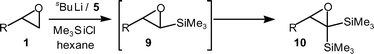 | ||
| Scheme 3 Epoxydisilanes 10via bis-silylation of epoxides 1. | ||
Methods to access epoxydisilanes 10 are of interest, as epoxydisilanes can be readily converted into acylsilanes and 1-halovinylsilanes.15 Acylsilanes in particular are finding increasing utility in synthesis.16 With this in mind it was decided to investigate the direct synthesis of epoxydisilanes 10 from readily available terminal epoxides 1.
Initially, the equivalents of Me3SiCl and sBuLi–sparteine complex were increased from the original 1.3 eq. to 3 or 5 eq. This led to a considerable increase in the formation of the epoxydisilane 10a, however epoxysilane 9a and traces of the starting terminal epoxide 1a were still observed by crude 1H NMR analysis (1a : 9a : 10a returned in ratios of 0.15 : 0.4 : 1 and 0.05 : 0.3 : 1 respectively). On the timescale of the reaction, and in the presence of excess reagents, competitive silylation of the sBuLi could contribute to incomplete disilylation. It was found that it is important to convert all of the epoxysilane 9 into the epoxydisilane 10, because of their similar mobility characteristics on column chromatography. Bearing this in mind, the reaction conditions were investigated in more detail.
To avoid observing epoxysilane 9 in the final product profile, it appeared advantageous to obtain complete conversion of the terminal epoxide 1 to the epoxysilane 9, before the addition of further reagents. To hopefully achieve this, the original reaction conditions (1.3 eq. of reagents at −90 °C, followed by warming to −50 °C over 1 h) were modified by then re-cooling back to −90 °C and adding a further 2 eq. of Me3SiCl and sBuLi and leaving for 15 min at −90 °C, then warming to room temperature over 18 h before work-up. Under this protocol, it was found that, as with the monosilylation procedure, hexane proved a better solvent than Et2O (1a : 9a : 10a returned in ratios of 0.3 : 1.3 : 1 and 2 : 3 : 1 respectively). To further improve this, instead of just sBuLi being added after warming to −50 °C and back to −90 °C, sBuLi–sparteine complex was added. This resulted in an encouraging improvement in the proportion of epoxydisilane 10a compared to the epoxysilane 9a (1a : 9a : 10a returned in ratio of 0 : 0.2 : 1 respectively). Indeed, warming to temperatures above −50 °C as part of a warm–cool cycle led to no epoxysilane 9a at all in the crude product; however, more noticeable alkenyl signals in the 1H NMR spectra of the crude products suggested that sBuLi-induced reductive alkylation of epoxides 1a and 9a had become a significant side-reaction.17 A similar trend to the monosilylation chemistry was shown in the effect of ligand with sBuLi–sparteine complex giving superior results to sBuLi–TMEDA complex (1a : 9a : 10a returned in ratio of 0 : 0.2 : 1 and 0.4 : 0.5 : 1 respectively).
Further insight into the reaction pathway occurring during the above warm–cool protocol was achieved by quenching with CD3OD at different stages, followed by GC-MS analysis to determine product ratios (after appropriate calibration graphs had been constructed) and D incorporations. The observation of D incorporation in 1 and/or 9 was expected to indicate the proportion of any oxiranyl anions 2 and 11 (Scheme 4) present at the time the reaction was quenched.
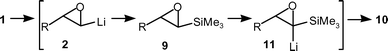 | ||
| Scheme 4 In-depth studies of the bis-silylation reaction. | ||
After only 15 min following the addition of Me3SiCl and sBuLi–sparteine complex (all 1.3 eq.) to the epoxide 1a (R = C10H21) at −90 °C a ratio of 1a : 9a of 0.5 : 1 with 13% D incorporation in 1a was obtained. It was established that complete reaction of the terminal epoxide 1a and 2a was achieved if the reaction was warmed to −50 °C over 5 min and then left at −50 °C for 1 h. As expected at that point the major compound present was epoxysilane 9a (traces of alkene signals were also observed by 1H NMR spectroscopy of the crude product). GC-MS analysis indicated a small amount of epoxydisilane 10a was present (9a : 10a ratio of 10 : 1), and a significant proportion of epoxysilane 9a was lithiated (29% D incorporation in 9a). The latter observations are likely to be due to the slight initial excess of sBuLi–sparteine complex employed. Following subsequent re-cooling to −90 °C and addition of further Me3SiCl and sBuLi–sparteine complex (all 2 eq.) and stirring for 15 min, there was a modest change to the product profile (9a : 10a ratio of 5 : 1, 22% of 9a deuterated). Thus, from epoxysilane 9a the second deprotonation and silylation do not occur as readily as from the terminal epoxide 1a since, although the oxiranyl anion 11a is stabilised by the α-silyl group, the proton that has to be removed to generate 11a occupies a relatively more hindered site.12 Compound 11a is also more hindered than 2a to subsequent electrophilic trapping. As the reaction warmed from −90 °C to 0 °C over 7 h, the proportion of epoxysilane 9a gradually fell relative to epoxydisilane 10a [after 2 h (−60 °C) 9a : 10a ratio of 2.5 : 1, with 46% of 9a deuterated; after 4 h (−30 °C) 9a : 10a ratio of 2 : 1, with 62% of 9a deuterated; after 7 h (0 °C) 9a : 10a ratio of 0.1 : 1, with 3% of 9a deuterated]. Therefore, the majority of the second silylation occurred between −30 °C and 0 °C. The stability observed for 11a at −30 °C under the current reaction conditions is noteworthy, given the generally accepted thermal lability of oxiranyl anions.2b However, Pulido and co-workers have shown that ethylene oxide bearing a tert-butyldiphenylsilyl group could be lithiated, with BuLi–TMEDA complex in THF followed by efficient trapping (93%) with MeI at −50 °C.18 At 0 °C, a lower yield of methylated epoxide (31%) together with alkene (48%) from reductive alkylation was obtained.
Application of the optimised disilylation conditions (warm to −50 °C over 5 min, maintain for 1 h then re-cool protocol) to a range of terminal epoxides gave the corresponding epoxydisilanes in good yields (Table 3). It was demonstrated that sBuLi–5 could be used to bis-silylate either enantiomer of epoxide 1a (entries 6 and 7).
| Entry | 1 | 10 | Yielda (%) | ||
|---|---|---|---|---|---|
| a Yield following chromatography (mean value of two runs for entries 1–5). | |||||
| 1 |

|
1a |

|
10a | 61 |
| 2 |

|
1b |

|
10b | 58 |
| 3 |

|
1c |

|
10c | 53 |
| 4 |

|
1d |

|
10d | 71 |
| 5 |

|
1e |

|
10e | 50 |
| 6 |

|
(S)-1a |

|
(S)-10a | 55 |
| 7 |

|
(R)-1a |

|
(R)-10a | 47 |
Interestingly, subjection of two 2,2-disubstituted-1-epoxides (1f and 1g, Table 2) to the reaction conditions led only to the corresponding monosilylated epoxysilanes 9 and it proved impossible to push these reactions past the monosilylation stage.
The introduction of two different electrophiles to a terminal epoxide in a one-flask operation was also possible, albeit in modest yield (37%) by initial silylation followed by addition of benzaldehyde at −30 °C (Scheme 5).
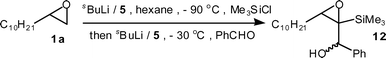 | ||
| Scheme 5 One-flask introduction of two different electrophiles. | ||
Having established methods for mono-silylation (Me3SiCl present during generation of the oxiranyl anion) and deuteration (CD3OD as external electrophile), attention returned to the reaction of oxiranyl anions with other (external) electrophiles, including C–C bond formation. In the original screen of diamine ligands (Table 1) the use of 3,7-dibutyl-3,7-diazabicyclo[3.3.1]nonane 6 (dibutylbispidine, DBB)–sBuLi9a,19 (2.5 eq. each) in the lithiation–deuteration of 1a in hexane at −90 °C for 15 min had afforded 82% recovered epoxide 1a with only 45% D incorporation (entry 5).
Compared to sBuLi–(−)-sparteine, sBuLi–DBB 6 was found to deprotonate 1 at a considerably slower rate, and as other bispidine-type ligands were more efficient, 6 was not studied in more detail at the time. However, further extensive investigations with 6 have revealed that 6 is the preferred ligand for lithiation–electrophile trapping of terminal epoxides.
Initially it was found that employing sBuLi–6 over a prolonged reaction time afforded surprising stabilisation of the otherwise labile oxiranyllithium 2a.2 Indeed, using sBuLi–6 (2.5 eq. each) in hexane at −90 °C for 16 h afforded epoxide 1a in good isolated yield (66%) with a high level of D incorporation (90%, as judged by 1H NMR), alongside alkene 3a and alcohol 4a in relatively small amounts (16% and 10%, respectively). This result was reproducible; under the same conditions a repeat experiment gave an almost identical result (65% yield of 1a with 90% D, alongside 12% of alkene 3a and 10% alcohol 4a). Subsequent studies showed that a good level of D incorporation could be achieved using sBuLi–6 (2.5 eq. each) in hexane at −90 °C after 4 h, accompanied by fairly small levels of alkene 3a and alcohol 4a by-products. Although the use of Et2O as solvent allowed an enhanced rate of epoxide deprotonation, this was at the expense of slightly increased levels of by-products 3a and 4a, while other solvents (cumene, tert-butylmethyl ether and THF) and different amounts (eq.) of sBuLi–6 gave inferior results.
Pleasingly, it was found that following lithiation of epoxide 1a using sBuLi–6 (2.5 eq. each) in hexane at −90 °C for 4 h, addition of Bu3SnCl (2.5 eq., added neat) afforded trans-α,β-epoxystannane 13a in good yield (73% after column chromatography on Florisil®). Since α,β-epoxystannanes are useful for indirect oxiranyl anion generation via transmetalation,4a and may also allow subsequent introduction of other electrophiles using cross-coupling techniques,20 an assessment of the stannylation scope was instigated using a range of epoxides.21 These studies revealed that conducting the reactions at a reduced reactant concentration (0.025 M for 1 rather than 0.050 M) together with using a shorter lithiation period (1–1.5 h) led to reduced by-product formation and returned the epoxystannane products 13 in good yield (Scheme 6, Table 4).
| Entrya | 1 | 13 | Yieldb (%) | ||
|---|---|---|---|---|---|
| a Solution of epoxide in hexane was added to sBuLi–6 (2.5 eq. each) in hexane at −90 °C, and then stirred at −90 °C for 1 h (1.5 h, entry 1; 4 h, entry 4) before addition of Bu3SnCl. b Yield following chromatography on Florisil® (neutral alumina, entry 4). c Epoxide added neat to sBuLi–6 (2.5 eq. each) at −100 °C, then reaction continued at −90 °C. | |||||
| 1 |

|
1a |

|
13a | 78 |
| 2 |

|
1b |

|
13b | 67 |
| 3 |

|
1c |

|
13c | 72 |
| 4 |

|
1f |

|
13f | 64 |
| 5 |

|
1h |

|
13h | 83 |
| 6 |

|
1i |

|
13i | 78 |
| 7 |

|
1j |

|
13j | 70 |
| 8 |

|
1k |

|
13k | 72 |
| 9 |

|
1l |

|
13l | 90 |
| 10 |

|
1m |

|
13m | 87 |
| 11 |

|
1n |

|
13n | 73 |
| 12 |

|
(S)-1a |

|
(1S,2S)-13a | 65 |
| 13c |

|
(S)-1o |

|
(1S,2S)-13o | 54 |
 | ||
| Scheme 6 Direct stannylation of terminal epoxides 1. | ||
Isopropyl and tert-butyl oxiranes gave the best yields of epoxystannanes (entries 9 and 10), but epoxides bearing side-chain functionality were also tolerated (entries 2, 3, 5, 6 and 8). Lithiation of a 2,2-disubstituted epoxide 1f (entry 4) was significantly slower than the monosubstituted epoxides, requiring a 4 h lithiation period. Commercially available (S)-propylene oxide was successfully stannylated (54%, entry 13), as was (S)-1,2-epoxydodecane (65%, entry 12).
An investigation of lithiation–electrophile trapping to achieve C–C bond formation was then undertaken. Lithiation of 1,2-epoxydodecane 1a for 1.5 h, followed by addition of benzaldehyde gave a separable mixture of diastereoisomeric epoxy alcohols22 (75% combined yield, dr 1 : 1, Table 5, entry 1). The lack of diastereoselectivity is consistent with that observed for other oxiranyl anion reactions.12 The methodology was extended to other aromatic aldehydes as the electrophile (Table 5, entries 2–-4). Non-enolisable aldehydes (cinnamaldehyde and pivaldehyde) were also trapped in good yields (73 and 70% respectively, entries 5 and 6). Potentially enolisable carbonyl electrophiles (isobutyraldehyde and 3-pentanone) were also incorporated, albeit in slightly lower yields (56 and 63% respectively, entries 7 and 8). Acylation using N,N-dimethylbenzamide also proceeded (50%, entry 9).23
| Entrya | Electrophile | Product b | Yieldc (%) | |
|---|---|---|---|---|
| a Solution of epoxydodecane 1a in hexane was added to sBuLi–6 (2.5 eq. each) in hexane at −90 °C, and then stirred at −90 °C for 1.5 h before addition of the electrophile. b Secondary alcohols obtained with dr 1 : 1. c Yield following chromatography (combined yields given for diastereomeric epoxy alcohols which, with the exception of entry 4, were largely chromatographically separable). | ||||
| 1 | PhCHO |

|
14a | 75 |
| 2 | 3-ClC6H4CHO |

|
14b | 72 |
| 3 | 4-MeOC6H4CHO |

|
14c | 57 |
| 4 | Furfural |

|
14d | 67 |
| 5 | Cinnamaldehyde |

|
14e | 73 |
| 6 | t BuCHO |

|
14f | 70 |
| 7 | i PrCHO |

|
14g | 56 |
| 8 | Et2CO |

|
14h | 63 |
| 9 | PhCONMe2 |

|
14i | 50 |
In conclusion, we have developed a methodology for the deprotonation–electrophile trapping of unfunctionalised terminal epoxides 1. This methodology provides a direct and stereocontrolled route to deuterated epoxides, epoxysilanes 9 and epoxystannanes 13, in addition to epoxydisilanes 10. The methodology has also been applied to C–C bond formation using carbonyl-based electrophiles. The wide availability of terminal epoxide starting materials, and the robustness of methods for their resolution to highly enantiomerically enriched form,11e renders the methodology described herein as particularly useful. Further research concerning direct epoxide functionalisation and the synthetic utility of the substituted epoxide products is currently underway in our laboratories.
Experimental
General
All reactions requiring anhydrous conditions were conducted in flame- or oven-dried apparatus under an atmosphere of argon. Ethers were distilled from sodium benzophenone ketyl under argon; hexane from CaH2 under argon. External reaction temperatures are reported unless stated otherwise. Reactions were monitored by TLC using commercially available (Merck) aluminium-backed plates, pre-coated with a 0.25 mm layer of silica containing a fluorescent indicator. Following aqueous work-up, organic layers were dried over MgSO4 unless stated otherwise. Light petroleum refers to the fraction with bp 40–60 °C. Specific rotation [α]D values were recorded at 25 °C and are given in 10−1 deg cm2 g−1. IR spectra were recorded as thin films unless stated otherwise. NMR spectra were recorded in CDCl3 unless stated otherwise with Bruker DPX 250, JEOL EX400 or Bruker AMX500 spectrometers. 1H NMR chemical shifts are reported relative to CHCl3 (δH 7.27), 13C NMR chemical shifts are reported relative to CDCl3 (δC [central line of three] 77.0) and 119Sn chemical shifts are reported relative to Bu4Sn as external reference. Coupling constants (J) are given in Hz.Silylation of terminal epoxides employing ligand 8
Silylation of terminal epoxides employing ligand 5
bis-Silylation of terminal epoxides
Stannylation of terminal epoxides: general procedure
To a solution of DBB 6 (162 mg, 0.68 mmol) in hexane (8.5 mL) at −90 °C was added dropwise sBuLi (1.40 M in cyclohexane, 0.48 mL, 0.67 mmol) and the mixture was allowed to warm to 0 °C over 5 min. On re-cooling to −90 °C, a solution of the appropriate epoxide 1 (0.27 mmol) in hexane (1 mL + 1 mL wash) was added dropwise over a period of 15 min and the mixture was stirred at −90 °C for 1 h. Tributyltin chloride (0.68 mmol) was then added neat in one portion and the mixture was manually agitated at −90 °C for 10 min before warming to 0 °C over 14 h. After quenching with aqueous 0.5 M H3PO4 (2.5 mL), the aqueous layer was extracted with Et2O (3 × 10 mL) and the combined organic phases were washed with H2O (5 mL), brine (5 mL), dried and concentrated in vacuo. Purification of the residue by flash chromatography (Florisil® unless otherwise stated, eluent, petrol–Et2O) gave the α,β-epoxystannane 13.Lithiation–electrophile trapping with carbonyl-based electrophiles:general procedure
To a solution of DBB 6 (162 mg, 0.68 mmol) in hexane (8.5 mL) at −90 °C was added dropwise sBuLi (1.40 M in cyclohexane, 0.48 mL, 0.67 mmol) and the mixture was allowed to warm to 0 °C over 5 min. On re-cooling to −90 °C, a solution of 1,2-epoxydodecane 1a (50 mg, 0.27 mmol) in hexane (1 mL + 1 mL wash) was added dropwise over a period of 15 min and the mixture was stirred at −90 °C for 1.5 h. The electrophile (0.68 mmol) was then added neat in one portion [or dropwise in Et2O (1 mL) for solids] and the mixture was manually agitated at −90 °C for 10 min before warming slowly to 0 °C (over 14 h). After quenching with aqueous 0.5 M H3PO4 (2.5 mL), the aqueous layer was extracted with Et2O (3 × 10 mL) and the combined organic phases were washed with H2O (5 mL), brine (5 mL), dried and concentrated in vacuo. Purification of the residue by column chromatography (SiO2 unless otherwise stated) gave the substituted epoxide 14.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1598, 1580, 1469, 1450, 1232, 1077, 877; δH (400 MHz) 8.02 (2H, d, J 7.5), 7.63 (1H, d, J 7.5), 7.51 (2H, d, J 7.5), 4.03 (1H, d, J 2.0), 3.15 (1H, td, J 5.5, 2.0), 1.84–1.66 (2H, m), 1.59–1.20 (16H, m), 0.89 (3H, t, J 7.0); δC (100 MHz) 194.7, 135.6, 133.8, 128.8, 128.3, 60.1, 57.4, 32.0, 31.9, 29.5, 29.4, 29.3, 25.8, 22.7, 14.1; MS (CI+, NH3) 289 ([M + H]+, 50%), 273 (100%), 159 (8%), 133 (5%), 120 (12%), 105 (58%); HRMS (CI+, NH3 for [M + H]+) found 289.2168, C19H29O2 requires 289.2168.
O), 1598, 1580, 1469, 1450, 1232, 1077, 877; δH (400 MHz) 8.02 (2H, d, J 7.5), 7.63 (1H, d, J 7.5), 7.51 (2H, d, J 7.5), 4.03 (1H, d, J 2.0), 3.15 (1H, td, J 5.5, 2.0), 1.84–1.66 (2H, m), 1.59–1.20 (16H, m), 0.89 (3H, t, J 7.0); δC (100 MHz) 194.7, 135.6, 133.8, 128.8, 128.3, 60.1, 57.4, 32.0, 31.9, 29.5, 29.4, 29.3, 25.8, 22.7, 14.1; MS (CI+, NH3) 289 ([M + H]+, 50%), 273 (100%), 159 (8%), 133 (5%), 120 (12%), 105 (58%); HRMS (CI+, NH3 for [M + H]+) found 289.2168, C19H29O2 requires 289.2168.
Acknowledgements
We thank the EPSRC for Research Grants (GR/N26302 and GR/S46789/01), the European Union for a Marie Curie Fellowship (HPMF-CT-1999-00264), the EPSRC and GlaxoSmithKline for a CASE award (to N.J.R.), and the EPSRC National Mass Spectrometry Service Centre for mass spectra. We also thank Dr P. O'Brien (York) for initial test samples of ligands 7 and 8.References
- I. Erden, in Comprehensive Heterocyclic Chemistry II, vol. 1A, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, 1996, pp. 97 Search PubMed.
- (a) T. Satoh, Chem. Rev., 1996, 96, 3303 CrossRef CAS; (b) D. M. Hodgson and E. Gras, Synthesis, 2002, 1625 CrossRef CAS; (c) D. M. Hodgson, K. Tomooka and E. Gras, in Topics in Organometallic Chemistry: Organolithiums in Enantioselective Synthesis, vol. 5, ed. D. M. Hodgson, Springer-Verlag, Heidelberg, 2003, pp. 217–250 Search PubMed; (d) F. Chemla and E. Vrancken, in The Chemistry of Organolithium Reagents, ed. Z. Rappoport and I. Marek, Wiley & Sons, New York, 2004, pp. 1165–1242 Search PubMed.
- J. J. Eisch and J. E. Galle, J. Am. Chem. Soc., 1976, 98, 4646 CrossRef CAS.
- (a) P. Lohse, H. Loner, P. Acklin, F. Sternfeld and A. Pfaltz, Tetrahedron Lett., 1991, 32, 615 CrossRef CAS; (b) T. Satoh, S. Kobayashi, S. Nakanishi, K. Horiguchi and S. Irisa, Tetrahedron, 1999, 55, 2515 CrossRef CAS.
- D. M. Hodgson, G. P. Lee, R. E. Marriott, A. J. Thompson, R. Wisedale and J. Witherington, J. Chem. Soc., Perkin Trans. 1, 1998, 2151 RSC.
- K. M. Morgan and S. Gronert, J. Org. Chem., 2000, 65, 1461 CrossRef CAS.
- Preliminary communications: (a) D. M. Hodgson and S. L. M. Norsikian, Org. Lett., 2001, 3, 461 CrossRef CAS; (b) D. M. Hodgson and E. H. M. Kirton, Synlett, 2004, 1610 CrossRef CAS; (c) D. M. Hodgson, N. J. Reynolds and S. J. Coote, Org. Lett., 2004, 6, 4187 CrossRef CAS.
- E. Doris, L. Dechoux and C. Mioskowski, Synlett, 1998, 337 CrossRef CAS.
- (a) K. M. Bertini Gross, Y. M. Jun and P. Beak, J. Org. Chem., 1997, 62, 7679 CrossRef; (b) J. R. Harrison, P. O'Brien, D. W. Porter and N. M. Smith, J. Chem. Soc., Perkin Trans. 1, 1999, 3623 RSC; (c) P. Scheiber and P. Nemes, Liebigs Ann. Chem., 1994, 1033 CrossRef CAS.
- (a) P. F. Hudrlik and A. M. Hudrlik, in Advances in Silicon Chemistry, vol 2, ed. G. L. Larson, JAI Press Inc., Greenwich, 1993, pp. 1 Search PubMed; (b) G. H. Whitham, in Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, vol. 4, ed. I. Fleming, Thieme, Stuttgart, 2001, pp. 633–646 Search PubMed.
- Epoxides 1 in Table 2 were commercially available or prepared according to (a) J. A. Elings, R. S. Downing and R. A. Sheldon, Eur. J. Org. Chem., 1999, 837 CrossRef CAS (entry 3); (b) L. H. Yang, A. E. Weber, W. J. Greenlee and A. A. Patchett, Tetrahedron Lett., 1993, 34, 7035 CrossRef CAS (entry 4); (c) I. Rothberg, L. Schneider, S. Kirsch and R. Ofee, J. Org. Chem., 1982, 47, 2675 CrossRef CAS (entry 5); (d) T. J. Michnick and D. S. Matteson, SYNLETT, 1991, 631 CrossRef CAS (entries 6–7); (e) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS (entries 9–10).
- G. A. Molander and K. Mautner, J. Org. Chem., 1989, 54, 4042 CrossRef CAS.
- (a) C. Burford, F. Cooke, G. Roy and P. Magnus, Tetrahedron, 1983, 39, 867 CrossRef (Table 2, entry 6); (b) J. D. Warren and Y. Shi, J. Org. Chem., 1999, 64, 7675 CrossRef CAS (Table 2, entry 7).
- (a) J. J. Eisch and J. E. Galle, J. Organomet. Chem., 1988, 341, 293 CrossRef CAS; (b) C. Courillon, J. C. Marie and M. Malacria, Tetrahedron, 2003, 59, 9759 CrossRef CAS.
- D. M. Hodgson, P. J. Comina and M. G. B. Drew, J. Chem. Soc., Perkin Trans. 1, 1997, 2279 RSC.
- (a) P. C. B. Page and M. J. McKenzie, in Science of Synthesis: Houben–Weyl Methods of Molecular Transformations, vol. 4, ed. I. Fleming, Thieme, Stuttgart, 2001, pp. 513 Search PubMed; (b) A. E. Mattson, A. R. Bharadwaj and K. A. Scheidt, J. Am. Chem. Soc., 2004, 126, 2314 CrossRef CAS; (c) X. Linghu, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070 CrossRef.
- (a) Y. Ukaji, A. Yoshida and T. Fujisawa, Chem. Lett., 1990, 157 CAS; (b) C. Agami, L. Dechoux and C. Mioskowski, Tetrahedron Lett., 1997, 38, 4071 CrossRef CAS.
- A. Barbero, P. Cuadrado, I. Fleming, A. M. González, F. J. Pulido and A. Sánchez, J. Chem. Soc., Perkin Trans. 1, 1995, 1525 RSC.
- D. M. Hodgson, T. J. Buxton, I. D. Cameron, E. Gras and E. H. M. Kirton, Org. Biomol. Chem., 2003, 1, 4293 RSC.
- J. R. Falck, R. K. Bhatt and K. M. Reddy, Synlett, 1997, 481 CAS.
- Epoxides 1 in Table 4 were either commercially available, prepared as described previously (ref. 11, above) or prepared according to: (a) 1,2-epoxy-5-(5,5-dimethyl-[1,3]dioxanyl)hexane was prepared in two steps from 5-hexen-2-one (entry 8); (b) cyclohexyl oxirane was prepared according to ref. 11d, above.
- Tentative stereochemical assignments for epoxy alcohol diastereoisomers 14 were made by comparison of NMR spectral data with those of structurally similar compounds in the following reports: (a) ref. 12 above; (b) T. Kawakami, I. Shibata, A. Baba and H. Matsuda, J. Org. Chem., 1993, 58, 7608 CrossRef CAS; (c) A. E. Lurain, P. J. Carroll and P. J. Walsh, J. Org. Chem., 2005, 70, 448 CrossRef.
- Trapping of other electrophiles (allyl bromide, MeI, EtOTf, PhSO2F) has so far been unsuccessful.
- J. A. Soderquist and C. Lopez, Tetrahedron Lett., 1991, 32, 6305 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |