Arginine magic with new counterions up the sleeve
Masamichi Nishiharaa, Florent Perreta, Toshihide Takeuchib, Shiroh Futakibc, Adina N. Lazard, Anthony W. Colemand, Naomi Sakai*a and Stefan Matile*a
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland
bInstitute for Chemical Research, Kyoto University, Japan
cPRESTO, JST, Kyoto, Japan
dInstitut de Biologie et Chimie des Protéines, CNRS UMR 5086, 7 passage du Vercors, F69367, Lyon, France. E-mail: naomi.sakai@chiorg.unige.ch; stefan.matile@chiorg.unige.ch; Fax: +41(0)22 379 3215; Tel: +41(0)22 379 6523
First published on 7th April 2005
Abstract
The elusive questions how arginine-rich sequences allow peptides and proteins to penetrate cells or to form voltage-gated ion channels are controversial topics of current scientific concern. The possible contributions of exchangeable counterions to these puzzling processes remain underexplored. The objective of this report is to clarify scope and limitations of certain counteranions to modulate cellular uptake and anion carrier activity of oligo/polyarginines. The key finding is that the efficiency of counteranion activators depends significantly on many parameters such as activator–membrane and activator–carrier interactions. This finding is important because it suggests that counteranions can be used to modulate not only efficiency but also selectivity. Specifically, activator efficiencies are found to increase with increasing aromatic surface of the activator, decreasing size of the transported anion, increasing carrier concentration as well as increasing membrane fluidity. Efficiency sequences depend on membrane composition with coronene > pyrene ≫ fullerene > calix[4]arene carboxylates in fluid and crystalline DPPC contrasting to fullerene > calix[4]arene ≈ coronene > pyrene carboxylates in EYPC with or without cholesterol or ergosterol. In HeLa cells, the efficiency of planar activators (pyrene) exceeds that of spherical activators (fullerenes, calixarenes). Polyarginine complexes with pyrene and coronene activators exhibit exceptional excimer emission. Decreasing excimer emission with increasing ionic strength reveals dominant hydrophobic interactions with the most efficient carboxylate activators. Dominance of ion pairing with the inefficient high-affinity sulfate activators is corroborated by the reversed dependence on ionic strength. These findings on activator–carrier and activator–membrane interactions are discussed as supportive of arene-templated guanidinium–carboxylate pairing and interface-directed translocation as possible origins of the superb performance of higher arene carboxylates as activators.
1 Introduction
Arginine-rich peptides and proteins seem to exist always in “functional symbiosis” with tightly bound but exchangeable counteranions. This “counteranion scavenging” could be the trick of oligoarginines to minimize intramolecular charge repulsion (Fig. 1A), because the weak acidity of the guanidinium cation hinders partial deprotonation under physiological conditions.1 More acidic oligoamines such as K-rich peptides do not show similar affinity to anions (Fig. 1B). Recently, we suggested that counteranion scavenging may explain some of the “mysterious” functions of arginine-rich peptides and proteins in biomembranes.1 The movement of R-rich cell-penetrating peptides (CPPs) like oligoarginines across bilayer membranes can, for example, be understood as repeated alterations in their solubility by multiple counteranion exchange to adapt to changing environments.1,2 In support of this hypothesis, it was not surprising to find that a) CPPs are anion carriers and b) that bilayer penetration by CPPs can be regulated with amphiphilic anions (Fig. 2).1,2 Counteranions (i.e., alignable dipoles of hydrophobic ion pairs) might contribute to the mechanism of voltage gating by R-rich motifs for the same reasons.1,3–8 | ||
| Fig. 1 To minimize repulsion between proximal charges, (A) guanidinium-rich oligomers scavenge anions X− to give complexes Rn·(X)m (Fig. 2), whereas (B) ammonium-rich oligomers release protons. | ||
 | ||
| Fig. 2 Counteranion activators Y (red) of oligo/polyarginine carriers/CPPs Rn (black) are thought to act by replacing scavenged hydrophilic anions X (blue) in complexes Rn·(X)m to produce complexes Rn·(X)m′·(Y)m″ (B) that act as carriers to release entrapped anions like CF (orange→green, B→G) or are directed by internal or external hydrophilic polyanions Xm (cyan) to accumulate inside (u, uptake) or outside a spherical bilayer membrane (i, inhibition).1,2 | ||
In our earlier studies, activator anions like EYPG are used as a component of the model membranes.1,2 Very recently, however, we found that externally added amphiphilic anions Y can activate oligo/polyarginine-anion complexes in live cells as well as otherwise inaccessible neutral EYPC bilayers (Fig. 3).9 Confirmed key characteristics of activators Y include one or more negative charge for ion pairing with guanidinium cations of the oligo/polyarginine.9 Moreover, all anion activators Y known so far are amphiphiles with large hydrophobic domains for favorable interactions with the bilayer membrane. Activators Y with hydrophobic domains composed of higher aromatics appear particularly promising because additional, more specific activator–carrier, activator–activator and activator–membrane interactions become possible (see below).
 | ||
| Fig. 3 Structure of potential anion activators Y (Fig. 2; anions 14, 15, 21 and 22 are inactive under the employed experimental conditions). | ||
Fig. 2 summarizes how activating and inactivating counteranions may contribute to the function of CPPs. In brief, R-rich CPPs Rn will always exist as complexes Rn·(X)m to minimize intramolecular charge repulsion (Fig. 1A). As an illustrative example for the need for counterions, we recently found that oligoarginines can scavenge inorganic phosphates that are present in trace amounts in the environment.10 Exchange of scavenged hydrophilic counteranions X such as inorganic phosphates in complexes Rn·(X)m with activators Y is expected to yield active complexes Rn·(X)m′·(Y)m″ at the membrane–water interface (Fig. 2B). The resulting supramolecular carriers Rn·(X)m′·(Y)m″ may then shuttle across the bilayer, pick up entrapped anions like CF, back-transfer and release unquenched CF (Fig. 2). Therefore, an efficient activator Y should have the ability to form stable but labile guanidinium–anion interactions. Hydrophilic high-affinity polyanions Xm like glycosaminoglycans (GAGs),1,2 oligonucleotides (RNA, DNA), micelles (SDS)1,2 and presumably also nucleotides like ATP1 may play the interesting role to regulate carrier activity and cell penetration. Namely, external Xm should inhibit both processes, whereas internal Xm should inhibit back transfer and thus facilitate the translocation. The possibly more complex role of interfacial polyelectrolytes, particularly GAGs, is a topic of current scientific concern.11–24 We reiterate that the synergistic carrier mechanism for CPP-activator complexes Rn·(X)m′·(Y)m″ outlined in Fig. 2 is assumed in this report based on results reported in references 1 and 2 that disfavor formation of ion channels and pores as well as the occurrence of more drastic events. Nevertheless, this assumption as well as its relevance for oligo/polyarginine uptake by live cells remains to be verified. Extensive experimental evidence from many groups in support of a carrier but also of other, competing or coinciding uptake mechanisms, particularly endocytosis, exists.7,8,11–24
The here envisioned role of CPP activators is reminiscent of the role of charge-reversal amphiphiles in gene delivery.25–27 The rare occurrence of intrinsic cationic activators in the biomembranes practically excludes spontaneous oligonucleotide translocation, a fact that naturally directs scientific attention to artificial countercation activators. In contrast, ubiquitous anionic activators in biomembranes may have shifted the focus of CPP research from counteranion activators to a mysteriously “spontaneous” uptake1,2,7,8,11–24 that is sometimes referred to as “arginine magic”.
This study focuses on nature and selectivity of the interactions between anion activators, polyarginine (pR) carriers and lipid bilayer membranes. The key question to address was why higher arenes and carboxylates emerge as privileged motifs in efficient activators. With regard to activator–membrane interactions, it is well known that many membrane-spanning proteins exhibit external surfaces with aromatic rings positioned to interact with the interfacial region of bilayer membranes.28–37 This interface contains the lipidic ester groups as well as their phosphates paired with counterions such as the ammonium cation in the case of phosphatidylcholine. Examples for membrane peptides and proteins with aromatic rings at the interface include β-helices with gramicidin A,36 α-helix bundles with potassium channels30 and β-barrels with maltoporin.28 Model studies confirmed preferential location of indoles at the interface.31–35 Hydrogen bonding to interfacial esters and cation–π interactions with ammonium cations (but not yet arene-templated ion pairing)38 have been considered beyond simple preference for intermediate dielectrics to explain interfacial accumulation of arenes.28–35
Model studies confirmed that appropriately positioned aromatics can stabilize transmembrane orientation of model helices.32–34 A similar anchoring function has been proposed for aromatics in synthetic ion channels and pores.36,37 Important for this study, it has been suggested that the interfacial preference of arenes may drive the translocation across bilayer membranes to interfaces at the other side.29 For carriers, preferential accumulation at the interface is essential for function because this is the place where the ions are exchanged. Indeed, we have recently found that interfacial preference is one of the distinguishing characteristics of octaarginine, an excellent CPP, compared to hexadecaarginine, a poor CPP that accumulates in the hydrophobic core of EYPC/EYPG membranes.2 Therefore, it was conceivable that fullerene, coronene and calixarene activators may preferentially accumulate at the interfacial region and, therefore, mediate interface-directed translocation of polyarginine carriers.
With regard to activator–carrier interactions, Thompson and Smithrud pointed out recently that arene-templated ion pairing occurs in nature and created an elegant synthetic model to secure experimental evidence for relevance of this recognition motif in water and in octanol.38 Important in the context of this study, the Thompson–Smithrud model identified guanidinium–carboxylate pairs39–45 as privileged motif; alkyl-templated ion pairing was not observed.38 Arene-templated ion pairing was explored in the context of α-helix recognition as well.45 We have proposed the same effect early on as a possible origin of activator–carrier interactions.1 With respect to the well-recognized cation–π interaction,46,47 arene-templated ion pairing may be considered as an extension that covers the counteranion and the resulting network of possible interactions between anion, cation and arene as well. Such an influence of a remote48 aromatic surface to the binding could be the key to achieve stable complexes pR·(X)m′·(Y)m″ with kinetic lability.
Taken together, these considerations suggested that the most efficient activators may bind to CPPs by arene-templated carboxylate–guanidinium pairing to subsequently mediate the interface-directing translocation of the resulting activator–carrier complexes. The studies on activator–membrane and activator–carrier interactions described in the following are in support of this hypothesis, although the involvement of the other mechanisms is likely.
2 Results and discussion
2.1 The efficiency sequence in EYPC membranes
Anions 1–22 were considered as activators Y of polyarginine (pR) to give functional activator–carrier complexes pR·(X)m′·(Y)m″ (Fig. 3). Fullerene (2) and coronene (4) sulfates were prepared in a few routine steps from commercial starting materials (see Experimental section). Fullerene (1) and coronene (3) carboxylates as well as calixarene (8–15) and naphthalene 19 activators were synthesized following reported procedures.9 Anions 5–7, 16–18, and 20–22 were commercially available.The classical CF assay, in which the release of self-quenched CF from EYPC-LUVs⊃CF is monitored continuously as an increase of fluorescence, was used to assess the ability of amphiphilic anions 1–22 to activate polyarginine carriers pR in otherwise inaccessible, neutral EYPC bilayers.9 Conventionally, the CF assay is used to identify large pores and membrane-disruptive detergents.36 However, recent mechanistic studies suggest that oligo-/polyarginines act as anion carriers under the selected conditions,1,2 although contributions from transient pores and other mechanisms cannot be excluded.36 In any case, elucidation of the transport mechanism of activator–polyarginine complexes pR·(X)m′·(Y)m″ is not the topic of this study.
Amphiphilic anions 1–22 stimulated pR-mediated CF release from EYPC-LUVs⊃CF in varying extent. Pyreneacetate49,505, on the one hand, formed complexes pR·(X)m′·(5)m″ that mediated up to 80% CF release at relatively high concentrations (Fig. 4C and 4D, ■). Complexes pR·(X)m′·(1)m″ with the Bingel fullerene27,51–561 as an activator, on the other hand, mediated up to 50% CF release at exceptionally low concentrations (Fig. 4B and 4D, ●). Laurate complexes pR·(X)m′·(16)m″,8 finally, mediated up to 10% CF release only at relatively high concentrations (Fig. 4A and 4D, □). These diverse, in part complementary characteristics of anion activators 1–22 were classified using two parameters: The maximal activity YMAX and the EC50 (Table 1). The effective activator concentration EC50 is defined as activator concentration needed to reach YMAX/2 (Table 1). As a general trend, EC50's decreased with decreasing YMAX. This was regrettable because the perfect activator should achieve high YMAX at low EC50. To illustrate this dilemma, activator efficiency E was defined as in eqn. (1) based on the roughly exponential YMAX–EC50 profile9 of the pyrene series 5–9. Namely,
| E = YMAX × pEC50/f | (1) |
| Entry | Anionb | YMAX (%)c,d | EC50/µMc,e | Ef |
|---|---|---|---|---|
| a From CF efflux from LUVs⊃CF (250 µM lipid), 250 nM pR, 10 mM NamHnPO4, 100 mM KCl, pH 6.5, 25 °C (if not otherwise indicated); part of the data in Table 1 has been communicated previously in ref. 9.b See Fig. 1 for structures.c From Hill analysis of dose response curves (e.g., Fig. 4D), see experimental part.d Maximal activity at saturation with activator.e Effective concentration of anions 1–22 to mediate 50% of maximal activity (YMAX/2).f Activator efficiency, eqn. (1).g No activity observed. | ||||
| 01 | 1 | 48 ± 1 | 0.051 ± 0.002 | 10.0 |
| 02 | 2 | 30 ± 2 | 0.11 ± 0.006 | 5.8 |
| 03 | 3 | 65 ± 2 | 4.3 ± 0.3 | 7.5 |
| 04 | 4 | 50 ± 3 | 7.5 ± 0.7 | 5.2 |
| 05 | 5 | 80 ± 3 | 86 ± 3 | 4.1 |
| 06 | 6 | 78 ± 2 | 44 ± 2 | 5.1 |
| 07 | 7 | 55 ± 4 | 9.3 ± 0.7 | 5.4 |
| 08 | 8 | 48 ± 3 | 6.7 ± 0.6 | 5.1 |
| 09 | 9 | 46 ± 2 | 2.2 ± 0.2 | 5.9 |
| 10 | 10 | 46 ± 3 | 1.2 ± 0.2 | 6.5 |
| 11 | 11 | 65 ± 2 | 5.3 ± 0.3 | 7.2 |
| 12 | 12 | 68 ± 2 | 3.1 ± 0.2 | 8.3 |
| 13 | 13 | 33 ± 3 | 5.1 ± 0.4 | 3.7 |
| 14 | 14 | —g | >100 | 0.0 |
| 15 | 15 | —g | >100 | 0.0 |
| 16 | 16 | 10 ± 1 | 34 ± 1 | 0.7 |
| 17 | 17 | 61 ± 3 | 19 ± 1 | 5.1 |
| 18 | 18 | 27 ± 1 | 16 ± 1 | 2.4 |
| 19 | 19 | 30 ± 1 | 18 ± 1 | 2.5 |
| 20 | 20 | 48 ± 1 | 9.3 ± 0.3 | 4.7 |
| 21 | 21 | —g | >100 | 0.0 |
| 22 | 22 | —g | >100 | 0.0 |
 | ||
| Fig. 4 Activators with poor efficiency (A), superb EC50 (B), and superb YMAX (C) exemplified with laurate 16 (A), fullerene malonate 1 (B) and pyrene acetate 5 (C), measured at 25 °C. (D) Dose response curves for 16 (□), 1 (●) and 5 (■) with curve fit to Hill equation. (A–C): Changes in CF emission I (λex 492 nm, λem 517 nm) as a function of time during addition of activator (at t = 60 s; with increasing intensity at t = 360 s, 0, 10, 20, 30, 40, 50, 60, 70, 80 and 100 µM 16 in A, 0, 5, 35, 50, 100, 200, 300 and 400 nM 1 in B, 0, 10, 50, 60, 75, 100, 150 and 200 µM 5 in C) and pR (at t = 120 s, 250 nM) to EYPC-LUVs⊃CF (250 µM EYPC), calibrated by final lysis (I = 100% with excess triton X-100). | ||
Only a few anion activators other than fullerene 1 (E = 10) exhibited efficiencies clearly above the pyrene threshold: calix[4]arenes5712 (E = 8.3), 11 (E = 7.2) and 10 (E = 6.5) as well as coronenebutyrate583 (E = 7.5). Amphiphilic carboxylates (fullerene ≫ calix[4]arene ≈ coronene > pyrene ≈ steroid > calix[6]arenes > naphthalene59,60 ≈ alkane) were clearly more efficient than the corresponding sulfates (Table 1) and alcohols (all inactive). Despite promising activity observed with alkyl phosphate (Table 1, entry 17), phosphate activators were not further investigated because of the inactivity of calix[4]arene 14 (Table 1, entry 14).
With 250 nM polymer, the average concentration of guanidinium cations calculated to 18 µM. This suggested that the EC50 of most activators was below stoichiometric ion pairing. Remarkably, the EC50 of fullerenes 1 (51 nM) and 2 (110 nM) was below the binding of one activator per carrier (250 nM). In other words, fullerenes activators appeared to act at “catalytic” concentrations. “Catalytic” activator efficiency demonstrated that activator–carrier complexes pR·(X)m′·(1)m″ form cooperatively as well as that they are active at extremely low concentrations.
Activator efficiency decreased with decreasing carrier concentration. The decrease in YMAX of coronene carboxylate 3 with decreasing pR concentration, for instance, contributed more to activator efficiency than the compensating decrease in EC50 (Fig. 5). The same trend was observed with the less efficient coronene sulfate activator 4 (Table 2).
| Entry | Anionb | pR/nMg | YMAX (%)c,d | EC50/µMc,e | Ef |
|---|---|---|---|---|---|
| a As in Table 1; measured at 25 °C.b As in Table 1.c As in Table 1.d As in Table 1.e As in Table 1.f As in Table 1.g pR, poly-L-arginine. | |||||
| 01 | 3 | 250 | 65 ± 2 | 4.3 ± 0.3 | 7.5 |
| 02 | 3 | 125 | 51 ± 2 | 3.3 ± 0.2 | 6.1 |
| 03 | 3 | 50 | 43 ± 2 | 2.6 ± 0.2 | 5.4 |
| 04 | 4 | 250 | 50 ± 3 | 7.5 ± 0.7 | 5.2 |
| 05 | 4 | 125 | 42 ± 1 | 7.6 ± 0.2 | 4.3 |
| 06 | 4 | 50 | 22 ± 1 | 4.6 ± 0.1 | 2.5 |
 | ||
| Fig. 5 Dependence of activator efficiency on carrier concentration at 25 °C. (A) Original curves for maximal activity at saturation with coronene activator 3 (added at t = 60 s) for (a) 50 nM, (b) 125 nM and (c) 250 nM polyarginine (added at t = 120 s, procedure as in Fig. 4). (B) Dose response curves for coronene activator 3 at polyarginine concentrations of 50 nM (+), 125 nM (×) and 250 nM (■). | ||
Transport of anions as large as CF by supramolecular carriers Rn·(X)m′·(Y)m″ was remarkable. However CPPs have been reported to mediate the uptake of even larger objects such as proteins, liposomes or even nanoparticles.1,2,7,8,11–24 For this reason, it was of interest to see whether or not complexes pR·(X)m′·(Y)m″ could mediate the release of anions larger than CF from EYPC LUVs. This question was addressed using CFD, i.e., CF-dextran, a classical probe in size exclusion experiments.61 CFD release from EYPC-LUVs⊃CFD by pR anion complexes was tested at concentrations above their EC50 for CF release from EYPC-LUVs⊃CF (Table 1). Overall poor efficiencies were found for the release of CFD compared to CF (Fig. 6). Highest efficiency to stimulate pR-mediated translocation of large anions was found for planar coronene and pyrene carboxylates 3 and 6, whereas amphiphilic sulfates were inactive. Highest size selectivity was observed for the more spherical calix[4]arene activator 10 and pyrene hexanoate 7 with poor efficiency to stimulate CFD release (Fig. 6) but acceptable efficiency with the smaller CF (Table 1). The ability of activators such as 3 and 6 to mediate together with polyarginine the release of the large CFD from vesicles suggested the involvement of mechanisms other than carrier type,36e.g., leakage through transient bilayer defects (compare with below DPPC experiments). The different kinetics observed for coronene and pyrene butyrates 3 and 6 hinted at the possibility that different activators may act by different mechanisms (Fig. 6Aa and c).
 | ||
| Fig. 6 (A) CF-dextran (CFD) efflux from EYPC-LUVs⊃CFD in the presence of pR (250 nM) and activators 3 (a, 20 µM), 4 (b, 10 µM), 6 (c, 100 µM), 7 (d, 15 µM), 8 (e, 20 µM), 9 (f, 6 µM), 10 (g, 5 µM) and 18 (h, 30 µM, procedure as in Fig. 4), measured at 25 °C. (B) Comparison of CF emission obtained for CF (□, Table 1) and CFD efflux (■, A) mediated by pR·(X)m′·(Y)m″ complexes at saturation with activators Y. | ||
2.2 Activator–membrane interactions
The main phase transition of DPPC-LUVs⊃CF occurs at 41.4 °C.62 Maximal efficiencies and EC50's of pertinent activators such as fullerene (1), coronene (3), pyrene (6) and calixarene (10, 12) carboxylates plus cholesterol sulfate 20 were determined below and above this temperature (Table 3). Efficiencies of all tested activators decreased with decreasing membrane fluidity. Whereas the EC50–T profile showed less significant changes (Fig. 7B), the YMAX-T profile for pyrene sulfate 9 as representative activator exhibited the expected continuous increase with increasing temperature (Fig. 7A).| Entry | Anionb | T/°Ci | YMAX (%)c,d | EC50 (µM)c,e | Ef |
|---|---|---|---|---|---|
| a As in Table 1.b As in Table 1.c As in Table 1.d As in Table 1.e As in Table 1.f As in Table 1.g As in Table 1.h DPPC: dipalmitoyl phosphatidylcholine.i T: temperature. | |||||
| 01 | 1 | 25 | —g | >100 | 0.0 |
| 02 | 1 | 60 | 40 ± 3 | 0.098 ± 0.007 | 7.8 |
| 03 | 3 | 25 | 70 ± 1 | 5.8 ± 0.1 | 7.6 |
| 04 | 3 | 60 | 83 ± 2 | 1.7 ± 0.1 | 11.2 |
| 05 | 6 | 25 | 65 ± 1 | 57 ± 1 | 3.9 |
| 06 | 6 | 60 | 91 ± 3 | 43 ± 2 | 6.0 |
| 07 | 9 | 25 | 18 ± 1 | 3.0 ± 0.3 | 2.2 |
| 08 | 9 | 60 | 48 ± 2 | 4.7 ± 0.6 | 5.4 |
| 09 | 10 | 25 | —g | >100 | 0.0 |
| 10 | 10 | 60 | 79 ± 2 | 0.80 ± 0.04 | 11.9 |
| 11 | 12 | 25 | 29 ± 1 | 38 ± 1 | 2.0 |
| 12 | 12 | 60 | 90 ± 1 | 15 ± 1 | 8.0 |
| 13 | 20 | 25 | 3 ± 1 | 26 ± 1 | 0.3 |
| 14 | 20 | 60 | 51 ± 3 | 13 ± 1 | 4.7 |
 | ||
| Fig. 7 (A) YMAX and (B) EC50 for pyrene sulfate 9 to activate pR (500 nM) in DPPC-LUVs⊃CF as a function of temperature. The dotted line indicates the phase transition of DPPC at 41.4 °C, data points were taken at 25, 35, 45, 50 and 60 °C. | ||
The efficiency sequences in sol-phase DPPC, in gel-phase DPPC and in sol-phase EYPC were not the same (Table 3). The sequence fullerene > calix[4]arene ≈ coronene > pyrene in fluid-phase EYPC at 25 °C (Table 1) changed to coronene > calix[4]arene > fullerene > pyrene carboxylates in fluid-phase DPPC at 60 °C (Table 3). The exceptional efficiencies of coronene activator 3 and calix[4]arene activators 10 and 12 originated mainly from maximal activities YMAX ≥ 80% (Table 3, entries 4 and 10). The EC50's of coronene 3 and calix[4]arene 10 in DPPC bilayers at 60 °C were slightly lower than those in EYPC bilayers at 25 °C.
Remarkably, the maximal activity of coronene-carrier complexes Rn·(X)m′·(3)m″ in crystalline DPPC at 25 °C (Table 3, entry 3) exceeded that in liquid-crystalline EYPC at 25 °C slightly (Table 1, entry 3). This distinct DPPC > EYPC selectivity of coronenebutyrate 3 was complementary to fullerene activator 1 with maximal EYPC > DPPC selectivity (Table 3, entries 1 and 2 versusTable 1, entry 1). Overall, the efficiency sequence in crystalline DPPC bilayers at 25 °C was coronene > pyrene > calix[4]arene > fullerene carboxylates. Calix[4]arene 10 and fullerene 1 were completely inactive (Table 3, entries 1 and 9). High activities of coronenes and pyrenes in DPPC vesicles might relate to the preference of polyaromatic hydrocarbons to partition into liquid ordered phase rather than liquid disordered phase of a bilayer.63,64 This sequence might also suggest that mechanisms other than a carrier mechanism36 contribute to the activity of complexes Rn·(X)m′·(Y)m″ with coronene and pyrene activators but, with all appropriate caution, presumably not to that of Rn·(X)m′·(Y)m″ carriers with fullerene and calixarene activators. This interpretation was compatible with the results from CF-dextran efflux (Fig. 6).
The synergistic activity of complexes Rn·(X)m′·(Y)m″ did not depend much on the presence of cholesterol (enriched in mammals) and ergosterol (enriched in fungi)65,66 in EYPC-LUVs⊃CF (Table 4). Activator efficiencies remained either nearly constant (calixarenes 10 and 12) or decreased slightly. Reduced efficiencies may, in part, originate from the increased order of sterol-rich membranes. With respect to alternative modes of membrane recognition, folate (21) was of interest because folate receptors occur enriched at the surface of tumor cells.67,68 No activity was found in sterol-free EYPC-LUVs⊃CF, presumably because the produced pR·(X)m′·(21)m″ complexes were too hydrophilic (Table 1, entry 21).
| Entry | Anionb | Sterolg | YMAX (%)c,d | EC50/µMc,e | Ef |
|---|---|---|---|---|---|
| a As in Table 1; measured at 25 °C.b As in Table 1.c As in Table 1.d As in Table 1.e As in Table 1.f As in Table 1.g chol: 10 mol% cholesterol, erg: 10 mol% ergosterol, total EYPC concentration constant. | |||||
| 01 | 1 | — | 48 ± 1 | 0.051 ± 0.002 | 10.0 |
| 02 | 1 | chol | 37 ± 2 | 0.077 ± 0.007 | 7.4 |
| 03 | 1 | erg | 36 ± 2 | 0.070 ± 0.006 | 7.3 |
| 04 | 3 | — | 65 ± 2 | 4.3 ± 0.3 | 7.5 |
| 05 | 3 | chol | 52 ± 3 | 14 ± 1 | 4.7 |
| 06 | 3 | erg | 51 ± 3 | 15 ± 1 | 4.5 |
| 07 | 6 | — | 78 ± 2 | 44 ± 2 | 5.1 |
| 08 | 6 | chol | 47 ± 1 | 39 ± 1 | 3.2 |
| 09 | 6 | erg | 59 ± 1 | 43 ± 2 | 3.9 |
| 10 | 9 | — | 46 ± 2 | 2.2 ± 0.2 | 5.9 |
| 11 | 9 | chol | 35 ± 1 | 1.6 ± 0.1 | 4.7 |
| 12 | 9 | erg | 33 ± 1 | 1.7 ± 0.1 | 4.4 |
| 13 | 10 | — | 46 ± 3 | 1.2 ± 0.2 | 6.5 |
| 14 | 10 | chol | 61 ± 4 | 3.1 ± 0.4 | 7.4 |
| 15 | 10 | erg | 60 ± 3 | 6.3 ± 0.8 | 6.4 |
| 16 | 12 | — | 68 ± 2 | 3.1 ± 0.2 | 8.3 |
| 17 | 12 | chol | 68 ± 1 | 5.5 ± 0.1 | 7.5 |
| 18 | 12 | erg | 70 ± 1 | 5.4 ± 0.1 | 7.7 |
| 19 | 20 | — | 48 ± 1 | 9.2 ± 0.3 | 4.7 |
| 20 | 20 | chol | 43 ± 3 | 8.6 ± 0.7 | 4.3 |
| 21 | 20 | erg | 41 ± 2 | 9.1 ± 0.5 | 4.1 |
Taken together, the efficiency of activator–carrier complexes depended on membrane fluidity rather than composition. This trend was more pronounced for spherical (fullerenes, calixarenes) than for planar activators (coronenes, pyrenes). These results are in agreement with the preferential interfacial partitioning of aromatic amphiphiles, rather than molecular recognition within the hydrophobic core of the bilayer.
The relevance of anion activation of oligo/polyarginines to penetrate live cells was demonstrated previously by incubating HeLa cells with pyrenebutyrate 6 before addition of R8-alexa, a fluorescently labeled octa-L-arginine.9 According to flow cytometry detection after removal of the cell surface-adsorbed peptides by trypsin treatment, CPP uptake increased up to 4-times in the presence of pyrenebutyrate 6. Measured under nearly identical conditions, the maximal increase of CPP uptake observed in the presence of less sterol-sensitive calix[4]arene 10 (maximal 1.5-fold increase, Fig. 8A) as well as the more sterol-sensitive, EYPC-selective fullerene carboxylate 1 (maximal 1.2-fold increase, Fig. 8B) were clearly below that of pyrenebutyrate 6. At high concentrations, both activators became inhibitors of CPP uptake. The attractive implication that spherical activators, particularly fullerene 1, may specifically mediate CPP-uptake in bacteria will be investigated in due course.
 | ||
| Fig. 8 Uptake ratios from flow cytometry for R8-alexa by HeLa cells as a function of the concentration of activator 10 (A) and 1 (B) relative to activator-free uptake. | ||
2.3. Activator–carrier interactions
In water, pyrenebutyrate complexes pR·(X)m′·(6)m″ exhibited characteristic excimer emission49,50,69–72 at 470 nm with a maximal excimer/monomer ratio of E/M = 0.07.1 This excimer emission increased for complexes pR·(X)m′·(7)m″ and pR·(X)m′·(8)m″ to a spectacular maximal E/M = 4.3 for complex pR·(X)m′·(9)m″ (Fig. 9 and Table 5).| Entry | Anione | E/Mf | EC50/nMg |
|---|---|---|---|
| a From fractional changes in emission intensities at high (pyrenes, 375 nm; coronenes, 427 nm) and low energy (pyrenes, 470 nm; coronenes, ∼500 nm) at constant anion concentration and varied pR concentration (107 mM NaCl, 10 mM NamHnPO4, pH 7.4, 25 °C).b Anion concentration 120 µM.c Anion concentration 12 µM.d Anion concentration 1.2 µM.e See Fig. 3 for structures.f Excimer/monomer emission ratio at saturation with pR (entry 1: Fig. 11, entry 6: Fig. 9).g Determined at minimal detectable anion concentration, from E/M-pR profiles, analyzed with eqns. (4)–(5), see experimental part.h No excimer emission observed. | |||
| 01 | 3 | 28.0c, 1.36d | 20.3 ± 1d |
| 02 | 5 | —h | —h |
| 03 | 6 | 0.07b | 920 ± 60b |
| 04 | 7 | 0.65b, 0.08c, | 150 ± 10c |
| 05 | 8 | 1.90b, 0.10c | 122 ± 4c |
| 06 | 9 | 4.27b, 3.91c, 1.88d | 20.0 ± 1d |
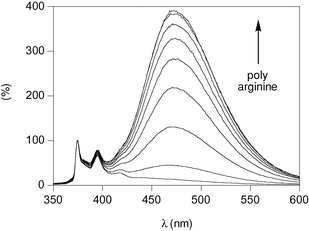 | ||
| Fig. 9 Emission spectra of anion 9 (12 µM, λex = 340 nm) in water in the presence of, with increasing intensity at 470 nm, 0, 50, 100, 150, 200, 250, 300, 350, and 400 nM pR (10 mM NamHnPO4, 107 mM NaCl, pH 7.4, 25 °C); normalized to monomer emission at 375 nm. | ||
E/M ratios as well as EC50's decreased with decreasing concentration of activator–pR-complexes. E/M–pR profiles were recorded at the lowest detectable anion concentration to obtain thermodynamic data (Table 5). Despite the difference in the experiments, the obtained EC50's at high dilution and the EC50's obtained from function (Table 1) showed the same trend (9 ≤ 8 < 7 < 6). Taking the multivalent nature of the carrier into account, EC50 values obtained from the excimer emission (referring to pR concentration) and from functional studies (referring to anion concentration) are in good agreement.
Contrary to that of pyrene carboxylate 6, the EC50 with respect to maximal excimer emission of pyrene sulfate 9 increased with increasing ionic strength (Fig. 10A). These differences suggested that ion pairing is dominant to the stability of the less efficient complexes with sulfate activators like pR·(X)m′·(9)m″, whereas hydrophobic interactions appear to contribute more significantly to the more efficient complexes with carboxylates like pyrene 6.
 | ||
| Fig. 10 Dependence of the formation of activator–pR complexes with pyrene carboxylate 6 (●), pyrene sulfate 9 (○) as well as coronene carboxylate 3 (■) on (A) ionic strength (at 25 °C) and (B) temperature (at 107 mM NaCl). EC50's and ΔG's were determined from Hill analysis of the changes in activator emission with increasing pR concentration as illustrated in Figs. 9 and 11. | ||
This interpretation was supported by the dependence of complex pR·(X)m′·(6)m″ on temperature (Fig. 10B). As expected for the exchange of high-affinity phosphate counteranions with carboxylates of lower affinity,39–45 complex formation was clearly endothermic (ΔH = +29.7 kJ mol−1). Overcompensation of these losses by massive entropy gains corroborated the importance of desolvation of ions as well as large aromatic surfaces (TΔS = +64.2 kJ mol−1 at 25 °C).
Compared to the pyrene series, the emission spectra of activator–pR complexes with coronene 3 exhibited quite strong excimer emission,69 reaching E/M ratios up to 28 (Fig. 11). Their “noisy” emission was indicative of scattering due to flocculation of the resulting complexes pR·(X)m′·(3)m″. Even stronger scattering was found in the emission spectra of the corresponding coronene sulfate complexes pR·(X)m′·(4)m″.
 | ||
| Fig. 11 Emission spectra of coronene carboxylate 3 (12 µM, λex = 340 nm) in water in the presence of, with increasing intensity at ∼500 nm, 0, 50, 100, 150, 200, 250, 500, 1200, 4000 and 5000 nM pR (10 mM NamHnPO4, 107 mM NaCl, pH 7.4), normalized to maximal monomer emission at 427 nm. Dotted spectrum: Magnified emission spectrum of 3 without pR (21×). | ||
According to their excimer emission, the EC50's of coronenebutyrate complexes pR·(X)m′·(3)m″ decreased with increasing ionic strength (Fig. 10A, ■), nicely corroborating results with the corresponding pyrenebutyrate complexes pR·(X)m′·(6)m″, this decrease implied dominance of hydrophobic interactions in complexes with comparably weak carboxylate–guanidinium pairs (Fig. 10A, ■ and ●). The values at high ionic strength may contain contributions from self-assembly of coronene 3 observed under these conditions. Compared to that of pyrene carboxylate complex pR·(X)m′·(6)m″, the lower EC50 of coronene carboxylate complex pR·(X)m′·(3)m″ corroborated the increased activator affinity with increasing aromatic surface area (Table 5, entry 1 versus entry 3).
The excimer emission of pyrene–polyarginine complexes allowed us to detect the formation of “invisible” anion–pR complexes as well. The excimer emission of pyrene complex pR·(X)m′·(8)m″, for instance, decreased in the presence of increasing concentrations of calixarene 12 (Fig. 12). This change in emission reflected the transformation of the “visible” complex pR·(X)m′·(8)m″ into the “invisible” complex pR·(X)m′·(12)m″. Competition experiments with other anion activators gave analogous results and could be used, if desired, to determine global dissociation constants of any activator-carrier complex of interest.73,74
![Counteranion exchange assay for adaptable fluorometric detection of activator–carrier complexes like calix[4]arene complex pR·(X)m′·(12)m″. Emission spectra of anion 8
(120 µM, λex
= 340 nm) in water with pR (2.5 µM) and, with decreasing intensity at 470 nm, 0, 1, 1.5, 1.75, 2, 2.5, 3, 5, 10, 15 and 30 µM calixarene 12
(10 mM NamHnPO4, 107 mM NaCl, pH 7.4); normalized to maximal monomer emission at 375 nm.](/image/article/2005/OB/b501472g/b501472g-f12.gif) | ||
| Fig. 12 Counteranion exchange assay for adaptable fluorometric detection of activator–carrier complexes like calix[4]arene complex pR·(X)m′·(12)m″. Emission spectra of anion 8 (120 µM, λex = 340 nm) in water with pR (2.5 µM) and, with decreasing intensity at 470 nm, 0, 1, 1.5, 1.75, 2, 2.5, 3, 5, 10, 15 and 30 µM calixarene 12 (10 mM NamHnPO4, 107 mM NaCl, pH 7.4); normalized to maximal monomer emission at 375 nm. | ||
Overall, the outcome of spectroscopic studies of complexes pR·(X)m′·(Y)m″ detected by excimer emission of pyrene and coronene activators confirmed that higher aromatics improve and high-affinity ion pairing weakens activator efficiency. The difference between the poor sulfate and the good carboxylate activators turned out to originate indeed from the dominant strong ion pairing which probably slows the anion exchange in the former, and the availability of supportive interactions such as hydrophobic contacts in the latter. This result supported the importance of arene-templated ion-pairing for function on the structural level. The increase in complex stability with increasing aromatic surface from pyrene to coronene revealed the importance of non-interfering remote contacts, i.e., interactions far from the active site,48 to support arene-templated ion pairing38 without loss in activity. The two carboxylates positioned above an extended π-surface of the best activator, i.e., the Bingel fullerene 1, appear perfectly preorganized for arene-templated ion pairing (Fig. 13B). Possibilities for remote contributions to complex stability include additional association of guanidinium cations on the extended aromatic surfaces,75 π–π interactions between proximal activators, and so on.
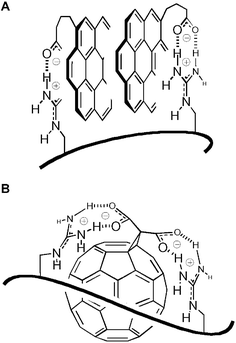 | ||
| Fig. 13 Arene-templated ion pairing exemplified with notional structures of activator–carrier complexes pR·(X)m′·(3)m″ (A) and pR·(X)m′·(1)m″ (B); additional possibilities like binding of phosphate–guanidinium pairs on π-surfaces are not depicted. | ||
3 Summary and conclusions
The objective of this study was to elaborate on the nature and selectivity of the interactions of selected anion activators with polyarginine (pR) carriers on the one hand and lipid bilayer membranes on the other hand. We found that activator–membrane interactions are dominated by the physical properties of the bilayer membrane with little recognition of specific components like ergosterol or cholesterol. This result was consistent with interface-directed translocation28,29 as one possible origin of the superb activator efficiency of higher aromatics like amphiphilic fullerenes, calixarenes and coronenes. Reduced dependence on membrane fluidity and on the size of the transported anion with planar coronene and pyrene compared to spherical fullerene and calix[4]arene activators suggested more significant contributions from mechanisms other than carriers with the former. In HeLa cells, planar pyrenes were clearly more efficient CPP-activators than spherical fullerenes and calixarenes, a trend that might be reversed in bacteria.Ideally, activators should bind tightly to carriers without sacrificing the anion exchange ability to retain carrier activities. Efficient activators are shown to fulfil this requirement with large aromatic surfaces for, presumably, arene-templated carboxylate–guanidinium pairing38 stabilized by multiple interactions remote48 from the active sites. Maybe accounting for interface-directed translocation as well, arene-templated ion pairing supported by remote contacts on large surfaces seems to reach perfection with fullerene 1 (Fig. 13B). The spectacular spectroscopic characteristics discovered for certain activator–carrier complexes open attractive perspectives with regard to sensing applications.
4 Experimental
4.1 Materials
Poly-L-arginine (HCl salt, MW 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, DP 72), sodium dodecyl sulfate (SDS), triton X-100, buffers and salts were purchased from Sigma, 4-pyrene-1-yl-butyric acid from Acros Organics, coronene from Aldrich, fullerene C60 from Alfa Aesar, egg yolk phosphatidylcholine (EYPC) and dipalmitoyl phosphatidylcholine (DPPC) from Avanti, cholesterol sulfate from Northern Lipids, mono-n-dodecylphosphate from Lancaster and reagents for synthesis, 5(6)-carboxyfluorescein (CF), 2-pyrene-1-yl-acetic acid, 6-pyrene-1-yl-caproic acid, cholesterol, ergosterol and lauric acid from Fluka. All reactions were performed under N2 or argon atmosphere. Column chromatography was carried out on silica gel 60 (Fluka, 40–63 µm), reverse phase column chromatography on ODS (Fluka Silicagel 100 C18-Reverse Phase), analytical thin layer chromatography (TLC) on silica gel 60 (Fluka, 0.2 mm) and preparative TLC (PTLC) on silica gel GF (Analtech, 1000 µm). Melting points (mp) were recorded on a heating table from Reichert (Austria). ESI-MS and APCI-MS were performed on a Finnigan MAT SSQ 7000 instrument. 1H and 13C spectra were recorded (as indicated) on either a Bruker 300 MHz, 400 MHz or 500 MHz spectrometer and are reported as chemical shifts (δ) in ppm relative to TMS (δ
= 0). Spin multiplicities are reported as a singlet (s), doublet (d), triplet (t), with coupling constants (J) given in Hz, or multiplet (m). 1H and 13C resonances were assigned with the aid of additional information from 2D NMR spectra (H,H-COSY, DEPT 135, HSQC and HMBC). UV-Vis spectra were measured on a Varian Cary 1 Bio spectrophotometer. Fluorescence measurements were preformed on either a FluoroMax-2 or a FluoroMax-3, Jobin Yvon-Spex. The Mini-Extruder with a polycarbonate membrane, pore size 100 nm, used for LUV preparation was from Avanti.
000, DP 72), sodium dodecyl sulfate (SDS), triton X-100, buffers and salts were purchased from Sigma, 4-pyrene-1-yl-butyric acid from Acros Organics, coronene from Aldrich, fullerene C60 from Alfa Aesar, egg yolk phosphatidylcholine (EYPC) and dipalmitoyl phosphatidylcholine (DPPC) from Avanti, cholesterol sulfate from Northern Lipids, mono-n-dodecylphosphate from Lancaster and reagents for synthesis, 5(6)-carboxyfluorescein (CF), 2-pyrene-1-yl-acetic acid, 6-pyrene-1-yl-caproic acid, cholesterol, ergosterol and lauric acid from Fluka. All reactions were performed under N2 or argon atmosphere. Column chromatography was carried out on silica gel 60 (Fluka, 40–63 µm), reverse phase column chromatography on ODS (Fluka Silicagel 100 C18-Reverse Phase), analytical thin layer chromatography (TLC) on silica gel 60 (Fluka, 0.2 mm) and preparative TLC (PTLC) on silica gel GF (Analtech, 1000 µm). Melting points (mp) were recorded on a heating table from Reichert (Austria). ESI-MS and APCI-MS were performed on a Finnigan MAT SSQ 7000 instrument. 1H and 13C spectra were recorded (as indicated) on either a Bruker 300 MHz, 400 MHz or 500 MHz spectrometer and are reported as chemical shifts (δ) in ppm relative to TMS (δ
= 0). Spin multiplicities are reported as a singlet (s), doublet (d), triplet (t), with coupling constants (J) given in Hz, or multiplet (m). 1H and 13C resonances were assigned with the aid of additional information from 2D NMR spectra (H,H-COSY, DEPT 135, HSQC and HMBC). UV-Vis spectra were measured on a Varian Cary 1 Bio spectrophotometer. Fluorescence measurements were preformed on either a FluoroMax-2 or a FluoroMax-3, Jobin Yvon-Spex. The Mini-Extruder with a polycarbonate membrane, pore size 100 nm, used for LUV preparation was from Avanti.Abbreviations. CF: 5(6)-carboxyfluorescein; DBU: 1,8-diaza-bicyclo[5.4.0]undec-7-ene; DIPEA: N,N′-diisopropylethylenediamine; DMAP: 4-dimethylaminopyridine; DMF: N,N-dimethylformamide; EYPC-LUVs: egg yolk phosphatidylcholine large unilamellar vesicles; TBDPS: tetrabutyldiphenylsilyl; THF: tetrahydrofuran.
4.2 Synthesis
Anion activators 1, 3, 8–15 and 19 were prepared following previously reported procedures.9,51,58,60 Stock solutions were prepared for all anions in buffer except for 1 and 2 (DMSO–water 1 : 1) and 3 and 4 (DMSO/water 9 : 1). The pH of all stock solutions was measured and accurately adjusted if necessary; concentrations were confirmed, if applicable, by UV/vis spectroscopy (if applicable). | ||
| Scheme 1 Conditions: a) TBDPSCl, DIPEA, CH2Cl2, rt, 2 h, 95%; b) 26, pyridine, DMAP, THF, rt, 12 h, 71%; c) 28, I2, DBU, toluene, rt, 12 h, 26%; d) AcOH, Bu4NF, THF, rt, 16 h, 62%; e) 1. ClSO3H, ether, 0 °C, 10 min, 2. NaOH, 80%. | ||
4-(tert-Butyl-diphenyl-silanyloxy)-butan-1-ol 23. To a solution of 24 (5.0 g, 55 mmol) in CH2Cl2 (10 ml) containing DIPEA (10 ml), TBDPSCl (5 ml, 18 mmol) was added dropwise at room temperature. The solution was stirred for 2 hours, concentrated in vacuo and purified by column chromatography (hexane–EtOAc 10 : 1, Rf = 0.8) to give 23 (5.63 g, 95%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.74 (d, 3J (H,H) = 7.8 Hz, 4H), 7.60–7.40 (m, 6H), 3.76 (t, 3J (H,H) = 5.8 Hz, 2H), 3.72 (t, 3J (H,H) = 5.8 Hz, 2H), 2.20 (broad s, 1H, exchange with D2O), 1.80–1.69 (m, 4H), 1.12 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 135.6 (d), 133.7 (s), 129.7 (d), 127.7 (d), 67.0 (t), 62.9 (t), 29.9 (t), 29.3 (t), 26.9 (q), 19.2 (s); MS (ESI, MeOH): m/z (%) 330 (100) [M + H]+.
Malonic acid bis-[4-(tert-butyl-diphenyl-silanyloxy)-butyl] ester 25. To a solution of 23 (2.0 g, 6.1 mmol), pyridine (520 µl, 6.4 mmol) and DMAP (4.0 mg, 33 µmol) in dried THF and malonyl chloride 26 (296 µl, 3.0 mmol) were added at room temperature. The mixture was stirred overnight. After evaporation of THF, CH2Cl2 (20 ml) was added, and the organic layer was washed with water (20 ml), dried over MgSO4, concentrated in vacuo and purified by column chromatography (CH2Cl2–MeOH 20 : 1, Rf = 0.7) to give 25 (1.56 g, 71%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.71 (d, 3J (H,H) = 7.6 Hz, 8H), 7.40–7.30 (m, 12H), 4.09 (t, 3J (H,H) = 6.6 Hz, 4H), 3.59 (t, 3J (H,H) = 6.3 Hz, 4H), 3.26 (s, 2H), 1.69 (tt, 3J (H,H) = 6.6 Hz, 3J (H,H) = 8.3 Hz, 4H), 1.53 (tt, 3J (H,H) = 6.3 Hz, 3J (H,H) = 8.3 Hz, 4H), 1.11 (s, 18H); 13C NMR (100 MHz, CDCl3): 166.7 (s), 135.6 (d), 133.9 (s), 129.7 (d), 127.7 (d), 65.5 (t), 63.3 (t), 28.9 (t), 28.3 (t), 26.9 (q), 25.1 (t), 19.3 (s); MS (ESI, MeOH): m/z (%) 726 (100) [M + H]+.
Fullerene 27. To a solution of 28 (50 mg, 69.4 µmol), 25 (55 mg, 76 µmol) and I2 (22 mg, 86 µmol) in dried toluene (100 ml), DBU (27 µl, 170 µmol) was added. The mixture was stirred for 12 hours at room temperature. After evaporation of toluene, the crude product was filtered through a short plug of silica, eluting first with toluene (to remove unreacted 28) and then with CH2Cl2. Purification by PTLC (petroleum ether–EtOAc 10 : 1, Rf = 0.5) gave pure compound 27 (26 mg, 26%) as a gummy oil. 1H NMR (400 MHz, CDCl3): δ 7.58 (d, 3J (H,H) = 7.6 Hz, 8H), 7.40–7.25 (m, 12H), 4.42 (t, 3J (H,H) = 6.6 Hz, 4H), 3.63 (t, 3J (H,H) = 6.6 Hz, 4H), 1.86 (tt, 3J (H,H) = 7.8 Hz, 3J (H,H) = 6.6 Hz, 4H), 1.63 (tt, 3J (H,H) = 6.6 Hz, 3J (H,H) = 7.8 Hz, 4H), 1.00–0.98 (m, 18H); 13C NMR (100 MHz, CDCl3): δ 162.6 (s), 144.3 (s), 144.2 (s), 144.1 (s), 144.1 (s), 143.9 (s), 143.7 (s), 143.6 (s), 142.9 (s), 142.0 (s), 142.0 (s), 142.0 (s), 141.1 (s), 140.9 (s), 137.9 (s), 134.5 (d), 132.7 (s), 128.6 (d), 126.7 (d), 70.6 (s), 66.3 (t), 62.1 (t), 51.3 (s), 27.9 (t), 25.9 (q), 24.2 (d), 18.2 (s); MS (ESI, CH2Cl2): m/z (%) 1443 (45) [M + H]+, 722 (100) [M + 2H]2+, 721 (10) [C60 + H]+.
Fullerene diol 29. A solution of 27 (25 mg, 17 µmol) in THF (3 ml) was treated successively with acetic acid (20 µl, 0.35 mmol) and tetrabutylammonium fluoride (1 M in THF, 350 µL, 0.35 mmol) at room temperature. After stirring for 16 hours, the reaction mixture was diluted with CHCl3–MeOH 1 : 1 (40 ml), washed with water (20 ml), dried over MgSO4, concentrated in vacuo and purified by column chromatography (CH2Cl2–MeOH 8 : 1 then 4 : 1, Rf = 0.6) to give pure 29 (10 mg, 62%) as a brown oil. 1H NMR (300 MHz, CD3OD–CDCl3): δ 4.05 (t, 3J (H,H) = 6.4 Hz, 4H), 3.55 (t, 3J (H,H) = 6.3 Hz, 4H), 1.70–1.55 (m, 4H), 1.55–1.40 (m, 4H); 13C NMR (75 MHz, CD3OD–CDCl3): δ 163.6 (s), 145.3 (s), 145.3 (s), 145.2 (s), 144.7 (s), 144.6 (s), 143.9 (s), 143.0 (s), 143.0 (s), 142.2 (s), 141.9 (s), 141.0 (s), 139.0 (s), 129.0 (s), 71.6 (s), 63.4 (t), 61.6 (t), 29.3 (t), 26.2 (d); MS (ESI, CH2Cl2): m/z (%) 967 (100) [M + H]+.
Fullerene disulfate 2 (disodium salt). Fullerene 29 (10 mg, 10 µmol) in anhydrous ether (4 ml) was added to chlorosulfonic acid (14 µl, 21 µmol) in anhydrous ether (2 ml) at 0 °C. The mixture was purged with nitrogen for an additional 30 minutes to remove HCl, diluted with water (4 ml), 1 M NaOH (to reach pH ∼7) and isopropanol (5 ml), washed with hexane (2 × 5 ml) and concentrated in vacuo. Purification by reverse phase column chromatography (MeOH, Rf = 0.5) yielded 2 (9 mg, 80%) as a yellowish powder. mp: >230 °C; 1H NMR (300 MHz, CD3OD): δ 3.91 (t, 3J (H,H) = 6.4 Hz, 4H), 3.79 (t, 3J (H,H) = 6.3 Hz, 4H), 2.14 (tt, 3J (H,H) = 6.4 Hz, 3J (H,H) = 7.3 Hz, 4H), 1.89 (tt, 3J (H,H) = 7.3 Hz, 3J (H,H) = 6.3 Hz, 4H); 13C NMR (75 MHz, CD3OD): δ 163.6 (s), 145.3 (s), 145.3 (s), 145.2 (s), 145.2 (s), 144.7 (s), 144.6 (s), 143.9 (s), 143.1 (s), 143.1 (s), 142.2 (s), 141.9 (s), 140.9 (s), 139.0 (s), 128.2 (s), 71.6 (s), 64.9 (t), 63.4 (t), 49.8 (s), 23.6 (t), 21.9 (d); MS (ESI negative mode, MeOH); m/z (%) 1147 (100) [M − Na]−, 562 (45), [M − 2Na]2−.
4-Coronenyl-1-butanol. A solution of 3 (240 mg, 0.62 mmol) in THF (16 ml) was transferred slowly to a solution of LiAlH4 (186 mg, 5.00 mmol) in THF (3 ml) at 0 °C. The mixture was stirred for 2 hours at 60 °C. The reaction mixture was cooled at 0 °C and water was added drop by drop. The reaction mixture was stirred for 20 min, filtered (washed with THF and CH2Cl2–MeOH) and concentrated in vacuo. Purification of the crude product by column chromatography (CH2Cl2, then CH2Cl2–EtOAc 10 : 1, Rf = 0.4) yielded 4-coronenyl-1-butanol (128 mg, 55%) as a yellow solid. Mp: 205.0–206.3 °C; IR: ν 3238 (m), 3019 (s), 1611 (s), 1461 (s), 1052 (s) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 9.24–9.00 (m, 10H), 8.93 (s, 1H), 4.46 (t, 3J (H,H) = 5.3 Hz, 1H, exchange with D2O), 3.79 (t, 3J (H,H) = 7.9 Hz, 2H), 3.57 (dd, 3J (H,H) = 6.4 Hz, 3J (H,H) = 5.3 Hz, 2H), 2.13 (tt, 3J (H,H) = 7.9 Hz, 3J (H,H) = 7.5 Hz, 2H), 1.77 (tt, 3J (H,H) = 7.5 Hz, 3J (H,H) = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 137.7 (s), 128.3 (s), 128.3 (s), 128.2 (s), 128.1 (s), 127.9 (s), 127.1 (s), 126.4 (d), 126.4 (d), 126.4 (d), 126.3 (d), 126.3 (d), 126.3 (d), 126.1 (d), 125.9 (d), 125.9 (d), 125.9 (d), 122.7 (d), 122.1 (s), 122.0 (s), 121.7 (s), 121.6 (s), 121.4 (s), 121.5 (s), 60.6 (t), 33.1 (t), 32.7 (t), 27.6 (t); MS (CI, DMF): m/z (%) 372 (100) [M]+.
4-Coronen-1-yl-butylsulfate 4 (sodium salt). A solution of 4-coronenyl-1-butanol (91 mg, 0.24 mmol) in DMF (1.5 ml) was transferred slowly to a solution of chlorosulfuric acid (35 mg, 0.30 mmol) in THF (1.5 ml) at 0 °C. The mixture was stirred for 1 hour at room temperature and purged with nitrogen to remove HCl. The mixture was diluted with water (2 ml), 1 M NaOH (to reach pH ∼7), water (50 ml) and 2-propanol (50 ml), washed with petroleum ether (2 × 30 ml) and concentrated in vacuo. Purification by reverse phase column chromatography (H2O–acetone 1 : 1 Rf = 0.6, first with H2O then to H2O–acetone 5 : 1 to 3 : 1) yielded pure 4 (70 mg, 63%) as a yellow solid. mp: >230 °C. IR: ν 3467 (m), 3016 (s), 1613 (s), 1216 (s) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 9.22–9.00 (m, 10H), 8.94 (s, 1H), 3.91 (t, 3J (H,H) = 6.5 Hz, 2H), 3.77 (t, 3J (H,H) = 7.8 Hz, 2H), 2.13 (tt, 3J (H,H) = 7.8 Hz, 3J (H,H) = 7.2 Hz, 2H), 1.87 (tt, 3J (H,H) = 7.2 Hz, 3J (H,H) = 6.5 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 137.6 (s), 128.3 (s), 128.3 (s), 128.2 (s), 128.1 (s), 127.4 (s), 127.1 (s), 126.4 (d), 126.4 (d), 126.4 (d), 126.3 (d), 126.3 (d), 126.3 (d), 126.2 (d), 126.2 (d), 126.1 (d), 125.9 (d), 122.8 (d), 122.1 (s), 122.0 (s), 121.8 (s), 121.6 (s), 121.4 (s), 120.5 (s), 65.4 (t), 33.0 (t), 29.4 (t), 27.7 (t); MS (ESI negative mode, DMF): m/z (%) 451 (100) [M − Na]−.
4.3 Vesicle preparation
Solutions of EYPC (for EYPC-LUVs), DPPC (for DPPC-LUVs), and mixtures of EYPC and either ergosterol (erg, for EYPC/erg-LUVs) or cholesterol (chol, for EYPC/chol-LUVs) (25 mg) in CHCl3–MeOH 1 : 1 (2 ml) were dried under a stream of N2 and then under vacuum (>2 h) to form thin films. The resulting films were hydrated with 1 ml buffer A (50 mM CF, 10 mM NamHnPO4, 10 mM NaCl, pH 7.4 (for LUVs⊃CF) or 7.4 mM CF-dextran, 7.4 mM HEPES, 37 mM NaCl, pH 7.4 (EYPC-LUVs⊃CFD)) for more than 30 min, subjected to freeze–thaw cycles (5×) and extrusions (15×, Mini-Extruder with two stacked polycarbonate membranes, pore size 100 nm). Extravesicular CF/CFD was removed by gel filtration (Sephadex G-50 for LUVs⊃CF, Sephacryl S300-HR for LUVs⊃CFD) with buffer B (10 mM NamHnPO4, 107 mM NaCl, pH 7.4). The LUV fractions were combined and diluted to 6 ml with buffer B. Lipid concentrations were estimated from amount of entrapped CF. The found values were in agreement with earlier results from phosphate analysis (due to 10 mM phosphate in the buffer, phosphate analysis did not give accurate results). The final stock solutions had the following characteristics: ∼1.3 mM lipid; inside: 50 mM CF, 10 mM NamHnPO4, 10 mM NaCl, pH 7.4, (LUVs⊃CF) or 7.4 mM CFD, 7.4 mM HEPES, 37 mM NaCl, pH 7.4 (LUVs⊃CFD); outside: 10 mM NamHnPO4, 107 mM NaCl, pH 7.4.4.4 Vesicle experiments
LUV stock solutions (1.3 mM lipid, above) were diluted with buffer (10 mM NamHnPO4, 107 mM NaCl, pH 7.4) to give ≈13 µM lipid, placed in a thermostated fluorescence cuvette (25 °C or as indicated) and gently stirred. CF efflux was followed at λem 517 nm (λex 490 nm) as a function of time during the addition of anion activators at varied concentration (1–22, 20–40 µl from stock solutions, final concentrations as indicated), polyarginine (250 nM) and aqueous triton X-100 at the end of each experiment for calibration (0.024%). Data were normalized to fractional emission intensity I using eqn. (2)| I = (Ft − F0)/(F∞ − F0) | (2) |
| Y = Y0 + (YMAX − Y0)/{1 + (EC50/cANION)n} | (3) |
4.5 Flow cytometry with HeLa cells
The influence of counteranion 1 and 10 on the uptake of R8-alexa by HeLa cells was determined using flow cytometry as described previously.9 In brief, R8-alexa was prepared by reacting H-(Arg)8-Gly-Cys-NH2 with 1.5 eq. alexa 488 C5 maleimide (Molecular Probes) in DMF–MeOH (1 : 1, 1.5 h), purified with RP-HPLC and characterized by MALDI-TOFMS (calcd. 2126.4, found 2127.2). Human cervical cancer-derived HeLa cells (1.5 × 105 cells in 1.5 ml of α-MEM containing 10% (v/v) calf serum) were cultured on 60 mm dishes (48 h) and washed twice with PBS after removal of the medium. After incubation of the cells in the presence or absence of fullerene 1 or calixarene 10 (300 µl PBS, 5 min, 37 °C), addition of R8-alexa (100 µl PBS, final peptide concentration 5 µM), incubation (15 min, 37 °C), washing (PBS, 3×), incubation with 0.01% trypsin (400 µl, 10 min, 37 °C), addition of PBS (600 µl) and centrifugation (2,000 rpm, 5 min), the cell pellet was suspended, washed (2 × 1 ml PBS) and finally resuspended in 1 ml of PBS for fluorescence analysis with a FACScalibur (BD Biosciences) flow cytometer using a 488 nm laser excitation and a 515–545 nm emission filter. Each sample was analyzed for 10000 events.4.6 Pyrene excimers
Solutions of pyrene anions 5–9 (0.12–120 µM) in buffer (2 ml, 10 mM NamHnPO4, 0–2 M NaCl, pH 7.4) were placed in a cuvette. Then, pR was added stepwise (concentrations appropriate to follow the appearance of excimer emission, see Table 5) and the solutions were gently mixed. Fluorescence emission spectra of the resulting solutions were measured with λex 340 nm at 25–60 °C. The obtained spectra were normalized to maximal monomer emission intensity at 375 nm. E/M ratios IEM = I470/I375 for given anions, NaCl concentrations and temperature were plotted as a function of pR concentration and analyzed using the eqn. (4)| IEM = I0EM + (I∞EM − I0EM)/[1 + (EC50/c)n] | (4) |
| ΔG = −RTln KD | (5) |
| ΔG = ΔH − TΔS. | (6) |
Data analysis was done with Kaleidagraph, version 3.5 (Synergy Software).
4.7 Pyrene excimers, anion competition assay
To solutions of pyrene sulfate 8 (120 µM) and anions 1, 2, 10–20 (appropriate concentration; c/µM) in buffer (2 ml, 10 mM NamHnPO4, 107 M NaCl, pH 7.4), pR (2.5 µM) was added. The solutions were gently mixed and the fluorescence emission spectra were measured with λex 340 nm at 25 °C (12, Fig. 12). The obtained spectra were normalized as above and analyzed using the eqn. (7)| IEM = I0EM + (I∞EM − I0EM)/[1 + (IC50/c)n] | (7) |
4.8 Coronene fluorescence
Solutions of coronene anion 3 (0.12–12 µM) in buffer (2 ml, 10 mM NamHnPO4, 100 mM NaCl, pH 7.4) were placed in a cuvette. Then, pR was added stepwise and the solutions were gently mixed. Fluorescence emission spectra of the resulting solutions were measured with λex 340 nm at 25 °C. The obtained spectra were normalized to maximal monomer emission intensity at 427 nm (Fig. 11) and analyzed as described for pyrenes.Acknowledgements
We thank D. Jeannerat, A. Pinto and J.-P. Saulnier for NMR measurements, P. Perrottet and the group of F. Gülaçar for MS, and the Swiss NSF (including National Research Program “Supramolecular Functional Materials” 4047-057496, SM), Japan Science and Technology Agency (JST) (SF), Delta Proteomics (ANL) and the CNRS (AWC) for financial support.References
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348–14356 CrossRef CAS.
- N. Sakai, T. Takeuchi, S. Futaki and S. Matile, ChemBioChem, 2005, 6, 114–122 CrossRef CAS.
- L. Monticelli, K. M. Robertson, J. L. MacCallum and D. P. Tieleman, FEBS Lett., 2004, 564, 325–332 CrossRef CAS.
- Y. Jiang, A. Lee, J. Chen, V. Ruta, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2003, 423, 33–41 CrossRef CAS.
- Y. Jiang, V. Ruta, J. Chen, A. Lee and R. MacKinnon, Nature, 2003, 423, 42–48 CrossRef CAS.
- R. B. Bass, P. Strop, M. Barclay and D. C. Rees, Science, 2002, 298, 1582–1587 CrossRef CAS.
- D. Terrone, S. Leung, W. Sang, L. Roudaia and J. R. Silvius, Biochemistry, 2003, 42, 13787–13799 CrossRef CAS.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS.
- F. Perret, M. Nishihara, T. Takeuchi, S. Futaki, A. N. Lazar, A. W. Coleman, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 1114–1115 CrossRef CAS.
- N. Sakai, N. Sordé, G. Das, P. Perrottet, D. Gerard and S. Matile, Org. Biomol. Chem., 2003, 1, 1226–1231 RSC.
- S. Futaki, Arginine-rich Peptides, Curr. Protein Pept. Sci., 2003, 4, 87–157 Search PubMed.
- G. Dom, C. Shaw-Jackson, C. Matis, O. Bouffioux, J. J. Picard, A. Prochiantz, M. P. Mingeot-Leclercq, R. Brasseur and R. Rezsohazy, Nucleic Acids Res., 2003, 31, 556–561 CrossRef CAS.
- P. E. G. Thorén, D. Persson, E. K. Esbjörner, M. Goksör, P. Lincoln and B. Nordén, Biochemistry, 2004, 43, 3471–3489 CrossRef CAS.
- A. Ziegler, X. L. Blatter, A. Seelig and J. Seelig, Biochemistry, 2003, 42, 9185–9194 CrossRef CAS.
- H. Binder and G. Lindblom, Biophys. J., 2003, 85, 982–995 CrossRef CAS.
- K. Takeshima, A. Chikushi, K. Lee, S. Yonehara and K. Matsuzaki, J. Biol. Chem., 2003, 278, 1310–1315 CrossRef CAS.
- S. D. Krämer and H. Wunderli-Allenspach, Biochim. Biophys. Acta, 2003, 1609, 161–169 CAS.
- S. M. Fuchs and R. T. Raines, Biochemistry, 2004, 43, 2438–2444 CrossRef CAS.
- P. E. G. Thorén, D. Persson, P. Isakson, M. Goksör, A. Onfelt and B. Nordén, Biochem. Biophys. Res. Commun., 2003, 307, 100–107 CrossRef CAS.
- D. Persson, P. E. G. Thorén, M. Herner, P. Lincoln and B. Nordén, Biochemistry, 2003, 42, 421–429 CrossRef CAS.
- J. L. Zaro and W. C. Shen, Biochem. Biophys. Res. Commun., 2003, 307, 241–247 CrossRef CAS.
- D. Seebach, K. Namoto, R. Yogesh, Y. R. Mahajan, P. Bindschädler, R. Sustmann, M. Kirsch, N. S. Ryder, M. Weiss, M. Sauer, C. Roth, S. Werner, H.-D. Beer, C. Munding, P. Walde and M. Voser, Chem. Biodiv., 2004, 1, 65–97 Search PubMed.
- T. B. Potocky, A. K. Menon and S. H. Gellman, J. Biol. Chem., 2003, 278, 50188–50194 CrossRef CAS.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS.
- G. Zuber, E. Dauty, M. Nothisen, P. Belguise and J.-P. Behr, Adv. Drug Delivery Rev., 2001, 52, 245–253 CrossRef CAS.
- A. D. Miller, Curr. Med. Chem., 2003, 10, 1195–1211 CrossRef CAS.
- E. Nakamura and H. Isobe, Acc. Chem. Res., 2003, 36, 807–815 CrossRef CAS.
- W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS.
- M. Schiffer, C. H. Chang and F. J. Stevens, Protein Eng., 1992, 5, 213–214 CrossRef CAS.
- R. MacKinnon, S. L. Cohen, A. Kuo, A. Lee and B. T. Chait, Science, 1998, 280, 106–109 CrossRef CAS.
- W. C. Wimley, Curr. Opin. Struct. Biol., 2003, 13, 404–411 CrossRef CAS.
- A. N. J. A. Ridder, S. Morein, J. G. Stam, A. Kuhn, B. de Kruijff and J. A. Killian, Biochemistry, 2000, 39, 6521–6528 CrossRef CAS.
- M. R. R. De Planque, J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe II, B. de Kruijff and J. A. Killian, J. Biol. Chem., 1999, 274, 20839–20846 CrossRef CAS.
- M. R. R. De Planque, B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts and J. A. Killian, Biochemistry, 2003, 42, 5341–5348 CrossRef CAS.
- J. M. Sanderson and E. J. Whelan, Phys. Chem. Chem. Phys., 2004, 6, 1012–1017 RSC.
- S. Matile, A. Som and N. Sordé, Tetrahedron, 2004, 60, 6405–6435 CrossRef CAS.
- G. W. Gokel and A. Mukhopadhyay, Chem. Soc. Rev., 2001, 30, 274–286 RSC.
- S. E. Thompson and D. B. Smithrud, J. Am. Chem. Soc., 2002, 124, 442–449 CrossRef CAS.
- K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67–114 CrossRef CAS.
- K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371–378 CrossRef CAS.
- M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3–15 CAS.
- P. Breccia, M. Van Gool, R. Perez-Fernandez, S. Martin-Santamaria, F. Gago, P. Prados and J. de Mendoza, J. Am. Chem. Soc., 2003, 125, 8270–8284 CrossRef CAS.
- M. Berger and F. P. Schmidtchen, J. Am. Chem. Soc., 1999, 121, 9986–9993 CrossRef CAS.
- X. Salvatella, M. W. Peczuh, M. Giarí, R. K. Jain, J. Sánchez-Quesada, J. de Mendoza, A. D. Hamilton and E. Giralt, Chem. Commun., 2000, 1399–1400 RSC.
- B. P. Orner, X. Salvatella, J. Sánchez-Quesada, J. de Mendoza, E. Giralt and A. D. Hamilton, Angew. Chem., Int. Ed., 2002, 41, 117–119 CrossRef CAS.
- R. A. Kumpf and D. A. Dougherty, Science, 1993, 261, 1708–1710 CrossRef CAS.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
- F. M. Menger, Biochemistry, 1992, 31, 5368–5373 CrossRef CAS.
- O. T. Jones and A. G. Lee, Biochemistry, 1985, 24, 2195–2202 CrossRef CAS.
- J. Martins and E. Melo, Biophys. J., 2001, 80, 832–840 CrossRef CAS.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- I. Lamparth and A. Hirsh, J. Chem. Soc., Chem. Commun., 1994, 1727–1728 RSC.
- S. Foley, C. Crowley, M. Smaihi, C. Bonfils, B. F. Erlanger, P. Seta and C. Larroque, Biochem. Biophys. Res. Commun., 2002, 294, 116–119 CrossRef CAS.
- C. Wang, L. A. Tai, D. D. Lee, P. P. Kanakamma, C. K.-F. Shen, T.-Y. Luh, C. H. Cheng and K. C. Hwang, J. Med. Chem., 1999, 42, 4614–4620 CrossRef CAS.
- H. Park, M. Chhowalla, Z. Iqbal and F. Sesti, J. Biol. Chem., 2003, 278, 50212–50216 CrossRef CAS.
- F. Rancan, S. Rosan, F. Boehm, A. Cantrell, M. Brellreich, H. Schoenberger, A. Hirsch and F. Moussa, J. Photochem. Photobiol. B, 2002, 67, 157–162 CrossRef CAS.
- E. Da Silva, A. N. Lazar and A. W. Coleman, J. Drug Delivery Sci. Technol., 2004, 14, 3–20 Search PubMed.
- Davenport, B. Shen, T. W. Joseph and M. P. Straher, Chem. Phys. Lipids, 2001, 109, 145–156 CrossRef.
- C. T. Miller, R. Weragoda, E. Izbicka and B. I. Iverson, Bioorg. Med. Chem., 2001, 9, 2015–2024 CrossRef CAS.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., in press Search PubMed.
- S. Rex, Biophys. Chem., 1996, 58, 75–85 CrossRef CAS.
- P. Yeagle, The Structure of Biological Membranes, CRC Press, Boca Raton, Florida, 1992 Search PubMed.
- T. Baumgart, S. T. Hess and W. W. Webb, Nature, 2003, 425, 821–824 CrossRef CAS.
- B. Hoff, E. Strandberg, A. S. Ulrich, D. P. Tieleman and C. Posten, Biophys. J., 2005, 88, 1818–1827 CAS.
- B. De Kruijff and R. A. Demil, Biochim. Biophys. Acta, 1974, 339, 57–70 CAS.
- S. C. Hartsel and J. Bolard, Trends Pharm. Sci., 1996, 17, 445–449 CrossRef CAS.
- J. Turk, D. J. Waters and P. S. Low, Cancer Lett., 2004, 213, 165–172 CrossRef.
- X. B. Zhao and R. J. Lee, Adv. Drug. Delivery Rev., 2004, 56, 1193–1204 CrossRef CAS.
- B. Stevens, Spectrochim. Acta, 1962, 18, 439–448 CAS.
- D. Y. Sasaki, Cell Biochem. Biophys., 2003, 39, 145–161 Search PubMed.
- B. C. Roy, B. Chandra, D. Hromas and S. Mallik, Org. Lett., 2003, 5, 11–14 CrossRef CAS.
- B.-B. Wang, X. Zhang, X.-R. Jia, Z.-C. Li, Y. Ji, L. Yang and Y. Wei, J. Am. Chem. Soc., 2004, 126, 15180–15194 CrossRef CAS.
- Y. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS.
- H. C. Cheng, Pharmacol. Res., 2004, 50, 21–40 CrossRef CAS.
- R. B. M. Ansem and L. T. Scott, J. Phys. Org. Chem., 2004, 17, 819–823 CrossRef.
| This journal is © The Royal Society of Chemistry 2005 |