Asymmetric hydrogenation using chiral Rh complexes immobilised with a new ion-exchange strategy
William P.
Hems
a,
Paul
McMorn
b,
Stewart
Riddel
a,
Simon
Watson
a,
Frederich E.
Hancock
a and
Graham J.
Hutchings
*b
aJohnson Matthey PCT, 28 Cambridge Science Park, Milton Road, Cambridge, UK CB4 0FP
bSchool of Chemistry, Cardiff University, Cardiff, UK. E-mail: hutch@cardiff.ac.uk; Fax: (+44)29 2087 4030; Tel: (+44)29 2087 4805
First published on 18th March 2005
Abstract
Rh diphosphine complexes using DuPhos and JosiPhos as chiral ligands have been immobilised by ion exchange into the mesoporous material MCM-41. When used as catalysts for the enantioselective hydrogenation of dimethyl itaconate and methyl-2-acetamidoacrylate, these heterogeneous catalysts give catalytic performance in terms of yield and enantioselection that are comparable to the corresponding homogeneous catalysts. Furthermore, the heterogeneous catalysts can be readily recovered and reused without loss of catalyst performance. A second immobilisation strategy is described in which [Rh(COD)2]+BF4− is initially immobilised by ion exchange and subsequently modified by the chiral diphosphine and this give comparable catalyst performance. This immobilisation strategy opens up the possibility of easy ligand-screening for parallel synthesis and libraries.
Introduction
The ability to synthesise pure enantiomers is of crucial importance for the preparation of modern pharmaceuticals and agrochemicals. Most early progress has been focussed on the design of molecular catalysts operating as homogeneous catalysts, work which culminated in the award of the Nobel Prize for Chemistry in 2001 to Noyori, Sharpless and Knowles.1 However, industry has been slow to take up these synthetic methodologies since separation of the catalyst from the reaction mixture can be difficult and hence catalyst recycle and reuse can be very difficult. For this reason, attention has recently focussed on the design of immobilised catalysts which can overcome these problems.2,3In general, progress in the design of robust heterogeneous enantioselective catalysts has proved to be much more elusive, and they tend to be very specific for particular reactions and classes of substrates, and there remain very few examples of heterogeneously catalysed reactions giving high enantioselection. Unfortunately, almost invariably, poorer enantioselection is achieved with the immobilised complex compared to the comparable homogeneous catalysts.4 However, there are a number of studies indicating that improved enantioselectivity can be achieved by immobilisation. In 1991, this was pioneered by the Corma and co-workers.5 In 2000 Raynor et al.6 showed that a carefully planned immobilisation strategy could result in enhanced enantioselection being observed with the immobilised catalyst when it is constrained within the ordered mesopores of MCM-41. They demonstrated this for the allylic amination of cinnamyl acetate using a catalyst based on 1,1′-bis(diphenylphosphino)ferrocene anchored to the inner wall of MCM-41 and coordinated to Pd2+. The approach was subsequently extended to the enantioselective reduction of ethyl nicotinate. The improved effect of immobilisation is noted in other studies7–9 and is considered to be due to a containment effect observed for the immobilised systems.10
We have used a different immobilisation strategy in which catalytically active cations are electrostatically immobilised within microporous and mesoporous materials by ion exchange and they are subsequently modified by chiral ligands.2 Until now, we have concentrated our attention on the heterogeneous asymmetric aziridination of alkenes11–14 using Cu2+ ion-exchanged into zeolite H-Y modified by chiral bis(oxazoline) ligands, but we have also used this approach to design heterogeneous enantioselective dehydration,15 epoxidation,16 Diels–Alder17 and imino–ene reactions.18 We have now used this immobilisation strategy to synthesise stable reusable catalysts for enantioselective hydrogenation.
Asymmetric hydrogenation reactions continue to demand attention using a whole library of chiral phosphine ligands, however, in many cases the enantioselectivity achieved with the immobilised complex can be significantly lower than those observed for the non-immobilised catalyst. It should be noted that other strategies for the stabilisation of homogeneous catalysts are being considered, for example the use of ionic liquids is producing interesting results,19–21 but most research effort has been directed towards immobilisation strategies. Progress can only considered to be made if the enantioselection or overall turnover numbers achieved with the stabilised or immobilised catalyst is comparable to the homogeneous catalyst, and to date there are only few examples of such improvement.6,18
There have been numerous previous studies concerning the design of immobilised asymmetric hydrogenation catalysts, due primarily to the central importance of hydrogenation as a unit process in industrial organic synthesis.22,23 Noyori et al.24 prepared an immobilised Ru-BINAP-diamine system that compared favourably with its homogeneous counterpart. Recent attention has focussed on the immobilisation of phosphine containing catalysts onto inorganic supports since their rigid structure prevent intermolecular aggregation of the active catalyst species which can lead to loss of activity in the polymer supported catalysts. Robust asymmetric catalysts using inorganic supports, such as MCM-41 have been reported by Raynor et al.,6 Hölderich and co-workers,25 de Rege et al.26 and by Augustine et al. for heteropolyacids.27 These researchers have shown that chiral Rh diphosphine complexes can be immobilised on MCM-41. However, these catalysts, although initially effective, tend to give lower enantioselection compared with their homogeneous counterparts and also typically required long reaction times. For example, Hölderich and co-workers25 immobilised Rh diphosphine complexes by adsorption onto MCM-41 and, although the catalysts were stable and could be reused, the ee obtained with the immobilised catalyst (85–92%) was significantly lower than that observed with the homogeneous catalyst (99%) and these catalysts required 24 h to achieve complete conversion. In this paper we demonstrate immobilisation strategies for enantioselective hydrogenation based on immobilisation of rhodium complexes into mesoporous materials. We demonstrate that comparably high enantioselection can be achieved with these supported catalysts using two immobilisation strategies using electrostatic forces. Furthermore, we demonstrate that the manner in which the immobilisation is achieved is of crucial importance in achieving high sustained enantioselectivity and reusability for these catalysts.
Results and discussion
To overcome the problems associated with the earlier immobilised enantioselective hydrogenation systems we considered that it is desirable to anchor the catalyst using electrostatic forces which does not require ligand modification that is often necessary for the preparation of immobilised catalysts using covalent linking of the ligand to the inorganic support.5,19,20 It is also more specific than just using physical adsorption, a method that is known to give extensive leaching of the active components, leading to poor stability.2 We have previously successfully used this approach for the immobilisation of copper bis(oxazoline) complexes,2 and now we extend this methodology to the supporting of chiral Rh phosphine complexes, using DuPhos 1 and Josiphos 2 ligands as representative examples.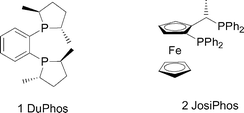
Our initial immobilisation methodology was carried out by direct immobilisation of the preformed [(R,R-MeDuPhos)Rh(COD)]+BF4− complex onto (H+)Al-MCM-41. Direct ion exchange of the acidic sites on MCM-41 was achieved by stirring a solution of the Rh complex with (H+)Al-MCM-41 for 1 h, filtration and washing of the yellow solid to give the supported catalyst [Rh(R,R-MeDuPhos)(COD)]–Al-MCM-41 3. This immobilised catalyst was then tested in the hydrogenation of dimethyl itaconate (Scheme 1).
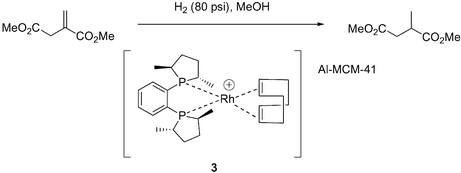 | ||
| Scheme 1 | ||
The results are shown in Table 1 for several successive experiments using a substrate : Rh molar ratio of 250 : 1 and 1 h reaction time at 20 °C. After the reaction the catalyst was allowed to settle and the liquid phase was removed and analysed. No rhodium was found in the liquid product and no activity was observed if fresh reactants were added to this solution. Fresh substrate was added to the recovered catalyst, with the same substrate : Rh molar ratio, and the experiment was repeated. Table 1 also shows the results for the homogeneous catalysts under the same reaction conditions and it is apparent that the immobilised catalysts give comparable enantioselection even after 8 reuses. Use of higher substrate : Rh molar ratio of 5000 : 1 is also possible, but some activity is lost on reuse possibly due to loss of the chiral ligand, but the ee is retained and is comparable to the homogeneous catalyst (Table 1).
| Substrate : Rh molar ratio | Number of times used | Time/h | Conversion (%) | Ee (%) |
|---|---|---|---|---|
| a Homogeneous catalyst [Rh(R,R-MeDuPhos)(COD)]BF4 used under the same conditions. | ||||
| 250 : 1 | 1 | 1 | >99 | >99 |
| 2 | 1 | >99 | >99 | |
| 3 | 1 | >99 | >99 | |
| 4 | 1 | >99 | >99 | |
| 5 | 1 | 99 | 98 | |
| 6 | 1 | 98 | 96 | |
| 7 | 1 | 95 | 95 | |
| 8 | 1 | 99 | 95 | |
| 250 : 1a | — | 1 | >99 | 99 |
| 5000 : 1 | 1 | 1 | >99 | 96 |
| 2 | 2.5 | 98 | 97 | |
| 3 | 12 | 97.5 | 94 | |
| 5000 : 1a | — | 1 | >99 | 99 |
In a subsequent set of experiments we used the immobilised catalyst 3 for the hydrogenation of methyl-2-acetomidoacrylate. The results (Table 2) show that this substrate can also effectively be asymmetrically hydrogenated and the catalyst can be readily recovered and reused.
Subsequently, we investigated the direct immobilisation of the preformed [(R,R-MeDuPhos)Rh(COD)]+BF4− complex onto (H+)Al-SBA-15. Direct ion exchange of the acidic sites on SBA-15 was achieved by stirring a solution of the Rh complex with (H+)Al-SBA-15 for 1 h, filtration and washing of the yellow solid to give the supported catalyst [Rh(R,R-MeDuPhos)(COD)]–Al-SBA-15 4. This immobilised catalyst was then tested in the hydrogenation of dimethyl itaconate and methyl-2-acetomidoacrylate. The initial usage of 4 as a heterogeneous catalyst gave reaction times for complete conversion and ee comparable to that observed for the homogeneous complex. However, the immobilised catalyst was not stable and significant quantities of Rh were leached during use, this may be due to the structure of Al-SBA-15 since the Al may have not been as accessible as in Al-MCM-41 due to the thickness of the wall structure. Consequently, the immobilised catalyst could not be readily reused and longer reaction times were required to achieve complete conversion and the ee also decreased significantly. For example, for the hydrogenation of methyl-2-acetomidoacrylate with 4, using the same conditions as in Table 2, for the first three catalyst uses the time for complete reaction increased from 1 h to 2.5 h and 12 h, and the ee decreased from 96–97% to 94%. This is in contrast to catalyst 3 prepared using MCM-41 but is in agreement with the very recent studies by Simons et al.28 in which similar Rh complexes were immobilised by ion exchange onto a new aluminosilicate AlTUD-1 which showed extensive leaching. These studies demonstrate the importance of the structure of the inorganic support with respect to catalyst stability and clearly immobilisation using the MCM-41 structure produces stable catalysts.
Following these initial successful experiments using the MCM-41 immobilised complex, a second, more flexible, immobilisation strategy was investigated which involved building the chiral Rh complex within the pores of the aluminosilicate. The Rh precursor, [Rh(COD)2]+BF4−, was initially ion-exchanged onto the (H+)Al-MCM-41 support, isolated by filtration and washed. Reaction of chiral ligands with this immobilised precursor allowed for a simple procedure for the preparation in situ of these complexes on the solid support. Initial treatment with R,R-MeDuPhos gave the immobilised catalyst 3. This catalyst was evaluated for the hydrogenation of dimethyl itaconate, as before, with a substrate : Rh molar ratio of 1000 : 1 at 20 °C and again 100% conversion with 98% ee was obtained. The material could be successfully reused several times without loss of Rh. Hence, no differences were observed in the catalytic performance or stability between the two immobilisation procedures. This demonstrates that the immobilised catalyst can be prepared either by direct immobilisation of the chiral complex or by formation of the chiral complex within the pores of the catalyst. We consider that this two step preparation strategy for immobilised catalysts offers great flexibility for catalyst design with a range of inorganic substrates particularly for use in high throughput studies since a range of ligands can be rapidly tested with a single immobilised catalyst precursor. We consider this novel strategy for immobilisation that significantly simplifies the strategy required for the synthesis of stable asymmetric immobilised catalysts.
The success of this approach allows for the simple immobilisation of other ligands without having to isolate the preformed catalyst. We have used an alternative chiral ligand, R,S-JosiPhos, to demonstrate the general applicability of our experimental methodology. [Rh(R,S-JosiPhos)(COD)]–Al-MCM-41 was prepared by reacting the free ligand 2 with the immobilised [Rh(COD)2]–Al-MCM-41 precursor. This catalyst was used for the hydrogenation of dimethyl itaconate using a substrate : Rh molar ratio of 500 : 1 at 20 °C with a reaction time of 15 min. Following reaction the catalyst was allowed to settle and the liquid was removed and analysed. As before fresh reagents were added using the same substrate : Rh molar ratio and condition. The results shown in Table 3 show that the catalyst can be reused several times without marked loss of catalyst performance.
Conclusions
Our results show that chiral Rh complexes can be easily immobilised by electrostatic interaction with negatively charged inorganic supports. Furthermore, the catalysts can be immobilised directly or the complex can be constructed within the pores of the support by first immobilising the active cation and subsequently modifying with the chiral ligand. We have found that the metal does not leach from the immobilised catalyst when MCM-41 is used as the support and that these catalysts can be readily recovered and reused effectively. The key observation is that high enantioselection and reactivity can be achieved using catalysts immobilised in this way which compare favourably with their homogeneous counterparts. In addition, the ion-exchange methodology is far simpler than the previously investigated immobilisation procedures using covalent linking of the chiral modifier to the inorganic support. Furthermore, the new two stage preparation strategy opens up the possibility of easy ligand-screening for parallel synthesis and libraries. Consequently, we consider this methodology provides a readily proven accessible pathway to highly enantioselective immobilised hydrogenation catalysts.Experimental
All experiments using metal complexes were performed under inert gas conditions using standard Schlenk techniques using anhydrous solvents.MCM-41 mesoporous aluminosilicate (Si : Al = 10 : 1, Si : H2O = 40 : 1) was prepared as follows. Tetramethylammonium hydroxide (101.21 g), cetyltrimethylammonium bromide (118.45 g), aluminium isopropoxide (26.56 g) and de-ionised water (860 ml) were stirred at 35 °C for 1 h. Fumed silica (78.0 g) was added and the mixture was stirred at room temperature for 1 h. The gel was then transferred to an autoclave, purged with nitrogen gas and heated at 150 °C for 48 h. After cooling to ambient temperature, the material was recovered by filtration and washed with de-ionised water (1 l) and ethanol (500 ml). The white solid was then oven-dried overnight (110 °C, 16 h) before being heated under nitrogen at 550 °C for 16 h. After this, the solid was calcined in air at 550° for 4 h.
SBA-15 mesoporous aluminosilicate (Si : Al = 8 : 1) was prepared as follows. Tetraethylorthosilicate (27 g), aluminium isopropoxide (3.9 g) and aqueous hydrochloric acid (30 ml, pH = 1.5) were stirred at 35 °C for 3 h. This was added to a second solution comprising poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol) tri-block copolymer (12 g, Mav 5800) dissolved in aqueous hydrochloric acid (450 ml, pH = 1.5). The mixture was stirred (25 °C, 1 h) and then transferred to an autoclave, purged with nitrogen gas and heated at 100 °C for 64 h. After cooling to ambient temperature, the material was recovered by filtration and washed with de-ionised water (1 l) and ethanol (500 ml). The white solid was then oven-dried overnight (110 °C, 16 h) before being heated under nitrogen at 550 °C for 4 h.
Preparation of [Rh(R,R-MeDuPHOS)(COD)]–Al-MCM-41 by direct ion-exchange was carried out as follows. The solid support H+Al-MCM-41, (0.2 g) and [Rh(R,R-MeDuPHOS)(COD)]+BF4− (0.020 g) in degassed methanol (5 ml) was heated at 55 °C for 1 h during which time the MCM-41 support became orange in colour. The mixture was filtered and the yellow–orange solid washed with dry degassed methanol (10 × 5 ml) to ensure that no uncomplexed Rh and ligand remained within the catalyst, the catalyst was then dried and stored under vacuum. [Rh(R,S-JosiPhos)(COD)]–Al-MCM-41 was prepared in an analogous manner.
[Rh-(R,R-MeDuPhos)(COD)]–Al-MCM-41 was prepared using a two step procedure as follows. The solid support H+Al-MCM-41 (Si/Al = 10, 0.2 g) and [Rh(COD)2]+BF4− (0.020 g, 0.05 mmol) were stirred in degassed methanol (5 cm3) at 50 °C for 16 h under a nitrogen atmosphere. The solid material became pale orange in colour. The liquid was decanted and a further portion of methanol added. The mixture was filtered and the solid [Rh(COD)2]–Al-MCM-41 was dried under vacuum. [Rh(R,R-MeDuPhos)(COD)]–Al-MCM-41 was then prepared by reacting [Rh(COD)2]–Al-MCM-41 (0.176 mg) with R,R-MeDuPhos (16 mg, 55 µmol) by stirring in degassed methanol (5 ml) at 50 °C for 1.5 h. The solid material changed in colour from pale orange to yellow, whilst the methanol solution also became a pale yellow colour. The mixture was cooled to 25 °C and then filtered. The yellow solid was washed thoroughly with dry degassed methanol several times to ensure that all non-complexed Rh and ligands were removed. The catalyst was then dried and stored under vacuum.
The general procedure for the hydrogenation reactions was as follows. The substrate and catalyst were weighed into a glass-liner that was placed inside a 50 ml autoclave to give the required substrate : catalyst molar ratio. The autoclave was sealed and flushed with nitrogen. The autoclave was then pressurised with hydrogen (to 80 psi) and then released (cycle repeated 5 times). Sufficient methanol was added to the autoclave to give an approximately 1 M solution and the 5 cycles of pressurising–releasing with hydrogen were repeated. Finally the autoclave was pressurised with H2 to 80 psi and left to stir. After the desired time the stirring was stopped and the H2 released slowly. The autoclave was flushed with nitrogen and the liquid phase removed by a syringe through the swagelok opening. A fresh aliquot of the substrate in solvent was added again through the swagelok opening and the autoclave was pressurised with hydrogen again. Products were analysed by chiral gas chromatography using a LIPODEX-E column
References
- Nobel lectures, Angew. Chem., Int. Ed., 2002, 41, 998 Search PubMed.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- L.-X. Dai, Angew. Chem., Int. Ed., 2004, 43, 5726 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS.
- A. Corma, M. Iglesias, C. Delpino and F. Sanchez, J. Chem. Soc., Chem. Commun., 1991, 1253 RSC.
- S. A. Raynor, J. M. Thomas, R. Raja, B. F. G. Johnson, R. G. Bell and M. D. Mantle, Chem. Commun., 2000, 1925 RSC.
- V. Ayala, A. Corma, M. Iglesias, J. A. Rincon and F. Sanchez, J. Catal., 2004, 224, 170 CrossRef CAS.
- M. J. Alcon, A. Corma, M. Iglesias and F. Sanchez, J. Organomet. Chem., 2002, 665, 134 CrossRef.
- A. Corma, M. Iglesias and F. Sanchez, Catal. Lett., 1995, 32, 313 CAS.
- J. M. Thomas, T. Maschmeyer, B. F. G. Johnson and D. S. Shephard, J. Mol. Catal. A, 1999, 141, 139 CrossRef CAS.
- G. J. Hutchings, Chem. Commun., 1999, 301 RSC.
- S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. Bullman Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin Trans. 2, 2001, 1714 RSC.
- S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. Bullman Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin Trans. 2, 2001, 1724 RSC.
- Y. Traa, D. M. Murphy, R. D. Farley and G. J. Hutchings, Phys. Chem. Chem. Phys., 2001, 3, 1073 RSC.
- S. Feast, D. Bethell, P. C. B. Pate, F. King, C. H. Rochester, M. R. H. Siddiqui, D. J. Willock and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1995, 2499 RSC.
- P. Piaggio, P. McMorn, D. Murphy, D. Bethell, P. C. Bullman Page, F. E. Hancock, C. Sly, O. J. Kerton and G. J. Hutchings, J. Chem. Soc., Perkin Trans. 2, 2000, 2008 RSC.
- Y Wan, P. McMorn, F. E. Hancock and G. J. Hutchings, Catal. Lett., 2003, 91, 145 CrossRef CAS.
- N. A. Caplan, F. E. Hancock, P. C. Bulman-Page and G. J. Hutchings, Angew. Chem., Int. Ed., 2004, 43, 1685 CrossRef CAS.
- A. Berger, R. F. de Souza, M. R. Delgado and J. Dupont, Tetrahedron: Asymmetry, 2001, 12, 1825 CrossRef CAS.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS.
- H. Olivier-Bourbigou and L. Magna, J. Mol. Catal. A, 2002, 182–183, 419 CrossRef CAS.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley-Interscience, New York, 1994 Search PubMed.
- H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale, Wiley-VCH, Weinheim, 2004 Search PubMed.
- T. Ohkuma, H. Takeno, Y. Honda and R. Noyori, Adv. Synth. Catal., 2001, 369 CrossRef CAS.
- H. H. Wagner, H. Hausmann and W. F. Hölderich, J. Catal., 2001, 203, 150 CrossRef CAS.
- F. M. de Rege, D. K. Morita, K. C. Ott, W. Tumas and R. D. Broene, Chem. Commun., 2000, 1797 RSC.
- R. Augustine, S. Tanielyan, S. Anderon and H. Yang, Chem. Commun., 1999, 1257 RSC.
- C. Simons, U. Hanefeld, I. W. E. Arends, R. A. Sheldon and T. Maschmeyer, Chem. Eur. J., 2004, 10, 5829 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |