Cyclohexane bis-urea compounds for the gelation of water and aqueous solutions†
Maaike
de Loos
a,
Arianna
Friggeri
b,
Jan
van Esch
*a,
Richard M.
Kellogg
a and
Ben L.
Feringa
*a
aDepartment of Organic and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: J.H.van.Esch@rug.nl; B.L.Feringa@rug.nl
bBiOMaDe Technology Foundation, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 21st March 2005
Abstract
A new class of efficient hydrogelators has been developed by a simple modification of the peripheral substituents of cyclohexane bis-urea organogelators with hydrophilic hydroxy or amino functionalities. These bis-urea hydrogelators were synthesised in two or three steps using an alternative procedure to the common isocyanate method. Gelation was obtained with organic solvents, water and strongly basic aqueous solutions like 25% ammonia. Hydrogelation was found to depend on a delicate balance between the hydrophobicity of the alkyl chains, hydrophilicity of the terminal substituents and the enantiomeric purity of the compound. The hydrogels consisted of a network of fibers, in which all urea groups are involved in intermolecular hydrogen bonding. Most likely, gelation is driven by hydrophobic interactions of the methylene units, whereas hydrogen bond formation between the urea groups provides the necessary anisotropy of the aggregation and the high thermal stability of the gels.
Introduction
Hydrogels find broad applications in, for instance, foods, pharmaceuticals, biomaterials, cosmetics and personal care products, and have therefore been studied extensively.1 Most of the hydrogels reported so far are based on polymers.1 However, as a result of recent developments in the field of low molecular weight (LMW) organogelators,2 the gelation of water and aqueous solutions by small molecules has been a topic of great current interest.3 Examples of LMW hydrogelators include compounds based on saccharides,4 bile acids,5 amino acids,6 nucleotides,7 nucleosides,8 gemini surfactants9 and dendritic compounds.10 Most of these compounds are derived from naturally occurring molecules and contain hydrophilic moieties together with aromatic groups or long hydrophobic alkyl chains. Owing to these structural properties, many of the hydrogelators reported so far are amphiphilic. Apart from a few examples, most of these hydrogelators were found by serendipity rather than by design.3b Focusing on a more rational approach, we decided to design a LMW hydrogelator, without the use of natural building blocks and clear-cut amphiphilic structures, by exploiting the self-assembling properties of the well-studied and highly efficient cyclohexane bis-urea organogelators.2b,11These compounds were developed based on the principle that gel formation is favoured by anisotropic self-assembly.2b Their molecular structure can roughly be divided into two parts: the cyclohexane bis-urea unit, designed to self-assemble in one-dimensional anisotropic stacks, and the peripheral substituents (Fig. 1). The peripheral substituents can be varied without disturbing the self-assembling ability and it is envisioned that these substituents partly determine the scope of gelated solvents. So far cyclohexane bis-urea compounds have only been reported to gelate organic solvents. However, if indeed the peripheral substituents determine the solvent scope, it should be possible to convert this typical organogelator into a hydrogelator by simply modifying these substituents with hydrophilic functionalities X, such as hydroxy groups, carboxylic acids or amines. A short hydrophobic spacer between the urea and the hydrophilic groups might facilitate the formation of intermolecular urea hydrogen bonds by shielding the urea from the aqueous phase.
 | ||
| Fig. 1 Design features for the conversion of a cyclohexyl bis-urea organogelator into a hydrogelator. | ||
In this article we report on the development of hydrogelators based on these design principles. The synthesis of cyclohexane bis-urea dialkanols and diaminoalkanes using a method alternative to the common isocyanate approach is described. The gelation behaviour of these compounds will be discussed as well as the properties of the corresponding gels.
Results and discussion
Synthesis
In general, cyclohexane bis-urea derivatives are prepared by reaction of trans-1,2-cyclohexanediamine with the corresponding isocyanate.11,12 The isocyanate in turn can be prepared from the carboxylic acid via a Curtius rearrangement.13 However, this synthesis strategy is not suitable for the preparation of cyclohexane bis-urea derivatives with terminal hydroxy or amino groups, since preparation of the required amino and hydroxy isocyanates at the reaction temperatures required for the Curtius rearrangement immediately leads to the formation of (poly-)carbamates and urea.12,14 Recently, the preparation of α,ω-isocyanato alkanols from amino alcohols has been reported.15 However the required reagent is not commercially available16 and the obtained isocyanate could not be isolated because in situ polymerisation into polycarbamates readily occurs. These problems could be circumvented by the use of protected alcohols or amines, however, this requires additional reaction steps.As an alternative to the reaction of 1,2-cyclohexanediamine with the desired isocyanato alcohol or amine, the reaction of an amino alcohol or diamine with trans-1,2-cyclohexanediisocyanate could also be considered. The synthesis of 1,2-cyclohexanediisocyanate under mild conditions has been reported, however, the desired diisocyanate was isolated in only 20% yield, whereas a large amount of a cyclic urea by-product is formed.15a Furthermore, the required reagent is not commercially available but has to be synthesised using the highly toxic gas phosgene.
To prevent the formation of the cyclic urea by-product, the isocyanates can be masked with alcohols in the form of their corresponding carbamates. It has been known for several decades that these form urea upon reaction with amines.17 Recently, an extensive study resulted in a mild reaction procedure in which phenyl carbamates and a variety of amines are employed with DMSO as solvent.17d Interestingly, the aminolysis of phenyl carbamates is compatible with numerous functionalities, including hydroxy groups. This offers the possibility of applying this reaction in a more direct synthesis of the desired 1,2-cyclohexane bis-urea alkanols and aminoalkanes.
Following this strategy, the desired cyclohexane bis-urea derivatives were synthesised in only two or three straightforward steps starting from commercially available compounds (Scheme 1). First the bis-phenyl carbamate 1 was prepared by reaction of trans-1,2-cyclohexanediamine with phenyl chloroformate. The modest yield is compensated by the possibility to perform the reaction on a large scale (>44 mmol) yielding multi-gram amounts of 1. Furthermore, compound 1 is very stable and can be stored at low temperatures for months. Subsequent reaction with the appropriate amino alcohols yielded cyclohexane bis-urea dialkanols 2 and 3. Unfortunately, the amino derivative 5 could not be obtained via direct reaction of the diamine with the carbamate, due to difficulties in the separation of the desired product from the excess diamine employed. However, the use of a commercially available mono-protected diamine, followed by one additional deprotection step, did yield bis-urea 5. For all bis-urea compounds the yields are good, isolation and purification of the products is straightforward and the reactions can easily be scaled up. The bis-urea compounds were obtained as white solids and were characterised by 1H NMR, 13C NMR and elemental analysis or mass spectrometry.
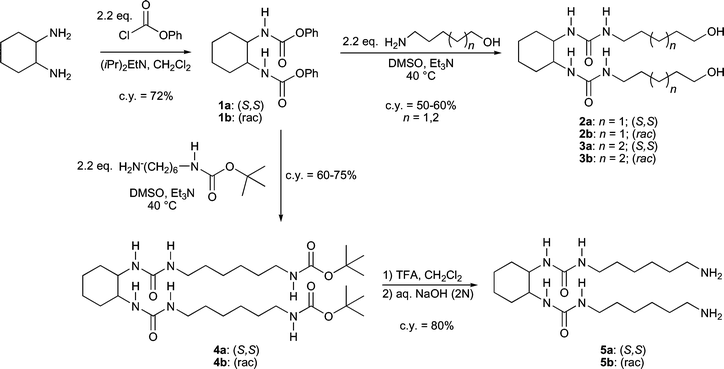 | ||
| Scheme 1 Synthesis of the cyclohexane bis-urea dialkanols and diaminoalkanes 2, 3 and 5. | ||
Gelation of organic solvents
At room temperature compounds 2–5, like many other organogelators, were found to be sparingly soluble in common organic solvents like ethanol, acetone or chloroform. The gelation properties were studied by first dissolving them by heating in the appropriate solvent, followed by cooling to room temperature. For several of the solid–solvent combinations, the formation of a gel was observed. The results are shown in Table 1, in which the solvents are arranged in order of increasing polarity according to their ET(30)-values.| Solvents | 2a | 2b | 3a | 3b | 4a | 4b | 5a | 5b |
|---|---|---|---|---|---|---|---|---|
| a Abbreviations used: digits: gel formation with minimal gelation concentration in mg ml−1; i: insoluble at solvent reflux temperature; p: precipitates; s: soluble at room temperature (solubility >20 mg ml−1); gp: gel-like precipitate; pg: partial gel; r: reaction with the solvent. b Precipitation above 20 mg ml−1. c Solubility below 1 mg ml−1. | ||||||||
| Hexadecane | i | i | i | i | i | i | i | i |
| p-Xylene | i | i | s | i | 10 | 10 | 5 | 5 |
| Tetraline | p | i | 10 | 10 | 20 | 20 | i | i |
| n-Butyl acetate | p | p | 5 | p | 5 | gp | pg | p |
| Cyclohexanone | p | p | p | p | pg | sb | r | r |
| 1,2-Dichloroethane | p | p | 5 | 10 | p | 20 | gp | i |
| DMSO | s | s | s | s | s | s | — | — |
| 1-Octanol | p | p | p | p | gp (>10) | p | sc | p |
| i-Propanol | p | p | s | s | gp (>10) | p | sc | p |
From Table 1, it can be seen that the gelation of organic solvents by the dialkanol compounds 2 and 3 exhibits straightforward behaviour: compound 2 (pentyl spacer) does not gelate any of the solvents tested but forms precipitates, whereas compound 3 (hexyl spacer) forms gels with solvents like tetraline and 1,2-dichloroethane. Furthermore, both compounds are soluble in DMSO, a solvent known to break up hydrogen bonds.18
Apparently, introduction of only one extra carbon atom in the side chains increased the compatibility of the compound with the organic solvents to such an extent that gel formation was obtained in some cases.19 The observed differences in gelation behaviour could also be due to an odd–even effect,20 however, preliminary results on dialkyl cyclohexane bis-amides are not in accord with such an effect.19 Compared to the cyclohexane bis-urea dialkyl compounds, the gelation ability for organic solvents has decreased as can be expected upon introduction of hydrophilic moieties.11
This is supported by the fact that the even more hydrophilic bis-aminoalkane compound 5 displays a decreased gelation ability compared to both the cyclohexane bis-urea dialkyl compounds11 and the dialkanol compound 3. In most solvents tested, the amino compounds were not soluble and only with p-xylene was a turbid gel formed. On the other hand, its precursor 4, which contains a hydrophobic t-butyl group as the peripheral substituent, was able to gelate aromatic solvents, n-butyl acetate and 1,2-dichloroethane. Furthermore, with the polar solvents, in most cases aggregation was observed and instead of a gel only a gel-like precipitate or a partial gel was formed. Compared to the cyclohexane bis-urea compounds the solvent scope of 4 has decreased and the minimal gelation concentrations have increased.11 This reduction in gelation ability can be explained by the bulkiness of the t-butyl groups, which will most likely disturb the packing of the molecules in the stacks. Furthermore, they will hinder the formation of additional hydrogen bonds between the carbamates, which would strengthen the aggregates.
Comparison of the gelation behaviour of the enantiomerically pure compounds (a) and the racemic compounds (b) does reveal slight differences depending on the solvent, but clear trends cannot be observed.
Gelation of water and aqueous solutions
At room temperature, compounds 2–5 were found to be insoluble in water and most aqueous solutions. The hydrogelation properties of compounds 2–5 were investigated as described for the gelation of organic solvents and the results are depicted in Table 2.| Solvents | 2a | 2b | 3a | 3b | 4a | 4b | 5a | 5b |
|---|---|---|---|---|---|---|---|---|
| a Abbreviations used, see Table 1; c: crystallization; g: gel formation. b Buffer system: K2HPO4/KH2PO4 (0.1 M). c Crystal formation in time. d Precipitation above 20 mg ml−1. e Gelation time of several months. | ||||||||
| Water (demi) | 10c | sd | c | 2–10 | i | i | 5 | 7 |
| Bufferb pH 8.4 | — | — | c | c | — | — | 10 | 10 |
| Bufferb pH 8.4 pH 7.8 | — | — | c | c | — | — | p | 20 |
| Bufferb pH 8.4 pH 6.8 | — | — | c | c | — | — | sc | s |
| Bufferb pH 8.4 pH 6.0 | — | — | c | c | — | — | s | s |
| 1N NaOH | — | — | — | — | — | — | ge | 5 |
| 25% Ammonia | — | — | — | — | — | — | 2 | 5 |
| 1N NaHCO3 | — | — | — | — | — | — | i | 10e |
At low concentrations, the enantiomerically pure compound 2a was found to form solutions in which a gel-like precipitate was formed after several weeks of standing. At a concentration of 10 mg ml−1 a weak, highly turbid gel was formed. A further increase in the concentration resulted in crystallisation of the compound. Its racemic counterpart, compound 2b, was found to be soluble in all tests up to concentrations of 15 mg ml−1 and at higher concentrations, precipitation was observed.
Elongation of the alkyl spacer with one carbon atom resulted in a different behaviour. The enantiomerically pure compound 3a formed microcrystals, which were unfortunately not suitable for X-ray crystallography. However, for its racemic counterpart 3b it was observed that at concentrations between 2–10 mg ml−1 a clear, stable gel was formed. The formation of these gels was very slow; four to eight weeks of standing at room temperature is necessary. At higher concentrations, crystalline or non-crystalline precipitates were formed, depending on the cooling rate. Gelation tests for compound 3 with a series of buffers of different pH revealed that neither the enantiomerically pure compound nor the racemic compound was able to form a gel with these buffer solutions. Instead, microcrystals were formed.
The minimal gelation concentrations for 2a and 3b are comparable to those reported for other hydrogelators.3 Remarkably, compound 3b was able to gelate both water and organic solvents, a property that was also observed for some other hydrogelators.4a,d,e,5a,6d,9a
The fact that dialkanols 2a and 3b only form hydrogels within a very narrow concentration range and exhibit long gelation times makes them inefficient. Furthermore, it seems that only water can be gelated and aqueous solutions of salts are not tolerated. Substitution of the hydroxy groups with more hydrophilic amino groups to increase the solvent compatibility is expected to improve the gelation ability for water.21 Indeed, compound 5 was found to be soluble in hot water and upon cooling, clear gels were formed within one hour both for the racemic mixture and the enantiomerically pure compound. Compared to the long gelation times of diol compounds 2 and 3, this is a major improvement of the gelation ability. Furthermore, compound 5 was able to gelate water at concentrations of 5 to at least 20 mg ml−1, which is a much broader range than those observed for the diol compounds. The minimal gelation concentrations of the diamine compounds are comparable to those obtained for other hydrogelators.3 The BOC-protected precursor 4 was found to be completely insoluble in water as expected from the lack of terminal hydrophilic substituents.
Encouraged by these results, a series of buffers and basic aqueous solutions was also tested. The tests revealed that at pH <7, solutions were obtained, but with several basic aqueous solutions and buffers, gels were formed (Table 2). Even with 25% ammonia, clear gels were formed at rather low concentrations. Interestingly, this is one of the very few examples of the gelation of such strongly basic solutions.10a Remarkably, in contrast to most LMW gelators,22 the racemic compound was found to gelate a broader range of aqueous solutions than the enantiomerically pure compound.
It should be mentioned that the pH of the obtained hydrogels did not match the original pH of the applied buffers and preliminary tests showed that the gels possess a pH of ∼11. To gain a better insight into the pH sensitivity of the minimal gelation concentrations, a series of tests was performed in which the pH of the hydrogel of 5b was decreased stepwise by addition of aqueous HCl (1N) until the gel turned into a solution. The pH at this transition point was plotted against the concentration of the hydrogel (Fig. 2).
 | ||
| Fig. 2 pH dependency of the gel to sol transition of the hydrogel of 5b. | ||
It can be seen that the pH of the gel to sol transition ranged from 11.2 to 10.1 in the measured concentration range. For each point the amount of neutral, monoprotonated and diprotonated molecules was calculated using the pKa values of both amino groups. It was assumed that the amino groups acted independently from each other, yielding pKa(1) = pKa(2) = pKa. Using a pKa value of 10.6,23 it was calculated that at c = 12.5 mM, the amount of neutral molecules was dominant, whereas at higher concentrations the diprotonated species was the most abundant molecule. Due to the presence of the double charge, this species is expected to be highly water soluble and not able to contribute to gel formation. However, at each point the total concentration of neutral and monoprotonated molecules corresponds to or exceeds the minimal gelation concentration (c = 12.5 mM). Therefore, it is proposed that both the neutral and the monoprotonated molecules participate in the gelation. For the enantiomerically pure gels, the pH sensitivity could be different although this was not investigated.
Infrared spectroscopy
The gelation of apolar organic solvents by cyclohexyl bis-urea compounds was found to be driven by the formation of intermolecular hydrogen bonds between the urea groups.11 The cyclohexyl bis-urea hydrogelators 2–5 possess the same aggregating unit and therefore it can be expected that hydrogelation by these compounds is also accompanied by the formation of intermolecular hydrogen bonds. In water, the detection of hydrogen bonds by FT-IR spectroscopy is strongly hindered. Therefore, to study hydrogen bond formation in the hydrogel, spectra were recorded of freeze dried hydrogels or gels of deuterated water. In the latter case, replacement of the hydrogens of the urea group by deuteriums causes a shift of the amide I and amide II bands to lower wavenumbers.24 The results for compounds 3 and 5 are shown in Table 3.First, a FT-IR spectrum of a solution of 3b in DMSO was measured. Absorptions were observed at 1669 and 1558 cm−1 for the amide I and amide II bands, respectively. The position of the amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band is characteristic for a non-hydrogen bonded urea group24 and is comparable to the amide I band observed for solutions of non-hydrogen bonded cyclohexane bis-urea organogelators.11 Thus it can be concluded that 3b does not form intermolecular hydrogen bonds in DMSO. However, the involvement of hydrogen bonding is more difficult to deduce from the position of the amide II (N–H) band, since it is located in between the values observed for non-hydrogen bonded (1540–1565 cm−1) and hydrogen bonded (1565–1590 cm−1) urea groups.11 In this respect, it should be taken into account that DMSO is able to act as a hydrogen bond acceptor and it can be expected that hydrogen bonds are formed involving the N–H protons of the urea groups. Then it is most likely that the position of the amide II band will be indicative of an urea group hydrogen bonded to DMSO. This is also observed for lysine derived hydrogelators.6g
O) band is characteristic for a non-hydrogen bonded urea group24 and is comparable to the amide I band observed for solutions of non-hydrogen bonded cyclohexane bis-urea organogelators.11 Thus it can be concluded that 3b does not form intermolecular hydrogen bonds in DMSO. However, the involvement of hydrogen bonding is more difficult to deduce from the position of the amide II (N–H) band, since it is located in between the values observed for non-hydrogen bonded (1540–1565 cm−1) and hydrogen bonded (1565–1590 cm−1) urea groups.11 In this respect, it should be taken into account that DMSO is able to act as a hydrogen bond acceptor and it can be expected that hydrogen bonds are formed involving the N–H protons of the urea groups. Then it is most likely that the position of the amide II band will be indicative of an urea group hydrogen bonded to DMSO. This is also observed for lysine derived hydrogelators.6g
Subsequently, spectra were recorded for the microcrystals formed by enantiomerically pure 3a during hydrogelation tests. Compared to the solution of 3b, the vibration bands of amide I and amide II bands were shifted and, together with the NH stretch band, observed at positions characteristic for the presence of hydrogen bonded urea groups.11,24 Thus, in these microcrystals the molecules are connected via hydrogen bonds between the urea groups.
The freeze dried racemic hydrogels formed by 3b and 5b also had vibration bands for the NH stretch and amide I and amide II at positions characteristic for hydrogen bonded urea groups. Thus, it can be concluded that also in the hydrogels of 3b and 5b, hydrogen bond formation between the urea groups contributes to the assembly of the molecules.
Additionally, spectra were recorded of D2O gels of the diamine compounds 5a and 5b.25 In both cases absorptions were observed at 1610 and 1510 cm−1 for the amide I and amide II bands. These positions are characteristic for hydrogen bonded urea groups in which the N–H protons are replaced by deuteriums.24
These FT-IR measurements strongly indicate that, despite the presence of water, the formation of a hydrogel is accompanied by the formation of intermolecular hydrogen bonds between the urea groups. The vibration bands in the freeze dried gels are observed at positions almost identical to the positions observed in bis-urea organogels (i.e. 3330 cm−1 for the NH stretch band and 1632 and 1590 cm−1 for the amide I and II bands),11 suggesting that the aggregates are similar. However, in view of results reported in the literature3 it is not very likely that hydrogen bond formation is the primary driving force for hydrogelation. Most likely, gelation will be driven by hydrophobic interactions of the methylene units, whereas urea hydrogen bonding will provide the necessary anisotropy of the aggregation and the high thermal stability of the gels (vide infra).26
Morphology of the gels
The morphology of the gels was studied using Transmission Electron Microscopy (TEM). Micrographs of tetralin gels of 3a and 3b and a hydrogel of 3b are shown in Fig. 3. The tetraline gels of the enantiomerically pure 3a (Fig. 3A) and racemic 3b (Fig. 3C) show profound differences. The enantiomerically pure compound displays an unusual morphology, consisting of large platelets together with tiny twisted fibres at the edges of these platelets. These fibres were typically 25 nm wide and exhibited a left-handed twist with a pitch of approximately 100–150 nm (Fig. 3B). The tetraline gel was found to precipitate overnight and it is possible that this instability is the origin of the observed large platelets. | ||
| Fig. 3 Electron micrographs of (A) a tetraline gel of 3a (c = 10 mg ml−1; 25 mM), (B) enlarged section of (A) showing twisted fibres, (C) a tetraline gel of 3b (c = 10 mg ml−1; 25 mM) and (D) the hydrogel of 3b (c = 2 mg ml−1; 5 mM). The bar represents 1 µm. | ||
The racemic tetralin gel of 3b displays a completely different morphology that consists of elongated, crystalline like, rigid fibres. The occurrence of tiny twisted fibres was not observed. The fibres were flat and could be up to 7 µm long and 80–500 nm wide. It was observed that the flat fibres were composed of several layers. No intertwining was observed, although the fibres were found to split occasionally. The formation of twisted fibres by an enantiomerically pure compound and flat sheet like fibres by a racemic compound has also been observed for other gelators.9a It indicates that the racemic compound does not separate into two enantiomeric chiral phases, but forms mixed aggregates. This is in agreement with results obtained for cyclohexane bis-urea derivatives in organic solvents11d and can be compared with the chiral bilayer effect as observed for amphiphiles.4a,27 The hydrogel formed by the racemic compound 3b displays a morphology that is somewhat similar to that of the corresponding tetralin gel (Fig. 3D). As for the tetralin gel when elongated, flat fibres are observed with a length of up to 11 µm and a width of 40–500 nm. However, these fibres appear to be less crystalline and rigid and a clear, fine structure can be observed. The fibres fuse and split regularly to form the gel network. Analogously to the tetralin gel, the racemic compound does not separate into two chiral phases.
The morphologies of the enantiomerically pure and racemic gels of the BOC-protected derivative 4 in p-xylene and of the free amine derivative 5 in p-xylene and water are illustrated in Fig. 4. It was observed that the p-xylene gels of the BOC-protected derivative displayed a somewhat similar morphology both for the enantiomerically pure compound 4a (Fig. 4A) and the racemic compound 4b (Fig. 4B). In both cases, the gel consists of rigid crystalline like fibres, which appear to be multi-layered. However, a difference can be found in the dimensions of the fibres. The fibres in the racemic gel exhibited a length of 2–9 µm and a width of 80–300 nm. In the enantiomerically pure gel, the fibres appeared to be shorter and narrower, with a length of 1–6 µm and a width of 50–200 nm. Furthermore, the fraction of long fibres had decreased and the fibres seem to be more brittle and fragmented. Possibly, this is related to the fact that an enantiomerically pure sample is used, which compared to the racemic gel might lead to an enhanced crystallinity. For both gels, twisting of the fibres was not observed.
 | ||
| Fig. 4 Electron micrographs of p-xylene gels of (A) 4a (c = 10 mg ml−1; 17 mM), (B) 4b (c = 10 mg ml−1; 17 mM), (C) 5a (c = 10 mg ml−1; 25 mM), (D) 5b (c = 10 mg ml−1; 25 mM) and hydrogels of (E) 5a and (F) 5b (c = 10 mg ml−1; 25 mM). The bar represents 1 µm. | ||
The p-xylene gels of the amine compounds 5a (Fig. 4C) and 5b (Fig. 4D) displayed morphologies very similar to each other. In both cases elongated, multi-layered sheets were observed, which did not intertwine. The length of the sheets was approximately 5–11 µm and the width 60–900 nm. Compared to their BOC-protected counterparts the aspect ratio of the fibres formed by 5a and 5b has clearly increased.
The hydrogels of the deprotected compounds 5a (Fig. 4E) and 5b (Fig. 4F) displayed completely different morphologies, also compared to the hydrogel of 3b. Instead of flat, sheet-like fibres, now in both cases elongated, thin, thread-like fibres are observed and it seems that these fibres are even more flexible than the fibres in the gel formed by 3b. The fibres in the enantiomerically pure gel formed by 5a were up to 10 µm long and 7–50 nm wide. Often a right-handed twisting of the thin fibres was observed. The thicker fibres are clearly built up from the thinner fibres. For the racemic gel the fibres appeared to be thicker with a length of up to 10 µm and a width of 12–300 nm. As observed in the gel of 5a, the thicker fibres were built up from thin fibres. Compared to the enantiomerically pure gel, the fibres in the racemic gel seemed to be somewhat flattened. Occasionally an irregular right-handed twist was observed. In both gels the fibres fuse and intertwine regularly to form a dense network.
Thermotropic properties
The thermotropic properties of the hydrogels of 3b, 5b and 5a have been investigated by the dropping ball method (Fig. 5). This method is used to determine the temperature at which the gel has lost its mechanical stability, i.e. has melted, and is not able to support a steel ball any more.28 At this point the network structure of the gel is disrupted, although large aggregates can still be present. The racemic hydrogel of the bis-urea diol 3b was found to melt at temperatures between 60 and 96 °C, depending on concentration. Such a concentration dependency is commonly observed for low molecular weight gelators.2 The melting temperatures for 3b are higher than the melting temperatures of the racemic hydrogel of the corresponding diamine 5b. Apparently, the gels formed by diol 3b are thermally more stable than the gels formed by diamine 5b. This can be explained by the fact that the dialkanol 3b is less hydrophilic than the diaminoalkane 5b and thus is less soluble in water. This will result in a shift of the gel-to-sol transition towards higher temperatures. This is consistent with the observed lower minimal gelation concentrations of 3b. | ||
| Fig. 5 Thermal behaviour of one year old hydrogels of 3b (-♦-) and fresh hydrogels of 5a (-●-) and 5b (-○-) as deduced by the dropping ball method. | ||
Interestingly, in contrast to most low molecular weight gelators, the hydrogel of racemic 5b melts almost independently of concentration. Furthermore, it was observed that the sample became more turbid upon heating. This suggests a change in the aggregation state upon increasing the temperature. After melting of the gel, a turbid sample remained, which turned into a clear solution at higher temperatures.
The enantiomerically pure hydrogels formed by the diamine 5a melt at the highest temperatures and thus are thermally the most stable. At concentrations above 17 mM, the melting of the hydrogel of the enantiomerically pure compound could not be observed because for safety reasons, the heating had to be stopped at 120 °C. At these concentrations the racemic compound had already melted at T ≅70 °C. Such differences between the racemic and the enantiomerically pure mixture can also be found for cyclohexane bis-urea organogelators.29
It was found that all gels showed thermoreversible behaviour, i.e. after cooling of the melted samples the gel phase was regained.
Compared to other hydrogelators,3 the hydrogels formed by the cyclohexane bis-urea compounds described here are among the most thermally stable hydrogels. This is most likely due to the presence of strong intermolecular hydrogen bonds between the urea groups.
Stereochemical aspects of the gelation ability for water
For the gelation of water, striking differences are observed between the enantiomerically pure and racemic compounds, especially for dialkanols 2 and 3. Both compounds are able to gelate water, however for 2 only the enantiomerically pure compound forms a gel, whereas for 3 only the racemic mixture forms a gel. For diaminoalkane 5 it is observed that both the enantiomerically pure and the racemic compound form a gel, although with higher minimal gelation concentrations and a much lower thermal stability for the latter together with a slightly broader scope. The behaviour of 2 and the higher gelation concentrations of 5b are in accordance with the general gelation behaviour observed for small molecules.22 However, the gelation behaviour of 3 and the broader scope of 5b contrasts with this. These results can be explained by considering the process of gelation in more detail (Scheme 2).2b,30
The gel is a metastable phase between the crystal phase and the solution phase, favoured by the presence of units that provide anisotropic self-assembly, and is balanced by gelator–gelator interactions and gelator–solvent interactions.2b Gelator–gelator interactions dictate self-assembly of the molecules into stacks and subsequently fibres leading to gel formation. Interactions that are too strong and lack of anisotropy result in the formation of a densely packed crystal phase or precipitate. Gelator–solvent interactions determine the compatibility of the compound with the solvent and which solvents are gelated, although gelator–solvent interactions that are too strong will prevent aggregation and the solution will remain.
The results suggest that compound 2 acts on the border between the gel phase and the solution phase. The gelator–solvent interactions are balanced by the hydrophilicity of the hydroxy group and the hydrophobicity of the alkyl chains and the cyclohexyl ring. Due to the hydroxy groups, compound 2 is compatible with water and heating results in dissolution. However due to the hydrophobic alkyl chains and cyclohexyl ring, the solubility is not infinite and the compounds tend to aggregate. Furthermore, the hydrogen bonding urea groups are shielded from the water by these hydrophobic groups. As a result of the shielding, hydrogen bond formation can provide anisotropy and together with the hydrophobic interactions of the alkyl chains the molecules can assemble into stacks.
At this point the enantiomeric purity of the compound becomes important. Most likely the racemic compound 2b will form stacks in which both the enantiomers are packed in an alternating fashion within the same stack (Scheme 2; top left).11d The resulting disorder prevents the stacks from assembling together to form large fibres and the compound will stay in solution (Table 2). The enantiomerically pure compound 2a will form translational aggregates, which results in more ordered stacks (Scheme 2; bottom left).11d These stacks can then assemble into fibres, which in turn form the gel. Apparently, the gelator–gelator interactions for this compound are not too strong and crystallisation is not observed.
For compound 3 the behaviour is completely different and it is observed that compound 3 acts on the border between the gel phase and the crystalline phase (see Table 1 and Table 2). As for compound 2 the hexyl spaced derivative 3 is compatible with water as can be concluded from the dissolution at elevated temperatures. However, since the alkyl chains are elongated with one carbon atom, the hydrophobic part of the compound is enlarged, resulting in weaker gelator–solvent interactions and stronger gelator–gelator interactions compared to 2. This will result in an increased aggregation ability, which is supported by the observation that upon cooling the compound is not soluble but forms a gel or crystals. Due to this enhanced aggregation ability, the gelation behaviour as a function of the stereochemistry changes. For the enantiomerically pure compound 3a regular translational aggregates will be formed which can be densely packed into fibres (Scheme 2).11d The increased gelator–gelator interactions most likely cause the fibres to be more crystalline in nature, ultimately resulting in the formation of crystals.
However, for the racemic mixture 3b, stacks are formed in which both the enantiomers are packed in an alternating fashion within the same stack (Scheme 2). The disorder in these stacks will prevent the formation of crystalline fibres and subsequently crystals. However, compared to compound 2 the gelator–gelator interactions are stronger and instead of a solution a gel is obtained.
For compound 5 the differences between the racemic and enantiomeric pure mixture are more subtle. Due to the increased hydrophilicity of the amino groups, the gelator–solvent interactions for 5 are somewhat stronger compared to 3. This is reflected in the lower gel-to-sol transition temperature, higher critical gelation concentrations and broader scope of gelated aqueous solutions. The expected disorder in stacks of racemic 5b results in the observed higher critical gelation concentrations and lower thermal stability compared to 5a. However for some aqueous solutions, the disorder of the stacks prevents the formation of highly crystalline fibres, like for compound 3b, resulting in an enlarged scope of gelated aqueous solutions.
Conclusions
In conclusion, a new class of efficient hydrogelators was developed based on design. The compounds were prepared by a simple modification of the peripheral substituents of the well-studied cyclohexane bis-urea organogelators with hydrophilic hydroxy and amino functionalities. For their synthesis an alternative method was used by applying phenyl carbamates instead of isocyanates. This resulted in a straightforward and easily applicable two or three step synthesis of the cyclohexane bis-urea dialkanols and diamines.The compounds are capable of gelating organic solvents, water and basic aqueous solutions. Their minimal gelation concentrations are comparable to those reported for other hydrogelators. The more hydrophilic cyclohexane bis-urea diaminoalkane compound is less efficient in the gelation of organic solvents than the cyclohexane bis-urea dialkanols. In contrast, the gelation ability for water had increased after substitution of the hydroxy groups with amines as can be expected from the increased hydrophilicity. Furthermore, diamine bis-urea belongs to the very few compounds able to gelate highly basic solutions like 25% aq. ammonia.10a
Remarkably, the enantiomeric purity of the compounds, together with the balance between the hydrophilicity of the hydroxy groups and the hydrophobicity of the alkyl spacers, had a pronounced effect on the hydrogelation by the dialkanol compounds.
TEM and FT-IR measurements showed that the hydrogels of 2–5 consisted of a network of fibers, comparable to the morphologies observed for cyclohexane bis-urea organogels,11 and all urea groups are involved in intermolecular hydrogen bonding. The latter finding indicates that water molecules do not interfere, presumably due to shielding of the urea groups from the water by the hydrophobic alkyl spacers. Most likely, gelation will be driven by hydrophobic interactions of the methylene units, whereas urea hydrogen bonding will provide the necessary anisotropy of the aggregation and is the origin of the high thermal stability of the gels.
These results confirm the initial considerations in the design of the cyclohexane bis-urea organogelators,2b,11i.e. an anisotropic self-assembling cyclohexane bis-urea unit combined with peripheral substituents that govern the solvent compatibility.
Experimental
General information
Melting points (uncorrected) were determined using a Stuart Scientific SMP1 melting point apparatus. FT-IR spectra were recorded on a Nicolet Nexus FT-IR apparatus. 1H NMR spectra were recorded on a Varian Gemini-200 (200 MHz) or a Varian VXR-300 (300 MHz) spectrometer operating at ambient temperature. Chemical shifts are denoted in δ-units (ppm) relative to the residual solvent peaks (CDCl3 = 7.26; DMSO-d6 = 2.49; MeOH-d4 = 3.31). J values are given in Hz. 13C NMR spectra were recorded on a Varian Gemini-200 (50.32 MHz) or a Varian VXR-300 (75.48 MHz) spectrometer. Chemical shifts are denoted in δ-units (ppm) relative to the solvent peaks (CDCl3 = 76.91; DMSO-d6 = 39.5; MeOH-d4 = 49.0) and converted to the TMS scale. The splitting patterns are designated as follows: s (singlet), bs (broad singlet), d (doublet), bd (broad doublet), t (triplet), q (quartet), m (multiplet) and bp (broad peak). Elemental analyses and mass spectrometry were performed at the analytical department of this laboratory.Phenyl chloroformate was purchased from Aldrich. Racemic and enantiomerically pure trans-1,2-cyclohexanediamine, 5-aminopentanol, 6-aminohexanol and tert-butyl 6-aminohexylcarbamate were purchased from Fluka.
Gelation experiments
In a typical gelation experiment, a weighed amount of the compound under investigation and 0.5 mL or 1.0 mL of the solvent were placed in a closed vial. The vial was heated using a heating gun or a heating block until the solid had dissolved, unless the solvent started to reflux prior to dissolution. The solution was allowed to cool to room temperature and was subsequently examined. Gelation was considered to have occurred when a homogeneous substance was obtained that exhibited no gravitational flow.Tests on pH dependent gelation
Gels with a volume of 0.5 mL or 0.25 mL were prepared as described above. To the gel was added a small amount (2–5 µL) of aqueous HCl (1N). The gel was melted by heating and was subsequently cooled to room temperature. The sample was examined to determine whether a gel or solution had formed. The sequence of addition of aliquots of acid, melting, cooling and examination was repeated until a solution was obtained. At this point, the pH of the sample was measured.Transmission electron microscopy
Gels were prepared as described above. Collidon and carbon coated 400 mesh copper grids were prepared following standard procedures. A piece of gel was carefully placed on a grid and shadowed with platinum (angle: 40°, distance: ∼15 cm). The samples were examined in a JEOL 1200 EX transmission electron microscope operating at 80 kV. First patches of gel were searched for, to be sure that the observed structures originate from the gel. Micrographs were taken from the periphery of the gel.Dropping ball measurements28
Gels with a volume of 1.0 mL were prepared as described above. A stainless steel ball (63 mg; ∅ 2.5 mm) was placed on top of the gel and the vial was closed. A series of these samples was placed in a heating block that was slowly heated (5 °C/h) while observing the positions of the balls with a video camera and simultaneously monitoring the temperature by means of a thermocouple placed in the heating block. Unless stated otherwise, the melting temperature of the gel was taken as the temperature at which the steel ball reached the bottom of the flask.References
- (a) Polymer gels: Fundamentals and Biomedical Applications, ed. D. DeRossi, K. Kajiwara, Y. Osada and A. Yamauchi, Plenum Press, New York, 1991 Search PubMed; (b) Hydrogels in Medicine and Pharmacy; Vol. III: Properties and Applications, ed. N. A. Peppas, CRC Press, Boca Raton, 1987 Search PubMed.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef; (b) J. van Esch, F. S. Schoonbeek, M. de Loos, E. M. Veen, R. M. Kellogg and B. L. Feringa, NATO ASI Ser., Ser C, 1999, 527, 233–259 CAS; (c) J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef.
- (a) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS; (b) M. de Loos, B. L. Feringa, J. van Esch, Eur. J. Org. Chem., submitted Search PubMed.
- Recent examples: (a) J. H. Fuhrhop, P. Schnieder, E. Boekema and W. Helfrich, J. Am. Chem. Soc., 1988, 110, 2861–2867 CrossRef CAS; (b) S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3504–3511 CrossRef CAS; (c) M. Amaike, H. Kobayashi and S. Shinkai, Chem. Lett., 2001, 620–621 CrossRef CAS; (d) O. Gronwald and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 2001, 1933–1937 RSC; (e) J. H. Jung, S. Shinkai and T. Shimizu, Chem. Eur. J., 2002, 8, 2684–2690 CrossRef CAS; (f) H. Kobayashi, A. Friggeri, K. Koumoto, M. Amaike, S. Shinkai and D. N. Reinhoudt, Org. Lett., 2002, 4, 1423–1426 CrossRef CAS.
- Recent examples: (a) P. Terech, W. G. Smith and R. G. Weiss, J. Chem. Soc., Faraday Trans., 1996, 92, 3157–3162 RSC; (b) U. Maitra, S. Mukhopadhyay, A. Sarkar, P. Rao and S. S. Indi, Angew. Chem., Int. Ed., 2001, 40, 2281–2283 CrossRef CAS; (c) N. M. Sangeetha, R. Balasubramanian, U. Maitra, S. Ghosh and A. R. Raju, Langmuir, 2002, 18, 7154–7157 CrossRef CAS.
- Recent examples: (a) S. Franceschi, N. de Viguerie, M. Riviere and A. Lattes, New J. Chem., 1999, 23, 447–452 RSC; (b) F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef; (c) L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 39, 3447–3450 CrossRef CAS; (d) J. Makarević, M. Jokić, B. Perić, V. Tomišić, B. Kojić-Prodić and M. Žinić, Chem. Eur. J., 2001, 7, 3328–3341 CrossRef CAS; (e) L. Frkanec, M. Jokić, J. Makarević, K. Wolsperger and M. Žinić, J. Am. Chem. Soc., 2002, 124, 9716–9717 CrossRef CAS; (f) T. Nakashima and N. Kimizuka, Adv. Mater., 2002, 14, 1113–1116 CrossRef CAS; (g) M. Suzuki, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Chem. Eur. J., 2003, 9, 348–354 CrossRef CAS; (h) K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS.
- R. Iwaura, K. Yoshida, M. Masuda, K. Yase and T. Shimizu, Chem. Mater., 2002, 14, 3047–3053 CrossRef CAS.
- S. M. Park, Y. S. Lee and B. H. Kim, Chem. Commun., 2003, 2912–2913 RSC.
- (a) R. Oda, I. Huc and S. J. Candau, Angew. Chem., Int. Ed., 1998, 37, 2689–2691 CrossRef CAS; (b) R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS.
- (a) G. R. Newkome, G. R. Baker, S. Arai, M. J. Saunders, P. S. Russo, K. J. Theriot, C. N. Moorefield, L. E. Rogers, J. E. Miller, T. R. Lieux, M. E. Murray, B. Phillips and L. Pascal, J. Am. Chem. Soc., 1990, 112, 8458–8465 CrossRef CAS; (b) C. Marmillon, F. Gauffre, T. Gulik-Krzywicki, C. Loup, A.-M. Caminade, J.-P. Majoral, J.-P. Vors and E. Rump, Angew. Chem., Int. Ed., 2001, 40, 2626–2629 CrossRef CAS; (c) M. McWatt and G.-J. Boons, Eur. J. Org. Chem., 2001, 2535–2545 CrossRef CAS.
- (a) K. Hanabusa, K. Shimura, K. Hirose, M. Kimura and H. Shirai, Chem. Lett., 1996, 885–886 CAS; (b) M. de Loos, J. van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675–12676 CrossRef CAS; (c) J. H. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem. Eur. J., 1999, 5, 937–950 CrossRef CAS; (d) M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 613–616 CrossRef CAS.
- J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn., 1992, p. 903 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn., 1992, pp. 1091–1092 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn., 1992, pp. 891–892 Search PubMed.
- (a) H. W. I. Peerlings and E. W. Meijer, Tetrahedron Lett., 1999, 40, 1021–1024 CrossRef CAS; (b) R. M. Versteegen, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1999, 38, 2917–2919 CrossRef CAS.
- According to ISIS/Base, Fluka, Acros and Aldrich.
- (a) D. G. Crosby and C. Niemann, J. Am. Chem. Soc., 1954, 76, 4458–4463 CrossRef CAS; (b) D. Martin and W. Mucke, Liebigs Ann. Chem., 1965, 682, 90–98 CrossRef CAS; (c) G. J. Durant, C. R. Ganellin, D. W. Hills, P. D. Miles, M. E. Parsons, E. S. Pepper and G. R. White, J. Med. Chem., 1985, 28, 1414–1422 CrossRef CAS; (d) B. Thavonekham, Synthesis, 1997, 1189–1194 CrossRef CAS.
- M. Mammen, E. E. Simanek and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 12614–12623 CrossRef CAS.
- Detailed studies on the effect of alkyl chain length on the gelation behaviour of cyclohexane bis-urea and bis-amide compounds will be reported separately.
- (a) N. Yamada, K. Okuyama, T. Serizawa and S. Oshima, J. Chem. Soc., Perkin Trans. 2, 1996, 2707–2713 RSC; (b) T. Shimizu and M. Masuda, J. Am. Chem. Soc., 1997, 119, 2812–2818 CrossRef CAS; (c) M. Kogiso, S. Ohnishi, K. Yase, M. Masuda and T. Shimizu, Langmuir, 1998, 14, 4978–4986 CrossRef CAS; (d) T. Ishi-I, R. Iguchi, E. Snip, M. Ikeda and S. Shinkai, Langmuir, 2001, 17, 5825–5833 CrossRef CAS.
- Generally primary amines are more water soluble than the corresponding alcohols: Handbook of Chemistry and Physics, CRC Press, Cleveland, 57th edn., 1976 Search PubMed.
- (a) K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1949–1951 CrossRef CAS; (b) K. Hanabusa, K. Okui, K. Karaki, M. Kimura and H. Shirai, J. Colloid Interface Sci., 1997, 195, 86–93 CrossRef CAS; (c) K. Hanabusa, M. Kobayashi, M. Suzuki, M. Kimura and H. Shirai, Colloid Polym. Sci., 1998, 276, 252–259 CrossRef CAS; (d) K. Hanabusa, Y. Maesaka, M. Kimura and H. Shirai, Tetrahedron Lett., 1999, 40, 2385–2388 CrossRef CAS; (e) X. Luo, B. Liu and Y. Liang, Chem. Commun., 2001, 1556–1557 RSC; (f) J. Makarević, M. Jokić, Z. Raza, Z. Štefanić, B. Kojić-Prodić and M. Žinić, Chem. Eur. J., 2003, 9, 5567–5580 CrossRef CAS.
- Based on the pKa values of linear monoamines: Handbook of Chemistry and Physics, CRC Press, Cleveland, 57th edn., 1976 Search PubMed.
- (a) Y. Mido, Spectrochim. Acta, 1972, 28A, 1503–1518 CrossRef; (b) Y. Mido, Spectrochim. Acta, 1973, 29A, 431–438 CrossRef.
- D2O gels of 3b were not prepared and measured for their elongated gelation time.
- J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. van Esch, Langmuir, 2000, 16, 9249–9255 CrossRef CAS.
- J.-H. Fuhrhop, P. Schnieder, J. Rosenberg and E. Boekema, J. Am. Chem. Soc., 1987, 109, 3387–3390 CrossRef CAS.
- A. Takahashi, M. Sakai and T. Kato, Polym. J., 1980, 12, 335–341 CAS.
- Unpublished results in our group showed that racemic organogels of trans-1,2-bis(dodecylureido)cyclohexane melted at lower temperatures than their enantiomerically pure counterparts.
- Attempts to support these differences in packing and thus gelation behaviour with FT-IR data and DSC measurements have been unsuccessful so far and SAXS measurements could not be performed due to the small size of the fibres.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for compounds 2a and b, 3a and b, 5a and b. See http://www.rsc.org/suppdata/ob/b5/b500837a/ |
| This journal is © The Royal Society of Chemistry 2005 |