New fluorescent probes reveal that flippase-mediated flip-flop of phosphatidylinositol across the endoplasmic reticulum membrane does not depend on the stereochemistry of the lipid†
Ram A.
Vishwakarma
*a,
Stefanie
Vehring
b,
Anuradha
Mehta
a,
Archana
Sinha
a,
Thomas
Pomorski
b,
Andreas
Herrmann
b and
Anant K.
Menon
*c
aBio-Organic Chemistry Laboratory, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi, 110067, India. E-mail: ram@nii.res.in; Fax: +91-11-26162125; Tel: +91-11-26717121
bInstitut für Biologie/Biophysik, Humboldt Universität zu Berlin, Invalidenstrasse 42, Berlin, Germany
cDepartment of Biochemistry, University of Wisconsin-Madison, 433 Babcock Drive, Madison, WI 53706-1569, USA. E-mail: menon@biochem.wisc.edu; Fax: +1-608-262-3453; Tel: +1-608-262-2913
First published on 1st March 2005
Abstract
Glycerophospholipid flip-flop across biogenic membranes such as the endoplasmic reticulum (ER) is a fundamental feature of membrane biogenesis. Flip-flop requires the activity of specific membrane proteins called flippases. These proteins have yet to be identified in biogenic membranes and the molecular basis of their action is unknown. It is generally believed that flippase-facilitated glycerophospholipid flip-flop across the ER is governed by the stereochemistry of the glycerolipid, but this important issue has not been resolved. Here we investigate whether the ER flippase stereochemically recognizes the glycerophospholipids that it transports. To address this question we selected phosphatidylinositol (PI), a biologically important molecule with chiral centres in both its myo-inositol headgroup and its glycerol–lipid tail. The flip-flop of PI across the ER has not been previously reported. We synthesized fluorescence-labeled forms of all four diastereoisomers of PI and evaluated their flipping in rat liver ER vesicles, as well as in flippase-containing proteoliposomes reconstituted from a detergent extract of ER. Our results show that the flippase is able to translocate all four PI isomers and that both glycerol isomers of PI flip-flop across the ER membrane at rates similar to that measured for fluorescence-labeled phosphatidylcholine. Our data have important implications for recent hypotheses concerning the evolution of distinct homochiral glycerophospholipid membranes during the speciation of archaea and bacteria/eukarya from a common cellular ancestor.
Introduction
Glycerophospholipids, the building blocks of cellular membrane bilayers, are made on the cytoplasmic face of biogenic (self-synthesizing) membranes such as the endoplasmic reticulum (ER) and must be flipped to the exoplasmic face for uniform bilayer expansion. Transbilayer flip-flop of glycerophospholipids is therefore an essential feature of membrane biogenesis.1–3 Although glycerophospholipids cannot flip-flop across synthetic liposomal membranes,3 in the ER membrane flip-flop is a rapid, ATP-independent, bi-directional process that is facilitated by a specific protein or flippase(s).4–11Despite the passage of almost 30 years since work in this area was first initiated,1–3 the ER glycerophospholipid flippase has yet to be identified and the molecular basis for its action remains a mystery. Here we consider two fundamentally different models of flippase action (Fig. 1). In the first model (model I), the flippase is envisaged as a transmembrane protein with two binding sites for glycerophospholipids, one located in each leaflet of the membrane bilayer. Phospholipids entering one site by lateral diffusion are specifically recognized, transferred to the opposite site and subsequently released into the opposing leaflet of the bilayer. Since the common glycerophospholipids are transported equally well by the flippase6,7,11 it is reasonable to suggest in this model that it is the glycerol moiety of the phospholipids that is the structural feature recognized by the flippase. In the second model (model II), amphipathic transmembrane helical domains of the flippase are envisaged to create a transbilayer hydrophilic conduit that accommodates the polar headgroup of transiting phospholipids, while allowing the lipid's acyl chains to remain in the hydrophobic interior of the bilayer.12 In a variation of this idea, it can also be imagined that transient defects generated by dynamic play in the transmembrane helices of the flippase would enable phospholipids to ‘slip’ into a mid-membrane transition state from which they could ‘pop’ into either leaflet of the membrane bilayer.1,13 Model II is distinct from model I in that it does not require specific recognition of phospholipids by the flippase.
 | ||
| Fig. 1 Models of flippase action. The flippase is illustrated in both models as a membrane protein composed of transmembrane domains (shaded cylinders) connected by extra-membrane loops (thick grey lines). Phospholipids are shown as an open circle (headgroup) to which two wavy lines (acyl chains) are attached. Two contrasting models of flippase action are illustrated. Model I envisages the flippase with phospholipid binding sites symmetrically placed with respect to the membrane. Phospholipids entering one site by lateral diffusion are specifically (stereochemically) recognized, transferred to the opposite site and subsequently released into the opposing leaflet of the bilayer. Model II proposes a dedicated flippase with a transbilayer conduit that provides a low energy (hydrophilic) path for the polar headgroup of transiting phospholipids, while allowing the lipid's acyl chains to remain in the hydrophobic interior of the bilayer. | ||
Phosphatidylinositol (PI) is a key cell signaling molecule that has surprisingly never been examined as a substrate for the ER flippase.14 It possesses two asymmetric moieties (D-myo-inositol substituted at the D-1 position and a D-glycerol moiety acylated at the sn-1 and sn-2 positions and phosphorylated at the 3-position in naturally occurring mammalian PI) that offer the opportunity to determine whether the ER flippase stereochemically recognizes the chiral hydrophilic headgroup and chiral lipophilic glycerol–lipid tail of the glycerophospholipids that it transports. We hypothesized that the ER flippase operating as described in model I would transport only the ‘natural’ stereoisomer of mammalian PI (containing D-myo-inositol and D-glycerol), whereas a flippase functioning as seen in model II would likely transport all four stereoisomers.
To examine this hypothesis we chemically synthesized all four possible diastereoisomers of PI in fluorescence-labeled form. These new fluorescent PI probes, containing different combinations of D- or L- myo-inositol or glycerol, as well as an NBD fluorophore attached to the sn-1 or sn-2-linked acyl chain, e.g., DD-NBD–PI containing D-myo-inositol and D-glycerol, DL-NBD–PI containing D-myo-inositol and L-glycerol etc., were tested for their ability to be flipped by the ER flippase in reconstituted systems as well as in ER vesicles. Our experiments indicate that all four stereochemical variants of PI are flipped in a protein-dependent, ATP-independent process and that the DD- and DL-NBD–PI's are flipped at similar rates in ER vesicles. These data (i) demonstrate flippase-mediated flip-flop of PI across the ER for the first time, (ii) support a model of flippase action (such as model II) that does not require specific stereochemical recognition of the glycerophospholipid by the flippase and (iii) have interesting implications for recent hypotheses concerning the origin of the three domains of cellular life (archaea, bacteria and eukarya) from a common cellular precursor.
Results and discussion
Synthesis of stereoisomers of acyl-NBD–PI
To address the questions posed in the Introduction we synthesized all four possible diasteroisomers of PI, one ‘natural’ with D-inositol and sn-1,2-glycerol and three ‘non-natural’ diasteroisomers (D-inositol with 2,3-sn-glycerol, L-inositol with sn-1,2-glycerol, and L-inositol with sn-2,3-glycerol).‡ The four new stereoisomers (compounds 1, 2, 3 and 4 (Fig. 2)) were prepared with a fluorescent NBD group appended to the sn-1-linked acyl chain. The second acyl chain in each case was stearate (C18:0) to mimic the acyl chain composition of naturally occurring mammalian PI.14 | ||
| Fig. 2 Structures of diastereoisomers of acyl-NBD-labeled phosphatidylinositol (PI) (compounds1–4) described in this paper. Compound 1 corresponds to the stereochemistry of PI that occurs naturally in mammalian cells, whereas compounds 3–4 represent non-natural stereoisomers of PI. | ||
In addition to these compounds, two further NBD-labeled-PIs bearing D-inositol, sn-1,2-glycerol or sn-2,3-glycerol, a myristoyl (C14:0) chain and a C6-NBD chain were also synthesized. These shorter-chain PI probes (compounds 5 and 6, Fig. 3) were necessary for use in the BSA back-extraction-based flippase assays described below. The combination of myristate and C6-NBD chains renders probes 5 and 6 more readily extractable by BSA8 than probes 1–4 that were designed to more closely mimic naturally occurring PI.
 | ||
| Fig. 3 Compounds 5 and 6. | ||
Our decision to synthesize acyl-NBD-labeled analogs of PI bears comment. It is known that membrane-bound acyl-NBD–phospholipid molecules adopt a conformation in which the polar NBD group is situated at the membrane–water interface.16–18 However, the extensive use of acyl-NBD–phospholipids as reporters of phospholipid flip-flop processes, together with the qualitative similarity between the data obtained with NBD–phospholipids, other phospholipid analogs (e.g., radiolabeled dibutyryl phospholipids or acyl-spin-labeled phospholipids)6,7,9 and natural phospholipids,7,10,11 justifies their choice for the flippase experiments we describe here.
Although the chemical synthesis of fluorescence labeled derivatives of phosphorylated PIs has been described by Prestwich,14 the synthesis of fluorescence-tagged PI itself and its diastereoisomeric variants has not. Here we describe a new and convergent approach to the synthesis of fluorescence-labeled PI diastereoisomers using optically pure myo-inositol and glycerol–lipid building blocks. All of the synthetic targets (1–6) required synthesis of D- and L-enantiomers of suitably protected 2,3,4,5,6-penta-O-benzyl-myo-inositol intermediates 11 and 12 (Fig. 4). The starting material, racemic 3,4,5,6-tetra-O-benzyl-myo-inositol (7), was prepared in two steps, then converted to racemic 2,3,4,5,6-penta-O-benzyl-myo-inositol intermediate (8) in three high yielding steps (regioselective 1-O-allylation, 2-O-benzylation and allyl removal from the 1-position). The racemate 8 was resolved into its enantiomers 11 and 12via the diastereoisomeric pair 9 and 10 of (−)-camphanic chloride (Fig. 4), followed by alkaline hydrolysis.
 | ||
| Fig. 4 Synthesis of the optical antipodes of protected myo-inositol (compounds 11 and 12). Reagents and conditions: (a) Bu2SnO, toluene, reflux, 4 h; allyl bromide, DMF, 80 °C, 4 h; NaH, BnBr, DMF; 5% Pd/C, EtOH, pTSA, H2O; (b) (−)-camphanic acid chloride, pyridine; (c) 1% NaOH in MeOH, reflux, 30 min. | ||
For the synthesis of compounds 1–4, we employed two chiral glycerol building blocks: 2-O-octadecanoyl-1-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (17) (Fig. 5A) and 2-O-octadecanoyl-3-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (22) (Fig. 5B). These were prepared from commercially available 1,2-isopropylidene-sn-glycerol (1S-2,2-dimethyl-1,3-dioxolane-4-methanol, 13) and 2,3-isopropylidene-sn-glycerol (1R-2,2-dimethyl-1,3-dioxolane-4-methanol, 18), respectively, by a multi-step synthesis as shown in Fig. 5.
![A: synthesis of 2-O-octadecanoyl-1-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 17). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 0 °C; stearic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. B: synthesis of 2-O-octadecanoyl-3-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 22). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 0 °C; stearic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h.](/image/article/2005/OB/b500300h/b500300h-f5.gif) | ||
| Fig. 5 A: synthesis of 2-O-octadecanoyl-1-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 17). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 0 °C; stearic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. B: synthesis of 2-O-octadecanoyl-3-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 22). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 0 °C; stearic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. | ||
Coupling of the D-myo-inositol intermediate 11 and 1,2-sn-glycero-H-phosphonate 17 in the presence of pivaloyl chloride followed by insitu oxidation with iodine provided the fully protected PI intermediate 23 (Fig. 6). Deprotection of all benzyl groups by Pd(OH)2-mediated hydrogenolysis and installation of the NBD fluorophore by reaction of the terminal amine at the sn-1 position with commercially available NBD-amino-caproic NHS ester yielded the first desired fluorescent-PI, 1D-myo-inositol 1-O-[1[6′[[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)amino]-hexanoyl]amino]-hexanoyl]-2-O-stearoyl-sn-glycer-3-yl]-phosphate (1). The remaining three isomers (2, 3 and 4) were synthesized by the above method from the corresponding chiral myo-inositol and glycerol–lipid intermediates. The fluorescent PIs were purified by preparative TLC and HPLC and were characterized by NMR and mass spectral analysis.
![Synthesis of 1d-myo-inositol 1-O-[1-[6′[[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)amino]-hexanoyl]amino]-hexanoyl]-2-O-stearoyl sn-glycer-3-yl]-phosphate (compound 1). Reagents and conditions: (a) pivaloyl chloride, pyridine, rt, 1 h; I2 in 95% aq. pyridine, 30 min; (b) Pd(OH)2, MeOH–CH2Cl2–H2O, H2, 12 h; (c) NBD-X, SE, DMF, Et3N, rt, 2 h.](/image/article/2005/OB/b500300h/b500300h-f6.gif) | ||
| Fig. 6 Synthesis of 1D-myo-inositol 1-O-[1-[6′[[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)amino]-hexanoyl]amino]-hexanoyl]-2-O-stearoyl sn-glycer-3-yl]-phosphate (compound 1). Reagents and conditions: (a) pivaloyl chloride, pyridine, rt, 1 h; I2 in 95% aq. pyridine, 30 min; (b) Pd(OH)2, MeOH–CH2Cl2–H2O, H2, 12 h; (c) NBD-X, SE, DMF, Et3N, rt, 2 h. | ||
The synthesis of the myristoyl–PI probes 5 and 6 required the chiral glycerol building blocks 1-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (27) (Fig. 7A) and 3-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (30) (Fig. 7B). These were prepared from commercially available 1,2-isopropylidene-sn-glycerol (1S-2,2-dimethyl-1,3-dioxolane-4-methanol, 13) and 2,3-isopropylidene-sn-glycerol (1R-2,2-dimethyl-1,3-dioxolane-4-methanol, 18), respectively, by synthesis as shown in Fig. 7.
![A: synthesis of 1-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 27). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) myristic acid, CH2Cl2, DCC, DMAP, 0 °C; 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. B: synthesis of 3-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 30). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) myristic acid, CH2Cl2, DCC, DMAP, 0 °C; 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h.](/image/article/2005/OB/b500300h/b500300h-f7.gif) | ||
| Fig. 7 A: synthesis of 1-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 27). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) myristic acid, CH2Cl2, DCC, DMAP, 0 °C; 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. B: synthesis of 3-O-myristoyl-2-O-[6-(N-carbobenzyloxyamino)-hexanoyl]-sn-glyceryl-H-phosphonate (compound 30). Reagents and conditions: (a) PMBCl, DMF, NaH, 1 h; pTSA, MeOH, 2 h; (b) myristic acid, CH2Cl2, DCC, DMAP, 0 °C; 6-Cbz-aminohexanoic acid, CH2Cl2, DCC, DMAP, 30 °C; (c) DDQ, CH2Cl2–H2O (99 : 1), 12 h; (d) PCl3, imidazole, triethylamine, toluene, −5 °C, 2 h. | ||
The coupling of the D-myo-inositol intermediate 11 with 1,2-sn-glycero-H-phosphonate 27 (Fig. 8) followed by oxidation, hydrogenation and reaction with NBD-chloride (bicarbonate buffer) provided 1D-myo-inositol 1-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)amino hexanoyl]-sn-glycer-3-yl]-phosphate (5). An identical three-step synthetic sequence starting from D-myo-inositol intermediate 11 and 2,3-sn-glycero-H-phosphonate 30 led to the corresponding glycerol isomer 1D-myo-inositol 3-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl) aminohexanoyl]-sn-glycer-1-yl]-phosphate (6) (Fig. 8).
![Syntheses of 1d-myo-inositol 1-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl) aminohexanoyl]-sn-glycer-3-yl]-phosphate (compound 5) and 1d-myo-inositol 3-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)aminohexanoyl]-sn-glycer-1-yl]-phosphate (compound 6). Reagents and conditions: (a) pivaloyl chloride, pyridine, rt, 1 h; I2 in 95% aq. pyridine, 30 min; (b) Pd(OH)2, MeOH–CH2Cl2–H2O, H2, 12 h; (c) NBD-chloride, sodium bicarbonate buffer, 2 h, rt.](/image/article/2005/OB/b500300h/b500300h-f8.gif) | ||
| Fig. 8 Syntheses of 1D-myo-inositol 1-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl) aminohexanoyl]-sn-glycer-3-yl]-phosphate (compound 5) and 1D-myo-inositol 3-O-myristoyl-2-O-[6-[(7-nitro-2-oxa-1,3-diazolobenz-4-yl)aminohexanoyl]-sn-glycer-1-yl]-phosphate (compound 6). Reagents and conditions: (a) pivaloyl chloride, pyridine, rt, 1 h; I2 in 95% aq. pyridine, 30 min; (b) Pd(OH)2, MeOH–CH2Cl2–H2O, H2, 12 h; (c) NBD-chloride, sodium bicarbonate buffer, 2 h, rt. | ||
Flippase assay in a reconstituted system
We previously reported that proteoliposomes prepared from a Triton X-100 extract (TE) of salt-washed rat liver ER (SWER) are capable of facilitating the transport of sn-1,2-dibutyroylphosphatidylcholine,9sn-1,2-dipalmitoylphosphatidylcholine10 and acyl-NBD derivatives of PC, PE and PS11 in an ATP-independent fashion requiring the participation of specific ER proteins. We now describe experiments to test whether the phospholipid flippase activity of proteoliposomes reconstituted from TE is capable of supporting the transbilayer movement of the acyl-NBD–PI isomers that we synthesized.Proteoliposomes were reconstituted from a Triton X-100-solubilized mixture consisting of egg phosphatidylcholine (ePC), ∼0.3 mol% of acyl-NBD–PI (compound 1, 2, 3 or 4) and TE. Liposomes (L) were reconstituted similarly, except that TE was omitted. Reconstitution was achieved by treating samples with detergent-adsorbing SM2 Bio-Beads™ to remove Triton X-100 (>99.9% of the detergent was removed as determined by spectroscopic measurement of a lipid extract of the vesicle preparations (data not shown)). Thin layer chromatographic analysis of lipid extracts of the reconstituted vesicles established that the NBD–PI tracers remained intact during the reconstitution process (data not shown). The vesicles formed by this procedure are unilamellar and typically ∼200–250 nm in diameter, as judged by electron microscopy and dynamic laser light scattering,9 implying that the inner and outer phospholipid monolayers have roughly the same surface area. As seen for other acyl-NBD–phospholipids,§ the acyl-NBD–PI probes are presumed to be symmetrically distributed between the inner and outer leaflet.
Flip-flop of the acyl-NBD–PI derivatives was assayed with dithionite (S2O42−), a membrane-impermeant dianion that reduces the NBD fluorophore to a non-fluorescent amine product.19,20Fig. 9A illustrates the principle of the dithionite-reduction assay using reconstituted proteoliposomes (P) or liposomes (L). On dithionite addition to liposomes, NBD–PI in the outer leaflet is reduced (Fig. 9A, left panel). NBD–PI molecules situated in the inner leaflet cannot be reduced since dithionite is not expected to permeate membranes significantly during the time-scale of the experiment (∼10 min). For proteoliposomes the situation is different. If the proteoliposomes possess an active flippase (as shown in Fig. 9A, right panel) and if the NBD–PI compound being tested is a substrate for the flippase, then NBD–PI molecules located in the inner leaflet of the proteoliposome will be flipped out and reduced when they reach the dithionite-accessible outer leaflet. This will result in a greater percent reduction than seen with liposomes. If the proteoliposomes lack an active flippase or if the NBD–PI isomer being tested is not a substrate, then the extent of reduction seen with the proteoliposome will be similar to that seen for liposomes. Thus, for liposomes ∼50% of the NBD–PI (corresponding to NBD–PI in the outer membrane leaflet) is expected to be reduced, whereas for flippase-competent proteoliposomes labeled with a transportable NBD–PI isomer all the fluorescence is expected to be reduced.
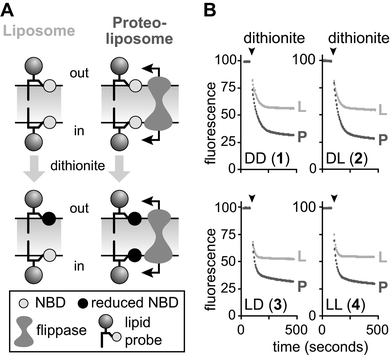 | ||
| Fig. 9 A: schematic illustration of the dithionite-reduction assay to measure phospholipid flip-flop in a reconstituted system. Proteoliposomes were reconstituted from a Triton X-100 extract of SWER (TE), egg phosphatidylcholine (ePC) and a trace quantity of one of the acyl-NBD–PI probes (compounds 1–4); liposomes were reconstituted similarly, except that TE was omitted. A section of each type of vesicle is illustrated, showing the NBD–PI probe symmetrically distributed between the inner and outer leaflet.§ On addition of dithionite, only the NBD–PI in the outer leaflet is reduced since dithionite does not permeate the membrane. However, if a flippase is present in the vesicle (as in the proteoliposome membrane shown) then NBD–PI located in the inner leaflet of the vesicle will be flipped out and reduced as well, resulting in a greater reduction of fluorescence. Theoretically, ∼50% of the fluorescence is expected to be reduced when NBD–PI-containing liposomes are treated with dithionite; ∼100% of the fluorescence is expected to be reduced in proteoliposome samples where every vesicle has a functional flippase. B: representative fluorescence traces. The traces correspond to dithionite reduction experiments with each of the four diastereoisomers of PI (compounds 1–4, labeled DD, DL, LD, LL to indicate stereochemistry at inositol and glycerol, respectively) reconstituted into liposomes (L) or proteoliposomes (P). Dithionite (3 mM final, added from a stock solution prepared in 1 M Tris base) was added at the indicated time (the pH of the solution remained at ∼7.5 throughout the experiment and no fluorescence change was seen when Tris base alone was added to the vesicles). The extent of fluorescence reduction was determined by comparing fluorescence prior to dithionite addition with the fluorescence signal observed 6 min after the addition of dithionite. | ||
Transbilayer movement of all four PI stereoisomers in proteoliposomes derived from a Triton X-100 extract of ER
Fig. 9B shows fluorescence traces obtained for dithionite reduction assays using liposomes and proteoliposomes individually reconstituted with compounds 1–4. The data show that when dithionite was added to liposomes prepared with any of the four test compounds, fluorescence was rapidly reduced by ∼40–45% of its starting value. In every case, a greater reduction was observed for proteoliposome samples, implying that NBD–PI molecules originally situated in the dithionite-inaccessible inner leaflet of the proteoliposomal membrane were able to flip out to the dithionite-accessible outer leaflet. For both liposomes and proteoliposomes, fluorescence dropped to baseline levels when 0.25% (w/v) Triton X-100 was added to permeabilize the membrane, indicating that the amount of dithionite used to probe the outer leaflet pool of NBD–PI was sufficient to reduce all the NBD–PI present in the sample. Since the fluorescence traces obtained on dithionite treatment of NBD–PI-containing proteoliposomes displayed a similar time-course to the trace for a corresponding liposome sample, we conclude that the rate of flipping of the NBD–PI isomers is similar to or faster than the rate of dithionite reduction of the NBD fluorophore. We conclude from these data that each of the four NBD–PI stereoisomers is a substrate for the ER phospholipid flippase activity that is reconstituted into proteoliposomes.Experiments of the type shown in Fig. 9B were repeated with a series of proteoliposome preparations containing different amounts of protein. The different proteoliposome samples were generated by varying the amount of TE used in the reconstitution mixture, while keeping the amount of egg PC and NBD–PI constant. Fig. 10A shows the extent of dithionite-mediated reduction as a function of protein content for vesicles reconstituted with each of the four test compounds 1–4. The figure shows that the four NBD–PI isomers display essentially identical protein dependence profiles, with the extent of reduction starting at a liposome-like level of ∼40–45% and increasing steadily to reach an apparent plateau at ∼70%. Data for NBD–PC generated in parallel are shown in Fig. 10B.
 | ||
| Fig. 10 A: plot showing percent reduction in fluorescence obtained on adding dithionite to reconstituted vesicle preparations containing DD-, DL-, LD-, or LL-NBD–PI (compounds 1–4) and different amounts of protein. The x-axis shows protein as the amount of TE (in µl) used for reconstitution, or the corresponding protein–phospholipid ratio (PPR; mg protein per mmol phospholipid) of the vesicles. The curved line is a monoexponential fit (Ymax = 69) to all the data points. The exponential fit is modeled as described in the text by two linear segments, one with positive slope obtained by linear regression of data points corresponding to TE <20 µl (PPR <17 mg mmol−1) and a second with zero slope corresponding to the Ymax of the monoexponential function. The two line segments intersect at TE ∼17 µl or PPR ∼13 mg mmol−1. B: data for 1-acyl-2-C6-NBD–PC. The data were acquired in parallel with the data shown in panel A; the two-component linear fit from panel A is superimposed on the data points. | ||
As discussed previously,9–11 when low amounts of TE are used for reconstitution the resulting proteoliposome preparation has a low protein–phospholipid ratio (PPR) and consists of a mixture of protein-free liposomes, proteoliposomes with membrane proteins other than the flippase and proteoliposomes with a mix of membrane proteins that includes a flippase. When higher TE amounts are used for the reconstitution, the PPR of the preparation is higher (PPR increases linearly with the amount of TE used).11 This increases the chance that an individual proteoliposome in the preparation contains a flippase and consequently increases the percent reduction obtained when the sample is treated with dithionite. As the PPR increases, a point is reached where all proteoliposomes in the sample contain a flippase. At this point it can be expected that the majority of the fluorescence will be destroyed in response to dithionite addition. We have modeled this scenario for the data shown in Fig. 10A by using two linear segments, a line of positive slope generated by linear regression of data points from the rising section of the graph and a plateau determined by single exponential curve fit of all the data. The point where the rising linear segment intersects the plateau (at TE ∼17 µl or PPR ∼13 mg mmol−1) is interpreted as the point where each vesicle contains a single functional flippase. The PPR value at the inflection point can be used together with data on vesicle size, phospholipid cross-sectional area and the assumption that ER membrane proteins have an average molecular mass of 50 kDa9–11 to deduce that functional flippases capable of flipping all PI stereoisomers represent ∼1% by weight of ER proteins in the TE. The two-component linear fit of the data for the four PI stereoisomers (Fig. 10A) can be readily superimposed on data generated in parallel for acyl-NBD–PC (Fig. 10B), indicating that all five lipids are likely to be flipped by the same flippase.
As discussed above, the linear regime of the dose–response plot in Fig. 10A corresponds to vesicle preparations in which each vesicle can be construed to contain either no or one functional flippase, with the proportion of flippase-bearing vesicles increasing with increasing PPR. The reason for the low final plateau of ∼70% (rather than 100%) is unclear. While more work needs to be done to understand this point, it is interesting to note that similar results have been reported previously in studies of outward translocation of NBD–PLs in membrane vesicles11,21,22 and in assays of the outward translocation of natural phospholipids.10
To establish further that the outward translocation of the NBD–PI analogs required the presence of ER proteins in the vesicle, we investigated the effect on transport of treating proteoliposomes with protease. Fig. 11 shows data for proteoliposomes prepared with trace amounts of DL-NBD–PI (compound 2). It is readily apparent that the reduction in fluorescence seen on adding dithionite to protease-treated proteoliposomes is significantly less than that seen with an untreated control sample (∼54.9% reduction for the protease-treated sample vs ∼64.9% and ∼45% for the untreated sample and liposomes, respectively). However, despite aggressive treatment with protease, the effect of proteolysis was incomplete with roughly half the population of flippase-active vesicles rendered inactive after protease treatment, whereas the remaining flippase-active vesicles retained activity after treatment. These data are consistent with a recent finding demonstrating that it is possible to reconstitute two functionally distinct populations of flippase in membrane vesicles.11 Thus, we propose that the flippase is a functionally symmetric molecule that can adopt either of two orientations during reconstitution, with only one of the orientations resulting in protease-sensitivity.
 | ||
| Fig. 11 Effect of proteinase K on dithionite-mediated reduction of DL-NBD–PI reconstituted in proteoliposomes. Liposomes and proteoliposomes were prepared using ∼0.3 mol% of compound 2. Fluorescence traces corresponding to the dithionite reduction assay are shown. Proteoliposomes were treated with ∼2 mg ml−1 proteinase K or mock-treated (control) at rt for 1 h before the assay. The extent of reduction 6 min after dithionite addition was 44.7% for the liposome sample and 64.9% and 53.8% for the control and proteinase K-treated proteoliposomes, respectively. The proportion of flippase-active proteoliposomes that was rendered inactive by proteinase K treatment is 45%. | ||
As an additional test, we confirmed the results described above with a different assay procedure that relies on using fatty acid-free bovine serum albumin (BSA) to extract NBD–phospholipids from the outer leaflet of the vesicle membrane. The quantum yield of the NBD fluorophore is reduced by ∼50% when the NBD–phospholipid is extracted from its membrane environment and sequestered in a hydrophobic binding pocket within BSA.11,23 For these experiments we used compounds 5 and 6, a matched pair of glycerol isomers of acyl-NBD–PI. The short myristoyl chain of these analogs compared with the acyl chains of compounds 1–4 is essential to allow efficient extraction by BSA. For proteoliposomes prepared with either compound 5 or 6, BSA extraction resulted in ∼43–48% reduction in fluorescence corresponding to essentially complete (∼86–96%) extraction of the analog from the membrane (data not shown). As discussed above, this could only occur if NBD–PI species from the inner leaflet of the vesicle are flipped into the outer leaflet and become available for BSA extraction. These data reinforce our conclusion that ER-derived proteoliposomes are capable of flipping both D- and L-glycerol isomers of NBD–PI.
Flipping kinetics measured in intact ER vesicles by stopped flow
Although the reconstitution experiments presented above clearly demonstrate that ER-derived proteoliposomes are capable of flipping all four stereoisomers of PI, they do not allow subtle distinctions to be made in the rate of flipping. In order to address this issue we analyzed the transbilayer transport of compounds 5 and 6 using BSA extraction in conjunction with stopped flow kinetic analysis. The experiments were carried out using SWER vesicles rather than reconstituted vesicle preparations. Briefly, SWER vesicles were incubated with either 5 or 6 for 10 min, a time period that was determined to be more than sufficient to permit all the NBD–phospholipid to integrate into and equilibrate across the SWER vesicle membrane (data not shown). NBD–PI-labeled SWER vesicles were then rapidly mixed with BSA in a stopped flow accessory linked to a fluorescence spectrophotometer and the fluorescence decay accompanying BSA extraction of the NBD–PIs was monitored. Multiple decay curves were combined and fit to a three pool model.8,23 Kinetic constants are summarized in Table 1. The data (Table 1) clearly show that 5 and 6 are flip-flopped at similar rates across SWER vesicles and that the rate of flipping resembles that seen for NBD–PC (this study and ref. 8) and NBD–PE.8| Sample | k +1/*10−3 s−1 | k −1/*10−3 s−1 | k +2/*10−2 s−1 |
|---|---|---|---|
| a k +1 and k−1 correspond to flop (translocation of lipid from the inner leaflet of the ER membrane bilayer to the BSA-accessible outer leaflet) and flip (outer leaflet to inner leaflet) of the NBD–phospholipid tracer and k+2 corresponds to extraction of NBD–phospholipid by BSA. The rate constant (k−2) corresponding to transfer of NBD–phospholipids from BSA back to the membrane is set to zero because of an excess of BSA in the assay. b M, myristate. c P, palmitate. | |||
| Experiment 1 | |||
| M-C6-NBD–PCb | 6.13 ± 0.69 | 2.89 ± 0.09 | 15.4 ± 0.7 |
| DL-NBD–PI (6) | 8.83 ± 0.33 | 3.20 ± 0.01 | 24.2 ± 0.7 |
| DD-NBD–PI (5) | 8.29 ± 0.39 | 4.36 ± 0.14 | 24.0 ± 1.7 |
| Experiment 2 | |||
| M-C6-NBD–PCb | 5.64 ± 1.86 | 2.65 ± 0.63 | 20.4 ± 4.9 |
| DL-NBD–PI (6) | 7.90 ± 0.35 | 3.14 ± 0.20 | 22.4 ± 1.5 |
| DD-NBD–PI (5) | 6.99 ± 0.35 | 4.92 ± 0.35 | 17.8 ± 1.2 |
| Data of Marx et al.8 | |||
| P-C6-NBD–PCc | 7.1 ± 0.6 | 4.7 ± 0.4 | 9.65 ± 1.3 |
| P-C6-NBD–PEc | 8.6 ± 0.9 | 11.2 ± 0.6 | 7.32 ± 1.1 |
Conclusion
We have synthesized a complete panel of fluorescently-labeled stereoisomers of PI and showed that they all undergo protein-mediated flip-flop in proteoliposomes reconstituted from a Triton X-100 extract of ER membrane proteins. We also tested the translocation of fluorescence-labeled glycerol isomers of PI using a stopped flow assay and showed that both isomers flip-flop at similar rates across the membrane of salt-washed ER vesicles. We conclude that the ER glycerophospholipid flippase is unspecific with regard to the stereochemistry of either the inositol or the glycerol moiety of PI. As the same flippase is presumed to transport all the major phospholipids6,7,11 we further conclude that the chirality of the glycerol moiety is immaterial to the selection of any glycerophospholipid for flippase-facilitated translocation across the ER membrane. Since glycerophospholipid flippase activity appears to be similar in all biogenic membranes studied thus far (E. coli inner membrane,12,23,24B. subtilis and B. megaterium cytoplasmic membrane,25,26Mycoplasma bovis cytoplasmic membrane (W.E. Watkins and A.K.M., unpublished data), yeast27 and mammalian ER4–11), our conclusions can be plausibly extended to glycerophospholipid flippases in all biogenic membranes.The literature describes a single previous attempt to explore the issue of glycerol stereochemistry in the context of phospholipid flipping in biogenic membranes. Bishop and Bell4 showed that the rate of uptake of sn-1,2-dibutyroyl–PC into rat liver ER vesicles could not be reduced in the presence of an excess of the non-natural isomer sn-2,3-dibutyroyl–PC, implying stereochemical recognition of the phospholipid's glycerol backbone by the flippase. We have since verified that diC4PC is a valid probe of flippase activity,9,12,25 yielding results that are similar to those obtained with other lipid probes. However, transport of dibutyroyl–PC into ER vesicles is a multi-step process involving association of the lipid with the vesicle membrane, followed by flipping across the bilayer and desorption from the inner leaflet into the intravesicular space. Since the flipping step in this instance is not rate limiting9,25 it cannot be readily concluded from the data of Bishop and Bell4 that the ER phospholipid flippase is stereochemically selective. It is also worth noting that the interpretation of competition studies of phospholipid flip-flop is far from straightforward since the phospholipid tracers being tracked must compete with a vast excess of natural, transportable phospholipids in the membrane system under study. Thus, it is more appropriate to carry out direct transport measurements such as those described in this paper.
The results described here allow us to distinguish between two models of flippase action (Fig. 1), both of which require that the polarity of the phospholipid headgroup is obscured during transbilayer transit with the acyl chains likely remaining in direct contact with the hydrophobic interior of the membrane. Model I is a gated transfer model in which phospholipids are specifically recognized by the flippase at one face of the membrane before being released after transfer to an equivalent recognition site on the opposite face. In contrast, model II presents the flippase as an ungated transbilayer conduit capable of admitting and transporting all glycerophospholipids irrespective of headgroup or glycerol stereochemistry. Our data are consistent with model II (Fig. 1).
Our discovery that the ER flippase is indifferent to the chirality of the glycerol moiety of the phospholipids that it transports has implications for models of cellular evolution. It is well known that bacterial and eukaryotic membranes are assembled from glycerophospholipids containing an sn-glycerol-3-phosphate backbone, whereas archaeal membranes are composed of glycerophospholipids containing exclusively sn-glycerol-1-phosphate.28 To explain this aspect of the speciation of archaea and bacteria/eukarya, it has been proposed29,30 that these distinct cell forms emerged from a common membrane-bound ancestral cell possessing a heterochiral glycerophospholipid membrane. Expansion of the membrane bilayer in this ancestral cell would presumably have required a non-stereospecific flippase. Since it seems unlikely that the flippase would have been under any selective pressure to evolve a stereospecific character after speciation of the common ancestor into cell types with more stable homochiral membranes, it can be anticipated—as we have found—that biogenic membrane flippases in modern cells would be insensitive to glycerol stereochemistry. The availability of the new functional PI probes described in this paper will aid in exploration of this important issue in our future studies.
Experimental
Synthesis of fluorescent phosphatidylinositol probes
Assays of flippase activity
For stopped flow measurements, 100 µl of NBD–phospholipid-labeled SWER membranes (the membranes were preincubated were mixed with an equal volume of fatty acid-free BSA dissolved in the same buffer by injecting into the stopped-flow chamber to give final concentrations of 2% (w/v) BSA and 0.25 mM membrane phospholipid containing 0.5 mol% of NBD–phospholipid, respectively. Fluorescence emission at 540 nm was monitored for 300 s (λex = 470 nm, slit widths 4 nm, resolution 0.2 s) and five successive fluorescence decay traces were recorded. In the course of a single experiment, this means that the first trace is recorded 15 min after initiating labeling of SWER with NBD–phospholipids while the last trace is recorded approximately 25 min later; all traces showed similar characteristics, indicative of the stability of the labeled membranes. The stopped-flow data were evaluated according to a three-compartment model as described previously,8,23 whereby three to five time traces in each case were fitted in order to derive rate constants and initial label distribution across the membrane.
Acknowledgements
This work was supported by the Department of Science and Technology (Government of India) (grant SR/S5/OC-11/2002 to R.A.V.), the NIH (grant GM55427 to A.K.M.), the Ruth and Milton Steinbach Fund, Inc. (award to A.K.M.) and the Deutsche Forschungsgemeinschaft (grant He1928-6 to A.H.). The Department of Biotechnology (Government of India) is acknowledged for provision of a core grant to the National Institute of Immunology for infrastructure support. A.K.M. acknowledges Kartikeya Menon for help with data collection, Kishen for general assistance, Qing-long Chang for TE and SWER preparations, Laura van der Ploeg for help with preparation of the figures and Alec Leamas (assistant librarian) for stimulation. A large part of this work was done while A.K.M. was on research leave in R.A.V.'s laboratory at the National Institute of Immunology, New Delhi. We thank David Moreira (Université Paris-Sud) for checking our interpretation of the membrane evolution model.References
- M. A. Kol, A. I. de Kroon, J. A. Killian and B. de Kruijff, Biochemistry, 2004, 43, 2673–2681 CrossRef CAS.
- T. Pomorski, J. C. Holthuis, A. Herrmann and G. van Meer, J. Cell Sci., 2004, 117, 805–813 Search PubMed.
- A. K. Menon, Trends Cell Biol., 1995, 5, 355–360 CrossRef CAS.
- W. R. Bishop and R. M. Bell, Cell, 1985, 42, 51–60 CrossRef CAS.
- J. M. Backer and E. A. Dawidowicz, Nature, 1987, 327, 341–343 CrossRef.
- A. Hermann, A. Zachowski and P. F. Devaux, Biochemistry, 1990, 29, 2023–2027 CrossRef.
- X. Buton, G. Morrot, P. Fellmann and M. Seigneuret, J. Biol. Chem., 1996, 271, 6651–6657 CrossRef CAS.
- U. Marx, G. Lassmann, H.-G. Holzhütter, D. Wüstner, P. Müller, A. Höhlig, J. Kubelt and A. Herrmann, Biophys. J., 2000, 78, 2628–2640 CrossRef CAS.
- A. K. Menon, W. E. Watkins and S. Hrafnsdóttir, Curr. Biol., 2000, 10, 241–252 CrossRef CAS.
- S. N. Gummadi and A. K. Menon, J. Biol. Chem., 2002, 277, 25337–25343 CrossRef CAS.
- Q. Chang, S. N. Gummadi and A. K. Menon, Biochemistry, 2004, 43, 10710–10718 CrossRef CAS.
- W. E. Watkins and A. K. Menon, Biol. Chem., 2002, 383, 1435–1440 CrossRef CAS.
- E. Fattal, S. Nir, R. A. Parente and F. C. Szoka, Biochemistry, 1994, 33, 6721–6731 CrossRef CAS.
- G. D. Prestwich, Chem. Biol., 2004, 11, 619–637 CrossRef CAS.
- IUPAC-IUB Commission on Biochemical Nomenclature; The Nomenclature of Lipids. J. Biol. Chem., 1967, 242, 4845–4849.
- A. Chattopadhyay and E. London, Biochemistry, 1987, 26, 39–45 CrossRef CAS.
- D. Huster, P. Müller, K. Arnold and A. Herrmann, Biophys. J., 2001, 80, 822–831 CrossRef CAS.
- P. F. Devaux, P. Fellmann and P. Hervé, Chem. Phys. Lipids, 2002, 116, 115–134 CrossRef CAS.
- J. W. Nichols, Semin. Cell Dev. Biol., 2002, 13, 179–184 CrossRef CAS.
- J. C. McIntyre and R. G. Sleight, Biochemistry, 1991, 30, 11819–11827 CrossRef CAS.
- J. M. Boon and B. D. Smith, J. Am. Chem. Soc., 1999, 121, 11924–11925 CrossRef CAS.
- R. A. Vishwakarma and A. K. Menon, Chem. Commun., 2005, 453–455 RSC.
- J. Kubelt, A. K. Menon, P. Müller and A. Herrmann, Biochemistry, 2002, 41, 5605–5612 CrossRef CAS.
- R. P. Huijbregts, A. I. P. M. de Kroon and B. de Kruijff, Biochim. Biophys. Acta, 1996, 1280, 41–50 CAS.
- S. Hrafnsdóttir and A. K. Menon, J. Bacteriol., 2000, 182, 4198–4206 CrossRef CAS.
- S. Hrafnsdóttir, J. W. Nichols and A. K. Menon, Biochemistry, 1997, 36, 4969–4978 CrossRef CAS.
- T. Nicolson and P. Mayinger, FEBS Lett., 2000, 476, 277–281 CrossRef CAS.
- M. Kates, The Biochemistry of Archaea (Archaebacteria), ed. M. Kates, D. J. Kushner and A. T. Matheson, Elsevier, Amsterdam, 1993, 261––295 Search PubMed.
- J. Peretó, L.-G. Purificación and D. Moreira, Trends Biochem. Sci., 2004, 29, 469–477 CrossRef CAS.
- G. Wächtershäuser, Mol. Microbiol., 2003, 47, 13–22 CrossRef CAS.
- S. N. Gummadi, S. Hrafnsdóttir, J. Walent, W. E. Watkins and A. K. Menon,Membrane Protein Protocols: Expression, Purification, and Crystallization, ed. B. S. Selinsky, Humana Press, Totowa, NJ, 2003, pp 271–279 Search PubMed.
- E. G. Bligh and W. J. Dyer, Can. J. Med. Sci., 1959, 37, 911–917 Search PubMed.
- G. Rouser, S. Fleischer and A. Yamamoto, Lipids, 1970, 5, 494–496 CAS.
- R. S. Kaplan and P. L. Pedersen, Methods Enzymol., 1989, 172, 393–399 CrossRef CAS.
- W. E. Watkins, M.S. thesis, University of Wisconsin-Madison, 2002.
Footnotes |
| † Electronic supplementary information (ESI) available: full experimental procedures and characterization data for the compounds 8–12, 14–17, 22, 25–27 and 30. See http://www.rsc.org/suppdata/ob/b5/b500300h/ |
| ‡ In view of the coexistence of two systems for nomenclature of glycerolipids, we have followed the IUPAC-IUB biochemical nomenclature of lipids described in reference 15. |
| § Preliminary collisional quenching experiments indicate that proteoliposomes reconstituted as described, with ∼0.3 mol% of NBD–PC, contain equivalent amounts of the fluorescent lipid in each membrane leaflet: ∼50% of the NBD fluorophores in acyl-NBD–PC-containing proteoliposome preparations were found to be inaccessible to quenching by iodide ions, consistent with a symmetric distribution of acyl-NBD–PC across the proteoliposomal membrane (I. Menon and A.K.M.; data not shown). |
| This journal is © The Royal Society of Chemistry 2005 |