Urea vs. thiourea in anion recognition
David Esteban
Gómez
,
Luigi
Fabbrizzi
*,
Maurizio
Licchelli
and
Enrico
Monzani
Dipartimento di Chimica Generale, Università di Pavia, via Taramelli 12, 27100, Pavia, Italy. E-mail: luigi.fabbrizzi@unipv.it; Fax: +39 0382 528544; Tel: +39 0382 987328
First published on 10th March 2005
Abstract
Neutral anion receptors (LH) form stable 1 : 1 H-bond [LH⋯X]− complexes with carboxylates, halides and phosphate (X−). Some of the [LH⋯X]− complexes, in presence of an excess of X−, release an HX fragment, with formation of [HX2]− and the deprotonated receptor L−. The tendency towards deprotonation increases with the acidity of the receptor and with the stability of the [HX2]− self-complex. Thus, the more acidic thiourea containing receptor deprotonates in the presence all the investigated anions except chloride, whereas the less acidic urea containing receptor undergoes deprotonation only in the presence of fluoride, due to the high stability of [HF2]−.
Introduction
Urea and thiourea subunits are currently used in the design of neutral receptors for anions, owing to their ability to act as H-bond donors.1–3 In particular, urea and thiourea can establish two directional H-bonds with the Y-shaped carboxylate group or chelate a spherical anion (e.g., halides). Thus, after the seminal papers by Wilcox4 and Hamilton5 on urea–anion interactions, a variety of anion receptors have been reported in which one or more urea/thiourea fragments are incorporated in an acyclic, cyclic or polycyclic framework.6–13 H-bond donating tendencies of a given donor group (e.g., the N–H fragment of urea/thiourea) are in some way related to its protonic acidity. Noticeably, hydrogen bonding has been defined as a ‘frozen’ proton transfer from the donor (the acid) to the acceptor (the base)14 and the more advanced the proton transfer, the stronger the H-bond interaction. Thiourea is a much stronger acid than urea (pKA = 21.1 and 26.9, respectively in DMSO),15 thus it is expected that thiourea containing receptors establish stronger H-bond interactions and form more stable complexes with anions than their urea containing counterparts. On the other hand, we have recently observed that urea derivatives (e.g., 1,3-bis-(4-nitro-phenyl)-urea) in an MeCN solution, in the presence of an excess of fluoride, undergo deprotonation of one of the N–H goups with formation of the [HF2]− anion.16 Anion induced deprotonation should be observed a fortiori in the case of receptors containing the more acidic thiourea fragment, even if evidence of such a behaviour has not yet been reported.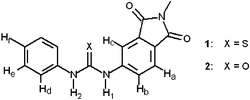
On this premise, we were interested in comparing the H-bond donor tendencies of urea and thiourea towards the most common inorganic and organic anions and to verify the occurrence of deprotonation processes in the presence of especially basic anions. We report here on the interaction of receptors 1 and 2 with halides and carboxylates in DMSO solution. In the two derivatives a phthalimide substituent, a classical chromophore, has been appended at the urea or thiourea subunit in order to provide an optical signal for the occurrence of the receptor–anion interaction.
Results and discussion
The binding tendencies of receptors 1 and 2 towards anions were investigated through UV-vis and 1H NMR titration experiments in DMSO solution. In particular, a solution of the receptor was titrated with a standard solution of the anion (as a tetrabutylammonium salt), up to a 5–10-fold excess. Data from UV-vis titrations were processed through a non-linear least-squares procedure17 in order to determine pertinent association constants K. Corresponding values of K are reported in Table 1.| LH | Equilibrium | F− | CH3COO− | C6H5COO− | H2PO4− | Cl− |
|---|---|---|---|---|---|---|
| 1 | LH + X− ⇌ [LH⋯X]− | 5.7 (1) | 6.02 (5) | 5.77 (5) | 5.44 (6) | 4.88 (1) |
| [LH⋯X]− + X− ⇌ L− + [HX2]− | 5.5 (3) | 3.23 (10) | 3.36 (10) | 0.55 (11) | — | |
| 2 | LH + X− ⇌ [LH⋯X]− | 4.86 (5) | 4.63 (3) | 4.18 (1) | 4.47 (1) | 4.38 (1) |
| [LH⋯X]− + X− ⇌ L− + [HX2]− | 1.83 (11) | — | — | — | — |
In general, the following two consecutive equilibria may take place in solution, involving the neutral receptor LH and the anion X−:
| LH + X− ⇌ [LH⋯X]− | (1) |
| [LH⋯X]− + X− ⇌ L− + [HX2]− | (2) |
In eqn. (1), characterised by the (association) constant K1, a genuine H-bond complex forms. In eqn. (2), characterised by K2, a second X− anion ‘abstracts’ an HX fragment from the complex to give [HX2]− and to leave the deprotonated derivative L−. All investigated anions undergo the first equilibrium (eqn. (1)), giving a more or less stable H-bond complex. Not all anions undergo the second equilibrium (eqn. (2)), whose occurrence is related to both the intrinsic acidity of LH and the stability of [HX2]−. In this context, it is useful to consider the overall equilibrium (eqn. (3)):
| LH + 2X− ⇌ L− + [HX2]− | (3) |
With a global constant β2
=
KA(LH)
×
β([HX2]−), where KA
=
[L−]
×
[H+]/[LH] and β([HX2]−)
=
[HX2]−/[H+]
×
[X−]2. Table 1 shows that receptor 2, containing the less acidic urea subunit, undergoes deprotonation and HX release only in presence of F−, to form the very stable [HF2]− self-complex. On the other hand, the more acidic thiourea containing receptor 1 undergoes deprotonation in presence of a greater number of anions, the value of K2 decreasing with the decreasing stability of the self-complex [HX2]− : F− > CH3COO− > C6H5COO− > H2PO4−. As an example, the hypothesised structure of the [HX2]− in the case of acetate is shown.

The above statements are based on extensive titration experiments, some examples of which are illustrated in the following.
Fig. 1 shows the UV-vis spectra taken over the course of the titration of a DMSO solution of the urea containing receptor 2 (5 × 10−6 M) with a standard solution of [Bu4N]CH3COO. Upon acetate addition the band at 330 nm, typical of the phthalimide chromophore and of charge transfer nature, undergoes red shift. A plot of the molar absorbance at significant wavelengths (in the inset of Fig. 1, the increasing band at 360 nm is considered as an example), indicates the formation of a 1 : 1 complex, as represented by eqn. (1). It is hypothesised that an H-bond complex forms, in which directional interactions between the two N–H fragments of 2 and the two oxygen atoms of acetate are established. More detailed pieces of information on the nature of the [2⋯CH3COO]− complex were obtained from 1H NMR titration experiments. Pertinent spectra are presented in Fig. 2 and Fig. 3.
![Spectra taken over the course of the titration of a DMSO solution of 2
(5 × 10−6 M) with a standard solution of [Bu4N]CH3COO. Inset: change in the molar absorbance at 360 nm upon acetate addition.](/image/article/2005/OB/b500123d/b500123d-f1.gif) | ||
| Fig. 1 Spectra taken over the course of the titration of a DMSO solution of 2 (5 × 10−6 M) with a standard solution of [Bu4N]CH3COO. Inset: change in the molar absorbance at 360 nm upon acetate addition. | ||
![1H NMR spectra taken over the course of the titration of a DMSO solution of 2
(5 × 10−3 M) with a standard solution of [Bu4N]CH3COO.](/image/article/2005/OB/b500123d/b500123d-f2.gif) | ||
| Fig. 2 1H NMR spectra taken over the course of the titration of a DMSO solution of 2 (5 × 10−3 M) with a standard solution of [Bu4N]CH3COO. | ||
 | ||
| Fig. 3 1H NMR spectra as in Fig. 2, zoomed on C–H protons. | ||
First (in Fig. 2), it is observed that both N–H protons undergo a downfield shift, which reflects the establishment of an H-bond interaction with acetate. In particular, Δδ values of both protons show a saturation profile which confirms a 1 : 1 stoichiometry. More subtle shifts of aromatic protons are shown in Fig. 3. In this context, it must be considered that hydrogen bond formation between the urea subunit and the anion can induce two distinct effects on the aromatic substituents: (i) it increases the electron density on the phenyl rings with a through-bond propagation, which generates a shielding effect and should produce an upfield shift of C–H protons; (ii) it induces the polarisation of the C–H bonds via a through-space effect, where the partial positive charge created onto the proton causes a deshielding effect and produces a downfield shift. The latter effect, of electrostatic nature, drastically decreases upon increasing the distance between the envisaged C–H proton and the site of H-bond interaction. It is seen in Fig. 3 that the electrostatic effect predominates for protons Hb and Hd, as indicated by the pronounced downfield shift. This reflects the rather strong through-space interaction with the N–H protons polarised by bonding with acetate. Protons Ha and He are too far away from the N–H protons to be subjected to any electrostatic effect. Thus, the shielding effect induced by the propagation of electron density associated with the formation of the H-bond complex is not (or is only poorly) contrasted and a significant upfield shift is observed. It may be surprising that the Hc protons, quite far away from the N–H fragment, undergo substantial downfield shift, which indicates a strong electrostatic effect. However, such a behaviour can be ascribed to the polarisation effect exerted by the rather close urea oxygen atom, which has acquired electron density following the N–H–anion interaction.
Let us consider now the interaction of the thiourea containing receptor 1 with acetate. Fig. 4 shows the UV-vis spectra taken over the course of the titration of a 5 × 10−6 M DMSO solution of 1 with [Bu4N]CH3COO. Upon acetate addition, first the charge transfer band centred at 360 nm undergoes red-shift, as observed in the titration of 2. Then, upon further acetate addition, a new band develops at 410 nm; something that had not been observed upon titration of the urea containing receptor 2.
![Spectra taken over the course of the titration of a DMSO solution of 1
(5 × 10−6 M) with a standard solution of [Bu4N]CH3COO. Inset: change of the concentration of the species at equilibrium upon acetate addition.](/image/article/2005/OB/b500123d/b500123d-f4.gif) | ||
| Fig. 4 Spectra taken over the course of the titration of a DMSO solution of 1 (5 × 10−6 M) with a standard solution of [Bu4N]CH3COO. Inset: change of the concentration of the species at equilibrium upon acetate addition. | ||
Best fitting of titration profiles was obtained on assuming the occurrence of two stepwise equilibria (eqn. (1) and eqn. (2)), to which the following K values corresponded: logK1 = 6.02 ± 0.05 and logK2 = 3.23 ± 0.10. The inset of Fig. 4 shows how the concentration of the species at the equilibrium, calculated from K1 and K2 values, varies upon acetate addition. First, the H-bond complex [1⋯CH3COO]− forms, as signalled by the shift of the band from 360 to 375 nm. Then, after the addition of the first anion eq., the new band at 410 nm develops, which corresponds to the deprotonated receptor L− and reflects the stabilisation of the charge transfer excited state upon N–H deprotonation.
Further insights regarding the nature of the species [1⋯CH3COO]− and L− were obtained from 1H NMR titration experiments. Spectra were recorded in CD3CN solution owing to the favourable solubility of 1 in such a solvent. Selected spectra taken over the course of the titration are shown in Fig. 5.
![1H NMR spectra taken over the course of the titration of a CD3CN solution of 1
(5 × 10−3 M) with a standard solution of [Bu4N]CH3COOH.](/image/article/2005/OB/b500123d/b500123d-f5.gif) | ||
| Fig. 5 1H NMR spectra taken over the course of the titration of a CD3CN solution of 1 (5 × 10−3 M) with a standard solution of [Bu4N]CH3COOH. | ||
It is seen that during the addition of 0→1 eq. of acetate, Hb and Hd protons undergo a downfield shift due to the dominating electrostatic effect present in the [1⋯CH3COO]− H-bond complex. Then, after the addition of the first anion eq., deprotonation of an N–H fragment occurs which induces delocalisation of electron density on the receptor's framework, causing nuclear shielding and an upfield shift of the Hb and Hd protons. The fact that such an effect is distinctly more pronounced for Hb (whose signal disappears after the addition of the second eq.) would suggest that deprotonation occurs at the N–H fragment close to the phthalimide substituent. However, receptor deprotonation can be more clearly followed upon titration with fluoride, due the higher value of β2 (and, in particular, of β([HF2]−)).
Fig. 6 shows the specta taken over the course of the titration of a DMSO solution of 1 (5 × 10−6 M) with a standard solution of [Bu4N]F. Fig. 7 (triangles) displays the titration profile based on the absorbance of the band centred at 410 nm.
![Spectra taken over the course of the titration of a DMSO solution of 1
(5 × 10−6 M) with a standard solution of [Bu4N]F. The increasing band at 410 nm signals deprotonation of the receptor.](/image/article/2005/OB/b500123d/b500123d-f6.gif) | ||
| Fig. 6 Spectra taken over the course of the titration of a DMSO solution of 1 (5 × 10−6 M) with a standard solution of [Bu4N]F. The increasing band at 410 nm signals deprotonation of the receptor. | ||
 | ||
| Fig. 7 UV-vis titration of 1 with F− (spectra of Fig. 6). Symbols: absorbance at 410 nm (normalised on the limiting value). Lines: how percent concentration of the species present at the equilibrium varies upon fluoride addition. | ||
Titration data are best fitted based on the assumption that the occurrence of the two consecutive equilibria (eqn. (1) and eqn. (2)). From the values of equilibrium constants (logK1 = 5.7 ± 0.1 and logK2 = 5.5 ± 0.3) the distribution diagram of the species present at the equilibrium, LH, [LH⋯F]− and L−, was calculated (lines in Fig. 7). It can be seen that, due to the especially high value of K2, the deprotonated receptor L− forms in the early stages of titration so that the band at 410 nm begins to develop upon the addition of the first subequivalents of fluoride. Notice also that under the conditions of the UV-vis titration experiment the highest concentration of the H-bond complex [LH⋯F]− is 35%, reached after the addition of 1 eq. of F−.
Fig. 8 shows the 1H NMR spectra taken over the course of the titration of 1 with fluoride. We first look at the Hb and Hd protons, which are especially sensitive to the H-bond interaction of the proximate N–H fragments. In the 0→1 eq. range, Hb undergoes a small upfield shift. This would indicate that the through-bond effect overcomes the through-space effect. The opposite occurs with the Hd proton, which undergoes a distinct downfield shift following up to 1 eq. addition. The interpretation is complicated by the fact that the [LH⋯F]− complex coexists since the beginning of the titration with L−, which exerts its own effects on the spectrum (vide infra). However, it appears that F− establishes unequivalent interactions with the two N–H fragments of the thiourea subunit, interacting with a single N–H group. A distinct downfield shift is observed for the Hc proton, which reflects the electrostatic interaction with the sulfur atom of the thiourea subunit. Then, on addition of excess fluoride and predominance of the L− species, the through-bond effect prevails for the Hd and Hc protons. In particular, the spectrum at 3 eq. of added fluoride should reasonably correspond to the limiting spectrum of the deprotonated receptor L−. In these circumstances, it has to be noticed that the upfield shift is more pronounced for Hb than for Hd. This would suggest that deprotonation occurs at the N–H fragment close to the phthalimide substituent. Such an event would be favoured by the electron-withdrawing properties of the phthalimide substituent, which allows delocalisation of the excess electron density according to a π-mechanism. This situation is illustrated by the resonance representation below (in which only two limiting formulae, a and b, are reported as an example).

![1H NMR spectra taken over the course of the titration of a CD3CN solution of 1
(5 × 10−3 M) with a standard solution of [Bu4N]F.](/image/article/2005/OB/b500123d/b500123d-f8.gif) | ||
| Fig. 8 1H NMR spectra taken over the course of the titration of a CD3CN solution of 1 (5 × 10−3 M) with a standard solution of [Bu4N]F. | ||
Thus, it appears that the more acidic receptor 1 undergoes deprotonation in the presence of an excess of the more basic anions (F−, carboxylates). In the case of less basic anions, a pronounced excess is required in order to produce a detectable amount of L−. This is the case for H2PO4−, as illustrated in Fig. 9, which displays the UV-vis spectra taken during titration. The band at 410 nm slowly develops only after the addition of the first eq. of H2PO4− and a detectable amount of L− can be observed in presence of a huge excess of phosphate .
![Spectra taken over the course of the titration of a DMSO solution of 1
(5 × 10−6 M) with a standard solution of [Bu4N]H2PO4. Inset: change in the concentration of the species at equilibrium upon phosphate addition.](/image/article/2005/OB/b500123d/b500123d-f9.gif) | ||
| Fig. 9 Spectra taken over the course of the titration of a DMSO solution of 1 (5 × 10−6 M) with a standard solution of [Bu4N]H2PO4. Inset: change in the concentration of the species at equilibrium upon phosphate addition. | ||
![1H NMR spectra taken over the course of the titration of a CD3CN solution of 1
(5 × 10−3 M) with a standard solution of [Bu3BzN]Cl.](/image/article/2005/OB/b500123d/b500123d-f10.gif) | ||
| Fig. 10 1H NMR spectra taken over the course of the titration of a CD3CN solution of 1 (5 × 10−3 M) with a standard solution of [Bu3BzN]Cl. | ||
Among the investigated anions, only chloride fails to induce the deprotonation of receptor 1. In particular, the H-bond complex that forms, [LH⋯Cl]−, definitively resists the addition of a large excess of chloride. Details on the nature of the complex have been obtained from the 1H NMR spectra recorded during the titration of chloride with a 5 × 10−3 M solution of 1 in CD3CN, as shown in Fig. 10.
Attention is first called to the downfield shift experienced by the Hb and Hd protons due to the predominance of the through-space electrostatic effect. Such a shift exhibits a saturation profile consistent with the formation of a 1 : 1 association complex. Moreover, the fact that Δδ(Hb) and Δδ(Hd) have nearly the same value suggests the formation of a bifurcate H-bond complex (in contrast to that observed with fluoride, where F− establishes a H-bond interaction with just one N–H fragment). As observed in the previously considered complexes, the thiourea–anion interaction induces an increase in the electron density on the sulfur atom, which causes a strong polarisation of the nearby C–Hc bond, as shown by the pronounced downfield shift. Due to the larger distance from the interaction site, the through-space effect on the Ha and Hd protons is outbalanced by the through-bond effect, which results in a slight upfield shift. On the other hand, the farthest Hf proton experiences very small or no electrostatic effect, so that the through-bond effect can induce a pronounced upfield shift.
The results of this study are summarised in the diagram presented in Fig. 11, in which the scale of pKA(LH) and that of logβ([HX2]−) are tentatively juxtaposed. The diagram is a graphical way to illustrate the concurrence of (i) the intrinsic acidity of LH and (ii) the stability of [HX2]− to favour the receptor's deprotonation, where the receptor, either 1 or 2, undergoes deprotonation in presence of those anions whose logβ([HX2]−) values are placed above the pertinent horizontal line.
![Matching of the acidity scale of the receptor LH and of the stability scale of [HX2]−. Each receptor deprotonates in presence of excess of anions whose logβ([HX2]−) values lie above the pertinent horizontal line (solid for 2 and dashed for 1).](/image/article/2005/OB/b500123d/b500123d-f11.gif) | ||
| Fig. 11 Matching of the acidity scale of the receptor LH and of the stability scale of [HX2]−. Each receptor deprotonates in presence of excess of anions whose logβ([HX2]−) values lie above the pertinent horizontal line (solid for 2 and dashed for 1). | ||
With regards to the H-bond complexes [LH⋯X]−, the more acidic receptor 1 forms, with a given anion, a more stable complex than the less acidic receptor 2. This behaviour is consistent with the view of hydrogen bonding as an incipient (and frozen) proton transfer from the receptor to the anion; where the more acidic the receptor, the stronger the H-bond with the anion.14 On the other hand, for a given receptor, the stability of the [LH⋯X]− complex increases with anion basicity. Comparison of the behaviour of fluoride and acetate is intriguing. CH3COO− forms a more stable complex than F−, due in part to the fact that acetate establishes with the receptor–bifurcate interaction whereas the small spherical fluoride ion interacts with a single N–H group. Thus, the release of HF in presence of F− seems to be favoured not only by the high stability of [HF2]−, but also by the relatively low stability of the monofurcated [LH⋯F]− complex.
Conclusion
Urea and thiourea are among the most frequently used fragments to design neutral receptors for the selective recognition of anions. Recognition implies the association of host and guest through relatively strong, and reversible, non-covalent interactions. In this sense, the choice of the receptor should be directed towards the most acidic H-bond donors. However, this work has demonstrated that a receptor which is too acidic, like thiourea, leads to the formation of complexes which are intrinsically stable but are unstable with respect to HX release and deprotonation. Moreover, fluoride recognition creates a particular problem, as the extreme stability of [HF2]− may induce deprotonation even of less acidic receptors, like urea. This usually leads to the development of an intense colour, an enjoyable and valuable event, which is not associated with the host–guest interaction, but derives from a charge transfer transition within the deprotonated receptor (while fluoride, as [HF2]−, moves far away in the solution and out of the control of supramolecular interactions).Experimental
General procedures and materials
All reagents for syntheses were purchased from Aldrich/Fluka and used without further purification. UV-vis spectra were recorded on a Varian CARY 100 spectrophotometer, with a quartz cuvette (path length: 1 or 0.1 cm). The cell holder was thermostatted at 25.0 °C, through circulating water. 1H NMR spectra were obtained on a Bruker AVANCE400 spectrometer (400 MHz), operating at 9.37 T. Spectrophotometric titrations were performed on 10−5–10−6 M solutions of 1 in DMSO (polarographic grade). Typically, aliquotes of a fresh alkyl-ammonium salt standard solution of the envisaged anion (F−, CH3COO−, C6H5COO− and H2PO4− as tetrabutylammonium; Cl− as tributylbenzylammonium) were added and the UV-vis spectra of the samples were recorded. All spectrophotometric titration curves were fitted with the HYPERQUAD program.17 Care was taken to ensure that in each titration the p parameter (p = [concentration of complex]/[maximum possible concentration of complex]) was lower than 0.8; a condition required for the safe determination of a reliable equilibrium constant.181H NMR titrations were carried out on DMSO-d6 solutions of 2 and on CD3CN solutions of 1 at 5 × 10−3 M concentration of the receptor (Scheme 1). | ||
| Scheme 1 Synthetic route to 1 and 2. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 3343, 3327 νas, νs(N–H); 1490, 1249 δas, δs(N–H).
O); 3372, 3313 νas, νs(N–H); 1460, 1240 δas, δs(N–H).
O); 3343, 3327 νas, νs(N–H); 1490, 1249 δas, δs(N–H).
O); 3372, 3313 νas, νs(N–H); 1460, 1240 δas, δs(N–H).
Acknowledgements
The financial support of the European Union (RTN Contract HPRN-CT-2000-00029) and the Italian Ministry of University and Research (PRIN-Dispositivi Supramolecolari; FIRB-Project RBNE019H9K) is gratefully acknowledged.References
- P. A. Gale, Coord. Chem. Rev., 2003, 240, 1–226 CAS.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS.
- P. J. Smith, M. V. Reddington and C. S. Wilcox, Tetrahedron Lett., 1992, 33, 6085–6088 CrossRef CAS.
- E. Fan, S. A. van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS.
- J. Scheerder, J. F. J. Engbersen, A. Casnati, R. Ungaro and D. N. Reinhoudt, J. Org. Chem., 1995, 60, 6448–6454 CrossRef CAS.
- M. P. Hughes, M. Shang and B. D. Smith, J. Org. Chem., 1996, 61, 4510–4511 CrossRef CAS.
- M. P. Hughes and B. D. Smith, J. Org. Chem., 1997, 62, 4492–4499 CrossRef CAS.
- R. J. Fitzmaurice, G. M. Kyne, D. Douheret and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2002, 841–864 RSC.
- A. J. Evans, S. E. Matthews, A. R. Cowley and P. D. Beer, Dalton Trans., 2003, 4644–4650 RSC.
- H. Miyaji, S. R. Collinson, I. Prokes and J. H. R. Tucker, Chem. Commun., 2003, 64–65 RSC.
- M. Barboiu, G. Vaughan and A. van der Lee, Org. Lett., 2003, 5, 3074–3076.
- T. Gunnlaugsson, A. P. Davis, G. M. Hussey, J. Tierney and M. Glynn, Org. Biomol. Chem., 2004, 2, 1856–1863 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- M. Boiocchi, L. Del Boca, D. Esteban Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507–16514 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- C. S. Wilcox, in Frontiers of Supramolecular Chemistry and Photochemistry, ed. H.-J. Schneider and H. Dürr, Wiley-VCH, Weinheim, 1991, pp. 123–143 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |