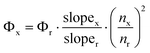DOI:
10.1039/B418964G
(Paper)
Org. Biomol. Chem., 2005,
3, 1013-1024
Synthesis and characterisation of highly emissive and kinetically stable lanthanide complexes suitable for usage ‘in cellulo’
Received
20th December 2004
, Accepted 19th January 2005
First published on 2nd March 2005
Abstract
The synthesis and photophysical characterisation are reported of a series of cationic, neutral and anionic europium and terbium complexes based on structurally related, nonadentate ligands based on the cyclen macrocycle. Each complex incorporates a tetraazatriphenylene moiety and overall absolute emission quantum yields are in the range 15–40% in aerated aqueous media. Dynamic quenching of the lanthanide excited state occurs with electron-rich donors, e.g. iodide, ascorbate and urate, and a mechanistic interpretation is put forward involving an electron transfer process. The cationic lanthanide complexes are taken up by NlH/3T3 cells and tend to localise inside the cell nucleus.
Introduction
Our understanding of the photophysical principles involved in devising emissive lanthanide probe complexes has developed to such an extent that we are now able to relate complex structure to spectral property with sufficient confidence that the development of practicable probes can be undertaken more assertively.1–3 This is evident in the increasing usage of luminescent europium cryptate complexes as donors in a variety of time-resolved homogeneous assays. These are of interest, for example, to bio-scientists using FRET techniques to study G-protein-coupled receptors and to chemists more interested in their application as luminescent tags in high-throughput technology.4 It has also driven the development of ‘responsive’ lanthanide complexes in which changes in the emission profile have been used to signal variations in the local environment, e.g. pH, pO2, pX and pM, allowing the definition of luminescent sensors.5,6 Most work has concentrated on the more emissive Eu and Tb complexes emitting in the red and green.
An essential step in continuing to extend their utility is to devise probes that may be used for live cell imaging and related ‘in vivo’ applications. The key issues to address are: complex stability/toxicity; excitation wavelength and the product of overall emission quantum yield and the extinction coefficient at that wavelength; absence of competitive quenching processes from endogenous ions/biomolecules. Thus, the lanthanide complex must possess sufficient kinetic stability to resist premature decomplexation or chemical degradation. Fortunately, there are obvious parallels to be drawn in the chemistry of gadolinium contrast agents in MRI and in radiolabelled lanthanide conjugates being used in clinical imaging and therapy.7 As lanthanide f–f transitions are parity-forbidden and exceedingly weak, an appropriate sensitiser needs to be engineered into the complex capable of absorbing light efficiently (high ε) and transferring its excited state energy to the lanthanide ion8
(Scheme 1). The excitation wavelength should preferably be in the range 340–420 nm. The lower limit marks the wavelength below which quartz optics are required and, also, includes the tail absorption wavelength for several common biomolecules. The upper limit is set by the limited number of chromophores with a small singlet–triplet energy gap that possess a triplet energy of more than 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1
(cf. the lowest energy 5D4 Tb ‘acceptor’ excited state at 20400 cm−1). Finally, each of the excited states along the photophysical reaction pathway (singlet and triplet states of the sensitiser and the lanthanide excited state itself) should resist the quenching processes (i.e.via electron or energy transfer) that may limit the overall emission quantum yield.5 Examples of such processes include singlet excited state quenching by halide anions or by Eu3+ ion (minimised by systems with a fast rate of inter-system crossing and a high oxidation potential); triplet quenching by oxygen (averted in systems with fast energy transfer from the triplet sensitiser to the lanthanide ion) and electron/charge transfer quenching of the long-lived lanthanide excited state by electron-rich species. The latter case is more likely to be prevalent in cationic complexes or with systems in which the ligand has a low-lying LUMO.
000 cm−1
(cf. the lowest energy 5D4 Tb ‘acceptor’ excited state at 20400 cm−1). Finally, each of the excited states along the photophysical reaction pathway (singlet and triplet states of the sensitiser and the lanthanide excited state itself) should resist the quenching processes (i.e.via electron or energy transfer) that may limit the overall emission quantum yield.5 Examples of such processes include singlet excited state quenching by halide anions or by Eu3+ ion (minimised by systems with a fast rate of inter-system crossing and a high oxidation potential); triplet quenching by oxygen (averted in systems with fast energy transfer from the triplet sensitiser to the lanthanide ion) and electron/charge transfer quenching of the long-lived lanthanide excited state by electron-rich species. The latter case is more likely to be prevalent in cationic complexes or with systems in which the ligand has a low-lying LUMO.
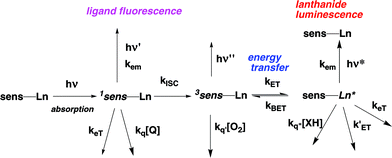 |
| | Scheme 1 Photophysical processes following light absorption. | |
The tetraazatriphenylene chromophore is an established sensitiser for lanthanide luminescence.9 It possesses a fast rate of inter-system crossing, so that the triplet excited state may be populated efficiently in polar media. It has a triplet energy of the order of 24000 cm−1
(singlet energy ca. 29000 cm−1) and, being electron-poor, it is difficult to oxidise. It may serve as a bidentate ligand and several examples of ruthenium (and related d-block) complexes incorporating this ligand have been examined as luminescent probes for nucleic acids.10,11 Indeed, examples of enantiopure cationic complexes have been postulated to bind with some selectivity to DNA, with contributions from electrostatic attraction, groove and intercalative binding. Analogous complexes of the ‘f’-block ions are virtually unexplored. The first examples were based on phenanthridinium conjugates12 but were of limited utility as the sensitiser singlet excited state was quenched by electron transfer, associated with intercalation with GC-rich DNA.
Here, we report the synthesis, and aspects of the characterisation, of an emerging family of enantiopure lanthanide complexes. A series of nine-coordinate ligands has been prepared based on the ‘cyclen’ ring skeleton. Each ligand includes a 2-substituted tetraazatriphenylene moiety. Some parts of this work have appeared in preliminary communications.13,14
Ligand and complex synthesis
The target series of complexes, [Ln.1]3+ to [Ln.7]3−, is based on structurally related nonadentate ligands, designed to form coordinatively saturated lanthanide complexes.
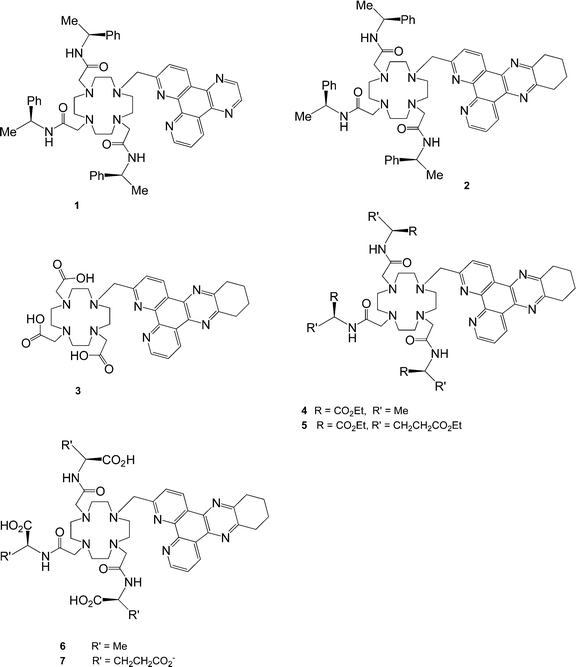
The variation of overall charge on the complex was considered to be an important issue as this affords (inter alia) a simple means of modulating the quenching effect of anionic or electron-rich species. The commonality of the 2-tetraazatriphenylene structural motif led to the initial requirement of an effective synthesis of the 2-halomethyl compounds, 8 and 9.
Reaction of 1,10-phenanthroline with one equivalent of methyl lithium, followed by oxidation with MnO2 in the presence of MgSO4 afforded the 2-methyl derivative in 51% yield. Oxidation of the 9,10-double bond with molecular bromine under strongly acidic conditions15 gave the dione, 10, in 77% yield. Condensation of 10 with 1,2-diaminoethane or 1,2-diaminocyclohexylamine was more effective in ethanol16 than THF9 and yielded 11
(2-Me dpq) and 12
(2-Me dpqC) in good yield, each of which was readily recrystallised from hot ethanol. Attempts to introduce a bromine or chlorine atom selectively at the α-Me site, by radical halogenation with NBS or NCS, were not very successful, and were compromised by competitive perhalogenation, as had been noted in the attempted functionalisation of 2,9-dimethyl-1,10-phenanthroline.17 Accordingly, an alternative route was used, involving stepwise oxidation with SeO216
(13, 14), reduction with sodium borohydride17 in ethanol (to give 15 and 16) and chlorination with PCl318 to yield 8 and 9.
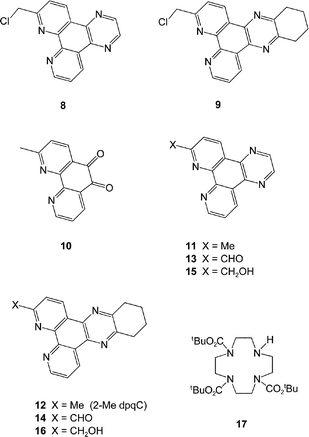
Selective mono-substitution of the cyclen ring was achieved via alkylation of tri-BOC-cyclen, 17,19 with 8 or 9 in acetonitrile to yield the carbonates 18 and 19. Deprotection of the BOC protecting groups (CF3CO2H/CH2Cl2) yielded the tetramines 20 and 21.
Alkylation with (
S)-
N-2-chloroethanoyl-2-phenylethylamine (MeCN, K
2CO
3, cat. KI) afforded the
triamides 1 and
2 which were purified by
chromatography on neutral
alumina. Similar procedures for
21 using the
N-2-chloroethanoyl derivatives of
ethyl alaninate and
diethyl glutamate, yielded the related
ligands 4 and
5. For the synthesis of the tris(carboxymethyl)
ligand,
3, an alternative procedure was used. Direct
alkylation of 1,4,7-tris(t-butoxycarbonylmethyl)-1,4,7,10-tetraazacylododecane,
22, with
9 under S
N2 conditions afforded the tri-ester
23. Deprotection with TFA gave the
ligand 3, and complexation with the LnCl
3 salt at pH 5.5 gave the neutral species, [Ln.
3], which was amenable to
purification by
column chromatography. For the cationic
triamide complexes, reaction of the
ligand with one equivalent of Ln(CF
3SO
3)
3 in dry MeCN gave the desired triflate salt. This could be converted to the more water soluble
chloride salt by
anion exchange chromatography. Finally, controlled base
hydrolysis of the cationic amide complexes [Ln.
4]
3+ and [Ln.
5]
3+ afforded the zwitterionic and anionic complexes, [Ln.
6] and [Ln.
7]
3−, which were purified by
ion exchange chromatography.
Complex characterisation
The absorption spectra of (RRR)- and (SSS)-[Ln.1]3+ in water were identical, with major bands at 262 (ε
= 19000 dm3 mol−1 cm−1), 305 (ε 10900 M−1 cm−1) and 340 nm (ε 3500 M−1 cm−1). For the lanthanide complexes of 2, and for each complex bearing a coordinated ‘dpqC’ chromophore, the absorption spectra were slightly red-shifted, with the longest wavelength band at 348 nm (ε 8300 M−1 cm−1). Mirror image circularly polarised luminescence (CPL) emission spectra were obtained for the enantiomeric Eu and Tb complexes of the ‘dpq’ ligand, 1
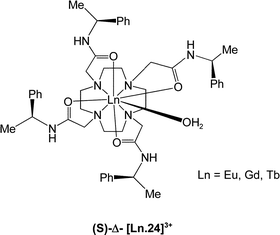
(Fig. 1). A comparison of the sign and magnitude of the observed CPL provides both information on the helicity about the metal centre and the structure of the complex.20,21 The sign and sequence of the transitions observed were the same as for the C-4 symmetric (S)-Δ and (R)-Λ Tb- and Eu-tetramide analogues [Ln.24]3+, whose absolute configuration has been verified by X-ray crystallography22
(Table 1).
The exception to this correlation was in the sign and form of the ΔJ
= 2 manifold for the europium complex. In this case, a sign reversal was noted. However, this electric-dipole-allowed transition (ΔJ
= 2) is particularly sensitive to the nature and polarisability of the donor atom that adopts an axial site in the mono-capped square anti-prismatic coordination environment.2,3 Indeed, a sign reversal was also observed when changing from a hard donor (e.g. OH2) to a more polarisable P–O bond (e.g. HMPA) with [Eu.23]3+. In [Ln.1]3+, it is very likely that the axial site in the putative nine-coordinate complex is occupied by a pyridyl N atom. Support for this premise comes from the 1H NMR assignment of [Eu.1]3+ in D2O or CD3OD. The proton α to this coordinated nitrogen (H-10) was assigned (COSY) at +31 ppm (243 K, CD3OD), consistent with its location within the large paramagnetic shielding effect associated with the ‘McConnell cone’. This is usually defined in relation to a principal (2) axis, passing through the apical substituent. A similarly large coordination shift for an analogous proton has been reported by Botta, Quici and co-workers, in a related 2-substituted phenanthridinyl complex23 based on cyclen.
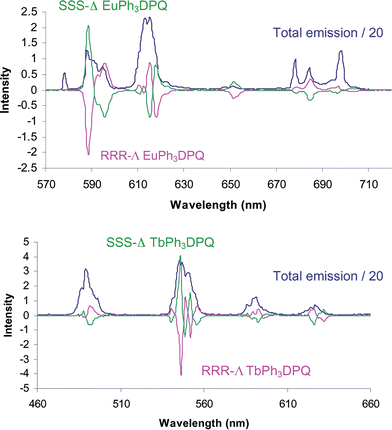 |
| | Fig. 1 Mirror image CPL spectra for (SSS)-Δ (green) and (RRR)-Λ (purple) europium and terbium complexes of 1. The blue trace represents the total emission spectrum (λexc = 340 nm, pH 7.4, 100 mM HEPES, 10 mM NaCl). | |
Table 1 Emission dissymmetry factors (±10%) for [Eu.1]3+, [Tb.1]3+ and related tetra-amide complexes, [Ln.23]3+ (H2O, 295 K, λexc = 340 nm)
| Complex |
g
588em
|
g
595em
|
g
614em
|
g
545em
|
| (SSS)-[Eu.1]3+ |
+0.18 |
−016 |
−0.04 |
|
| (S)-Δ-[Eu.23]3+ |
+0.09 |
−0.09 |
+0.08 |
|
| (SSS)-[Tb.1]3+ |
|
|
|
+0.12 |
| (S)-Δ-[Tb.23]3+ |
|
|
|
+0.27 |
The number of coordinated water molecules in [Ln.1]3+ and [Ln.2]3+ was assessed by measuring the radiative rate constants associated with depopulation of the Eu 5D0 and Tb 5D4 excited states in H2O and D2O and applying an established analysis that allows for the quenching effect of second-sphere waters and amide N–H oscillators.24 Values obtained (Table 2) are consistent with the absence of any bound water molecule, supporting the hypothesis of a nine-coordinate structure.
Table 2 Hydration states, q,24 and rate constants (kobs/ms−1; ±10%) for depopulation of the lanthanide excited state for [Ln.1]3+ (λexc = 340 nm) and [Ln.2]3+ (λexc = 350 nm) (pH 7.4, 100 mM HEPES, 10 mM NaCl)
| Complexa |
k
H2O
|
k
D2O
|
q
|
|
Similar sets of values were obtained for [Ln.3], [Ln.4]3+, [Ln.5]3+, [Ln.6] and [Ln.7]3− and were unchanged in deoxygenated samples.
|
| [Eu.1]3+ |
0.95 |
0.61 |
0 |
| [Eu.2]3+ |
0.96 |
0.63 |
0 |
| [Tb.1]3+ |
0.54 |
0.42 |
0.3 |
| [Tb.2]3+ |
0.64 |
0.58 |
0 |
The stability of the complex with respect to acid-catalysed dissociation or degradation was assessed in two series of experiments. First, TFA was added to aqueous solutions of [Eu.1]3+ and [Eu.3], and the pH varied between 6 and 2 (298 K, 10 mM NaCl). No observable change was found in the absorption and emission spectra of each complex over a two week period, consistent with the absence of protonation on the heterocyclic chromophore and the resistance to decomplexation. Similar stability studies were undertaken using [Tb.2]3+ over a 48 h period at pH 4 (0.2 M acetate buffer) and in reconstituted human serum (pH 7.2). In each case less than a 5% time-dependent change was observed, consistent with high complex stability.
Emission quantum yields
Overall emission quantum yields, ϕem (eqn (1), where ϕT is the quantum yield for formation of the triplet state, ηET is the efficiency of the energy transfer process, k0 is the natural radiative constant of the complex and τobs is the observed luminescence lifetime of the lanthanide ion, i.e. the inverse of the radiative decay constant), were measured in water,25 using an excitation wavelength corresponding to the lowest energy maximum in the absorption/excitation spectrum. The values found (Table 3) are amongst the highest observed in aqueous media,26,27 consistent with the high triplet quantum yield of the ‘dpq’ and ‘dpqC’ chromophore9
(no measurable fluorescence) and the high efficiency of the energy transfer step. This latter aspect is undoubtedly related to the minimisation of the lanthanide/chromophore distance and the observed insensitivity of the emission quantum yield and the rate of decay of the Ln excited state to dissolved oxygen. The triplet energy of the ‘dpq’ chromophore was measured for [Gd.1]3+ at 77 K in a frozen glass (MeOH–EtOH 4 : 1). The shortest wavelength transition (0–0 band)
(Fig. 2) was observed at 420 nm (23800 cm−1), sufficiently higher than the Eu (17200 cm−1) and Tb (20400 cm−1) emissive excited state levels to preclude any back energy transfer process at ambient temperature.
Table 3 Absolute emission quantum yieldsa for aqueous solutions of Eu and Tb complexes (λexc = 340 for [Eu.1]3+ and 348 nm for others)
| Complex |
φ
H2O
|
φ
D2O
|
|
Determined as described earlier25 with reference to p-cresyl violet in MeOH (ϕ
= 0.54) and rhodamine-101 (ϕ
= 1.0) in ethanol.
|
| [Eu.1]3+ |
0.21 |
0.27 |
| [Tb.1]3+ |
0.36 |
0.46 |
| [Eu.2]3+ |
0.16 |
0.20 |
| [Tb.2]3+ |
0.40 |
0.48 |
| [Eu.3] |
0.18 |
|
| [Tb.3] |
0.33 |
|
| [Eu.6] |
0.18 |
|
| [Tb.6] |
0.36 |
|
| [Eu.7]3− |
0.15 |
|
| [Tb.7]3− |
0.34 |
|
![Phosphorescence spectrum of [Gd.1]3+ (CF3SO3)3 in a frozen glass (77 K, MeOH–EtOH 4 : 1, λexc = 340 nm), compared to that of 7-methyl-tetraazatriphenylene, 11 (dashed line).](/image/article/2005/OB/b418964g/b418964g-f2.gif) |
| | Fig. 2 Phosphorescence spectrum of [Gd.1]3+ (CF3SO3)3 in a frozen glass (77 K, MeOH–EtOH 4 : 1, λexc = 340 nm), compared to that of 7-methyl-tetraazatriphenylene, 11 (dashed line). | |
Dynamic quenching of the lanthanide excited state
In the photophysical pathway that leads to lanthanide emission, there are three salient excited states to consider: the singlet and triplet states of the sensitising chromophore and the lanthanide excited state itself. The facility of the S1 to T1 transition with the dpq and dpqC chromophores, coupled with the absence of any overt triplet quenching process, allows – for the first time – an assessment of the quenching of the lanthanide excited state by a charge transfer mechanism. Prior studies have focused on the quenching by X–H energy-matched oscillators (X = O, N, C).6,9,24 Here, the absence of a bound water molecule and the subsequent minimisation of quenching by vibrational energy transfer to OH oscillators makes these complexes well suited for this analysis.
The Tb and Eu excited states lie at 2.52 and 2.13 eV respectively and this free energy can be used to ‘drive’ the charge transfer process in quenching by electron-rich species. The Weller equation [eqn. (2)]28 can be used to assess the facility of this process,
| | | ΔGET
=
nF[(Eox
−
Ered)
−
ELn*
−
e2/εr] J mol−1 | (2) |
where
Eox is the oxidation potential (
versus NHE) of the
electron donor (quencher, Q),
Ered is the reduction potential of the acceptor (
ligand or metal based) in the complex,
ELn* is the energy of the lanthanide excited state, and
e2/
εr is a Coulombic attraction correction term associated with the formation of the transient ion pair (or radical/ion). It is usually a small term (<0.2 eV). As an example of this approach, consider the quenching of [Eu.
1]
3+ by iodide anions, which has been reported (in our preliminary work
13) to reduce the emission intensity and the lifetime, with a Stern–Volmer quenching constant [
eqn. (3)],
KSV−1, of 9 mM.
| | 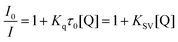 | (3) |
Given that the iodide oxidation potential is +0.54 V and that both the reduction of related cationic Eu(
III) tetra-amide complexes
5,25
(
metal-centred at −1.1 V) and of related [Rudpq(dpqC)
2]
2+ complexes (
ligand-centred at −1.07 V)
10 occur at a similar potential, then [
eqn. (4)] applies, giving a favourable free energy change of −65 kJ mol
−1.
| | ΔGET
= 96.5![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) [(0.54 + 1.07)
− 2.13 − 0.15]
=
−65 kJ mol−1 [(0.54 + 1.07)
− 2.13 − 0.15]
=
−65 kJ mol−1 | (4) |
This analysis is in line with the insensitivity of the europium emission intensity/lifetime (±10%) to added chloride ions (up to 100 mM). The chloride
oxidation potential is +1.36 V, rendering the overall free energy change positive and unfavourable. The magnitude of the second-order rate constant that defines the iodide quenching process is of the order of 10
5 M
−1 s
−1. This is a rather low value compared to the values of >10
9 M
−1 s
−1 that characterise dynamic quenching of
aryl triplet states by O
2. This probably reflects the inhibition to collisional encounter on diffusion (
i.e. the longer range nature of the process) associated with the relative inaccessibility of the encapsulated lanthanide to the quenching species.
Stern–Volmer plots (Fig. 3) were obtained for a range of complexes (Table 4). Quenchers included selected halide anions, urate [serum concentration is typically 0.3 mM, with an oxidation potential (in serum) of +0.42 V],29 ascorbate and glutathione. The latter three species are well-established low molecular-weight anti-oxidants.29 The concentration of glutathione in serum or in selected cells is typically much lower than that of ascorbate, which averages 60 μM in human serum,30 but is found at much higher concentrations (0.8 to 2 mM typically) in oxygen-sensitive tissues such as the eye, adrenal glands, and in the heart, liver and brain.31
![Stern–Volmer plots showing the differing sensitivity of complexes to quenching by iodide (pH 7.4, 100 mM HEPES). Data for [Tb.2]3+ (■), [Tb.3] (▲), [Tb.6]3− (×) and [Eu.2]3+ (◆) are illustrated with KSV−1 values of 0.9, 2.1, 2.5 and 27 mM, respectively.](/image/article/2005/OB/b418964g/b418964g-f3.gif) |
| | Fig. 3 Stern–Volmer plots showing the differing sensitivity of complexes to quenching by iodide (pH 7.4, 100 mM HEPES). Data for [Tb.2]3+ (■), [Tb.3] (▲), [Tb.6]3− (×) and [Eu.2]3+ (◆) are illustrated with KSV−1 values of 0.9, 2.1, 2.5 and 27 mM, respectively. | |
Table 4 Stern–Volmer quenching constants (KSV−1/mM, ±100%, pH 7.4, 100 mM HEPES) defining the deactivation of the lanthanide excited state,a,b and relevant oxidation potential for quenching species (Eox vs. NHE)
| Oxidation potential/V |
+1.36 |
+1.07 |
+0.54 |
+0.42 |
+0.48 |
+0.19 |
+0.36 |
| Quenching species |
Cl− |
Br− |
I− |
Urate |
Ascorbate |
Glutathione |
Fe(CN)64− |
| |
| Complex |
K
SV
−1
(I−)/mM |
K
SV
−1
(urate)/mM |
K
SV
−1
(ascorbate)/mM |
|
K
SV
−1 values for glutathione were: [Eu.2]3+, 24 mM; [Tb.2]3+, 20 mM.
Data were analysed by examining the change in emission lifetime; analysis of emission intensity variation gave values within 15% of those stated above.
Value was 10 mM for the Λ-complex; in the presence of poly(dGdC)
(4 base pairs per complex), KSV−1 for the Δ-complex was 50 mM but was 9 mM for the Λ-complex, consistent with preferential inhibition to I− encounter in the diastereoisomeric complex with the Δ-isomer.
Exhibited slight curvature in the Stern–Volmer plot at [I−] > 10 mM.
|
| Δ-[Eu.1]3 |
9.0c |
|
|
| Δ-[Eu.2]3+ |
27.0d |
0.93 |
0.39 |
| Δ-[Tb.2]3+ |
0.9 |
0.25 |
0.18 |
| [Tb.3] |
2.1d |
0.006 |
0.30 |
| [Tb.6]3− |
2.5 |
0.012 |
0.38 |
Analysis of the information in Table 4 reveals some common trends. First, quenching of the Tb emission is greater than for the Eu analogue, in line with the higher excited state energy of the 5D4 Tb state. Second, the sequence of sensitivity to quenching generally follows the ease of oxidation of the quenching anion, i.e. ascorbate ≈ urate >> I− >>> Br−/Cl−. Glutathione does not fall readily into this sequence but is only partially ionised under the pH conditions used (thiol pKa is 8.66). Third, the facility of quenching decreases as the overall complex charge varies from positive to neutral and anionic, consistent with the sequence of enhanced Coulombic repulsion in formation of the encounter complex. Given that the overall emission intensity with [Eu.2]3+ and [Tb.2]3+ in 50% human serum decreased by 40 and 70% respectively, compared to a 10 mM HEPES buffer standard solution, it seems very likely that ascorbate/urate are the major quenching species.
Earlier work had revealed that both [Tb.1]3+ and [Eu.1]3+ bind to poly(dGdC) and to calf-thymus DNA. Similar behaviour was noted with [Ln.3]3+, and DNA-binding was characterised by a decrease in the lifetime of the lanthanide emission, with the effect being bigger for Tb compared to Eu, and for poly(dGdC) with respect to poly(dAdT).13 Such behaviour accords with the charge transfer mechanism postulated above, with the electron-rich guanines the likely reductant. Competitive quenching experiments with I− were performed with Δ- and Λ-[Eu.1]3+ in the presence of polynucleotides, in order to deduce information concerning their relative sensitivity to dynamic quenching. Addition of iodide to the poly(dAdT)-bound complex reduced the apparent KSV values by about 50% for both the Δ- and the Λ-complex. In the presence of poly(dGdC), the Stern–Volmer constant was unchanged for Λ-[Eu.1]3+, but KSV−1 changed from 9 to 50 mM for the Δ-isomer. It appears that quenching by iodide is suppressed by the DNA-binding interaction and that the Λ-isomer must be equally exposed to I− quenching in the free and DNA-bound states. This is likely to occur only if it is neither intercalated nor ‘groove’-bound. In the diastereoisomeric complex with the Δ-isomer, the strong inhibition of I− quenching suggests a binding interaction in which the dpq chromophore is shielded much more effectively from iodide encounter.
Preliminary microscopy experiments
Some preliminary experiments have been carried out monitoring the spatio-temporal localisation of [Tb.2]3+ in mouse skin fibroblasts (NlH/3T3 cells). Cultured cells were exposed to a growth medium containing between a 50 μM and 1 mM solution of the terbium complex. The living cells were then examined by fluorescence microscopy at time intervals in the range of 30 min to 48 h, monitoring the green terbium luminescence selectively, with the aid of appropriate excitation and emission cut-off filters. Selected cell populations were exposed to the complex for periods of up to 24 h, then the growth medium was replaced (after washing with phosphate buffered saline) to allow the examination of the localised complex in the absence of a concentration gradient.
Representative images are shown in Figs. 4, 5 and 6. The first image, taken after 4 h, for a cell exposed to a 0.3 mM solution complex, highlights the distribution of the complex through the cytoplasm, with brighter regions near the centre of the cell. The cell nucleus remains dark. As the cell remains exposed to the complex, most of the luminescence is observed in the cell nucleus and around the cell membrane. Similar results were obtained for the analogous Eu complex. For example, in Fig. 5, an image is shown (1 mM complex in medium, 24 h) clearly highlighting the localisation of the complex inside the nucleus of the cell and in certain compartments within the nucleus. The nuclear membrane is also delineated, with a residual diffuse luminescence in the cytosol. In the absence of complex in the external medium, the luminescence in the cell nucleus becomes progressively less intense and the complex was again to be found distributed mostly outside the cell nucleus. Thus, 24 h after sequential incubation with complex followed by removal of the medium containing the complex, the images obtained (Fig. 6) revealed the complex in the cytosol once more. The slightly ‘spotty’ nature of the image suggested some preferential localisation in a particular cell organelle. This might be the cellular mitochondria, which possess a negative surface potential and contain DNA. Co-localisation studies are underway to verify this hypothesis, using a known mitochondrial stain, such as ‘Mitotracker Red’.
![Fluorescence microscope image highlighting the egress of the [Tb.2]3+ complex out of the cell nucleus and back into the extra-nuclear compartments, 24 h after removal of the external concentration gradient.](/image/article/2005/OB/b418964g/b418964g-f6.gif) |
| | Fig. 6 Fluorescence microscope image highlighting the egress of the [Tb.2]3+ complex out of the cell nucleus and back into the extra-nuclear compartments, 24 h after removal of the external concentration gradient. | |
These experiments confirm that the complex is able to penetrate first the cell membrane and then the nuclear membrane, and can localise inside the cell nucleus, in the presence of a sufficient external concentration gradient. Transport in and out of the cell nucleus appears to be reversible. The neutral [Tb.3] and anionic [Tb.6]3− complexes were examined in a parallel manner, but were very poorly taken up by the cells, even at the highest concentration of added complex. The complex localises inside the cell rapidly (within 15–30 min), and this rate of ingress appeared to be similar, in an incubation carried out at 4 °C, monitored after 30 mins, 1 h and 3 h. Such behaviour is typical of an uptake mechanism involving a channel or pore in the cell membrane rather than by an activated process. Certainly, these preliminary findings indicate that these emissive lanthanide complexes offer much scope as luminescent probes in biological systems, and are suitable for further study ‘in cellulo’.
Experimental
Reactions requiring anhydrous conditions were carried out using Schlenk-line techniques under an atmosphere of dry argon. Solvents were dried from an appropriate drying agent when required and water was purified by the ‘PuriteSTILL plus’ system. Thin-layer chromatography was carried out on neutral alumina plates (Merck Art 5550) or silica plates (Merck 5554) and visualised under UV (254 nm) or by staining with iodine. Preparative column chromatography was carried out using neutral alumina (Merck Aluminium Oxide 90, activity II–III, 70–230 mesh), pre-soaked in ethyl acetate, or silica (Merck Silica Gel 60, 230 ± 400 mesh).
1H and 13C NMR spectra were recorded on a Varian Mercury 200 (1H at 199.975 MHz, 13C at 50.289 MHz), a Varian Unity 300 (1H at 299.908 MHz, 13C at 75.412 MHz), a Varian VXR 400 (1H at 399.968 MHz, 13C at 100.572 MHz), or a Bruker AMX 500 spectrometer. Spectra were referenced internally to the residual protio-solvent resonances and are reported relative to TMS. Electrospray mass spectra were recorded on a VG Platform II (Fisons instrument), operating in positive or negative ion mode as stated, with methanol as the carrier solvent. Accurate mass spectra were recorded at the EPSRC Mass Spectrometry Service at the University of Wales, Swansea. UV-Visible absorbance spectra were recorded on a Perkin Elmer Lambda 900 UV/Vis/NIR spectrometer and emission spectra and lifetimes were measured on a Perkin Elmer LS55 luminescence spectrometer. Lifetimes were measured by excitation of the sample by a short pulse of light (348 nm) followed by monitoring the integrated intensity of light (545 nm for terbium, 620 nm for europium) emitted during a fixed gate time, tg, and a delay time, td, later. At least 20 delay times were used covering three or more lifetimes. A gate time of 0.1 ms was used, and the excitation and emission slits were set to 10 nm band-pass. The obtained decay curves were fitted to the equation below using Microcal Origin or Microsoft Excel,
| I
=
A0
+
A1exp(−kt) |
where
I
= intensity at time
t after the flash,
A0
= intensity after the decay has finished,
A1
= pre-exponential factor,
k
= rate constant for decay of the excited state.
Quenching constants were determined by measuring the emissive lifetime, τ, of each of the complexes with varying concentrations of the quenching species (Q). A plot of τ0/τversus
[Q] (as seen previously in Fig. 3) was used to determine the Stern–Volmer quenching constant, KSV, as the slope of this plot. In each case, a 10 μM solution of the complex buffered to pH 7.4 with HEPES (100 mM) in the presence of NaCl (10 mM) was used.
Quantum yield measurements were made relative to two standards as reported earlier.25 Five solutions with an absorbance of between 0.02 and 0.1 were used. For each of these solutions the absorbance at the excitation wavelength and the total integrated emission was determined. A plot of total integrated emission against absorbance gives a straight line with slope E/A. The unknown quantum yield can thus be calculated from the following equation.
Since values for both the sample (x) and reference (r) compounds were recorded in
water the refractive index term (
n) cancels out.
Errors in quantum yield determinations can arise due to the inner filter effect or errors in the amount of absorbed light. This effect was minimised by only using samples with an absorbance below 0.1. Errors in the amount of light absorbed by each sample were minimised by choosing an excitation wavelength on a relatively flat area of the absorption curve and by using a small band-pass for excitation.
Mouse skin fibroblasts (NIH-3T3 cells) were cultured to 50% confluence on a cover slip in a DMEM (Dulbeccos Modified Eagles Medium) solution supplemented with 10% foetal bovine serum and a 1% penicillin/streptomycin mixture. The cells were washed with phosphate buffered saline and the lanthanide complexes added in a clean medium. The incubation time was varied from 30 min to 48 h, and the concentration of added complex was examined in the range 0.05 to 1 mM, For examination by fluorescence microscopy, the cells were washed with phosphate buffered saline and mounted onto slides (Zeiss Axiovert 200 M). For the imaging of the terbium complexes, the excitation wavelength was 348 nm.
Ligand and complex synthesis
2-Methyl-1,10-phenanthroline.
A solution of 1,10-phenanthroline (10 g, 55 mmol) in dry THF (100 ml) was added dropwise to a stirred solution of methyl lithium (1.6 mol dm−3 in Et2O, 34.5 ml, 55 mmol) in dry THF (150 ml) at 0 °C under argon. The mixture was stirred overnight at room temperature under argon to give a dark green solution. The reaction was quenched by careful addition of water (100 ml) at 0 °C. THF was removed under reduced pressure using a rotary evaporator and the product extracted into diethyl ether (3 × 150 ml). Activated manganese dioxide (53 g, 0.61 mol) was added and the solution stirred for 1.5 h; magnesium sulfate was added and the solution stirred for a further 2 h. Filtration through a Celite plug yielded a clear yellow solution. The solvent was evaporated to dryness to yield the crude product, which was purified by recrystallisation from ethyl acetate/hexane to give a white product (5.50 g, 28 mmol, 51%). 1H NMR (300 MHz, CDCl3): δ 2.97 (3H, s, CH3), 7.54 (1H, d, J
= 8.1 Hz, H3), 7.63 (1H, dd, J
= 8.1, 4.2, H8), 7.75 (1H, d, J
= 8.7, H6), 7.79 (1H, d, J
= 8.7, H5), 8.16 (1H, d, J
= 8.1, H4), 8.26 (1H, dd, J
= 8.1, 1.5, H7), 9.22 (1H, dd, J
= 4.2, 1.2, H9). 13C NMR (75 MHz, CDCl3): δ 25.9 (CH3), 122.8 (C8), 123.7 (C3), 125.5 (C6), 126.5 (C5), 126.7 (q Ar), 128.8 (q Ar), 136.1 (C7), 136.2 (C4), 145.7 (q Ar), 146.0 (q Ar), 150.3 (C9), 159.5 (C2). m/z
(ES+) 195 (MH+), 217 (MNa+), 411 (M2Na+); mp 75–76 °C (lit.32 78 °C). Elemental analysis: found C 79.5, H 5.44, N 14.5; C13H10N2·0.17H2O requires C 79.2, H 5.23, N 14.2%.
2-Methyl-1,10-phenanthroline-5,6-dione, 10.
A mixture of 2-methyl-1,10-phenanthroline (2.00 g, 10.29 mmol) and potassium bromide (12.25 g, 102.9 mmol) was placed in an ice bath. Cold sulfuric acid (43 ml) was added carefully to the flask in a dropwise fashion, which turned the solution to a dark reddish colour. Concentrated nitric acid (21.5 ml) was then added in a similar way and the solution was boiled under reflux overnight. The mixture was cooled, poured into water (500 ml) and neutralised by addition of sodium hydroxide pellets. The product was extracted into dichloromethane (3 × 300 ml), which was dried over K2CO3 and removed under reduced pressure to yield a yellow solid. Purification was achieved by chromatography on silica [1.79 g, 7.98 mmol, 77%; gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2, Rf
= 0.34 (5% MeOH–CH2Cl2)]. 1H NMR (300 MHz, CDCl3): δ 2.86 (3H, s, CH3), 7.44 (1H, d, J
= 7.8 Hz, H3), 7.58 (1H, dd, J
= 7.8, 4.8, H8), 8.40 (1H, d, J
= 8.1, H4), 8.50 (1H, dd, J
= 7.8, 2.1, H7), 9.14 (1H, dd, J
= 4.8, 1.8, H9). 13C NMR (75 MHz, CDCl3): δ 26.2 (CH3), 125.6 (C8), 125.9 (C3), 126.2 (q Ar), 128.3 (q Ar), 137.5 (C7), 137.7 (C4), 152.7 (q Ar), 153.2 (q Ar), 156.6 (C9), 167.4 (C2), 178.7 (C5), 179.2 (C6). m/z
(ES+) 246 (MNa+), 279 (MNa+
+ MeOH), 471 (M2Na+), 503 (M2Na+
+ MeOH), 535 (M2Na + 2MeOH). IR (KBr): 3074 (C–H Ar), 1681 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1568 (py ring vibration), 1436 and 1301 (CC py), 1216, 1123, 1013, 925, 829, 739 cm−1; mp 98–100 °C. Elemental analysis: found, C 66.3, H 3.46, N 12.2; C13H8N2O2·0.67H2O requires C 66.1, H 3.94, N 11.9%.
O), 1568 (py ring vibration), 1436 and 1301 (CC py), 1216, 1123, 1013, 925, 829, 739 cm−1; mp 98–100 °C. Elemental analysis: found, C 66.3, H 3.46, N 12.2; C13H8N2O2·0.67H2O requires C 66.1, H 3.94, N 11.9%.
7-Methyldipyrido[3,2-f:2′,3′-h]quinoxaline, 11.
A solution of ethylenediamine (0.23 ml, 3.48 mmol) in THF (15 ml) was added in a dropwise fashion to a solution of the dione (0.78 g, 3.48 mmol) in THF (400 ml) and the resulting mixture was boiled under reflux overnight. The solvent was removed by evaporation and the dark red solid taken into CH2Cl2
(100 ml) and washed with H2O (3 × 100 ml). The organic layer was dried over K2CO3. Removal of the solvent under reduced pressure afforded a yellow solid (0.82 g, 0.33 mmol, 96%). Further purification was achieved by chromatography on silica (gradient elution: CH2Cl2 to 1% CH3OH containing 0.5% NH3–CH2Cl2, Rf
= 0.18, 10% CH3OH containing 0.5% NH3–CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 2.96 (3H, s, CH3), 7.64 (1H, d, J
= 8.4 Hz, H6), 7.74 (1H, dd, J
= 8.1, 4.5, H11), 8.91 (2H, m, H2, H3), 9.28 (1H, dd, J
= 4.5, 1.8, H10), 9.31 (1H, d, J
= 8.4, H5), 9.44 (1H, dd, J
= 8.4, 1.8, H12). 13C NMR (75 MHz, CDCl3): δ 26.1 (CH3), 123.9 (C11), 124.7 (C6), 124.9 (q Ar), 127.1 (q Ar), 133.3 (C12), 133.4 (C5), 140.2 (q Ar), 140.8 (q Ar), 144.2 (C2), 144.6 (C3), 147.1 (q Ar), 147.5 (q Ar), 152.5 (C10), 162.1 (C7). m/z
(ES+) 247 (30%, MH+), 269 (33%, MNa+), 515 (100%, M2Na+); mp 192–194 °C. Elemental analysis: found C 71.8, H 3.99, N 22.7; C15H10N4·0.25H2O requires C 71.9, H 4.19, N 22.4%.
3-Methyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine, 12.
To a solution of 2-methyl-1,10-phenanthroline-5,6-dione (0.65 g, 2.90 mmol) in absolute ethanol (50 ml) was added trans-1,2-diaminocyclohexane (0.33 g, 2.90 mmol) and the resulting mixture boiled under reflux for 5 h. The solution was allowed to cool and the solvent was removed under reduced pressure. The product was recrystallised from ethanol (0.81 g, 2.90 mmol, 93%), mp >220 °C (decomp). 1H NMR (300 MHz, CDCl3): δ 2.09 (4H, m, H10, 13), 2.98 (3H, s, H15), 3.24 (4H, m, H11, 12), 7.63 (1H, d, J
= 8.3 Hz, H2), 7.73 (1H, dd, J
= 8.2, 4.4, H7), 9.26 (1H, dd, J
= 4.4, 1.8, H6), 9.35 (1H, d, J
= 8.3, H1), 9.47 (1H, dd, J
= 8.2, 1.8, H8). 13C NMR (75 MHz, CDCl3): δ 22.7 (C11), 22.8 (C12), 25.7 (CH3), 32.7 (C10), 32.8 (C13), 123.4 (C7), 124.2 (C2), 124.8 (q Ar), 151.5 (C6), 153.2 (q Ar), 153.6 (q Ar), 160.9 (q Ar). HRMS (+
m/z): [M + H]+ calculated for C19H16N4Na, 323.1273; found, 323.1269.
Dipyrido[3,2-f:2′,3′-h]quinoxaline-7-carboxyaldehyde, 13.
Selenium dioxide (0.77 g, 6.97 mmol) was added to a solution of the 3-methyl phenazine (0.78 g, 3.17 mmol) in dioxane (100 ml). The mixture was heated under reflux for 2 h, then filtered through Celite. Evaporation of the solvent afforded a white solid. Some more product was obtained by washing the Celite with CH2Cl2
(0.77 g, 2.96 mmol, 93%). 1H NMR (300 MHz, CDCl3): δ 7.89 (1H, dd, J
= 8.1, 4.5 Hz, H11), 8.46 (1H, d, J
= 8.4, H5), 9.06 (1H, d, J
= 1.8, H2), 9.08 (1H, d, J
= 1.8, H3), 9.39 (1H, dd, J
= 4.2, 1.8, H10), 9.57 (1H, dd, J
= 8.1, 1.8, H12), 9.73 (1H, dd, J
= 8.4, 0.73, H6), 10.61 (1H, d, J
= 0.73, CHO). 13C NMR (75 MHz, CDCl3): δ 121.0 (C5), 124.8 (C11), 127.8 (q Ar), 130.2 (q Ar), 133.7 (C12), 135.1 (C6), 140.0 (q Ar), 141.6 (q Ar), 145.2 (C2), 145.8 (C3), 147.1 (q Ar), 147.7 (q Ar), 152.9 (C10), 154.1 (C7), 194.0 (CHO). m/z
(ES+) 283 (60%, MNa+), 315 (100%, MNa+
+ MeOH), 543 (60%, M2Na+), 575 (10%, M2Na+
+ MeOH), 607 (40%, M2Na+
+ 2MeOH). IR (KBr): 3074, 3029 (C–H Ar), 2865 (C–H in CHO), 1703 (CO), 1566, 1467 and 1449 (CC py), 1391 (C–H CHO), 1362, 1207, 1077, 767 and 748 cm−1; mp > 220 °C (decomp.). Elemental analysis: found C 67.3, H 3.08, N 20.9; C15H8N4O·0.33H2O requires C 67.7, H 3.27, N 21.0%.
3-Carboxaldehyde-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine, 14.
Selenium dioxide (0.96 g, 8.66 mmol) was added to a solution of 3-methyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine (1.30 g, 4.33 mmol) in dioxane (200 ml) and the mixture boiled under reflux for 4 h. The solution was cooled to room temperature and filtered through a Celite plug to afford a crude product that was used directly in the next step. 1H NMR (300 MHz, CDCl3): δ 2.11 (4H, m, H10, 13), 3.28 (4H, m, H11, 12), 7.83 (1H, dd, J
= 8.2, 4.4 Hz, H7), 8.41 (1H, d, J
= 8.3, H1), 9.33 (1H, dd, J
= 4.5, 1.8, H6), 9.53 (1H, dd, J
= 8.2, 1.7, H8), 9.68 (1H, dd, J
= 8.3, 0.9, H2), 10.60 (1H, d, J
= 0.9, CHO). 13C NMR (75 MHz, CDCl3): δ 22.5 (C11), 22.6 (C12), 32.7 (C10), 32.8 (C13), 120.2 (C7), 124.1 (c1), 127.5 (q Ar), 130.0 (q Ar), 133.0 (c8), 134.1 (C2), 136.6 (q Ar), 138.3 (q Ar), 146.2 (q Ar), 146.7 (q Ar), 151.7 (C6), 153.1 (q Ar), 154.4 (q Ar), 155.1 (q Ar), 193.9 (CHO). EI (+
m/z): [M+] 314.
7-(Hydroxymethyl)dipyrido[3,2-f:2′,3′-h]quinoxaline, 15.
Sodium borohydride (54 mg, 1.43 mmol) was added to a solution of the precursor aldehyde (300 mg, 1.15 mmol) in CH2Cl2–EtOH 7 : 1 (120 ml). The mixture was heated at reflux for 2 h. The solvent was evaporated and the orange solid taken into a saturated solution of Na2CO3
(50 ml) and extracted into CH2Cl2
(3 × 70 ml). The organic layer was dried over K2CO3. Removal of the solvent under reduced pressure afforded a yellow solid (207 mg, 0.79 mmol, 68%), which was used without further purification. 1H NMR (300 MHz, CDCl3): δ 5.20 (2H, s, CH2), 7.82 (1H, dd, J
= 8.1, 4.5 Hz, H11), 7.87 (1H, d, J
= 8.7, H6), 9.00 (2H, s, H2, H3), 9.27 (1H, dd, J
= 4.5, 1.8, H10), 9.51 (1H, d, J = 8.4, H5), 9.54 (1H, dd, J
= 8.1, 1.8, H12). 13C NMR (75 MHz, CDCl3): δ 65.4 (CH2), 121.5 (C11), 124.0 (C6), 126.0 (q Ar), 127.3 (q Ar), 133.5 (C12), 133.9 (C5), 140.2 (q Ar), 140.5 (q Ar), 144.4 (C2), 144.7 (C3), 146.3 (q Ar), 147.0 (q Ar), 152.0 (C10), 163.0 (C7). m/z
(ES+) 262 (100%, M+), 284 (30%, MNa+), 546 (20%, M2Na+).
7-(Chloromethyl)dipyrido[3,2-f:2′,3′-h]quinoxaline, 8.
Phosphorus trichloride (0.82 ml, 9.36 mmol) in CHCl3
(5 ml) was added in a dropwise fashion to a stirred solution of the hydroxymethyl compound (206 mg, 0.78 mmol) in CHCl3
(100 ml). The mixture was heated at reflux for 4 h. The solution was neutralised by addition of a saturated aqueous Na2CO3 solution (100 ml). The organic layer was separated and dried over K2CO3. Removal of the solvent under reduced pressure afforded a pink solid. Purification by chromatography on silica gel (gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2, Rf
= 0.47, 10% CH3OH–CH2Cl2) gave a colourless solid (95 mg, 0.34 mmol, 43%). 1H NMR (200 MHz, CDCl3): δ 5.14 (2H, s, CH2), 7.81 (1H, dd, J
= 8.2, 4.4 Hz, H11), 8.08 (1H, d, J
= 8.4, H6), 8.99 (2H, s, H2, H3), 9.32 (1H, dd, J
= 4.4, 1.8, H10), 9.51 (1H, dd, J
= 8.2, 1.8, H12), 9.54 (1H, d, J
= 8.4, H5). 13C NMR (75 MHz, CDCl3): δ 47.1 (CH2), 123.1 (C11), 124.9 (C6), 126.1 (q Ar), 127.0 (q Ar), 133.0 (C12), 134.2 (C5), 139.8 (q Ar), 140.1 (q Ar), 144.4 (C2, C3), 146.2 (q Ar), 146.7 (q Ar), 152.2 (C10), 159.2 (C7). m/z
(ES+) 268 (20%, MNa+
− Cl), 302 (95%, MNa+), 548 (50%, M2Na+
− Cl), 582 (100%, M2Na+); mp 194–196 °C (decomp.). Accurate mass: found MH+ 281.0593; C15H10ClN4 requires 281.0594.
3-Hydroxymethyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine, 16.
Sodium borohydride (164 mg, 4.33 mmol) was added to a solution of 3-carboxaldehyde-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine (1.36 g, 4.33 mmol) in CHCl3–EtOH (200 ml). The mixture was heated under reflux for 2 h. The solvent was evaporated and the orange solution taken into a saturated solution of Na2CO3
(200 ml) and extracted with CH2Cl2
(3 × 150 ml) then CHCl3
(1 × 150 ml). The organic phase was dried over Na2SO4 filtered and the solvent removed under reduced pressure to afford a yellow solid (0.89 g, 2.81 mmol, 65%); mp 210 °C (decomp.). 1H NMR (300 MHz, CDCl3): δ 2.10 (4H, m, H10, 13), 3.25 (4H, m, H11, 12), 5.18 (2H, s, CH2OH), 7.75 (1H, dd, J
= 8.2, 4.4 Hz, H7), 7.80 (1H, d, J
= 8.3, H2), 9.20 (1H, dd, J
= 4.4, 1.7, H6), 9.47 (1H, d, J
= 8.3, H1), 9.49 (1H, dd, J
= 8.3, 1.7, H8). 13C NMR (75 MHz, CDCl3): δ 22.7 (C11, 12), 32.8 (C10, 13), 65.7 (CH2), 121.0 (C7), 123.6 (C2), 125.9 (q Ar), 127.2 (q Ar), 132.9 (C8), 133.4 (C1), 137.4 (q Ar), 151.1 (C6), 153.5 (q Ar), 153.8 (q Ar), 162.3 (q Ar). HRMS (+
m/z): [M + Na]+ calculated for C19H16N4ONa, 339.1222; found, 339.1219.
3-Chloromethyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine, 9.
To a solution of 3-hydroxymethyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine (0.84 g, 2.66 mmol) in CHCl3
(450 ml) was added phosphorus trichloride (1.46 g, 10.6 mmol). The mixture was heated under reflux for 4 h. The solution was neutralised by addition of a saturated aqueous solution of Na2CO3
(500 ml). The organic layer was separated and the aqueous layer extracted with CH2Cl2
(2 × 400 ml) and CHCl3
(1 × 400 ml). The combined organic layers were dried over K2CO3 and the solvent evaporated to afford a yellow solid. Purification by chromatography on silica (gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2) gave a pale yellow solid (Rf 0.38, 5% MeOH–CH2Cl2; 0.75 g, 2.25 mmol, 84%), mp 207–208 °C. 1H NMR (300 MHz, CDCl3): δ 2.05 (4H, m, H10, 13), 3.17 (4H, m, H11, 12), 5.10 (2H, s, CH2Cl), 7.70 (1H, dd, J
= 8.2, 4.4 Hz, H7), 7.97 (1H, d, J
= 8.3, H2), 9.23 (1H, dd, J
= 4.5, 1.8, H6), 9.37 (1H, dd, J
= 8.2, 1.8, H8), 9.40 (1H, d, J
= 8.3, H1). 13C NMR (75 MHz, CDCl3): δ 22.7 (C11, 12), 32.2 (C10, 13), 47.0 (CH2), 122.3 (C7), 123.2 (C2), 125.7 (q Ar), 126.7 (q Ar), 128.9 (q Ar), 130.1 (q Ar), 132.2 (C8), 133.4 (C1), 145.4 (q Ar), 145.9 (q Ar), 151.1 (q Ar), 153.3 (C6), 157.9 (q Ar). HRMS (+
m/z): [M + Na]+ calculated for C19H15N4NaCl, 357.0883; found, 357.0885.
1-(7′-Methyldipyrido[3,2-f:2′,3′-h]quinoxaline)-4,7,10-tris(tert-butoxycarbonyl)-1,4,7,10-tetraazacyclododecane, 18.
K2CO3
(218 mg, 1.58 mmol) and a catalytic amount of KI were added to a solution of 1,4,7-tris(tert-butoxycarbonyl)-1,4,7,10-tetraazacyclododecane, 17, (148 mg, 0.31 mmol) in CH3CN (5 ml). The mixture was heated at 60 °C and a solution of the3-chloromethylphenazine (88 mg, 0.31 mmol) in CH2Cl2
(5 ml) was added. The reaction mixture was boiled under reflux under argon overnight. The solution was filtered and the salts washed with CH2Cl2. Evaporation of the solvent afforded a crude residue, which was purified by chromatography on silica (gradient elution: CH2Cl2 to 3% CH3OH–CH2Cl2, Rf
= 0.23, 10% CH3OH–CH2Cl2) to yield a colourless oil (160 mg, 0.22 mmol, 71%). 1H NMR (300 MHz, CDCl3): δ 1.29 (27H, m, CH3), 2.78 (4H, br, ring CH2), 3.50 (12H, m, ring CH2), 4.27 (2H, s, CH2), 7.75 (1H, dd, J
= 8.1, 4.5 Hz, H11), 7.89 (1H, br, H6), 8.94 (2H, m, H2, H3), 9.25 (1H, d, J
= 3.0, H10), 9.39 (1H, d, J
= 6.6, H5), 9.46 (1H, dd, J
= 8.1, 1.5, H12). m/z
(ES+) 716 (3%, M+), 739 (100%, MNa+), 1455 (5%, M2Na+).
1-(7′-Methyldipyrido[3,2-f:2′,3′-h]quinoxaline)-1,4,7,10-tetraazacyclododecane, 20.
Trifluoroacetic acid (10 ml) was added to a solution of the tricarbamate (160 mg, 0.22 mmol) in CH2Cl2
(3 ml). The mixture was stirred for 2 h at room temperature. The solvent was evaporated and the residue re-dissolved in CH2Cl2 three times to facilitate elimination of excess acid and tert-butyl alcohol. The resultant orange oil was taken into a KOH solution (5 ml) and the product extracted into CH2Cl2
(3 × 5 ml). The organic layer was dried over K2CO3. Removal of the solvent under reduced pressure afforded a very pale orange solid (65 mg, 0.16 mmol, 71%). 1H NMR (300 MHz, CDCl3): δ 2.57 (4H, m, ring CH2), 2.70 (8H, s, ring CH2), 2.80 (4H, m, ring CH2), 4.20 (2H, s, CH2), 7.71 (1H, dd, J
= 8.1, 4.5 Hz, H11), 8.07 (1H, d, J
= 8.4, H6), 8.87 (1H, d, J
= 2.1, H2), 8.88 (1H, d, J
= 2.1, H3), 9.25 (1H, dd, J
= 4.5, 1.8, H10), 9.41 (1H, dd, J
= 8.4, 1.8, H12), 9.43 (1H, d, J
= 8.4, H5). 13C NMR (75 MHz, CDCl3): δ 45.1, 46.4, 47.1, 52.0 (ring CH2), 61.9 (CH2), 123.1 (C11), 123.5 (C6), 125.8 (q Ar), 126.9 (q Ar), 133.0 (C12), 133.8 (C5), 140.0 (q Ar), 140.5 (q Ar), 144.0 (C2), 144.4 (C3), 146.4 (q Ar), 147.2 (q Ar), 152.2 (C10), 163.5 (C7). m/z
(ES+) 228 (10%, MCa2+), 417 (100%, MH+), 439 (10%, MNa+), 833 (3%, M2H+); mp 175–178 °C (decomp.). Accurate mass: found MH+ 417.2515; C23H29N8 requires 417.2515.
(RRR)-1-(7′-Methyldipyrido[3,2-f:2′,3′-h]quinoxaline)-4,7,10-tris[1-(1-phenyl)ethylcarbamoylmethyl]-1,4,7,10-tetraazacyclododecane, 1.
K2CO3
(83 mg, 0.6 mmol) and a catalytic amount of KI were added to a solution of 1-(7′-methyldipyrido[3,2-f:2′,3′-h]quinoxaline)-1,4,7,10-tetraazacyclododecane (50 mg, 0.12 mmol) in CH3CN (4 ml). The mixture was heated at 60 °C and a solution of compound 7
(71 mg, 0.36 mmol) in CH2Cl2
(4 ml) was added. The reaction mixture was boiled under reflux under argon overnight. The solution was filtered and the salts washed with CH2Cl2. Evaporation of the solvent afforded a crude residue, which was purified by chromatography on alumina (gradient elution: CH2Cl2 to 2% CH3OH–CH2Cl2, Rf
= 0.31, 10% CH3OH–CH2Cl2) to yield a colourless solid (50 mg, 0.06 mmol, 50%). 1H NMR (300 MHz, CDCl3): δ 1.46 (H, d, J
= 2.7 Hz, CH3), 1.48 (H, d, J
= 2.7, CH3), 2.64 (16H, m, ring CH2), 2.90 (2H, d, J
= 6, CH2), 2.96 (4H, d, J
= 6, CH2), 4.08 (2H, s, CH2-dpq), 5.14 (3H, m, J
= 7.5, CH), 6.88 (2H, br, NH), 7.05 (1H, br, NH), 7.16–7.37 (15H, m, Ph), 7.80 (1H, dd, J
= 8.1, 4.2, H11), 7.91 (1H, d, J
= 8.4, H6), 9.01 (2H, m, H2, H3), 9.28 (1H, dd, J
= 4.5, 1.8, H10), 9.41 (1H, d, J
= 8.4, H5), 9.54 (1H, dd, J
= 8.1, 1.8, H12). 13C NMR (75 MHz, CDCl3): δ 21.4 (CH3), 21.9 (CH3), 48.2 (CH), 48.3 (CH), 52.8 (CH2N), 53.3 (CH2N), 53.6 (CH2N), 58.3 (NCH2CO), 58.9 (NCH2CO), 62.3 (CH2-dpq), 123.3 (C11), 123.9 (C6), 126.2 (o-Ar), 127.3 (p-Ar), 128.6 (m-Ar), 133.2 (C12), 133.6 (C5), 143.1 (q Ar), 144.6 (C2, C3), 140.4 (q-dpq), 152.3 (C10), 161.0 (C7), 169.5 (CO), 170.4 (CO). m/z
(ES+) 469 (40%, MCa2+), 899 (5%, M+), 922 (100%, MNa+); mp 136–138 °C. Accurate mass: found MH+ 900.5035; C53H62N11O3 requires 900.5037.
The (SSS) ligand was synthesised with the same procedure.
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(tert-butoxycarbonyl)-1,4,7,10-tetraazacyclododecane, 19.
Potassium carbonate (124 mg, 0.716 mmol) and a catalytic amount (2 mg) of KI were added to a solution of 1,4,7-tris-tert-butoxycarbonyl-1,4,7,10-tetraazacyclododecane, 17, (77 mg, 0.179 mmol) in CH3CN (3 ml). The mixture was heated at 60 °C and a solution of 3-chloromethyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine (60 mg, 0.179 mmol) in CH2Cl2
(3 ml) was added. The reaction mixture was boiled under reflux under argon overnight. The solution was filtered and the salts washed with CH2Cl2. The residue was purified by chromatography on silica gel (gradient elution: CH2Cl2 to 3% CH3OH–CH2Cl2, Rf
= 0.44, 10% CH3OH–CH2Cl2) to yield a pale yellow solid (105 mg, 0.136 mmol, 76%), mp 114–116 °C. 1H NMR (300 MHz, CDCl3): δ 0.92–0.71 (27H, br m, CH3), 2.04 (4H, m, H10, H13), 2.42–2.99 (4H, m, CH2 ring), 3.07–3.87 (16H, br m, CH2 ring + H11, H12), 4.24 (2H, s, CH2-dpqC), 7.67 (1H, dd, J
= 8.2, 4.4 Hz, H7), 7.72–7.97 (1H, br s, H2), 9.71 (1H, br s, H6), 9.26–9.44 (2H, br m, H8, H1). 13C NMR (75 MHz, CDCl3): δ 22.7 (C10, C13), 28.3 (CH3), 28.5 (CH3), 32.7 (C11, C12), 46.2–51.3 (C ring), 60.0–61.2 (CH2-dpqC), 79.3 (CHN), 123.3 (C7), 123.8 (C Ar), 125.9 (C Ar), 127.0 (C2), 129.3 (C Ar), 130.4 (C Ar), 130.5 (C Ar), 137.6 (C Ar), 142.2 (C Ar), 146.4 (C Ar), 146.8 (C Ar), 151.3 (C Ar), 153.3 (C Ar), 154.7–157.0 (CO). HRMS (+
m/z): [M + Na]+ calculated for C42H58N8O6Na, 793.4377; found, 793.4379.
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecane, 21.
Trifluoroacetic acid (10 ml) was added to a solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(tert-butoxycarbonyl)-1,4,7,10-tetraazacyclododecane (104.7 mg, 0.136 mmol) in CH2Cl2
(3 ml). The mixture was stirred for 2 h at room temperature. The solvent was evaporated and the residue re-dissolved three times to facilitate elimination of excess acid and tert-butyl alcohol. The residue was taken into a 1 M KOH solution (5 ml) and the product extracted into CH2Cl2
(3 × 5 ml). The organic layer was dried over K2CO3. Removal of the solvent under reduced pressure yielded an orange solid, mp > 240 °C (52.5 mg, 0.112 mmol, 82%). 1H NMR (300 MHz, CDCl3): δ 2.09 (4H, m, H10, 13), 2.60–3.00 (16H, m, CH2 ring), 3.24 (4H, m, H11, H12), 4.16 (2H, s, CH2-dpqC), 7.69 (1H, d, J
= 8.0 Hz, H2), 7.76 (1H, dd, J
= 8.2, 4.4, H7), 9.26 (1H, dd, J
= 4.4, 1.4, H6), 9.40 (1H, d, J
= 8.0, H1), 9.46 (1H, dd, J
= 8.2, 1.4, H8). 13C NMR (75 MHz, CDCl3): δ 22.6 (CH2), 32.6 (CH2), 45.2, 46.3, 47.1, 52.0 (CH2 ring), 61.7 (CH2), 122.8 (C7), 123.2 (C2), 125.9 (q Ar), 127.0 (q Ar), 132.7 (C8), 133.5 (C1), 137.1 (q Ar), 146.0 (q Ar), 146.8 (q Ar), 151.3 (C6), 153.3 (q Ar), 153.6 (q Ar), 162.3 (q Ar).
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(N-acetyl-L-glutamic acid diethyl ester)-1,4,7,10-tetraazacyclododecane, 5.
Potassium carbonate (129 mg, 0.935 mmol) and a catalytic amount of KI were added to a solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-1,4,7,10-tetraazacyclododecane (87.8 mg, 0.187 mmol) in CH3CN (7 ml). The reaction mixture was heated at 60 °C and a solution of N-chloroacetyl-L-glutamic acid diethyl ester (157 mg, 0.561 mmol) in CH2Cl2
(7 ml) was added. The reaction mixture was boiled under reflux overnight, under argon. The solution was filtered and the salts washed with CH2Cl2. The product was purified by chromatography on neutral alumina (gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2, Rf
= 0.54, 7.5% CH3OH–CH2Cl2) to yield a pale yellow solid (105 mg, 0.089 mmol, 48%), mp 82–84 °C. 1H NMR (300 MHz, CDCl3): δ 0.96–1.34 (18H, m, CH3), 1.74–2.56 (16H, m, CH2CH3, H10, H13), 2.82–3.42 (20H, br m, CH2 ring, H11, H12), 3.72–4.64 (21H, m, CH2CO, CH2-dpqC, CHCH2CH2, CHCH2CH2, CHCH2CH2), 7.68 (1H, m, H2), 7.76 (1H, br m, H7), 7.95 (1H, br m, H6), 9.35 (1H, m, H1), 9.45 (1H, m, H8). 13C NMR (75 MHz, CDCl3): δ 14.1 (CH3), 14.2 (CH3), 22.8 (C10, C13), 26.2 (CH2), 30.4 (CH2), 30.6 (CH2), 32.9 (C11, C12), 50.4–60.7 (CH2 ring, CH2CO, CH2CH3), 61.6 (CH2-dpqC), 124.2 (C2), 124.7 (C Ar), 126.7 (C Ar), 127.7 (C7), 133.2 (C8), 134.1 (C1), 137.4 (C Ar), 146.3 (C Ar), 146.9 (C Ar), 151.9 (C6), 154.2 (C Ar), 157.5 (C Ar), 160.9 (C Ar), 169.4 (CO), 171.3 (CO), 171.6 (CO), 172.4 (CO), 172.8 (CO). m/z
(ES+): 620 (MCa2+), 632 (MCu2+), 1223 (MNa+).
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(N-acetyl-L-alanine diethyl ester)-1,4,7,10-tetraazacyclododecane, 4.
Potassium carbonate (116 mg, 0.840 mmol) and a catalytic amount of KI were added to a solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-1,4,7,10-tetraazacyclododecane (79.1 mg, 0.168 mmol) in CH3CN (6 ml). The reaction mixture was heated at 60 °C and a solution of N-chloroacetyl-L-alanine ethyl ester (97.6 mg, 0.504 mmol) in CH2Cl2
(6 ml) was added. The reaction mixture was boiled under reflux overnight, under argon. The solution was filtered and the salts washed with CH2Cl2. The product was purified by chromatography on neutral alumina (gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2, Rf
= 0.55, 7.5% CH3OH–CH2Cl2)
(94 mg, 0.10 mmol, 59%). 1H NMR (400 MHz, CDCl3): δ 0.90–1.58 (18H, m, CH2CH3, CHCH3), 2.06 (4H, m, H10, H13), 2.24–3.18 (16H, br m, CH2 ring), 3.21 (4H, m, H11, H12), 3.42–4.45 (17H, m, CH2CO, CH2-dpqC, CH), 7.60 (1H, m, H2), 7.77 (1H, br m, H7), 7.89 (1H, br m, H6), 9.24 (1H, m, H1), 9.43 (1H, m, H8). 13C NMR (75 MHz, CDCl3): δ 14.1 (CH3), 14.2 (CH3), 17.1 (CH3), 17.1 (CH3), 17.5 (CH3), 22.8 (C10, C13), 32.9 (C11, C12), 48.3–57.4 (CH2 ring, CH2CO, CH2CH3), 61.3 (CH2-dpqC), 124.2 (C2), 124.9 (C Ar), 126.7 (C Ar), 127.6 (C7), 133.2 (C8), 134.1 (C1), 137.4 (C Ar), 146.4 (C Ar), 146.9 (C Ar), 151.6 (C6), 154.3 (C Ar), 157.5 (C Ar), 160.9 (C Ar), 169.0 (CO), 172.5 (CO), 172.7 (CO). m/z
(ES+): 491 (MCa2+), 508 (MCu2+) 965 (MNa+).
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris[(S)-1-(1-phenyl)ethylcarbamoylmethyl]-1,4,7,10-tetraazacyclododecane, 2.
Potassium carbonate (138 mg, 0.995 mmol) and a catalytic amount of KI were added to a solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-1,4,7,10-tetraazacyclododecane (94.1 mg, 0.199 mmol) in CH3CN (7 ml). The reaction mixture was heated at 60 °C and a solution of (S)-N-(1-phenyleth-1-yl)chloroacetamide (118 mg, 0.597 mmol) in CH2Cl2
(7 ml) was added. The reaction mixture was boiled under reflux overnight, under argon. The solution was filtered and the salts washed with CH2Cl2. The product was purified by chromatography on neutral alumina (gradient elution: CH2Cl2 to 1% CH3OH–CH2Cl2, Rf
= 0.55, 7.5% CH3OH–CH2Cl2) to yield a colourless solid (91 mg, 0.096 mmol, 48%), mp 148–150 °C. 1H NMR (300 MHz, CDCl3): δ 1.35 (6H, d, J
= 6.7 Hz, CH3), 1.56 (3H, d, J
= 6.7, CH3), 2.06 (4H, m, CH2), 2.16–3.12 (16H, br m, CH2 ring), 3.21 (4H, d, J
= 4.0, CH2), 3.42–4.45 (8H, br m, CH2CO, CH2-dpqC), 4.57 (1H, br m, CH), 4.75 (2H, br m, CH), 6.70–7.49 (18H, m, Ar, NH), 7.53 (1H, d, J
= 8.3, H2), 7.96 (1H, br m, H7), 8.30 (1H, br m, H6), 9.37 (1H, d, J
= 8.3, H1), 9.48 (1H, d, J
= 8.2, H8). 13C NMR (75 MHz, CDCl3): δ 22.2 (CH3), 22.7 (C10, C13), 22.8 (CH3), 32.7 (C11, C12), 49.5 (CHN), 49.8–52.4 (CH2 ring + CH2CO), 60.4 (CH2-dpqC), 123.4 (C2), 125.1 (C7), 126.3 (C Ar), 126.4 (C Ar), 126.8 (C Ar), 126.9 (C Ar), 127.4 (C Ar), 128.1 (C Ar), 128.2 (C Ar), 128.4 (C Ar), 133.2 (C8), 134.1 (C1), 137.0 (C Ar), 137.5 (C Ar), 143.5 (C Ar), 144.2 (C Ar), 145.6 (C Ar), 146.2 (C Ar), 153.6 (C Ar), 153.9 (C Ar), 154.1 (C Ar), 159.9 (C Ar), 169.1 (CO), 170.7 (CO). HRMS (+
m/z): [M + H]+ calculated for C57H68N11O3, 954.5506; found, 954.5497.
EuPh3dpq (CF3SO3)3, [Eu.1].
A solution of ligand 1
(28 mg, 0.03 mmol) and Eu(OTf)3
(19 mg, 0.03 mmol) in dry CH3CN (2 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added dropwise to diethyl ether (100 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was redissolved in CH3CN and the process repeated. A colourless solid was obtained (35 mg, 0.02 mmol, 75%). 1H NMR (300 MHz, CD3OD, −10 °C) partial assignment: δ 37.9 (1H, s, Hax), 37.3 (1H, s, Hax), 31.9 (1H, s, Hax), 27.7 (1H, s, Hax), 26.9 (H10-dpq), 15.7 (dpq), 14.9 (dpq), 11–5 (dpq and Ph signals), −1.8 (6H, s, CH3), −2.3 (3H, s, CH3), −3.5 (4H, m, Heq), −7.2 (2H, s, Hax), −7.6 (2H, s, Hax), −9.7 (1H, s, Heq), −10.2 (1H, s, Heq), −15.7 (1H, s, Heq), −15.8 (1H, s, Heq), −16.4 (1H, s, NCH2CO), −17.0 (1H, s, NCH2CO), −17.6 (1H, s, NCH2CO), −17.9 (1H, s, NCH2CO), −24.0 (2H, s, NCH2CO). m/z
(ES+) 350 (60%, M3+), 525 (80%, M2+), 534 (50%, M2+
+ H2O), 600 (30%, M3+
+ CF3SO3−), 1348 (3%, M3+
+ 2CF3SO3−); mp 176–179 °C. Accurate mass spectrum: found (M + 2CF3SO3)+ 1350.3208; C55H61N11O9EuS2F6 requires 1350.3202. λmax
(H2O) 340 nm (ε
= 3500 M−1 cm−1); τ(H2O)
= 1.05 ms; τ(D2O)
= 1.64 ms, ϕ(H2O)
= 21%, ϕ(D2O)
= 27%.
TbPh3dpq (CF3SO3)3, [Tb.1].
The ligand 1
(15 mg, 0.02 mmol) and Tb(Otf)3
(10 mg, 0.02 mmol) were dissolved in dry CH3CN (1 ml). The procedure followed was identical to that described for the europium complex above A colourless solid was obtained (20 mg, 0.01 mmol, 78%). m/z
(ES+) 352 (100%, M3+), 603 (60%, M3+
+ CF3SO3−), 1356 (5%, M3+
+ 2CF3SO3−); mp 177–179 °C. Accurate mass spectrum: found (M + CF3SO3)2+ 603.6862; C54H61N11O6TbSF3 requires 603.6866. λmax
(H2O) 340 nm (ε
= 3500 M−1 cm−1); τ(H2O)
= 1.85 ms; τ(D2O)
= 2.38 ms, ϕ(H2O)
= 36%, ϕ(D2O)
= 46%.
GdPh3dpq (CF3SO3)3, [Gd.1].
Ligand 1
(10 mg, 0.01 mmol) and Gd(OTf)3
(7 mg, 0.01 mmol) were dissolved in dry CH3CN (1 ml). The procedure followed was identical to that described above. A colourless solid was obtained (7 mg, 0.02 mmol, 42%). m/z
(ES+) 352 (100%, M3+), 603 (60%, M3+
+ CF3SO3−), 1354 (5%, M3+
+ 2CF3SO3−); mp 210–212 °C. Accurate mass spectrum: found (M + 2CF3SO3)+ 1355.3240; C55H61N11O9GdS2F6 requires 1355.3251.
1-(3-Methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris-tert-butoxycarbonylmethyl-1,4,7,10-tetraazacyclododecane, 23.
Potassium carbonate (401 mg, 2.90 mmol) and a catalytic amount of KI were added to a solution of 1,4,7-tris-tert-butoxycarbonylmethyl-1,4,7,10-tetraazacyclododecane, 22, (299 mg, 0.580 mmol) in CH3CN (15 ml). The mixture was heated at 60 °C and a solution of 3-chloromethyl-10,11,12,13-tetrahydrodipyrido-[3,2a:2′,3′-c]phenazine (223 mg, 0.580 mmol) in CH2Cl2
(20 ml) was added. The reaction mixture was boiled under reflux overnight, under argon. The solution was filtered and the salts washed with CH2Cl2. Evaporation of the solvent afforded the crude residue. The product ester was purified by chromatography on silica gel (gradient elution: CH2Cl2 to 3.3% CH3OH–CH2Cl2, Rf
= 0.47, 10% CH3OH–CH2Cl2) to yield a very pale yellow solid (68 mg, 0.084 mmol, 15%), mp 137–139 °C. 1H NMR (300 MHz, CDCl3): δ 1.08 (18H, s, CH3), 1.41 (9H, s, CH3), 2.05 (4H, m, H10, H13), 2.25–3.58 (28H, br m, CH2 ring, H11, H12), 7.68 (1H, m, H7), 7.75 (1H, m, H2), 8.76 (1H, dd, J
= 4.2, 1.2 Hz, H6), 9.42 (1H, m, H1), 9.48 (1H, m, H8). 13C NMR (100 MHz, CDCl3): δ 27.9 (CH3), 50.9–60.4 (CH2 ring), 81.6–82.7 (CH2CO), 123.6 (C2), 123.9 (C Ar), 126.2 (C Ar), 127.7 (C Ar), 133.5 (C Ar), 133.9 (C Ar), 137.2 (C8), 137.5 (C1), 145.8 (C Ar), 146.4 (C Ar), 151.0 (C6), 154.2 (C Ar), 154.5 (C Ar), 160.4 (C Ar), 172.5 (CO), 172.9 (CO). m/z
(ES+): 836 (MNa+).
EuDO3AdpqC, [Eu.3].
Trifluoroacetic acid (4 ml) was added to a solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris-tert-butoxycarbonylmethyl-1,4,7,10-tetraazacyclododecane (33.8 mg, 0.042 mmol) in CH2Cl2
(2 ml). The mixture was stirred overnight at room temperature. The solvent was evaporated and the residue re-dissolved in CH2Cl2 three times to facilitate elimination of excess acid and tert-butyl alcohol. The product, 3, was checked by 1H NMR to ensure complete ester hydrolysis, and was used for complexation immediately. 1H NMR (300 MHz, CD3OD): δ 2.06 (4H, m, H10, H13), 2.90–3.96 (28 H, br m, CH2 ring, CH2, CH2-dpqC, H11, H12), 8.21 (2H, m, H7, H2), 9.19 (1H, m, H6), 9.38 (1H, d, H1), 9.72 (1H, m, H8). The ligand was dissolved in a mixture of methanol and water (1 : 1, 10 ml) and the pH raised to 5.5 by addition of a 1 M solution of KOH. EuCl3·6H2O (15.4 mg, 0.042 mmol) was added and the mixture heated at 90 °C overnight. After being allowed to cool to room temperature the pH of the mixture was raised to 10.0 using dilute KOH solution and the mixture further stirred for 1 h. The solid in the suspension was removed by syringe filtration and the pH of the resulting clear solution again reduced to 5.5. The solution was freeze dried to yield a white solid product that was purified by chromatography on neutral alumina, eluting with CH2Cl2–MeOH (70 : 30), to give a colourless complex. λmax
(H2O) 348 nm (ε
= 6500 M−1 cm−1); τ(H2O)
= 1.06 ms; ϕ(H2O)
= 18%.
TbDO3AdpqC, [Tb.3].
This complex was prepared and purified as for the Eu complex above. λmax
(H2O) 348 nm (ε
= 8300 M−1 cm−1); τ(H2O)
= 1.46 ms; ϕ(H2O)
= 33%.
Eu(Glu-Et)3dpqC (CF3SO3)3, [Eu.5].
A solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(N-acetyl-L-glutamic acid diethyl ester)-1,4,7,10-tetraazacyclododecane (25.4 mg, 0.021 mmol) and Eu(OTf)3
(12.5 mg, 0.021 mmol) in dry CH3CN (5 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added in a dropwise manner to diethyl ether (ca. 20 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was re-dissolved in CH3CN and the process repeated to yield an off-white solid product (36 mg, 0.02 mmol, 94%). m/z
(ES+): 451 (M3+), 751 (MOTf2+).
Eu(Ala-Et)3dpqC (CF3SO3)3, [Eu.4].
A solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris(N-acetyl-L-alanine diethyl ester)-1,4,7,10-tetraazacyclododecane (27.7 mg, 0.029 mmol) and Eu(OTf)3
(17.4 mg, 0.029 mmol) in dry CH3CN (5 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added in a dropwise manner to diethyl ether (ca. 20 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was redissolved in CH3CN and the process repeated to yield an off-white solid product (23 mg, 0.015 mmol, 51%). m/z
(ES+): 365 (M3+), 621 (MOTf2+).
EuPh3dpqC (CF3SO3)3, [Eu.2].
A solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazine)-4,7,10-tris[(S)-1-(1-phenyl)ethylcarbamoylmethyl]-1,4,7,10-tetraazacyclododecane (31.8 mg, 0.033 mmol) and Eu(OTf)3
(20.0 mg, 0.033 mmol) in dry CH3CN (5 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added in a dropwise manner to diethyl ether (ca. 20 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was re-dissolved in CH3CN and the process repeated to yield an off-white solid product (39 mg, 0.025 mmol, 76%). m/z
(ES+): 628 (MOTf2+), 1402 (MOTf2+). This complex was converted to the more water soluble chloride salt by ion exchange chromatography using Amberlite IRA resin. λmax
(H2O) 348 nm (ε
= 8300 M−1 cm−1); τ(H2O)
= 1.04 ms; τ(D2O)
= 1.59 ms, ϕ(H2O)
= 16%, ϕ(D2O)
= 20%. 1H NMR (CD3OD): δ 37.51, 36.88, 31.43, 27.03, 26.45 (axial ring protons +
ortho aromatic), 15.17, 14.39, 10.27, 9.56, −2.06, −2.62, −3.78, −4.14, −7.40, −7.71, −8.08, −15.71, −16.24, −16.84, −17.13, −17.85, −18.49, −24.32. (m/z, ESMS): [M + CF3SO3]2+ calculated for C58H68N11O6F3SEu, 627.7081; found, 627.7080.
Tb(Glu-Et)3dpqC (CF3SO3)3, [Tb.5] and Tb(Ala-Et)3dpqC (CF3SO3)3, [Tb.4].
Each of these complexes was prepared by a similar method.
A solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazinyl)-4,7,10-tris(N-acetyl-L-alanine diethyl ester)-1,4,7,10-tetraazacyclododecane, 4, (25.4 mg, 0.027 mmol) and Tb(OTf)3
(16.4 mg, 0.027 mmol) in dry CH3CN (5 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added in a dropwise manner to diethyl ether (ca. 20 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was re-dissolved in CH3CN and the process repeated to yield the product (14.8 mg, 0.0957 mmol, 35%). m/z
(ES+): 367 (M3+), 625 (MOTf2+), 1399 (MOTf2+).
TbPh3dpqC (CF3SO3)3, [Tb.2].
A solution of 1-(3-methyl-10,11,12,13-tetrahydrodipyrido[3,2-a:2′,3′-c]phenazinyl)-4,7,10-tris[(S)-1-(1-phenyl)ethylcarbamoylmethyl]-1,4,7,10-tetraazacyclododecane (30.9 mg, 0.032 mmol) and Tb(OTf)3
(19.6 mg, 0.032 mmol) in dry CH3CN (5 ml) was boiled under reflux under argon overnight at 80 °C. The solution was then added in a dropwise manner to diethyl ether (ca. 20 ml) with stirring, the precipitate centrifuged and the solvent decanted. The solid was re-dissolved in CH3CN and the process repeated to yield the product (39.5 mg, 0.0253 mmol, 79%). m/z
(ES+): 371 (M3+), 631 (MOTf2+), 1411 (MOTf2+). λmax
(H2O) 348 nm (ε
= 8300 M−1 cm−1); τ(H2O)
= 1.56 ms; τ(D2O)
= 1.72 ms, ϕ(H2O)
= 40%, ϕ(D2O)
= 48%. 1H NMR (CD3OD): δ 118.57, 93.49, 73.02, 67.19, 64.91, 63.33, 54.31, 37.96, −2.26, −7.03, −7.28, −8.11, −9.07, −9.35, −15.62, −17.19, −41.84, −73.53, −79.19, −110.18. ESMS (m/z): [M + CF3SO3]2+ calculated for C58H68N11O6F3STb, 630.7101; found, 630.7099.
Other complexes reported were prepared and purified by analogous methods.
Acknowledgements
We thank EPSRC for studentship support (R. P.), the ONE-NE Durham County Partnership (STBE), EMIL (EC network of Excellence) and COST Action D-18 for partial support and the EPSRC Mass Spectroscopy Service (Swansea) for assistance with MS spectral measurements.
References
- R. S. Dickins, S. Aime, A. S. Batsanov, A. Beeby, M. Botta, J. I. Bruce, J. A. K. Howard, C. S. Love, D. Parker, R. D. Peacock and H. Puschmann, J. Am. Chem. Soc., 2002, 124, 1697.
- D. Parker, Chem Soc. Rev., 2004, 33, 156 RSC.
- R. S. Dickins, D. Parker, J. I. Bruce and D. J. Tozer, Dalton Trans., 2003, 1264 RSC.
- G. Mathis, Chin. Chem., 1995, 41, 1391 Search PubMed ; see also www.htrf-assays.com.
- D. Parker, Coord. Chem. Rev., 2000, 205, 109 CrossRef CAS; S. Blair, R. Kataky and D. Parker, New J. Chem., 2002, 26, 530 RSC; S. Blair, M. P. Lowe, C. E. Mathien, D. Parker and P. K. Senanayake, Inorg. Chem., 2001, 40, 5860 CrossRef CAS.
- R. S. Dickins, C. Crossland, D. Parker, H. Puschmann and J. A. K. Howard, Chem. Rev., 2002, 102, 1977 CrossRef CAS.
- B. Stolz, G. Weckbecker, P. M. Smith-Jones, R. Albert, F. Ranlf and C. Bruns, Eur. J. Nucl. Med., 1998, 25, 668 CrossRef CAS; A. Heppeler, S. Froidevaux, A. N. Eberle and H. R. Maecke, Curr. Med. Chem., 2000, 7, 791 ; for MRI, see: P. Caravan, J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 Search PubMed.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613 RSC.
- B. H. Bakker, M. Goes, N. Hoebe, H. J. van Ramesdonk, J. W. Verhoeven, M. H. V. Werts and J. W. Hofstraat, Coord. Chem. Rev., 2000, 208, 3 CrossRef CAS; E. B. van der Tol, H. J. Ramesdonk, J. W. Verhoeven, F. J. Skeemers, E. G. Kever, W. Verboom and D. N. Reinhoudt, Chem. Eur. J., 1998, 4, 1935.
- S. Delaney, M. Pascaly, P. K. Bhattacharya, K. Hain and J. K. Barton, Inorg. Chem., 2002, 41, 1966 CrossRef CAS.
- J. C. Collins, J. R. Aldrich-Wright, I. D. Greguric and P. A. Pellegrini, Inorg. Chem., 1999, 38, 5502 CrossRef CAS; C. G. Coates, J. Olofsson, M. Coletti, J. J. McGarvey, B. Onfelt, P. Lincoln, B. Norden, E. Tuite, P. Matousek and A. W. Parker, J. Phys. Chem. B, 2001, 105, 12653 CrossRef CAS.
- G. Bobba, R. S. Dickins, S. D. Kean, C. E. Mathieu, D. Parker, R. D. Peacock, G. Siligardi, M. J. Smith, J. A. G. Williams and C. F. G. C. Geraldes, J. Chem. Soc., Perkin Trans. 2, 2001, 1729 RSC; G. Bobba, S. D. Kean, D. Parker, A. Beeby and G. Baker, J. Chem. Soc., Perkin Trans. 2, 2001, 1738 RSC.
- G. Bobba, J.-C. Frias and D. Parker, Chem. Commun., 2002, 890 RSC.
- J.-C. Frias, G. Bobba, M. J. Cann, D. Parker and C. J. Hutchinson, Org. Biomol. Chem., 2003, 1, 905 RSC.
- M. Yamonda, Y. Tanaka, Y. Yoshimoto, S. Kuroda and I. Shimao, Bull. Chem. Soc. Jpn., 1992, 65, 1006 CAS ; care needs to be exercised during basification to avoid an exotherm that may promote decarboxylation to give a substituted diazafluorenone: G. A. Shabir and N. J. Forrow, J. Pharm. Biomed. Anal., 2003, 33, 219 Search PubMed.
- A. M. S. Garas and R. S. Vagg, J. Heterocycl. Chem., 2000, 37, 151 CrossRef CAS.
- C. J. Chandler, L. W. Deady and J. A. Reiss, J. Heterocycl. Chem., 1981, 18, 599 CrossRef CAS.
- G. R. Newkome, K. J. Theriot, V. K. Gupta, F. R. Fronczek and G. R. Baker, J. Org. Chem., 1989, 54, 1766 CrossRef CAS.
- S. Brandes, C. Gros, F. Dendt, P. Pullumbi and R. Guilard, Bull. Soc. Chim. Fr., 1996, 133, 65 CAS.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS.
- J. I. Bruce, D. Parker, S. Lopinski and R. D. Peacock, Chirality, 2002, 14, 562 CrossRef CAS.
- R. S. Dickins, J. A. K. Howard, C. L. Maupin, J. M. Moloney, J. P. Riehl, G. Siligardi and J. A. G. Williams, Chem. Eur. J., 1999, 5, 1095 CrossRef CAS.
- S. Quici, G. Marzani, M. Cavazinni, P. L. Anelli, M. Botta, E. Gianolio, G. Accorsi, N. Armaroli and F. Barigeletti, Inorg. Chem., 2002, 41, 2777 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- I. M. Clarkson, A. Beeby, J. I. Bruce, L. J. Govenlock, M. P. Lowe, C. E. Mathieu, D. Parker and K. Senanayake, New J. Chem., 2000, 24, 377 RSC.
- S. Petoud, S. M. Cohen, J. C. G. Bunzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324 CrossRef CAS.
- N. Weibel, L. J. Charbonniere, M. Guardigli, A. Roda and R. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888 CrossRef CAS.
- A. Weller, Pure Appl. Chem., 1968, 16, 115 CrossRef CAS.
- S. Chevion, E. M. Berry, N. Kitrosky and R. Kohen, Free Radical Biol. Med., 1997, 22, 411 CrossRef CAS; J. M. Zen and C. T. Hsu, Talanta, 1998, 46, 1363 CrossRef CAS; J. Chen, L. Gorton and B. Ahessan, Anal. Chim. Acta, 2002, 474, 137 CrossRef CAS.
- H. Reiber, M. Ruff and M. Uhr, Chin. Chim. Acta, 1993, 217, 163 Search PubMed.
- B. Frei, L. England and B. N. Ames, Proc. Natl. Acad. Sci., 1989, 86, 6377 CAS.
- M. Jastrzebskaglapa and J. Mlochowski, Roczniki Chemii, 1976, 987 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here. 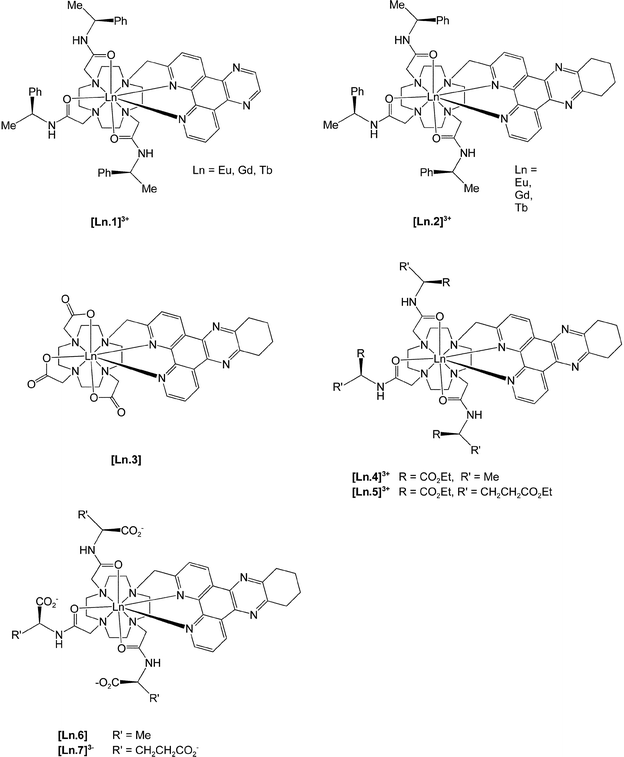
![Fluorescence microscope image showing a live NIH/3T3 cell stained with [Tb.2]3+ (0.3 mM in growth medium), 4 h post-incubation.](/image/article/2005/OB/b418964g/b418964g-f4.gif)
![Fluorescence microscope image revealing the localisation of [Tb.2]3+ (1 mM in medium, 24 h post-incubation) in the cell nucleus and defining the nuclear cell membrane.](/image/article/2005/OB/b418964g/b418964g-f5.gif)