Unexpected Z-stereoselectivity in the Ramberg–Bäcklund reaction of diarylsulfones leading to cis-stilbenes: the effect of aryl substituents and application in the synthesis of the integrastatin nucleus
Jonathan S.
Foot
a,
Gerard M. P.
Giblin
b,
A. C.
Whitwood
a and
R. J. K.
Taylor
*a
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: rjkt1@york.ac.uk; Tel: 01904 432606
bDepartment of Medicinal Chemistry, Neurology and GI CEDD, GlaxoSmithKline, New Frontiers Science Park North, Harlow, UK CM19 5AW
First published on 9th February 2005
Abstract
With certain substituent patterns, benzyl benzyl sulfone systems have been found to give unexpectedly high Z-stereoselectivity (up to E : Z = 1 : 16) in the Meyers variant of the Ramberg–Bäcklund reaction. A range of sulfones, bearing various aryl substituents, were explored to rationalize this unprecedented selectivity for Z-stilbene systems. This high level of double bond stereocontrol has also been utilized in the synthesis of integrastatin nucleus, the core of two highly bioactive anti-HIV compounds.
Introduction
Stilbene systems are found throughout nature and are present in many man-made, biologically active compounds, the most recognizable of which is the widely prescribed drug Tamoxifen®1 (Fig. 1), used for the treatment of breast cancer.1 Other interesting examples include combretastatin A-4 2, an anti-tumour compound,2 and N-(1-mercaptomethyl-2-phenylethyl)-2,3-diphenylacrylamide 3, which has been found to inhibit the endothelin converting enzyme (ECE), an enzyme involved in vascoconstriction.3 All three of these molecules are based on cis-stilbenes. | ||
| Fig. 1 Tamoxifen®1, combretastatin 2 and E-N(R)-(1-mercaptomethyl-2-phenylethyl)-2,3-diphenylacrylamide 3. | ||
During investigations into the synthesis of the HIV-1 integrase-inhibiting integrastatin natural products (4 and 5, Fig. 2),4 we found that sulfone 6 underwent the Ramberg–Bäcklund reaction (RBR), using Meyers' conditions,5 to give the key olefin intermediate 7 in good yield and in a 1 : 1 ratio of E and Z-isomers (Scheme 1).
 | ||
| Fig. 2 The integrastatins. | ||
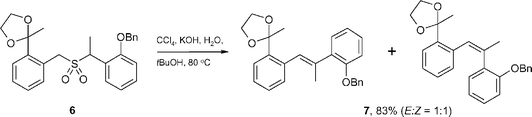 | ||
| Scheme 1 Exposure of sulfone 6 to Meyers' conditions. | ||
At first glance this reaction seemed unremarkable, but a comparison with literature stilbene syntheses highlighted two major discrepancies. The first of these was the fact that any cis-product had been formed in a RBR synthesis. In general, benzyl benzyl sulfone systems undergo the RBR to afford solely trans-stilbene products, as has been shown by Bordwell and Cooper (Scheme 2(a)),6 Neureiter,7 Meyers et al. (Scheme 2(b)),5 and more recently, Chan et al.(Scheme 2(c)).8
 | ||
| Scheme 2 Previous benzyl benzyl sulfone RBR studies. | ||
Although it is widely accepted that in the course of the reaction the cis-episulfone intermediate 9 is formed preferentially (Scheme 3), deuterium-labelling experiments by Tokura et al.9 have shown that phenyl substituents are sufficiently anion-stabilizing to ensure a fast epimerization of the episulfone thus affording exclusively the trans-stilbene product (also Scheme 3).
 | ||
| Scheme 3 Reaction pathway of the RBR in stilbene systems. | ||
The second unexpected factor in our findings was that KOH–tBuOH was employed as the base system, a method that has been shown, even in aliphatic systems, to promote the epimerization of the intermediate episulfone to give the trans-products. In-depth base vs. selectivity studies by both Neureiter7 and Scholz and Burtscher10 have highlighted this property, and it has been used to great effect in aliphatic systems (where the cis-products, e.g.12, usually predominate) as shown in Scheme 4.
 | ||
| Scheme 4 Treatment of α-chloroethyl ethyl sulfone with different bases.7 | ||
The “high” Z-selectivity that had been observed with sulfone 6 was consequently of great interest, and further work was undertaken to try to rationalize this result.
Results and discussion
In the published examples of RBR-formed stilbene systems, most possessed unsubstituted aryl groups. It was decided, therefore, to investigate the effect of altering the ketal group in sulfone system 6, and to this end ketone 13 and alcohol 14 were synthesized and exposed to the same RBR conditions (Scheme 5). | ||
| Scheme 5 RBR of sulfones 13 and 14. | ||
Surprisingly, these sulfones produced even larger proportions of the cis-stilbene products, affording the olefins 15 and 16 in 1 : 8 and 1 : 16 E : Z ratios, respectively. The absolute stereochemistry of the olefin double bond was confirmed through NOE analysis of 15. To our knowledge, this observed cis-selectivity is the largest of any reported for the synthesis of stilbenes by the RBR.11
Initial theories as to the reason for this cis-preference centered on the electronics of the aromatic rings. One suggestion was that the sulfones might be undergoing π-stacking to minimize the interaction between the surfaces of the molecules with the solvent molecules, and thus directing a cis-alignment from the beginning (Fig. 3).
 | ||
| Fig. 3 Proposed π-stacking for sulfone 14. | ||
To investigate this theory further, a range of sulfones were synthesized (20 and 27–33, Scheme 6) through coupling of benzyl halides 21–23 with thiols 18 and 24–26 using the standard procedures outlined in Scheme 6. Of the coupling partners, compounds 22, 23 and 26 were commercially available, whilst compounds 21 and 25 were synthesized via literature procedures.12,13 Novel thiols 18 and 24 were prepared by treating the known compounds, 1-(2-benzyloxyphenyl)-ethanol (17)14 and 1-(2-methoxyphenyl)-ethanol15 respectively, with Lawesson's reagent.
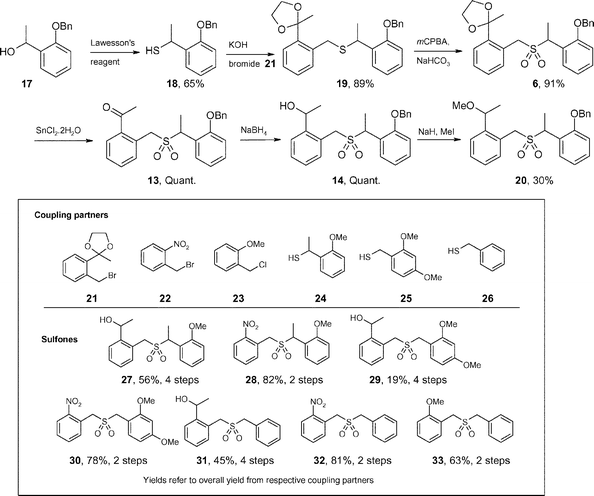 | ||
| Scheme 6 Synthetic route to the sulfones. | ||
These sulfones were then treated with potassium hydroxide and carbon tetrachloride in aqueous tert-butanol under standard Meyers conditions. The stereochemical outcomes of these reactions are shown in Table 1.









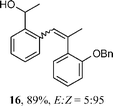












Table 1 suggests that the observed selectivity is not solely due to π-stacking. Comparison of sulfones 27 and 29 (entries [iv] and [vii]) with sulfones 28 and 30 (entries [v] and [viii]) shows a much higher selectivity for the cis-isomer when the hydroxyethyl moiety is present. If, indeed, π-stacking was the reason for the cis-selectivity, then it should be expected that the sulfones with the most powerful electron withdrawing group (the nitro group) would afford the cis-olefins in the greatest proportions, providing an electron donating group is also present on the opposing aromatic ring. This is not the case, and the only nitro-containing sulfone to give any appreciable amount of the cis-isomer is sulfone 28 (entry [v]), with a ratio of E : Z = 1 : 1.
When there are only two substituents present on the sulfone, and disubstituted stilbenes are formed (entries [vii]–[xi]), the effect of the hydroxyethyl group is again apparent, with the sulfones bearing this group (29 and 31) the only systems to afford any of the cis-olefin product. The other three sulfones, 30, 32 and 33, yield solely the trans-isomer of the corresponding olefin, as would be expected from the literature studies.
Furthermore, comparison of entries [vii] and [ix] shows equal selectivity for the cis-product (E : Z = 2 : 3) from sulfones 29 and 31. These results highlight the apparent unimportance of the methoxy substituents of the second aryl ring.
This apparent reliance on the hydroxyethyl group for the high cis-selectivities suggests a revised mechanism for the RBR in our stilbene systems:
Assuming that the formation of the cis-episulfone is favoured in our systems, as is predicted,5–10 then both the acidity of the benzylic protons and the strength of the base should favour fast epimerization to the trans-isomer. We propose that the observed cis-selectivity in these studies is due to an intramolecular base-promoted extrusion of SO2, which is taking place before full epimerization of the intermediate episulfone 42 occurs, therefore retaining the cis-configuration in the product. This process would proceed by way of a 5-exo-tet ring breaking process to give 43,16 which would then break down to afford Z-16 (Scheme 7).
 | ||
| Scheme 7 Proposed intramolecular base-promoted SO2 extrusion from episulfone 42. | ||
This proposed mechanism can now be used to explain all of the observed results. The highest proportion of cis-product is consistently seen when the free hydroxyethyl group is present. The reduction in selectivity when forming disubstituted, as opposed to trisubstituted olefins, is likely to be a statistical effect, due to the presence of twice as many epimerisable protons in the intermediate episulfone, leading to a higher proportion of trans-product.
The high Z-stereoselectivity apparent when the methyl ketone functionality is present (sulfone 13, entry [ii]) can be explained by considering the enolate form of the ketone, which it is presumed will be present in some quantity under the highly basic reaction conditions to effect the SO2 extrusion process. This can also go through a five-membered transition state (analogous to 14), selectively affording the cis-product, in only a slightly lower proportion than 14.
The reasonable Z-stereoselectivity showed with sulfones 6 and 20 (entries [i] and [vi]) can also be explained by this theory, with the oxygen lone pairs on the ortho-alkoxy substituent (rather than a formal anion) responsible for the intramolecular extrusion. The higher selectivity observed with 20 over 6 is possibly due to the reduced steric hindrance of the methyl ether compared to the ketal group.
The only other example in which the cis-product is observed is a nitro-example where an α-methyl group is also present (entry [v]). It is thought that the cis-selectivity in this case may be due to nitro-group involvement in sulfur dioxide extrusion.
Exploiting the selectivity in synthesis
The importance of this work has been highlighted in the synthesis of the integrastatin nucleus 45 (Scheme 8). We demonstrated that only the cis-isomer 15 undergoes a Lewis acid-promoted cyclisation, which is thought to proceed by using the olefin double bond to hold the starting olefin in the reactive conformation (impossible with the trans-isomer), to give tetracycle 44 (confirmed by X-ray crystallography17). Various benzylic oxidation trials on this tetracycle have shown that by utilization of a PDC–tBuOOH complex formed on Celite® at low temperature, the integrastain nucleus 45 can be formed in a yield of 61% from the sulfone 14.17 | ||
| Scheme 8 Synthesis of the integrastatin nucleus. | ||
The structure of the integrastatin nucleus has been further ascertained through X-ray analysis of the crystalline product (Fig. 4).

Conclusions
Through investigations into various benzyl sulfone systems, we have been able to demonstrate a remarkable cis-selectivity in the Ramberg–Bäcklund reaction leading to disubstituted and particularly trisubstituted stilbenes. The Z-stereochemistries attained are the highest yet recorded in the literature for stilbenes (up to a 95% composition of the cis-isomer has been observed), and have been achieved through manipulation of aryl groups remote from the central sulfone system.These results extend the scope of the RBR in synthetic chemistry, as shown by the synthesis of the integrastatin nucleus 45.
Experimental
(±)-1-(2-Benzyloxyphenyl)-ethanethiol, 18 and (±)-1-(2-methoxyphenyl)-ethanethiol, 24
(a) To a stirred solution of 1-(2-benzyloxyphenyl)-ethanol (4.36 g, 19.0 mmol) in dichloromethane (300 mL) under nitrogen was added Lawesson's reagent (4.64 g, 11.0 mmol). The reaction mixture was stirred at room temperature for 28 hours and the solvent was then removed in vacuo to give a yellow gum (6.20 g). Purification by flash chromatography, eluting with 9 : 1 petroleum ether–ethyl acetate, afforded the title compound18 (3.00 g, 65%) as a pale yellow oil; RF [petroleum ether–ethyl acetate (4 : 1)] 0.44; νmax (neat)/cm−1 2968, 2560 (SH), 1241; δH (CDCl3, 270 MHz) 1.67 (3 H, d, J 7.0 Hz, CH3), 2.15 (1 H, d, J 6.2 Hz, SH), 4.69 (1 H, dq, J 7.0, 6.2 Hz, CH), 5.14 (2 H, s, CH2), 6.94–6.97 (2 H, m, 2 ArH), 7.18–7.44 (7 H, m, 7 ArH); δC (CDCl3, 67.9 MHz) 24.6 (CH3), 31.8 (CH), 70.3 (CH2), 112.1 (ArH), 121.3 (ArH), 126.5 (ArH), 127.4 (2 ArH), 128.1 (2 ArH), 128.8 (2 ArH), 137.2 (C), 155.2 (C)—1 quaternary carbon signal not observed; m/z (CI) 262 (MNH4+, 25%), 211 (100); [Found MNH4+, 262.1268 (error = 0.8 ppm). C15H16OS requires: MNH4+, 262.1266].(b) Thiol 24 was prepared in an identical procedure to that for 18 and isolated in a 58% yield as a colourless oil; RF [petroleum ether–ethyl acetate (4 : 1)] 0.47; νmax (neat)/cm−1 2965, 1491, 1244, 753; δH (CDCl3, 400 MHz) 1.65 (3 H, d, J 6.8 Hz, CH3), 2.12 (1 H, d, J 6.0 Hz, SH), 3.87 (3 H, s, OCH3), 4.61 (1 H, dq, CH), 6.87 (1 H, d, J 7.6 Hz, ArH), 6.96 (1 H, dd, J 7.6, 7.2 Hz, ArH), 7.18–7.21 (1 H, m, ArH), 7.40 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 24.9 (CH3), 31.9 (CH), 55.8 (OCH3), 111.0 (ArH), 121.2 (ArH), 126.6 (ArH), 128.3 (ArH), 134.5 (C), 156.3 (Ar(C)O); m/z (CI) 186 (MNH4+, 24%), 135 (100); [Found MNH4+, 186.0950 (error = 1.2 ppm). C9H12OS requires: MNH4+, 186.0953].
(±)-2-{2-[1-(2-Benzyloxyphenyl)-ethylsulfanylmethyl]-phenyl}-2-methyl-[1,3]dioxolane, 19
To a stirred solution of 18 (3.06 g, 12.5 mmol) in ethanol (60 mL) was added powdered potassium hydroxide (0.74 g, 13.2 mmol). The resultant mixture was cooled to 0 °C and a solution of 21 (3.39 g, 13.2 mmol) in ethanol (40 mL) added dropwise over 10 minutes. The reaction was allowed to warm to room temperature and stirred for 18 hours. The solvent was then removed in vacuo and the residue extracted with dichloromethane, washing with water and saturated sodium chloride solution, before drying over magnesium sulfate. Filtration and removal of the solvent in vacuo gave a clear gum. Purification by flash chromatography, eluting with 19 : 1 petroleum ether–ethyl acetate, afforded the title compound19 (4.67 g, 89%) as a clear oil; RF [petroleum ether–ethyl acetate (4 : 1)] 0.31; νmax (neat)/cm−1 2970, 1450, 1239, 1038; δH (CDCl3, 400 MHz) 1.56 (3 H, d, J 7.0 Hz, CH3), 1.62 (3 H, s, CH3), 3.68–3.71 (2 H, m, 2 CH), 3.90 (2 H, s, CH2), 3.93–3.96 (2 H, m, 2 CH), 4.72 (1 H, q, J 7.0 Hz, CH), 5.10 (2 H, s, CH2), 6.95 (1 H, d, J 6.2 Hz, ArH), 7.02 (1 H, dd, J 7.7, 7.7 Hz, ArH), 7.10–7.12 (2 H, m, 2 ArH), 7.22–7.26 (2 H, m, 2 ArH), 7.30–7.45 (5 H, m, 5 ArH), 7.51 (1 H, d, J 7.6 Hz, ArH), 7.59 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 22.0 (CH3), 27.6 (CH3), 33.5 (CH2), 37.2 (CH), 64.3 (2 CH2), 70.4 (CH2), 109.3 (C), 112.0 (ArH), 121.4 (ArH), 126.5 (ArH), 126.7 (ArH), 127.4 (2 ArH), 127.9 (ArH), 128.0 (ArH), 128.1 (ArH), 128.2 (ArH), 128.7 (2 ArH), 131.6 (ArH), 132.8 (C), 136.4 (C), 140.5 (C), 155.9 (Ar(C)O)—1 quaternary carbon signal not observed; m/z (CI) 421 (MH+, 100%); [Found MH+, 421.1837 (error = 0.1 ppm). C26H28O3S requires: MH+, 421.1837].(±)-2-{2-[1-(2-Benzyloxyphenyl)-ethanesulfonylmethyl]-phenyl}-2-methyl-[1,3]dioxolane, 6
To a stirred solution of 19 (1.14 g, 2.7 mmol) and sodium hydrogen carbonate (0.94 g, 10.2 mmol) in dichloromethane (30 mL) and water (5 mL) at 0 °C, was added meta-chloroperbenzoic acid (1.40 g, 8.1 mmol). The reaction mixture was allowed to stir for a further 30 minutes at 0 °C, then warmed to room temperature and stirred for 16 hours. Saturated sodium metabisulfite solution was then added and the organic layer extracted with dichloromethane, washing with saturated sodium hydrogen carbonate solution and saturated sodium chloride solution, before drying over sodium sulfate. Filtration and removal of the solvent in vacuo and purification by flash chromatography, eluting with 3 : 1 petroleum ether–ethyl acetate, afforded the title compound6 (1.11 g, 91%) as a white solid; RF [petroleum ether–ethyl acetate (4 : 1)] 0.15; mp 137–140 °C; (Found C, 69.3; H, 6.3. C26H28O5S requires: C, 69.0; H, 6.2%); νmax (nujol)/cm−1 2725, 1297, 1043; δH (CDCl3, 400 MHz) 1.42 (3 H, s, CH3), 1.75 (3 H, d, J 6.6 Hz, CH3), 3.67–3.70 (2 H, m, 2 CH), 3.81–3.83 (2 H, m, 2 CH), 4.55 (2 H, d, J 7.6 Hz, CH2), 5.11 (1 H, q, J 6.6 Hz, CH), 5.14 (2 H, d, J 6.4 Hz, CH2), 7.04–7.07 (2 H, m, 2 ArH), 7.23–7.27 (2 H, m, 2 ArH), 7.34–7.47 (5 H, m, 5 ArH), 7.56 (2 H, d, J 7.8 Hz, 2 ArH), 7.64 (2 H, d, J 7.8 Hz, 2 ArH); δC (CDCl3, 67.9 MHz) 13.5 (CH3), 28.0 (CH3), 53.0 (CH2), 55.6 (CH), 64.3 (2 CH2), 70.8 (CH2), 108.9 (C), 112.2 (ArH), 121.7 (ArH), 123.7 (C), 125.1 (C), 126.9 (ArH), 127.7 (2 ArH), 128.2 (ArH), 128.4 (ArH), 128.5 (ArH), 128.9 (3 ArH), 129.9 (ArH), 130.1 (ArH), 132.7 (C), 143.0 (C), 156.2 (Ar(C)O); m/z (CI) 453 (MH+, 25%), 211 (100); [Found MH+, 453.1737 (error = 0.2 ppm). C26H28O5S requires: MH+, 453.173].(±)-1-{2-[1-(2-Benzyloxyphenyl)ethanesulfonylmethyl]-phenyl}-ethanone, 13
To a stirred solution of 6 (1.30 g, 2.9 mmol) in dichloromethane (30 mL) was added tin(II) chloride dihydrate (1.30 g, 5.8 mmol) and the reaction stirred for 8 hours. The reaction mixture was then filtered through Celite® and the solvent removed in vacuo to give a colourless gum. Purification by flash chromatography, eluting with 3 : 2 petroleum ether–ethyl acetate, afforded the title compound13 (1.18 g, 100%) as a colourless oil; RF [petroleum ether–ethyl acetate (2 : 1)] 0.23; νmax (neat)/cm−1 1734, 1693 (CO), 1312 (SO2), 1133 (SO2); δH (CDCl3, 400 MHz) 1.71 (3 H, d, J 7.0 Hz, CH3), 2.56 (3 H, s, CH3), 4.44 (1 H, d, J 12.9 Hz, CH2), 4.68 (1 H, d, J 12.9 Hz, CH2), 5.03 (1 H, q, J 7.0 Hz, CH), 5.11 (1 H, d, J 11.6 Hz, CH2), 5.21 (1 H, d, J 11.6 Hz, CH2), 7.04–7.09 (2 H, m, 2 ArH), 7.22–7.25 (1 H, m, ArH), 7.32–7.50 (8 H, m, 8 ArH), 7.60–7.63 (2 H, m, 2 ArH); δC (CDCl3, 100 MHz) 13.5 (CH3), 29.3 (CH3), 52.2 (CH2), 55.4 (CH), 70.8 (CH2), 112.3 (ArH), 121.7 (ArH), 123.2 (C), 125.1 (C), 127.6 (2 ArH), 128.2 (ArH), 128.5 (2 ArH), 128.8 (2 ArH), 129.4 (ArH), 130.0 (ArH), 131.0 (ArH), 133.7 (ArH), 136.4 (C), 140.5 (C), 156.1 (Ar(C)O), 202.5 (ketone); m/z (CI) 426 (MNH4+, 60%), 228 (95), 211 (100); [Found MNH4+, 426.1742 (error = 0.6 ppm). C24H24O4S requires: MNH4+, 262.1266].(±)-1-{2-[1-(2-Benzyloxyphenyl)ethanesulfonylmethyl]-phenyl}-ethanol, 14
To a stirred solution of 13 (1.17 g, 2.9 mmol) in ethanol (15 mL) at room temperature was added sodium borohydride (0.22 g, 5.8 mmol). The reaction was stirred at room temperature for 6 hours. The solvent was then removed in vacuo and the residue extracted with ethyl acetate, washing with water and brine and drying over sodium sulfate. Filtration and removal of the solvent in vacuo afforded a 1 : 1 mixture of diastereomers of the title compound14 (1.15 g, 98%) as a colourless gum; RF [petroleum ether–ethyl acetate (1 : 1)] 0.26; νmax (neat)/cm−1 3490 (OH), 1734, 1245; δH (CDCl3, 400 MHz) 1.33 (3 H, d, J 6.4 Hz, CH3), 1.38 (3 H, d, J 6.4 Hz, CH3), 1.76 (6 H, m, 2 CH3), 2.77 (2 H, brs, 2 OH), 4.05 (1 H, d, J 13.6 Hz, CH2), 4.12 (1 H, d, J 13.6 Hz, CH2), 4.23 (1 H, d, J 13.6 Hz, CH2), 4.33 (1 H, d, J 13.6 Hz, CH2), 4.71 (1 H, q, J 6.4 Hz, CH), 4.82 (1 H, q, J 6.4 Hz, CH), 5.06–5.19 (6 H, m, 2 CH2), 7.07–7.18 (8 H, m, 8 ArH), 7.34–7.50 (16 H, m, 16 ArH), 7.62 (1 H, d, J 8.0 Hz, ArH), 7.64 (1 H, d, J 8.0 Hz, ArH); δC (CDCl3, 100 MHz) 13.6 (CH3), 14.9 (CH3), 22.7 (CH3), 23.1 (CH3), 51.3 (2 CH2), 55.4 (2 CH), 71.4 (2 CH2), 74.3 (CH), 74.4 (CH), 112.4 (2 ArH), 122.1 (2 ArH), 124.3 (2 C), 126.4 (2 ArH), 127.4 (2 ArH), 127.5 (2 ArH), 127.8 (4 ArH), 128.7 (6 ArH), 128.8 (2 ArH), 129.8 (2 C), 130.3 (2 ArH), 132.4 (2 ArH), 135.3 (2 C), 143.4 (2 C), 156.4 (2 Ar(C)O); m/z (CI) 428 (MNH4+, 6%), 211 (100); [Found MNH4+, 428.1896 (error = 0.1 ppm). C24H26O4S requires: MNH4+, 428.1896].(±)-1-(2-Benzyloxyphenyl)-ethylsulfonylmethyl-[2-(1-methoxyethyl)]-phenyl, 20
To a stirred solution of 14 (0.28 g, 0.7 mmol) in THF (8 mL) at 0 °C under nitrogen was added sodium hydride (60% w/w in mineral oil, 0.03 g, 0.8 mmol). The reaction was stirred for 30 minutes at 0 °C before addition of methyl iodide (50 µL, 0.8 mmol). The mixture was then warmed to room temperature and allowed to stir for 3 hours. The solvent was removed in vacuo, and the residue extracted with ethyl acetate, washing with water and brine and drying over magnesium sulfate. Filtration and removal of the solvent in vacuo gave a yellow oil (0.21 g). Purification by flash chromatography, eluting with 3 : 1 petroleum ether–ethyl acetate, afforded a 1 : 1 mixture of diastereomers of the title compound20 (0.09 g, 30%) as a yellow oil; RF [petroleum ether–ethyl acetate (1 : 1)] 0.43; νmax (neat)/cm−1 2977, 1600, 1312, 1134 (SO2); δH (CDCl3, 400 MHz) 1.18 (3 H, d, J 6.4 Hz, CH3), 1.26 (3 H, d, J 6.4 Hz, CH3), 1.76 (6 H, d, J 7.0 Hz, 2 CH3), 3.05 (3 H, s, OCH3), 3.09 (3 H, s, OCH3), 4.03 (1 H, d, J 6.7 Hz, CH2), 4.06 (1 H, d, J 6.7 Hz, CH2), 4.10 (1 H, d, J 6.7 Hz, CH2), 4.14 (1 H, d, J 6.7 Hz, CH2), 4.26 (1 H, q, J 6.4 Hz, CH), 4.36 (1 H, q, J 6.4 Hz, CH), 5.05–5.14 (6 H, m, 2 CH2; 2 CH), 7.06–7.19 (8 H, m, 8 ArH), 7.31–7.48 (16 H, m, 16 ArH), 7.66 (2 H, d, J 6.8 Hz, ArH); δC (CDCl3, 100 MHz) 13.2 (CH3), 14.3 (CH3), 22.6 (CH3), 23.0 (CH3), 52.3 (2 CH2), 55.3 (CH), 55.4 (CH), 56.4 (2 OCH3), 71.0 (2 CH2), 75.4 (CH), 75.5 (CH), 112.3 (2 ArH), 122.0 (2 ArH), 124.3 (2 C), 126.3 (2 ArH), 127.4 (2 ArH), 127.5 (2 ArH), 127.7 (4 ArH), 128.6 (6 ArH), 128.9 (2 ArH), 129.8 (2 C), 130.3 (2 ArH), 132.4 (2 ArH), 136.3 (2 C), 144.2 (2 C), 156.0 (2 Ar(C)O); m/z (CI) 442 (MNH4+, 100%); [Found MNH4+, 442.2055 (error = 0.3 ppm). C25H28O4S requires: MNH4+, 442.2052].In a similar manner to the procedures described above sulfones 27–33 were prepared.
(±)-1-{2-[1-(2-Methoxyphenyl)-ethanesulfonylmethyl]-phenyl}-ethanol, 27
Isolated as a 1 : 1 mixture of diastereomers in an overall yield of 56% (4 steps) as a creamy white oil; RF [petroleum ether–ethyl acetate (4 : 1)] 0.08; νmax (neat)/cm−1 3355 (OH), 1265, 739; δH (CDCl3, 400 MHz) 1.36 (3 H, d, J 6.4 Hz, CH3), 1.47 (3 H, d, J 6.4 Hz, CH3), 1.76 (6 H, 2 d, J 7.7 Hz, 2 CH3), 2.82 (1 H, s, OH), 2.91 (1 H, s, OH), 3.89 (3 H, s, OCH3), 3.92 (3 H, s, OCH3), 4.11 (2 H, m, CH2), 4.29 (1 H, d, J 13.4 Hz, CH2), 4.36 (1 H, d, J 13.4 Hz, CH2), 4.75 (1 H, q, J 6.4 Hz, CH), 4.97 (1 H, q, J 6.4 Hz, CH), 5.04–5.07 (2 H, m, 2 CH), 6.94–6.97 (2 H, m, ArH), 7.08–7.26 (6 H, m, 6 ArH), 7.37–7.41 (4 H, m, 4 ArH), 7.50–7.53 (2 H, m, 2 ArH), 7.60 (1 H, d, J 7.6 Hz, ArH), 7.65 (1 H, d, J 7.6 Hz, ArH); m/z (CI) 352 (MNH4+, 3.5%), 152 (45), 135 (100); [Found MNH4+, 352.1583 (error = 1.3 ppm). C18H22O4S requires: MNH4+, 352.1578].Due to the complex nature of the diastereomeric mixture, it proved impossible to fully assign the 13C-NMR spectrum.
1-(2-Methoxyphenyl)-ethylsulfonylmethyl-(2-nitro)-phenyl, 28
Isolated in an overall yield of 82% (2 steps) as a white solid; RF [petroleum ether–ethyl acetate (4 : 1)] 0.19; mp 155–158 °C; νmax (nujol)/cm−1 1461, 1377 (SO2), 1115; δH (CDCl3, 400 MHz) 1.73 (3 H, d, J 7.2 Hz, CH3), 3.92 (3 H, s, OCH3), 4.35 (1 H, d, J 13.3 Hz, CH2), 4.83 (1 H, d, J 13.3 Hz, CH2), 5.06 (1 H, q, J 7.2 Hz, CH), 6.96 (1 H, d, J 8.0 Hz, ArH), 7.07 (1 H, dd, J 8.0, 8.0 Hz, ArH), 7.35–7.38 (2 H, m, 2 ArH), 7.53–7.55 (3 H, m, 3 ArH), 8.03 (1 H, d, J 8.0 Hz, ArH); δC (CDCl3, 100 MHz) 13.4 (CH3), 53.0 (CH2), 56.1 (OCH3), 56.3 (CH), 111.1 (ArH), 122.0 (ArH), 127.7 (C), 122.8 (C), 126.0 (ArH), 129.8 (ArH), 130.1 (ArH), 130.7 (ArH), 133.4 (ArH), 134.4 (ArH), 150.3 (Ar(C)NO2), 157.0 (Ar(C)O); m/z (CI) 353 (MNH4+, 3.5%), 135 (100); [Found MNH4+, 353.1176 (error = 1.3 ppm). C16H17NO5S requires: MNH4+, 353.1171].(±)-1-[2-(2,4-Dimethoxyphenylmethanesulfonylmethyl)-phenyl]-ethanol, 29
Isolated in an overall yield of 19% (4 steps) as a white solid; RF [petroleum ether–ethyl acetate (4 : 1)] 0.12; mp 101–104 °C; δH (CDCl3, 400 MHz) 1.45 (3 H, d, J 6.4 Hz, CH3), 2.96 (1 H, s, OH), 3.85 (3 H, s, OCH3), 3.88 (3 H, s, OCH3), 4.33–4.39 (4 H, m, 2 CH2), 4.96 (1 H, q, J 6.4 Hz, CH), 6.53 (1 H, s, ArH), 6.57 (1 H, d, J 8.0 Hz, ArH), 7.23–7.27 (2 H, m, 2 ArH), 7.37–7.41 (2 H, m, 2 ArH), 7.53 (1 H, d, J 8.0 Hz, ArH); this compound was immediately subjected to the RBR due to problems with stability.(2,4-Dimethoxy)-phenylmethanesulfonylmethyl-(2-nitro)-phenyl, 30
Isolated in an overall yield of 78% (2 steps) as an orange glass; RF [petroleum ether–ethyl acetate (4 : 1)] 0.16; mp 119–122 °C; νmax (nujol)/cm−1 1613, 1348 (SO2), 1157 (SO2); δH (CDCl3, 400 MHz) 3.84 (3 H, s, OCH3), 3.89 (3 H, s, OCH3), 4.38 (2 H, s, CH2), 4.71 (2 H, s, CH2), 6.53–6.57 (2 H, m, 2 ArH), 7.34 (1 H, d, J 8.4 Hz, ArH), 7.49–7.60 (3 H, m, 3 ArH), 8.06 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 54.0 (CH2), 54.8 (CH2), 55.9 (OCH3), 56.1 (OCH3), 99.3 (ArH), 105.7 (ArH), 108.8 (C), 122.7 (C), 126.0 (ArH), 130.2 (ArH), 133.5 (ArH), 133.8 (ArH), 134.5 (ArH) 150.5 (Ar(C)NO2), 158.8 (Ar(C)O), 162.3 (Ar(C)O); m/z (CI) 369 (MNH4+, 15%), 151 (100); [Found MNH4+, 369.1125 (error = 1.3 ppm). C16H17NO6S requires: MNH4+, 369.1120].(±)-1-(2-Phenylmethanesulfonylmethylphenyl)-ethanol, 31
Isolated in an overall yield of 45% (4 steps) as a clear gum; RF [petroleum ether–ethyl acetate (4 : 1)] 0.15; νmax (neat)/cm−1 3320 (OH), 1122; δH (CDCl3, 400 MHz) 1.46 (3 H, d, J 6.4 Hz, CH3), 2.80 (1 H, s, OH), 4.31 (2 H, s, CH2), 4.37 (2 H, d, J 5.6 Hz, CH2), 4.99 (1 H, q, J 6.4 Hz, CH), 7.25–7.28 (2 H, m, 2 ArH), 7.40–7.44 (6 H, m, 6 ArH), 7.56 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 23.8 (CH3), 54.9 (CH2), 59.8 (CH2), 67.2 (CH), 123.9 (C), 127.6 (ArH), 127.9 (C), 128.2 (ArH), 129.5 (2 ArH), 129.6 (ArH), 130.2 (ArH), 131.2 (2 ArH), 132.9 (ArH), 146.2 (C); m/z (CI) 308 (MNH4+, 5%), 273 (100); [Found MNH4+, 308.1325 (error = 1.6 ppm). C16H18O3S requires: MNH4+, 308.1320].(2-Nitro)-phenylmethanesulfonylmethylphenyl, 32
Isolated in an overall yield of 81% (2 steps) as a white solid; RF [petroleum ether–ethyl acetate (4 : 1)] 0.30; mp 132–135 °C; νmax (nujol)/cm−1 1460, 1308 (SO2), 1121 (SO2); δH (CDCl3, 400 MHz) 4.32 (2 H, s, CH2), 4.72 (2 H, s, CH2), 7.40–7.48 (5 H, m, 5 ArH), 7.54–7.57 (3 H, m, 3 ArH), 8.09 (1 H, d, J 8.0 Hz, ArH); δC (CDCl3, 100 MHz) 54.8 (CH2), 60.7 (CH2), 122.7 (C), 126.2 (ArH), 127.5 (C), 129.5 (2 ArH), 129.7 (ArH), 130.6 (ArH), 131.2 (2 ArH), 133.9 (ArH), 134.6 (ArH)–Ar(C)NO2 signal not observed; m/z (CI) 309 (MNH4+, 20%), 262 (60), 108 (100); [Found MNH4+, 309.0908 (error = 0.3 ppm). C14H13NO4S requires: MNH4+, 309.0909].(2-Methoxy)-phenylmethanesulfonylmethylphenyl, 33
Isolated in an overall yield of 63% (2 steps) as a white solid; RF [petroleum ether–ethyl acetate (4 : 1)] 0.26; mp 92–95 °C; νmax (nujol)/cm−1 1461, 1303 (SO2), 1157 (SO2); δH (CDCl3, 400 MHz) 3.88 (3 H, s, OCH3), 4.12 (2 H, s, CH2), 4.36 (2 H, s, CH2), 6.95 (1 H, d, J 8.4 Hz ArH), 7.02 (1 H, dd, J 7.6, 7.6 Hz, ArH), 7.34–7.39 (6 H, m, 6 ArH), 7.44 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 53.5 (CH2), 56.0 (OCH3), 58.3 (CH2), 111.4 (ArH), 117.0 (C), 121.6 (ArH), 127.6 (C), 129.1 (2 ArH), 129.2 (ArH), 131.0 (ArH), 131.5 (2 ArH), 133.1 (ArH), 157.8 (Ar(C)O); m/z (CI) 294 (MNH4+, 6%), 277 (MH+, 2), 121 (100); [Found MNH4+, 294.1163 (error = 0.2 ppm). C15H16O3S requires: MNH4+, 294.1164].Representative RBR procedure
Z-7: RF [petroleum ether–ethyl acetate (4 : 1)] 0.48; νmax (neat)/cm−1 2886, 1596, 1039; δH (CDCl3, 400 MHz) 1.59 (3 H, s, CH3), 1.97 (3 H, s, CH3), 3.63–3.65 (2 H, m, 2 CH), 3.90–3.92 (2 H, m, 2 CH), 5.05 (2 H, s, CH2), 6.89–6.94 (2 H, m, 2 ArH), 6.90 (1 H, s, vinyl CH), 7.16–7.37 (10 H, m, 10 ArH), 7.51 (1 H, d, J 8.0 Hz, ArH); δC (CDCl3, 100 MHz) 19.0 (CH3), 26.6 (CH3), 64.4 (2 CH2), 70.6 (CH2), 109.7 (C), 112.8 (ArH), 121.4 (ArH), 126.3 (ArH), 126.9 (ArH), 127.6 (2 ArH), 127.9 (ArH), 128.1 (ArH), 128.8 (2 ArH), 130.1 (ArH), 130.4 (vinyl CH), 131.7 (ArH), 135.8 (C), 136.6 (C), 136.7 (C), 137.8 (C), 141.1 (C), 156.4 (Ar(C)O)—1 ArH signal not observed; m/z (CI) 387 (MH+, 90%), 325 (100); [Found MH+, 387.1959 (error = 0.4 ppm). C26H26O3 requires: MH+, 387.1960].
E-7: RF [petroleum ether–ethyl acetate (4 : 1)] 0.48; νmax (neat)/cm−1 2886, 1237, 1040; δH (CDCl3, 400 MHz) 1.70 (3 H, s, CH3), 2.18 (3 H, s, CH3), 3.703.73 (2 H, m, 2 CH), 3.96–3.99 (2 H, m, 2 CH), 5.02 (2 H, s, CH2), 6.68 (1 H, dd, J 8.0, 8.0 Hz, ArH), 6.74–6.79 (4 H, m, 4 ArH), 6.94–6.97 (1 H, m, ArH), 7.03 (1 H, dd, J 8.0, 8.0 Hz, ArH), 7.12 (1 H, brs, vinyl CH), 7.20–7.25 (5 H, m, 5 ArH), 7.41 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 26.4 (CH3), 26.9 (CH3), 64.6 (2 CH2), 70.1 (CH2), 109.9 (C), 112.5 (ArH), 121.2 (ArH), 125.7 (ArH), 126.2 (vinyl CH), 127.3 (2 ArH), 127.5 (ArH), 127.6 (ArH), 128.0 (ArH), 128.2 (ArH), 128.9 (2 ArH), 130.4 (ArH), 131.4 (ArH), 132.3 (C), 136.4 (C), 136.6 (C), 137.8 (C), 140.2 (C), 156.4 (Ar(C)O); m/z (CI) 387 (MH+, 100%), 325 (85); [Found MH+, 387.1968 (error = 2.1 ppm). C26H26O3 requires: MH+, 387.1960].
In a similar manner to the procedure described above, olefins 15 (see later for corresponding data), 16 and 34–41 were prepared.
Due to the complex nature of the isomeric mixture, it proved impossible to fully assign the 13C-NMR spectrum.
Due to the complex nature of the isomeric mixture, it proved impossible to fully assign the 13C-NMR spectrum.
Z-37: RF [petroleum ether–ethyl acetate (4 : 1)] 0.45; νmax (neat)/cm−1 3412 (OH), 2967, 1608; δH (CDCl3, 400 MHz) 1.43 (3 H, d, J 6.4 Hz, CH3), 1.85 (1 H, brs, OH), 3.73 (3 H, s, OCH3), 3.77 (3 H, s, OCH3), 5.15 (1 H, q, J 6.4 Hz, CH), 6.18 (1 H, d, J 8.8 Hz, ArH), 6.40 (1 H, s, ArH), 6.66 (1 H, d, J 12.0 Hz, vinyl CH), 6.78 (2 H, m, 1 ArH and 1 vinyl CH), 7.08–7.13 (2 H, m, ArH), 7.22–7.25 (1 H, m, ArH), 7.53 (1 H, d, J 7.6 Hz, ArH); δC (CDCl3, 100 MHz) 24.2 (CH3), 55.7 (OCH3), 55.8 (OCH3), 67.5 (CH), 98.7 (ArH), 104.5 (ArH), 118.7 (C), 125.3 (ArH), 126.3 (vinyl CH), 126.9 (vinyl CH), 127.5 (ArH), 127.8 (ArH), 129.8 (ArH), 130.7 (ArH), 136.3 (C), 143.7 (C), 158.7 (Ar(C)O), 160.6 (Ar(C)O); m/z (CI) 285 (MH+, 15%) 267 (100), 151 (25); [Found MH+, 285.1488 (error = 0.9 ppm). C18H20O3 requires: MH+, 285.1491].
E-37: RF [petroleum ether–ethyl acetate (4 : 1)] 0.41; νmax (neat)/cm−1 3412 (OH), 2968, 1608; δH (CDCl3, 400 MHz) 1.51 (3 H, d, J 6.4 Hz, CH3), 1.86 (1 H, brs, OH), 3.83 (3 H, s, OCH3), 3.86 (3 H, s, OCH3), 5.31 (1 H, q, J 6.4 Hz, CH), 6.47 (1 H, s, ArH), 6.52 (1 H, d, J 8.4 Hz, ArH), 7.22–7.27 (3 H, m 2 ArH and 1 vinyl CH), 7.34 (1 H, d, J 16.4 Hz, vinyl CH), 7.49 (1 H, d, J 8.4 Hz, ArH), 7.51–7.54 (1 H, m, ArH), 7.58–7.61 (1 H, m, ArH); δC (CDCl3, 100 MHz) 24.7 (CH3), 55.8 (OCH3), 55.9 (OCH3), 67.5 (CH), 99.0 (ArH), 105.4 (ArH), 120.1 (C), 124.3 (vinyl CH), 125.1 (ArH), 126.3 (vinyl CH), 126.4 (ArH), 127.8 (ArH), 127.9 (ArH), 128.0 (ArH), 136.3 (C), 143.1 (C), 158.5 (Ar(C)O), 161.0 (Ar(C)O); m/z (CI) 285 (MH+, 10%), 267 (100), 151 (20); [Found MH+, 285.1491 (error = 0.1 ppm). C18H20O3 requires: MH+, 285.1491].
Due to the complex nature of the isomeric mixture, it proved impossible to fully assign the 13C-NMR spectrum.
Data consistent with literature.19
Data consistent with literature.20
CCDC reference number 219622. See http://www.rsc.org/suppdata/ob/b4/b418426b/ for crystallographic data in .cif or other electronic format.
CCDC reference number 257403. See http://www.rsc.org/suppdata/ob/b4/b418426b/ for crystallographic data in .cif or other electronic format.
Acknowledgements
We would like to thank the EPSRC and GlaxoSmithKline for their financial support (J.S.F) and Dr Julie Quayle (GSK) for assistance with the NOE experiments.References
- For a recent review on the therapeutic properties of Tamoxifen, see: ; J. Schwartz, Front. Biosci., 2004, 9, 2827 Search PubMed and references therein.
- M. Cushman, D. Nagarathnam, D. Gopal, H.-M. He, C. M. Lin and E. Hamel, J. Med. Chem., 1992, 35, 2293 CrossRef CAS.
- P. J. Kukkola, N. A. Bilci, W. X. Kozak, P. Savage and A. Y. Yeng, Bioorg. Med. Chem. Lett., 1996, 6, 619 CrossRef CAS.
- S. B. Singh, D. L. Fink, D. S. Quamina, F. Pelaez, A. Teran, P. Felock and D. J. Hazuda, Tetrahedron Lett., 2002, 43, 2351 CrossRef CAS.
- C. Y. Meyers, A. M. Malte and W. S. Matthews, J. Am. Chem. Soc., 1969, 91, 7510 CrossRef CAS.
- F. G. Bordwell and G. D. Cooper, J. Am. Chem. Soc., 1951, 73, 5187 CrossRef CAS.
- N. P. Neureiter, J. Am. Chem. Soc., 1966, 88, 558 CrossRef CAS.
- T.-L. Chan, S. Fong, Y. Li, T.-O. Man and C.-D. Poon, J. Chem. Soc., Chem. Commun., 1994, 1771 RSC.
- N. Tokura, T. Nagai and S. Matsumura, J. Org. Chem., 1966, 31, 349 CrossRef CAS.
- D. Scholz and P. Burtscher, Liebigs Ann. Chem., 1985, 517 CrossRef CAS.
- G. Casy and R. J. K. Taylor, Org. React., 2003, 62, 357; L. Paquette, Org. React., 1977, 25, 1 CAS.
- E. Reimann and H. Renz, Arch. Pharm., 1993, 326, 253 CAS.
- S. Vetter, Synth. Commun., 1998, 28, 3219 CrossRef CAS.
- G. W. Rewcastle, G. J. Atwell, B. C. Baguley, M. Boyd and L. L. Thomsen, J. Med. Chem., 1991, 34, 2864 CrossRef CAS.
- T. Hayashi, Y. Matsumoto, I. Yonetatsu and Y. Ito, Tetrahedron Asymmetry, 1991, 2, 601 CrossRef CAS.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC ; for a more detailed discussion dealing with the structurally similar episulfonium ion systems, see: P. Bird, J. Eames, A. G. Fallis, R. V. H. Jones, M. Roddis, C. F. Sturino, S. O'Sullivan, S. Warren, M. S. Westwell and J. Worrall, Tetrahedron Lett., 1995, 36, 1909 Search PubMed and references therein.
- J. S. Foot, G. M. P. Giblin and R. J. K. Taylor, Org. Lett., 2003, 23, 4441 CrossRef.
- Crystal data for 45: Empirical formula = C17H14O3; Formula weight = 266.28; Unit cell dimensions: a
= 9.2508(9)
Å, α
= 100.325(2)°; b
= 9.3130(9)
Å, β
= 90.360(2)°; c
= 15.5363(15)
Å, γ
= 99.592(2)°; Volume = 1297.5(2)
Å3; Temperature/Space group = 115(2) K/P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; Z
= 4; Linear absortion coefficient = 0.093 mm−1; Reflections collected = 10629; Independent collections = 4551 [R(int)
= 0.0382]; Final R indices [I > 2σ(I)]
=
R1 = 0.0427, wR2 = 0.0917; R indices (all data)
=
R1 = 0.0713, wR2 = 0.1019. CCDC reference number 257403. See http://www.rsc.org/suppdata/ob/b4/b418426b/ for crystallographic data in .cif or other electronic format..
; Z
= 4; Linear absortion coefficient = 0.093 mm−1; Reflections collected = 10629; Independent collections = 4551 [R(int)
= 0.0382]; Final R indices [I > 2σ(I)]
=
R1 = 0.0427, wR2 = 0.0917; R indices (all data)
=
R1 = 0.0713, wR2 = 0.1019. CCDC reference number 257403. See http://www.rsc.org/suppdata/ob/b4/b418426b/ for crystallographic data in .cif or other electronic format.. - J. Wang, Y. Fu and Y. Hu, Angew. Chem., Int. Ed., 2002, 2757 CrossRef CAS.
- P. S. Kendurkar and R. S. Tewari, J. Organomet. Chem., 1975, 85, 173 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |