Structure–activity relationships of galabioside derivatives as inhibitors of E. coli and S. suis adhesins: nanomolar inhibitors of S. suis adhesins†
Jörgen Ohlssona, Andreas Larssonb, Sauli Haatajac, Jenny Alajääskic, Peter Stenlundd, Jerome S. Pinknere, Scott J. Hultgrene, Jukka Finne*c, Jan Kihlberg*b and Ulf J. Nilsson*a
aOrganic Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: ulf.nilsson@organic.lu.se; Fax: (+46) 46 222 82 09; Tel: (+46) 46 222 82 18
bOrganic Chemistry, Department of Chemistry, Umeå University, SE-901 87 Umeå, Sweden
cDepartment of Medical Biochemistry and Molecular Biology, University of Turku, FI-20520 Turku, Finland
dBiochemistry, Department of Chemistry, Umeå Universitet, SE-901 87 Umeå, Sweden
eDepartment of Molecular Biology, Washington University School of Medicine, St. Louis, Missouri 63110, USA
First published on 4th February 2005
Abstract
Four collections of Galα1-4Gal derivatives were synthesised and evaluated as inhibitors of the PapG class II adhesin of uropathogenic Escherichia coli and of the PN and PO adhesins of Streptococcus suis strains. Galabiosides carrying aromatic structures at C1, methoxyphenyl O-galabiosides in particular, were identified as potent inhibitors of the PapG adhesin. Phenylurea derivatisation at C3′ and methoxymethylation at O2′ of galabiose provided inhibitors of the S. suis strains type PN adhesin with remarkably high affinities (30 and 50 nM, respectively). In addition, quantitative structure–activity relationship models for E. coli PapG adhesin and S. suis adhesin type PO were developed using multivariate data analysis. The inhibitory lead structures constitute an advancement towards high-affinity inhibitors as potential anti-adhesion therapeutic agents targeting bacterial infections.
Introduction
The alarming increase in the resistance of bacteria to traditional antibiotics1–3 makes it imperative to develop alternative ways of treating bacterial infections. The majority of infectious diseases are initiated by adhesion of pathogenic organisms to host tissue and in many cases glycoconjugates present on the mammalian cell surface, e.g. glycoproteins and glycolipids, act as receptors for a wide variety of extracellular bacterial proteins termed adhesins.4,5 This carbohydrate–protein interaction is often a prerequisite for the later stages of bacterial infection and inhibitors of this recognition process are potential pharmaceutical agents. Bacterial resistance towards such anti-adhesive drugs is believed to evolve slowly because the infecting bacteria are not killed and are consequently not under selection pressure.Two well-known examples of pathogenic bacteria adhering to glycoconjugates are uropathogenic Escherichia coli, which is the main cause of urinary tract infections, and Streptococcus suis, which causes meningitis in pig and man. The majority of E. coli bacteria causing pyelonephritis (kidney infection) adhere via proteinaceous appendices, termed P-pili. These pili are terminated with an adhesin, PapG, that binds to the Galα1-4Gal (galabiose)6–8 moiety present in the globoseries of glycolipids on uroepithelial cells and erythrocytes. Three different classes of the PapG adhesin (classes I–III)9,10 have been identified based on different erythrocyte agglutination patterns. Pyelonephritis in both children and adult women is associated with PapG class II,11,12 while class III is associated with cystitis in adult women.13 In addition to anchoring the bacteria to the host cell, the adhesion of PapG induces the release of ceramides14 that are important second messenger molecules, and results in up-regulation of, and eventual secretion of, several immunoregulatory cytokines from host cells.15Streptococcus suis is a frequent colonizer of the pig respiratory tract. Galα1-4Gal terminating oligosaccharides have been shown to be optimal receptors for S. suis. Systematic competitive inhibition studies characterized the key hydroxyl groups that are required for binding to Galα1-4Gal and also classified the adhesion activities into two types, PN and PO.16
Most natural carbohydrate ligands bind lectins with low affinity (Kd normally in the 0.1–1 mM range). One attractive strategy to overcome this problem is to use a small key saccharide as the core structure and attach substituents that interact with the lectin in a favourable manner. It has been shown that for the PapG class II adhesin the galabiose disaccharide is such a key structure17 necessary for recognition and that galabioside derivatives substituted at C1 and C3′ often display enhanced affinity for the PapG adhesins.18 Furthermore, the crystal structure of the class II PapG adhesin in complex with the globotetraose tetrasaccharide has been solved19 and it confirmed that the galabiose disaccharide unit is the most critical structural element for the formation of the complex. The complex structure revealed an extended surface composed of H1, H2 and H6 of Galα, H1, H3, H4, H5 and H6 of Galβ, and H2 and H4 of Glc, that make hydrophobic contact primarily with the Trp107 sidechain. The adhesin additionally forms four hydrogen bonds to HO4, O5 and HO6 of the non-reducing GalNAc residue, seven to HO2, O3, HO4 and HO6 of the Galα residue, four to HO3, O5 and HO6 of the Galβ, and three to HO2 and HO3 of the Glc residue. Apparently, the two residues flanking galabiose, GalNAc and Glc, are involved via hydrophobic contacts and hydrogen bonding (e.g. Lys172 to GalNAc and Arg170 and Trp107 to Glc), but to a lesser extent.
The galabiose disaccharide has also been shown to be the key recognition element for adhesins from S. suis strains of both type PN and PO. The two adhesins from S. suis, however, display differences in the sub-molecular details of galabiose recognition.16 Multivalent galabiose derivatives have been reported to display greatly enhanced affinity.20 However, although multivalent galabiosides provide potent inhibitors, they all share the disadvantages of large size and high polarity resulting in poor bioavailability.
The present paper describes an attempt to improve the affinity of inhibitors for the PapG class II adhesins and the two S. suis adhesins by the synthesis of four collections of galabiosides modified at C1 and C3′ in four different ways. In order to position the substituents at C1 as close as possible to the galabiose core structure, either amide formation (galabioside collection I), glycosylation of alcohols (collection II), or of thiols (collection III) were employed. At C3′, amide formation was used to create structural diversity on a p-methoxyphenyl galabioside core structure (collection IV). Recently reported synthesis and evaluation of earlier generations of galabiose derivatives has shown that the p-methoxyphenyl galabioside itself is an excellent inhibitor of the PapG adhesins.18,21 The further evaluation of these earlier generations of galabiose derivatives against the two S. suis adhesins is also reported herein. In addition, quantitative structure–activity relationship models for E. coli PapG adhesin and S. suis adhesin type PO were developed using multivariate data analysis.
Results and discussion
Synthesis of galabiose collections I–IV (Table 1)
For the synthesis of the collection I galabiosides, the galabiosyl bromide 122 was converted to the β-azide 4 (Scheme 1) via treatment with trimethylsilyl azide and tetrabutylammonium fluoride,23 (3, 92%) followed by conventional deacylation with methanolic sodium methoxide to give 4 in 96% yield. | ||
| Scheme 1 a) TMSN3, Bu4NF, THF, 92%. b) NaOMe, MeOH, 96%. For c) and d) see Table 1. | ||
| Compound/conditionsa/yield (%) |
| Kd/µM E. coli PapG II | IC50/µM S. suis PN/PO | ||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| a A) i4, H2 (1 atm), Pd/C (10%), MeOH, iiRC(O)Cl, Na2CO3, THF. B) i4, H2 (1 atm), Pd/C (10%), MeOH, iiRC(O)Cl, DMAP, pyridine. C) i10, R′-OH, TMSOTf, CH2Cl2, iiNaOMe, MeOH. D) i1, NaH, R′-OH, DMF, ii NaOMe, MeOH. E) i22, H2 (1 atm), Pd/C (10%), MeOH, iiAc2O, Na2CO3, THF. E) i1, NaH, R–SH, DMF, ii1, NaOMe, MeOH. F) 32, H2 (1 atm), HCl (aq), Pd/C (10%), MeOH. G) 33, RC(O)Cl, RC(O)OC(O)R, or RNCO, Na2CO3, THF. | |||||
| Collection I | |||||
| 5/A/90 | NHC(O)Ph | OH | H | 2340 | 7.8/5.0 |
| 6/A/61 |  | OH | H | 1030 | 2.5/2.5 |
| 7/A/51 |  | OH | H | 1640 | 2.5/2.5 |
| 8/A/54 |  | OH | H | 1190 | 2.5/2.5 |
| 9/A/48 | NHC(O)Et | OH | H | 3030 | 2.5/1.25 |
| 10/B/28 | 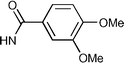 | OH | H | 1320 | 0.98/7.8 |
| Collection II | |||||
| 11/C/53 |  | OH | H | 150 | 2.5/1.25 |
| 12/C/74 |  | OH | H | 176 | 7.8/5.0 |
| 13/C/66 |  | OH | H | 170 | 0.63/0.31 |
| 14/C/80 |  | OH | H | 191 | 0.63/0.63 |
| 15/C/62 |  | OH | H | 217 | 0.31/0.63 |
| 16/C/44 |  | OH | H | 217 | 0.63/0.63 |
| 17/C/78 |  | OH | H | 512 | 0.63/0.63 |
| 18/C/83 |  | OH | H | 269 | 0.63/0.63 |
| 19/C/65 |  | OH | H | 373 | 0.63/0.31 |
| 20/D/54 |  | OH | H | 229 | 0.63/0.63 |
| 21/D/36 |  | OH | H | 235 | 7.8/2.5 |
| 22/D/70 |  | OH | H | 189 | 0.63/0.63 |
| 23/E/68 |  | OH | H | 180 | 0.63/0.31 |
| Collection III | |||||
| 24/E/76 |  | OH | H | 605 | 0.63/0.31 |
| 25/E/79 |  | OH | H | 321 | 0.63/0.31 |
| 26/E/85 |  | OH | H | 428 | 5.0/0.31 |
| 27/E/88 |  | OH | H | 333 | 2.5/1.25 |
| Collection IV | |||||
| 33/F/93 |  | NH2 | H | 4110 | 15.6/2.5 |
| 34/G/74 | | CH3C(O)NH | H | 23500 | 0.63/15.6 |
| 35/G/79 | | CH3CH2C(O)NH | H | 30500 | 1.25/62.5 |
| 36/G/78 | | HO2C(CH2)2C(O)NH | H | 500000 | 0.63/31.3 |
| 37/G/70 | | PHC(O)NH | H | 34500 | 0.63/62.2 |
| 38/G/66 | |  | H | 14000 | 0.63/31.3 |
| 39/G/61 | |  | H | 99000 | 0.31/31.3 |
| 40/G/83 | |  | H | 11400 | 1.25/62.5 |
| 41/G/69 | |  | H | 76700 | 0.63/62.5 |
| 42/G/55 | |  | H | 141000 | 1.25/62.5 |
| 43/G/49 | | CH3CH2NHC(O)NH | H | 10100 | 0.31/62.5 |
| 44/G/84 | | PhNHC(O)NH | H | 27200 | 0.04/62.5 |
| Known galabioside | |||||
| 45 | | OH | H | 14021 | 0.31/0.31 |
| 46 | OCH2CH2Si(Me)3 | OH | H | 64621 | 0.63/0.63 |
| 47 | OMe | OH | H | 30621 | 0.63/0.63 |
| 48 | | OH | Me | 15021 | 0.63/3.9 |
| 4935 | NHC(O)Ph | OH | Me | n.d. | 5.0/31.3 |
| 50 | | OH | Pr | 100021 | 0.31/5.0 |
| 51 | | OH | MeOCH2 | 49021 | 0.08/15.6 |
| 5218 | |  | H | n.d. | 2.5/1.25 |
| 53 | |  | H | 19921 | 2.5/3.9 |
| 54 | |  | H | 17521 | 2.5/1.25 |
| 5518 | | HO2CCH2O | H | n.d. | 1.25/5.0 |
| 5618 | | MeO2CCH2O | H | n.d. | 7.8/31.3 |
| 5718 |  |  | H | n.d. | 15.6/15.6 |
| 5818 |  | | H | n.d. | 15.6/31.3 |
| 5918 | OEt | PrO | H | n.d. | 0.63/15.6 |
| 6018 | |  | H | n.d. | 1.25/15.6 |
| 6118 | | HO(CH2)2S(CH2)3O | H | n.d. | 7.8/7.8 |
| 6218 |  |  | H | n.d. | 5.0/2.5 |
| 6318 | | MeO2CCH2S(CH2)3O | H | n.d. | 5.0/31.3 |
| 6418 | | | H | n.d. | 5.0/31.3 |

Hydrogenation of 4 over Pd/C in methanol gave an intermediate amine, which was immediately converted to amides by treatment with five different acid chlorides in the presence of sodium carbonate in THF, affording compounds 5–9 in 48–90% overall yields and β/α ratios of ∼15 : 1. No product was observed under these reaction conditions with a hindered acid chloride (i.e.10). However, acylation with the acid chloride, pyridine, and a catalytic amount of DMAP, gave compound 10 in a moderate 28% yield. Attempts to increase the yields for compounds 6–9 with these latter reaction conditions were unsuccessful. Furthermore, different reaction conditions were unsuccessfully evaluated in attempts to improve the β-selectivities (i.e. PtO2 as a hydrogenation catalyst, one-pot azide reduction and acylation, hydrogenation in the presence of HCl, and the use of different solvents). It is likely that the anomerisation of the amine occurs faster than the acylation, because similar α/β ratios were obtained under all conditions tried. No α-anomer was detected when fully acylated lactosyl azide24 was reacted under the same reaction conditions, suggesting that the resulting anomeric mixtures obtained in the case of the galabiose derivatives 5–10 are probably due to the inherent properties of galactose (or galabiose).
Collection II galabiose derivatives were prepared from either the galabiosyl α-trichloroacetimidate 2 (compounds 11–18) or from the galabiosyl bromide 1 (compounds 19–22). Trimethylsilyl trifluoromethanesulfonate (TMSOTf)-promoted glycosylation of aromatic and aliphatic alcohols with the trichloroacetimidate 2, followed by deacylation in methanolic sodium methoxide, furnished compounds 11–18 in 44–83% yield (β/α ∼15 : 1). Under these conditions, phenols carrying electron withdrawing groups typically gave low yields and poor α/β-selectivities. Instead, nucleophilic displacement of the galabiosyl bromide 1 with the corresponding sodium phenolates fortunately afforded compounds 19–22 in 36–70% yield after deacylation. Complete β-selectivity was observed for compounds 20–22, while compound 19 had a β/α-ratio of about 10 : 1. Compound 23 was prepared from 22 by catalytic hydrogenation followed by acylation under conditions similar to those described for the preparation of collection I.
Galabioside collection III was prepared by nucleophilic displacement of the galabiosyl bromide 1 with thiophenolates, followed by deacylation, to furnish β-galabiosides 24–27 in 76–88% yield.
Synthesis of collection IV required the introduction of a handle (amine) at C3′ of p-methoxyphenyl galabioside (Scheme 2). Henceforth, the known galactoside 2825 was deacylated and benzylated to give the galactosyl donor 29. α-Galactosylation of the acceptor 30,22 using N-iodosuccinimide-trimethylsilyl trifluoromethanesulfonate as promoter,26,27 gave the protected 3′-azido galabioside 31 in 93% yield. Debenzoylation in methanolic sodium methoxide gave 32 in 80% yield, the key starting material for the synthesis of galabioside collection IV.
 | ||
| Scheme 2 a) iNaOMe, MeOH, iiNaH, BnBr, DMF, 86%. b) NIS, TMSOTf, CH2Cl2–Et2O (1 : 2), −50 °C, 93%. c) NaOMe, MeOH, 80%. d) See Table 1. | ||
The azido group of galabioside 32 was reduced to the amine 33 under reaction conditions similar to those described for the preparation of collection I, with the exception that 2 equivalents of HCl were added to ensure complete hydrogenolysis of the benzyl groups. The 3′-amino galabioside 33 was treated with sodium carbonate and either an acyl chloride (34–35 and 37–41), acid anhydride (36 and 42), or isocyanate (43–44) to give galabiosides 34–44 in 49–83% yield.
Binding of the E. coli PapG class II adhesin to galabioside derivative collections I–IV
Binding of galabiose derivative collections I–IV by an N-terminal 196 amino acid truncate of the class II PapG adhesin19 was determined by surface plasmon resonance as recently described.21 The binding data were analysed using the software Scrubber28 (Fig. 1, Table 1). | ||
| Fig. 1 Equilibrium isotherms fit to a 1 : 1 interaction model for 45 (△), 25 (■), 46 (□) and 36 (○) binding to PapGII immobilized to a CM5 surface plasmon resonance biosensor surface. The binding responses at equilibrium were normalised against maximum binding (Rmax). In cases where the affinity of the galabioside was too low (36) to saturate an expected binding isotherm, the Rmax was determined using a reference substance and the galabioside’s molecular weight. | ||
The presence of amides at C1 of galabiose (collection I) turned out to be detrimental to binding. The benzamido derivatives (5–8, and 10) displayed Kd of 1.0–2.3 mM, which is much worse than that of the known reference compound p-methoxyphenyl galabioside 4518 (Kd 140 µM21). Virtually no interaction was seen with an aliphatic amide at C1 (9). The amide-functionality at C1 probably positions the aromatic rings of 5–8 and 10 in non-favourable positions relative to the side chains of Trp107 and Arg170 of PapGII. In contrast, the p-methoxyphenyl glycoside of galabiose 45 positions the aromatic ring to interact favourably with Trp107 and Arg170, resulting in a Kd as low as 140 µM.21
Collection II (O-galabiosides 11–23) turned out to be more successful in providing ligands for the class II PapG adhesin. All compounds showed higher affinities for the adhesin than those observed for aliphatic galabiosides (i.e. the 2-(trimethylsilyl)ethyl galabioside 4618). The positions of methoxy groups on the aromatic aglycons had some impact on the affinity. p-Methoxyphenyl galabioside 45 and m-methoxyphenyl galabioside 11 had virtually the same Kd. However, the Kd (176 µM) for o-methoxyphenyl galabioside 12 was somewhat higher than 45 and close to that of the phenyl galabioside 13 (170 µM). Thus, the methoxyphenyl group interacts favourably with the adhesin when positioned in the m- or p-position. Exchange of the p-methoxy for a p-methyl group (i.e.14) resulted in an increase in Kd in the same range as removal of the p-methoxy group did. This suggests that the oxygen atom of the p-methoxy group is important for affinity to the adhesin. Introduction of other groups at the p-position of a phenyl aglycon (i.e. methylester 20, nitro 22, or acetamido 23) resulted in galabiosides with the same Kd as the p-methylphenyl galabioside 14, as did introduction of a methylester at the o-position (21). In contrast, the introduction of fluorine (19) resulted in a large increase in Kd to 373 µM, i.e. 2.5 times of that observed for phenyl galabioside 13. Increasing the size of the aromatic substituent (i.e. naphthyl 15 or indolyl 16) or moving the aromatic substituent away from the galabiose C1 (i.e. benzyl galabioside 17), resulted in lowered affinity for the adhesin compared to phenyl galabioside 13, further demonstrating the sensitivity towards the exact position of the aromatic substituents at C1. Furthermore, cyclohexyl galabioside 18 (Kd 269 µM) was a less potent inhibitor than phenyl galabioside 13, suggesting that aromatic aglycons are beneficial. Most likely, phenyl aglycons of galabiosides stabilise complex formation via interactions with the aromatic side chain of Trp107 and with the guanidino group of Arg170 in the PapGII adhesin.21
Exchange of the anomeric oxygen atom for sulfur results in more hydrolytically stable galabiosides. However, the phenyl thio-galabiosides 24–27 (collection III) had Kd values in the 320–600 µM range, i.e. about one third of the affinity of their O-glycosidic counterparts. An altered conformation of phenyl thio-galabiosides, compared to the corresponding O-glycosides,29 most likely explains the higher Kd values observed for these compounds. The aromatic aglycons of 24–26 are folded back onto the galabiose disaccharide moiety, which causes a conformational change in the α(1–4) disaccharide linkage of galabiose. Hence, the phenyl thio-galabiosides 24–26 must adopt a high-energy conformation in order to be recognised by the class II PapG adhesin.
Exchange of a hydroxyl for an amino group at C3′ (33) turned out to be detrimental for binding; the Kd value for 33 was 29 times higher than that of p-methoxyphenyl galabioside 45. Functionalisation of the amine at C3′ (34–44) resulted in even worse inhibitors (Kd 10–500 mM). The reason for the low affinity could be that the interaction between the Lys172 and O3′ seen in the crystal structure between the adhesin and the Gb4 tetrasaccharide is lost when an amine, amide or urea replaces the hydroxyl at C3′ of the galabiose disaccharide.
Binding of the S. suis adhesins type PN and type PO to galabioside derivative collections I–IV (5–44) and to known galabioside derivatives 45–64
Binding of galabiose derivatives 5–64 by S. suis types PN and PO was determined by hemagglutination inhibition essentially as previously described.30 The galabiose substituents of 5–64 clearly interact with the two S. suis adhesins as the structures of the substituents exert large influences on their inhibitory powers (Table 1). Furthermore, the two adhesins display different preferences with regard to the substituents’ position and structure, which is consistent with earlier observations.16 The recognition patterns of the type PN and PO adhesins are also markedly different to that of the E. coli class II adhesin. For S. suis, galabiosyl amides 5–10 (collection I) were poor inhibitors suggesting that O-galabiosides are preferred, possibly because the adhesins donate hydrogen bonds to galabiose O1. Hydrophobic (aromatic) galabioside aglycons (11–27, collection II) had, in general, a minor influence on the inhibitory powers compared to the reference methyl galabioside 47, except for phenyl aglycons carrying an ortho-substituent (12 and 21) or an m-methoxy group (11) which were detrimental to inhibition. Introducing amides at galabiose C3′ (34–42) was also detrimental to inhibition of the type PO adhesin, while it was well tolerated by the type PN adhesin. Ureas at C3′ (43–44) were, not surprisingly, poor inhibitors of the type PO adhesin. However, the ureas 43–44 were potent inhibitors of the type PN adhesin. This correlates with the previously suggested Gal/GalNAc binding pocket at this site, which differentiates the PN adhesin from the PO adhesin.16 Galabiose derivatives modified at O2′ with alkyl groups (48–51) appeared to be accepted by the type PN adhesin. Interestingly, O2′-methoxymethyl substitution (51) provided one of the two best inhibitors against this adhesin, indicating the proximity of a hydrogen bond donor in the adhesin. Galabiosides carrying alkoxy-substituents at C3′ (52–64) were all relatively poor inhibitors of both S. suis adhesins.The screening experiments against the two S. suis adhesins were further confirmed by selecting the ten best inhibitors and eight poorest inhibitors against each S. suis adhesin for refined evaluations in triplicate (Table 2). The refined evaluation established the IC50 values of the two best inhibitors, the C3′-phenylurea 44 and the O2′-methoxymethyl 51, against the type PN adhesin to be 30 and 50 nM, respectively, which is up to one order of magnitude better than the parent unsubstituted p-methoxyphenyl galabioside 45 (IC50 310 nM) and significantly better than the previously reported best small-molecule inhibitor against this adhesin, the natural globotriose trisaccharide (IC50 190 nM).16 The high affinities of 44 and 51 are extraordinary within the field of small-molecule inhibition of lectins. The synthesis of further galabiose collections modified with O-alkyl or alkoxymethyl substituents at O2′ or with ureas at C3′ thus emerges as an attractive route towards improved inhibitors. An obvious extension of this result would also be to combine the substituents of 44 and 51 into one single novel inhibitor, which would be significantly more potent provided that the affinity-enhancing effects of each substituent are additive. Furthermore, displaying 44 or 51, or a combination of these two inhibitors, on a multivalent scaffold would most likely result in more powerful inhibitors as multivalent inhibitors are known to be particularly efficient against the S. suis adhesins.20
| IC50/µM S. suis PN | Range | IC50/µM S. suis PO | Range | |
|---|---|---|---|---|
| 5 | 5.2 | 3.9–7.8 | ||
| 12 | 4.4 | 2.0–7.8 | ||
| 13 | 0.42 | 0.31–0.63 | ||
| 15 | 0.20 | 0.16–0.31 | ||
| 19 | 0.31 | 0.31 | ||
| 22 | 0.42 | 0.31–0.63 | ||
| 23 | 0.31 | 0.31 | ||
| 24 | 0.42 | 0.31–0.63 | ||
| 25 | 0.21 | 0.16–0.31 | ||
| 26 | 0.31 | 0.31 | ||
| 33 | 15.6 | 15.6 | ||
| 35 | 54.7 | 31.3–62.5 | ||
| 37 | 52.1 | 31.3–62.5 | ||
| 38 | 20.8 | 15.6–31.3 | ||
| 39 | 0.16 | 0.08–0.31 | ||
| 40 | 62.5 | 62.5 | ||
| 41 | 0.32 | 0.16–0.63 | 41.7 | 31.3–62.5 |
| 42 | 62.5 | 62.5 | ||
| 43 | 0.18 | 0.08–0.31 | 62.5 | 62.5 |
| 44 | 0.03 | 0.02–0.04 | ||
| 45 | 0.18 | 0.08–0.31 | 0.31 | 0.31 |
| 47 | 0.63 | 0.63 | 0.63 | 0.63 |
| 49 | 31.3 | 31.3 | ||
| 50 | 0.18 | 0.08–0.31 | ||
| 51 | 0.05 | 0.04–0.08 | ||
| 56 | 6.5 | 3.9–7.8 | ||
| 57 | 10.4 | 7.8–15.6 | ||
| 58 | 15.6 | 15.6 | ||
| 61 | 7.8 | 7.8 | ||
| 64 | 10.4 | 7.8–15.6 |
The results with the type PO adhesin were less impressive as only marginal affinity enhancements, compared to the known references 46–47, were obtained. Clearly, other strategies have to be considered for the development of inhibitors against this adhesin. From a drug development perspective, it would of course be desirable to find one inhibitor with high affinity against both S. suis adhesins. However, this appears to be a formidable challenge in light of the results reported herein. Possibly, chemical modifications at positions other than C1 and C3′ of the galabiose disaccharide will be required to find an efficient inhibitor against both adhesins.
Structure–activity relationships for the E. coli class II PapG adhesin and the adhesin from S. suis type PO
In order to develop a quantitative structure–activity relationship using multivariate data analysis, the four carbohydrate collections were characterised with molecular descriptors using the MOE software.31 The 2D molecular descriptors included described properties such as size, lipophilicity, flexibility and hydrogen-bonding capabilities (see Table 3). The affinity of the carbohydrates as measured by log Kd and log IC50 for the E. coli adhesin PapGII and the S. suis type PO adhesin, respectively, were related to the various molecular descriptors by means of partial least-squares projections to latent structures (PLS).32| Abbreviation | Descriptors | S. suis type PO | E. coli PapG | |||
|---|---|---|---|---|---|---|
| C1 | C3′ | C1 | C3′ | |||
| 1 | diameter | Molecular diameter | 0.030 | |||
| 2 | radius | Molecular radius | 0.016 | |||
| 3 | VDistEq | Vertex distance equation | 0.013 | |||
| 4 | VDistMa | Vertex distance magnitude | ||||
| 5 | weinerPath | Weiner path number | −0.029 | |||
| 6 | weinerPol | Weiner polarity number | ||||
| 7 | a_aro | Number of aromatic atoms | 0.052 | |||
| 8 | b_ar | Number of aromatic bonds | 0.053 | |||
| 9 | b_rotN | Number of rotatable bonds | −0.058 | |||
| 10 | b_rotR | Fraction of rotatable bonds | −0.050 | 0.082 | −0.061 | 0.092 |
| 11 | chi0v | Atomic valence connectivity index | ||||
| 12 | chi0v_C | Carbon valence connectivity index | ||||
| 13 | chi1v | Atomic valence connectivity index | ||||
| 14 | chi1v_C | Carbon valence connectivity index | ||||
| 15 | Weight | Molecular weight | ||||
| 16 | chi0 | Atomic connectivity index | −0.033 | |||
| 17 | chi0_C | Carbon connectivity index | ||||
| 18 | chi1 | Atomic connectivity index | ||||
| 19 | chi1_C | Carbon connectivity index | 0.053 | |||
| 20 | FCharge | Sum of formal charges | 0.045 | |||
| 21 | VAdjEq | Vertex adjacency equation | ||||
| 22 | VAdjMa | Vertex adjacency magnitude | ||||
| 23 | zagreb | Zagreb index | ||||
| 24 | balabanJ | Balaban connectivity index | −0.033 | −0.078 | ||
| 25 | Q_PC+ | Total positive partial charge | 0.029 | 0.058 | −0.030 | |
| 26 | Q_PC- | Total negative partial charge | −0.029 | −0.062 | 0.029 | |
| 27 | Q_RPC+ | Relative positive partial charge | ||||
| 28 | Q_RPC- | Relative negative partial charge | −0.010 | −0.059 | 0.021 | |
| 29 | Q_VSA_FHYD | Fractional hydrophobic van der Waals surface area | 0.008 | −0.008 | ||
| 30 | Q_VSA_FNEG | Fractional negative van der Waals surface area | 0.062 | |||
| 31 | Q_VSA_FPNE | Fractional polar negative van der Waals surface area | −0.002 | 0.064 | ||
| 32 | Q_VSA_FPOL | Fractional polar van der Waals surface area | −0.008 | 0.008 | ||
| 33 | Q_VSA_FPOS | Fractional positive van der Waals surface area | −0.062 | |||
| 34 | Q_VSA_FPPO | Fractional polar positive van der Waals surface area | −0.042 | −0.016 | −0.096 | |
| 35 | Q_VSA_HYD | Total hydrophobic van der Waals surface area | ||||
| 36 | Q_VSA_NEG | Total negative van der Waals surface area | ||||
| 37 | Q_VSA_PNEG | Total polar negative van der Waals surface area | −0.038 | 0.039 | ||
| 38 | Q_VSA_POL | Total polar van der Waals surface area | −0.104 | −0.052 | ||
| 39 | Q_VSA_POS | Total positive van der Waals surface area | −0.071 | |||
| 40 | Q_VSA_PPOS | Total polar positive van der Waals surface area | −0.099 | −0.188 | −0.200 | |
| 41 | Kier1 | Kappa shape index | −0,039 | |||
| 42 | Kier2 | Kappa shape index | −0.043 | |||
| 43 | Kier3 | Kappa shape index | −0.018 | |||
| 44 | KierA1 | Alpha modified shape index | −0.054 | |||
| 45 | KierA2 | Alpha modified shape index | ||||
| 46 | KierA3 | Alpha modified shape index | ||||
| 47 | KierFlex | Flexibility index | −0.018 | −0.052 | 0.007 | |
| 48 | apol | Atomic polarizabilities | ||||
| 49 | bpol | Atomic polarizabilities | ||||
| 50 | mr | Molecular refractivity | ||||
| 51 | a_acc | Number of hydrogen bond acceptors | −0.051 | 0.116 | 0.044 | 0.058 |
| 52 | a_acid | Number of acidic atoms | −0.008 | −0.074 | ||
| 53 | a_base | Number of basic atoms | ||||
| 54 | a_don | Number of hydrogen bond donors | −0.109 | −0.356 | −0.140 | −0.197 |
| 55 | a_hyd | Number of hydrophobic atoms | ||||
| 56 | vsa_acc | van der Waals surface areas of hydrogen bond acceptors | −0.044 | −0.006 | ||
| 57 | vsa_acid | van der Waals surface areas of acidic atoms | −0.008 | −0.074 | ||
| 58 | vsa_base | van der Waals surface areas of basic atoms | ||||
| 59 | vsa_don | van der Waals surface areas of hydrogen bond donors | −0.105 | −0.171 | −0.140 | −0.174 |
| 60 | vsa_hyd | van der Waals surface areas of hydrophobic atoms | 0.025 | |||
| 61 | vsa_other | van der Waals Surface Areas of other atoms | −0.058 | −0.217 | −0.210 | |
| 62 | vsa_pol | van der Waals surface areas of polar atoms | ||||
| 63 | SlogP | Log of the octanol/water partition coefficient | 0.109 | 0.008 | 0.100 | 0.116 |
| 64 | SMR | Molecular refractivity | ||||
| 65 | TPSA | Total polar surface area | −0.080 | −0.052 | −0.050 | −0.096 |
| 66 | density | Molecular mass density | −0.049 | |||
| 67 | vdw_area | van der Waals surface area | −0.029 | |||
| 68 | vdw_vol | van der Waals volume | ||||
| 69 | logP(o/w) | Log of the octanol/water partition coefficient | 0.074 | 0.009 | 0.084 | 0.085 |
A PLS model was calculated including compounds 5, 8, 10–17, 19–20, 22–24, 26–27, 33–35, 39–48, 51 and 53–54 using all descriptors and log Kd for the PapGII adhesin as the response. After variable selection leaving 34 molecular descriptors as predictor variables (X), the model explained 78% (R2Y = 0.78) of the total variation in the response data (Y) and was able to predict 68% (Q2 = 0.68) of the response variation according to cross-validation. The predictive properties of the model were further validated using an independent test set which included three carbohydrates from collection I (6, 7, 9), three from collection II (18, 21, 23), one from collection III (25), three from collection IV (36–38) and one C2′ substituted compound 50 (Fig. 2a). The model was able to predict the affinity of the test set compounds in an excellent way with a root mean square prediction error (RMSEP) of 0.49. Only one compound (23) was poorly predicted by the model. It was predicted to have a rather low affinity with a Kd of 350 µM, while the experimentally determined value was 150 µM.
 | ||
| Fig. 2 Calculated response values for galabiosides (■) using the two different QSAR versus the experimental values for a) the binding affinity to E. coli adhesin PapG type II expressed as −log Kd (R2Y = 0.78, Q2 = 0.68) and b) the inhibition of S. suis adhesin type PO expressed as −log IC50 (R2Y = 0.89, Q2 = 0.75). Both models were validated with an independent set of diverse galabiosides (◊); for chemical structures see Table 1. | ||
To further evaluate two of the positions that were varied, C1 and C3′, two local PLS-models were created using compounds 5–27 and 45–47 for the anomeric position and 33–44 and 53–54 for the C3′ position. The number of substances with variation in the C2′ position were too few (48, 50–51) to be able to create a representative model. Variables not related to the response were removed by means of filtering. Two models with 20 and 23 important factors were retrieved for the anomeric and the C3′ position, respectively (see Table 3). For the anomeric position it could be further verified that aromatic substituents are important for affinity to the PapGII adhesin since regression coefficients for variables describing aromaticity and lipophilicity (a_aro, b_ar, SlogP and logP(o/w)) were positively correlated with the response. Coefficients related to flexibility (KierFlex, b_rotR) were negatively correlated indicating that groups with a high degree of freedom are unfavourable for binding (cf.9 and 46). In addition, it could be seen that the presence of either hydrogen bond donors or acceptors on the anomeric substituent was strongly correlated with the affinity. This could be seen in the poor affinity for inhibitors containing an amide functionality with hydrogen bond donation capacities adjacent to C1 (collection I) in comparison with the relatively high affinity for inhibitors with only hydrogen bond accepting properties at the same position (collection II and III). For the C3′ position it could be confirmed that replacing the ether functionality with a hydrogen bond donating amide is detrimental to binding ability since the presence of hydrogen bond acceptors was positively correlated and the presence of donors was negatively correlated with the response. Furthermore, positively correlated coefficients for flexibility indicate that a higher degree of freedom might be necessary in order to achieve the correct positioning of the substituent.
A schematic summary of the structure–activity relationships for the PapGII adhesin is shown in Fig. 3a. The shallow pocket formed by Arg170, Trp107, and Asp108, which is seen in the crystal structure of PapGII together with a tetrasaccharide,19 could explain the increase in affinity provided by C1 substituents with low flexibility. The preference for aromatic groups in the same pocket could derive from Π-stacking or cation-Π interactions from Trp107 and Arg170. The hydrogen bonding properties seen at both O1 and O3′ indicate the presence of important hydrogen bonds from Lys172 to O3′, as seen in the crystal structure, and from either residue Arg170 or Trp107 to the neighbouring O1. Flexibility in inhibitors is not normally beneficial for entropic reasons and the prediction by the model that the inhibitors should have flexible substituents at C3′ probably reflects that the geometric requirements on rigid substituents are higher, as rigid substituents are less adaptable to the steric requirements of the protein binding site. Presumably, a rigid substituent properly designed to sterically match the binding site of the PapGII adhesin would improve the affinity.
 | ||
| Fig. 3 a) Graphic summary of the structure–activity relationship of galabioses 5, 8, 10–17, 19–20, 22–24, 26–27, 33–35, 39–48, 51 and 53–54 in binding to the PapGII adhesin. Amino acids shown are in the proximity of the galabiose substituents according to the crystal structure of the adhesin–globotetraose complex.19 b) Graphic summary of the structure–activity relationship of galabiose inhibitors 5–9, 11, 14–17, 19, 21–26, 34–36, 38–48, 50–53, 56 and 59 of the S. suis PO adhesin. | ||
Relating the aforementioned molecular descriptors for compounds 5–9, 11, 14–17, 19, 21–26, 34–36, 38–48, 50–53, 56 and 59 to IC50 values (from the refined measurements in Table 2 when applicable) for S. suis adhesins PO and PN by using PLS gave, after variable selection, a prediction model for inhibition of the adhesin type PO with 38 variables describing 89% of the total variation in the response and Q2 = 0.75. The predictive properties of the model were further validated using an independent test set, which included one compound from collection I (10), four from collection II (12–13, 18, 20), one from collection III (27), two from collection IV (33 and 37), one C2′ substituted compound (49) and two known galabiosides (54 and 55), Fig. 2b. The prediction of the test set gave good results, with the exception of compound 33, with an RMSEP of 0.66. The galabioside 33, however, is the only basic amine in the collection tested, which could explain the prediction difficulties for this compound. Excluding that object from the test set gave excellent prediction results, RMSEP = 0.44. No significant model for the adhesin type PN could be retrieved, maybe due to insufficient variation in the response. The galabiosides 57–58 and 60–64 were excluded, since the long and flexible side-chains in both of the positions varied made them difficult to model with the 2D descriptors that were used.
Local PLS models on galabiosides 5–27, 45–47 and 33–44, 52–56 for the anomeric and C3′ positions, respectively, resulted in two models with 22 important variables. The model for the anomeric position could further verify that substituents with low flexibility and high lipophilicity such as aromatic rings are beneficial for affinity. Both hydrogen bond accepting and donating capabilities are negatively correlated to the response, donors to a larger extent. The negatively correlated term for hydrogen bond donors is probably related to the C1 linkage position since good activity can be observed in all cases when donor substituents are present elsewhere (16, 23). The opposite can be said of the negatively correlated term for hydrogen bond acceptors where it seems that acceptors on positions other than the C1 linkage are unfavourable, as can be seen in the galabiosides with methoxy substituted aromatic rings (e.g.11, 12). The C3′ position model clarifies the importance of having the ether linkage intact. Amides with hydrogen bond donor coefficients are strongly negatively correlated, whereas hydrogen bond acceptor terms are positively correlated with affinity (cf.52–55 with collection IV). A schematic summary of the structure–activity relationships is shown in Fig. 3b. This proposes that a good inhibitor of the S. suis adhesin type PO should be a galabioside with large aromatic and highly lipophilic aglycons, e.g. naphthyl galabiosides. In addition, a highly flexible group should be attached to a hydrogen bond acceptor at C3′. However, the prediction that a flexible group at C3′ is beneficial could be misleading, as discussed for the PapGII adhesin above, and a rigid substituent properly designed to sterically match the binding site of the PO adhesin could improve the affinity.
Two PLS models were obtained with the ability to predict the affinity of new galabiosides for the E. coli adhesin PapGII and the S. suis adhesin type PO in an excellent fashion. In addition, local models for each position varied and provided quantitative structure–activity relationships for both adhesins. These relationships may be used to optimise the substituents further and constitute a base for future designed libraries where all positions will be varied at the same time in order to reveal interaction effects between the different substituents.
Conclusions
Four collections of galabiosides derivatised at C1 and C3′ have been synthesised aiming at enhanced affinity, compared to the parent galabioside disaccharide, for the E. coli class II PapG adhesin and the type PN and type PO adhesins from S. suis. The present study clearly shows that the E. coli class II PapG adhesin recognizes m- or p-methoxyphenyl galabiosides 11 and 45 with high affinity and specificity. Although various approaches towards galabiose derivatives carrying aromatic structures at C1 were evaluated (i.e. replacing the anomeric oxygen with an amido group or a sulfur atom or modifying the aromatic structure) no improvements in affinity for the adhesin were revealed. Furthermore, structural modifications at C3′ did not provide any significant affinity enhancements for the PapGII adhesin. Hence, the m- or p-methoxyphenyl galabiosides 11 and 45 constitute lead structures in future work towards agents targeting urinary tract infections. Remarkably potent inhibitors were discovered against one of the S. suis adhesins (type PN, IC50 down to 30 nM), which provide an ideal starting point for the further development of mono-, as well as multivalent, inhibitors of this adhesin. The PapGII preferentially bound to substituted phenyl galabiosides, while the PO preferentially bound p-methoxyphenyl galabiosides substituted with hydrogen bond accepting alkyl groups at O2′ or phenyl ureas at C3′. PLS models could accurately predict affinity-enhancing effects of substituents in binding to the E. coli PapGII and S. suis type PO adhesins, which is of value for the design of further improved inhibitors.Experimental
Synthesis
| δH(300 MHz; CD3OD) | m/z (FAB) (M+ + Na) required/found | |
|---|---|---|
| a Virtual long-range couplings. | ||
| 5 | 7.91 (m, 2H, Ar), 7.58–7.45 (m, 3H, Ar), 5.13 (d, 1H, J 8.9, H-1), 5.03 (d, 1H, J 3.6, H-1′), 4.31 (t, 1H, J 6.2, H-5′) | 468.1482/468.1482 |
| 6 | 7.89 (m, 2H, Ar), 7.00 (m, 2H, Ar), 5.11 (d, 1H, J 8.8, H-1), 5.03 (d, 1H, J 3.4, H-1′), 4.31 (t, 1H, J 6.8, H-5′) | 498.1587/498.1583 |
| 7 | 7.55 (m, 1H, Ar), 5.08 (ma, 1H, H-1), 5.02 (d, 1H, J 3.7, H-1′), 4.23 (t, 1H, J 6.0, H-5′) | 540.1105/540.1107 |
| 8 | 7.53 (m, 2H, Ar), 7.18 (m, 1H, Ar), 5.10 (d, 1H, J 8.9, H-1), 5.03 (d, 1H, J 3.8, H-1′), 4.29 (t, 1H, J 6.8, H-5′) | 504.1293/504.1300 |
| 9 | 5.01 (d, 1H, J 3.8, H-1′), 4.88 (d, 1H, J 8.3, H-1), 4.26 (t, 1H, J 5.6, H-5′), 2.29 (m, 2H, CH2), 1.14 (t, 3H, J 7.6, CH3) | 420.1481/420.1501 |
| 10 | 8.01 (m, 1H, Ar), 6.67 (m, 2H, Ar), 5.12 (ma, 1H, H-1), 5.02 (d, 1H, J 2.6, H-1′), 4.34 (t, 1H, J 6.7, H-5′) | 528.1693/528.1697 |
| 11 | 7.17 (m, 1H, Ar), 6.69 (m, 2H, Ar), 6.60 (m, 1H, Ar), 5.02 (ma, H-1′), 4.93 (d, 1H, J 7.4, H-1), 4.33 (t, 1H, J 5.9, H-5′) | 471.1478/471.1471 |
| 12 | 7.16 (m, 1H, Ar), 7.02 (m, 2H, Ar), 6.90 (m, 1H, Ar), 5.02 (d, J 1.7, H-1′), 4.93 (d, 1H, J 7.6, H-1), 4.34 (t, 1H, J 6.4, H-5′) | 471.1478/471.1482 |
| 13 | 7.29 (m, 2H, Ar), 7.10 (m, 2H, Ar), 7.02 (m, 1H, Ar), 5.01 (ma, 1H, H-1′), 4.96 (d, 1H, J 7.4, H-1), 4.33 (t, 1H, J 5.8, H-5′) | 441.1373/441.1359 |
| 14 | 7.08 (m, 2H, Ar), 6.99 (m, 2H, Ar), 5.01 (ma, 1H, H-1′), 4.90 (d, 1H, J 7.5, H-1), 4.32 (t, 1H, J 5.8, H-5′) | 455.1529/455.1520 |
| 15 | 7.78 (m, 3H, Ar), 7.49–7.28 (m, 4H, Ar), 5.12 (d, 1H, J 7.5, H-1), 5.03a (m, 1H, H-1′), 4.34 (t, 1H, J 5.6, H-5′) | 491.1529/491.1525 |
| 16 | 7.34–7.21 (m, 3H, Ar), 6.97 (m, 1H, Ar), 6.37 (m, 1H, Ar), 5.02 (d, 1H, J 2.8, H-1′), 4.87 (d, 1H, J 7.5, H-1), 4.36 (t, 1H, J 6.6, H-5′) | 480.1482/480.1490 |
| 17 | 7.45–7.26 (5, 2H, Ar), 4.97 (d, 1H, J 1.7, H-1′), 4.91 (AB, 1H, J 11.7, CH2), 4.69 (AB, 1H, J 11.7, CH2), 4.40 (ma, 1H, H-1), 4.31 (t, 1H, J 6.5, H-5′) | 455.1529/455.1526 |
| 18 | 4.97 (ma, 1H, H-1′), 4.40 (d, 1H, J 7.5, H-1), 4.34 (t, 1H, J 6.6, H-5′), 1.96 (m, 2H, CH2), 1.77 (m, 2H, CH2), 1.55 (m, 1H, CH2), 1.47–1.20 (m, 5H, CH2) | 447.1842/447.1849 |
| 19 | 5.01 (d, 1H, J 2.8, H-1′), 4.93 (d, 1H, J 7.5, H-1), 4.29 (t, 1H, J 6.4, H-5′) | 531.0901/531.0901 |
| 20 | 7.97 (m, 2H, Ar), 7.16 (m, 2H, Ar), 5.06 (d, 1H, J 7.5, H-1), 5.01 (d, 1H, J 3.0, H-1′), 4.31 (t, 1H, J 5.9, H-5′) | 499.1428/499.1432 |
| 21 | 7.78 (m, 1H, Ar), 7.54 (m, 1H, Ar), 7.37 (m, 1H, Ar), 7.14 (m, 1H, Ar), 5.01 (d, 1H, J 1.8, H-1′), 4.92 (d, 1H, J 7.5, H-1), 4.32 (t, 1H, J 6.6, H-5′) | 499.1428/499.1426 |
| 22 | 8.22 (m, 2H, Ar), 7.25 (m, 2H, Ar), 5.11 (d, 1H, J 7.5, H-1), 5.01 (d, 1H, J 3.4, H-1′), 4.30 (t, 1H, J 6.1, H-5′) | 486.1224/486.1222 |
| 23 | 7.45 (m, 2H, Ar), 7.06 (m, 2H, Ar), 5.01 (d, 1H, J 1.8, H-1′), 4.90 (d, 1H, J 7.5, H-1), 4.32 (t, 1H, J 5.9, H-5′) | 498.1587/498.1584 |
| 24 | 7.55 (m, 2H, Ar), 6.90 (m, 2H, Ar), 4.87 (d, 1H, J 3.8, H-1′), 4.38 (d, 1H, J 9.3, H-1) | 487.1250/487/1251 |
| 25 | 7.59 (m, 2H, Ar), 7.30 (m, 3H, Ar), 4.93 (d, 1H, J 3.8, H-1′), 4.58 (d, 1H, J 9.0, H-1) | 457.1144/457.1137 |
| 26 | 7.49 (m, 2H, Ar), 7.15 (m, 2H, Ar), 4.90 (d, 1H, J 3.8, H-1′), 4.49 (d, 1H, J 9.2, H-1) | 471.1301/471.1294 |
| 27 | 7.91 (m, 1H, Ar), 7.74 (m, 1H, Ar), 7.59 (m, 1H, Ar), 7.42 (m, 1H, Ar), 4.95 (d, 1H, J 2.9, H-1′), 4.89 (d, 1H, J 9.6, H-1), 4.11 (t, 1H, J 6.5) | 515.1199/515.1208 |
| 33 | 5.06 (d, 1H, J 3.6, H-1′), 4.85 (d, 1H, J 7.1, H-1), 4.43 (t, 1H, J 6.3, H-5′), 3.53 (dd, 1H, J 1.7, 10.8, H-3′) | 470.1638/470.1642 |
| 34 | 5.03 (d, 1H, J 3.8, H-1′), 4.83 (d, 1H, J 7.6, H-1), 4.44 (t, 1H, J 6.5, H-5′), 4.23 (dd, 1H, J 2.9, 11.4, H-3′), 2.02 (s, 3H, NHAc) | 512.1744/512.1744 |
| 35 | 5.08 (d, 1H, J 3.8, H-1′), 4.86 (d, 1H, J 7.6, H-1), 4.44 (t, 1H, J 6.1, H-5′), 4.23 (dd, 1H, J 2.9, 11.4, H-3), 2.29 (q, 2H, J 7.7, CH2), 1.15 (t, 3H, J 7.6, CH3) | 526.1900/526.1893 |
| 36 | 5.04 (d, 1H, J 3.7, H-1′), 4.82 (d, 1H, J 7.6, H-1), 4.43 (t, 1H, J 6.8, H-5′), 4.23 (dd, 1H, J 2.9, 11.4, H-3), 2.60 (m, 4H, J 7.7, CH2) | 570.1799/570.1786 |
| 37 | 7.89 (m, 2H, Ar–H), 7.50 (m, 3H, Ar–H), 5.10 (d, 1H, J 3.7, H-1′), 4.84 (d, 1H, J 7.7, H-1), 4.49 (m, 2H, H-3′, H-5′) | 574.1900/574.1895 |
| 38 | 7.86 (m, 2H, Ar–H), 6.98 (m, 2H, Ar–H), 5.09 (d, 1H, J 3.7, H-1′), 4.84 (d, 1H, J 7.6, H-1), 4.51 (t, 1H, J 5.8, H-5′), 4.45 (dd, 1H, J 2.9, 11.4, H-3′), 3.85 (s, 3H, OMe) | 604.2006/604.2021 |
| 39 | 7.06 (m, 2H, Ar–H), 6.64 (m, 1H, Ar–H), 5.09 (d, 1H, J 3.7, H-1′), 4.84 (d, 1H, J 7.6, H-1), 4.52 (t, 1H, J 6.1, H-5′), 4.44 (dd, 1H, J 2.9, 11.4, H-3′) | 634.2112/634.2101 |
| 40 | 7.54 (m, 2H, Ar–H), 7.15 (m, 1H, Ar–H), 5.09 (d, 1H, J 3.8, H-1′), 4.84 (d, 1H, J 7.6, H-1), 4.52 (t, 1H, J 5.9, H-5′), 4.43 (dd, 1H, J 2.9, 11.4, H-3′) | 610.1712/610.1699 |
| 41 | 8.78 (m, 1H, Ar–H), 8.40 (m, 1H, Ar–H), 8.28 (m, 1H, Ar–H), 7.73 (m, 1H, Ar–H), 5.10 (d, 1H, J 3.8, H-1′), 4.84 (d, 1H, J 7.7, H-1), 4.53 (t, 1H, J 6.7, H-5′), 4.49 (dd, 1H, J 2.9, 11.4, H-3′) | 619.1751/619.1774 |
| 42 | 7.89 (m, 1H, Ar–H), 7.52 (m, 3H, Ar–H), 5.06 (d, 1H, J 3.7, H-1′), 4.83 (d, 1H, J 7.6, H-1), 4.50 (t, 1H, J 6.1, H-5′), 4.38 (dd, 1H, J 2.8, 11.3, H-3′) | 618.1799/618.1791 |
| 43 | 5.02 (d, 1H, J 3.7, H-1′), 4.81 (d, 1H, J 7.5, H-1), 4.40 (t, 1H, J 5.7, H-5′), 4.10 (dd, 1H, J 3.0, 11.2, H-3′), 3.15 (q, 2H, J 7.2, CH2), 1.09 (t, 3H, J 7.2, CH2CH3) | 541.2009/541.2011 |
| 44 | 7.35 (m, 2H, Ar–H), 7.23 (m, 2H, Ar–H), 6.95 (m, 1H, Ar–H), 5.06 (d, 1H, J 3.8, H-1′), 4.83 (d, 1H, J 7.6, H-1), 4.45 (t, 1H, J 5.6, H-5′), 4.49 (dd, 1H, J 3.0, 11.2, H-3′) | 589.2009/589.1995 |
Surface plasmon resonance experiments
Surface plasmon resonance studies of binding of compounds 5–46 to the class II PapG adhesin were performed as described earlier.21 The binding data were analysed using the software Scrubber.28Hemagglutination inhibition experiments
Bacteria were grown overnight at 37 °C at 5% CO2-incubator. The bacteria were harvested by centrifugation, 5000 × g, 15 min, +4 °C, and washed twice with PBS. The hemagglutination activities were titrated and the lowest bacterial densities causing agglutination were used for the inhibition studies. Twofold dilutions of the inhibitors were tested and the results were observed after incubation on ice for two hours. A first screen was done once with strain 628 (type PN) and strain 836 (type PO). The hemagglutination type was verified by using galactose and N-acetylgalactosamine as inhibitors. Ten strong inhibitors and eight poor inhibitors were selected for refined evaluation in triplicate. The results are presented as the average of three individual determinations and the range of the inhibitory values is shown.Computational methods
The two prediction models were validated using an independent test set. The local PLS models were validated using a permutation test where the order of the response (Y) was randomly permutated 30 times. By plotting the explanatory power (R2) and the predictive power (Q2) of the mutated models as a function of the correlation coefficient between the original and predicted values, the degree to which these values rely on chance is reflected by the intercept with the y-axis. A model is generally considered valid if the intercept is negative for Q2 and below 0.3 for R2.34
Acknowledgements
This work was supported by grants from the Swedish Research Council, from the program “Glycoconjugates in Biological Systems” sponsored by the Swedish Foundation for Strategic Research, and from the Finnish Academy.References
- V. Morell, Science, 1997, 278, 575–576.
- R. J. Williams and D. L. Heymann, Science, 1998, 279, 1153–1154 CrossRef CAS.
- C. Walsh, Nature, 2000, 406, 775–781 CrossRef CAS.
- D. Mirelman and I. Ofek, Microbial Lectins and Agglutinins, ed. D. Mirelman, Wiley, New York, 1986, p. 1–19 Search PubMed.
- K.-A. Karlsson, Curr. Opin. Struct. Biol., 1995, 5, 622–635 CrossRef CAS.
- H. Leffler and C. Svanborg-Edén, FEMS Lett., 1980, 8, 127–134 Search PubMed.
- G. Källenius, R. Möllby, S. B. Svensson, J. Winberg, A. Lundblad, S. Svensson and B. Cedergren, FEMS Lett., 1980, 7, 297–302 Search PubMed.
- K. Bock, M. E. Breimer, A. Brignole, G. C. Hansson, K.-A. Karlsson, G. Larsson, H. Leffler, B. E. Samuelsson, N. Strömberg, C. Svanborg-Edén and J. Thurin, J. Biol. Chem., 1985, 260, 8545–8551 CAS.
- N. Strömberg, B.-I. Marklund, B. Lund, D. Ilver, A. Hamers, W. Gaastra, K.-A. Karlsson and S. Normark, EMBO J., 1990, 9, 2001–2010 CAS.
- N. Strömberg, P.-G. Nyholm, I. Pascher and S. Normark, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9340–9344 CAS.
- I. M. Johansson, K. Plos, B.-I. Marklund and C. Svanborg, Microb. Pathog., 1993, 15, 121–129 CrossRef.
- G. Otto, T. Sandberg, B.-I. Marklund, P. Ulleryd and C. Svanborg, Clin. Infect. Dis., 1993, 17, 448–456 CAS.
- J. R. Johnson, T. A. Ruso, J. J. Brown and A. Stapleton, J. Infect. Dis., 1998, 177, 97–101 CrossRef CAS.
- M. Hedlund, M. Svensson, A. Nilsson, R. D. Duan and C. Svanborg, J. Exp. Med., 1996, 1843, 1037–1044 CrossRef.
- M. Hedlund, C. Wachtler, E. Johansson, L. Hang, J. E. Somerville, R. P. Darveeau and C. Svanborg, Mol. Microbiol., 1999, 33, 693–703 CrossRef CAS.
- S. Haataja, K. Tikkanen, U. Nilsson, G. Magnusson, K.-A. Karlsson and J. Finne, J. Biol. Chem., 1994, 269, 27466–27472 CAS.
- R. Striker, U. Nilsson, A. Stonecipher, G. Magnusson and S. J. Hultgren, Mol. Microbiol., 1995, 16, 1021–1029 CrossRef CAS.
- J. Ohlsson, J. Jass, B. E. Uhlin, J. Kihlberg and U. J. Nilsson, ChemBioChem, 2002, 3, 772–779 CrossRef CAS.
- K. W. Dodson, J. S. Pinkner, T. Rose, G. Magnusson, S. J. Hultgren and G. Waksman, Cell, 2001, 105, 733–743 CrossRef CAS.
- (a) H. C. Hansen, S. Haataja, J. Finne and G. Magnusson, J. Am. Chem. Soc., 1997, 119, 6974–6979 CrossRef CAS; (b) during the preparation of this paper dendritic galabiose compounds with low nanomolar affinity for Streptococcus suis adhesins were reported: J. A. F. Joosten, V. Loimaranta, C. C. M. Appeldoorn, S. Haataja, F. A. E. Maate, R. M. J. Liskamp, J. Finne and R. J. Pieters, J. Med. Chem., 2004, 47, 6499–6508 Search PubMed.
- A. Larsson, J. Ohlsson, K. W. Dodson, S. J. Hultgren, U. J. Nilsson and J. Kihlberg, Bioorg. Med. Chem., 2003, 11, 2255–2261 CrossRef CAS.
- J. Ohlsson and G. Magnusson, Carbohydr. Res., 2000, 329, 49–55 CrossRef CAS.
- E. D. Soli and P. DeShong, J. Org. Chem., 1999, 64, 9724–9726 CrossRef CAS.
- C. Peto, G. Batta, Z. Gyorgydeak and F. Sztariskai, Liebigs Ann. Chem., 1991, 505–507 CrossRef CAS.
- P. Sörme, Y. Qian, P.-G. Nyholm, H. Leffler and U. J. Nilsson, ChemBioChem, 2002, 3, 183–189 CrossRef CAS.
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS.
- G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS.
- Scrubber 1.1f, 2000, BioLogic Software Pty Ltd, Unit 30 116 Blamey Crescent, Campell, ACT 2612, Australia.
- J. Ohlsson, A. Sundin and U. J. Nilsson, Chem. Commun., 2003, 384–385 RSC.
- S. Haataja, K. Tikkanen, J. Liukkonen, C. François-Gerard and J. Finne, J. Biol. Chem., 1993, 268, 4311–4317 CAS.
- MOE 2003.02, 2003, Chemical Computing Group Inc., 1010 Sherbrooke St. West Suite 910 Montreal, Canada H3A 2R7.
- S. Wold, in Chemometric methods in molecular design, ed. H. van de Waterbeemd, Wiley, Weinheim, 1995, p. 195–218 Search PubMed.
- Simca-P 10.02′, 2002, Umetrics, Box 7960, S-907 19 Umeå, Sweden.
- L. Eriksson, E. Johansson and S. Wold, in Quantitative Structure-Activity Relationships in Environmental Sciences-VII, ed. F. Chen and G. Schüürmann, 1997, SETAC Press, Pensacola, FL, p. 381-397 Search PubMed.
- J. Ohlsson and U. J. Nilsson, Tetrahedron Lett., 2003, 44, 2785–2787 CrossRef CAS.
Footnote |
| † Jörgen Ohlsson, Andreas Larsson and Sauli Haataja contributed equally. |
| This journal is © The Royal Society of Chemistry 2005 |