Thermotropic liquid–crystalline peptide derivatives: oligo(glutamic acid)s forming hydrogen-bonded columns
Masayuki
Nishii
,
Toru
Matsuoka
,
Yuko
Kamikawa
and
Takashi
Kato
*
Department of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kato@chiral.t.u-tokyo.ac.jp; Fax: (+81) 3-5841-8661
First published on 2nd February 2005
Abstract
We report on new thermotropic liquid–crystalline oligo(amino acid) derivatives forming columnar structures. These are based on branched oligo(glutamic acid)s and 2-(3,4-dialkyloxyphenyl)ethyl moieties. An oligo(glutamic acid) derivative, α,γ-bis(L-glutamoyl) L-glutamic acid tetra[2-(3,4-dioctadecyloxyphenyl)ethyl]ester, shows a hexagonal columnar phase, whilst a glutamic acid derivative, α,γ-bis[2-(3,4-dioctadecyloxyphenyl)ethyl] L-glutamate, does not show a mesophase. Hydrogen bonds formed by the oligo(glutamic acid) moieties should contribute to the induction of the columnar liquid–crystalline properties. In addition, we have examined the effects of the molecular chirality of the oligo(glutamic acid) parts and the functionalisation at the focal position of the taper shaped molecules on the liquid–crystalline properties of the compounds.
Introduction
Intensive work has focused on molecular self-assembled materials that can be used for nanoscience and advanced technologies.1,2 Amino acids have been used as building blocks for self-assembled materials such as liquid crystals,3–8 fibres,9 micelles10 and tubes.11 For thermotropic liquid crystals, poly(amino acids)s such as poly(γ-benzyl L-glutamate)5 and poly(γ-alkyl L-glutamate)s6 have been shown to exhibit liquid–crystalline properties. Liquid–crystalline peptides bearing mesogens in the side chains were also reported to show mesophases.7 However, examples of liquid crystals in which the amino acid parts play critical roles for mesomorphism are rare, whilst a large number of sugar derivatives have shown thermotropic and lyotropic liquid–crystalline properties.12Our intention is to explore new liquid crystals composed of amino acids, forming supramolecular structures. We previously developed liquid–crystalline folic acid derivatives containing oligo(glutamic acid) moieties.3,4 These folic acid derivatives exhibit thermotropic smectic, columnar and cubic phases. For these materials, the glutamic acid moieties play a critical role in the self-assembly and the induction of supramolecular chirality. Glutamic acid has two carboxylic acids and one amino group. We consider that this molecular structure can function as a new useful component for the molecular design of liquid crystals.
A number of dendritic and taper shaped liquid crystals having no rod-like mesogens have been designed and synthesised,3,4,13 whilst a large number of dendrimers14 and the dendrimers having rod-like mesogens15 have been reported. Tschierske et al.13a–c showed gluconamide derivatives with di- or trialkyloxybenzoyl groups exhibit columnar and cubic phases. Percec and coworkers13d,e developed a series of aromatic taper shaped dendrons having alkyl chains that exhibit thermotropic liquid–crystalline properties. Recently, Stupp et al.13f have reported liquid crystals with rod-dendron block molecular structures of biphenyl-based segments and polyether dendrons. The liquid–crystalline properties of these materials are induced by nanoscale segregation and specific molecular interactions as well as by specific molecular shape.1d,e,2–4,13,15
Here we report on new thermotropic liquid–crystalline compounds consisting of oligo(amino acid)s as shown in Fig. 1. We focus on glutamic acid as a building block for thermotropic liquid crystals. We have designed taper shaped molecular structures based on glutamic acid. Oligo(glutamic acid) derivatives 1–4 were prepared and their liquid–crystalline properties were examined. Compounds 1–4 are made up of two molecular components, the oligo(glutamic acid) parts capable of hydrogen bonding and the lipophilic alkyl chains. Compounds 4 have a benzoyl group (4a) and alkanoyl groups (4b, 4c) attached to the inner amino groups.
 | ||
| Fig. 1 Structures of oligo(glutamic acid) derivatives. | ||
Results and discussion
Synthesis of oligo(glutamic acid) derivatives 1–4
Glutamic acid derivatives 1 and oligo(glutamic acid) derivatives 2–4 were synthesised according to previously reported methods .3,4N-Fmoc-L-glutamic acid was condensed with 2-(3,4-dialkyloxyphenyl)ethanol. The deprotection of the Fmoc groups by piperidine gave α,γ-bis[2-(3,4-dialkyloxyphenyl)ethyl] L-glutamates 1a–c. These procedures were repeated for 1a–c to obtain α,γ-bis(L-glutamoyl) L-glutamic acid tetra[2-(3,4-dialkyloxyphenyl)ethyl]esters 2a–c. Oligo(glutamic acid) 3 was synthesised from N-Fmoc-D-glutamic acid and 1a. Compound 2b was further reacted with benzoyl chloride, decanoic acid and nonadecanoic acid to obtain 4a–c, respectively. The thermal properties and liquid–crystalline behaviour of 1–4 were examined by differential scanning calorimetry (DSC), polarising optical microscopy and X-ray diffraction measurements.Thermotropic liquid–crystalline behaviour of oligo(glutamic acid) derivatives 1–4
The thermal properties of glutamic acid derivatives 1 and oligo(glutamic acid) derivatives 2–4 are listed in Table 1. For a series of glutamic acid derivatives 1a–c, only 1b shows liquid–crystalline behaviour. All of the oligo(glutamic acid) derivatives of 2a–c however, show thermotropic hexagonal columnar phases. For 2a, having hexyloxy chains, a columnar phase is seen from 51 to 69 °C. In contrast, 1a is an isotropic liquid at room temperature. For 1a, the transition to a glassy state is observed below room temperature upon cooling. Compound 1b, having undecyloxy chains, shows a hexagonal columnar phase at around room temperature, whilst oligo(glutamic acid) derivative 2b exhibits a columnar phase from 74 to 88 °C. The isotropisation temperature of 2b is 51 °C higher than that of 1b. Fig. 2 shows the DSC thermograms of 1c and 2c on the second heating. Compound 1c melts from the crystalline state to the isotropic liquid state at 65 °C. For 2c, the endothermic peaks corresponding to crystalline–columnar and columnar–isotropic transitions are seen at 71 and 78 °C, respectively. On cooling from the isotropic liquid state, fan-like textures characteristic of the columnar phases are observed for 2c under a polarising optical microscope, as shown in Fig. 3.| X-Ray results | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | Phase transition behavioura | T/°C | Intercolumnar distances/Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Transition temperatures (°C) and enthalpy changes (kJ mol−1 in parentheses) were determined by DSC on the second heating. G: glassy; Cr: crystalline; Colh: hexagonal columnar; Iso: isotropic. b A monotropic hexagonal columnar phase is seen from 45 to 34 °C. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1a | G | −49 | Iso | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1b | Cr | 26 (63) | Colh | 37 (2.8) | Iso | 30 | 46.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1c | Cr | 65 (119) | Iso | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2a | Cr | 51 (26) | Colh | 69 (4.5) | Iso | 60 | 37.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2b | Cr | 74 (97) | Colh | 88 (26) | Iso | 80 | 42.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2c | Cr | 71 (172) | Colh | 78 (26) | Iso | 75 | 50.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Cr1 | 75 (16) | Cr2 | 90 (62) | Iso | 40b | 37.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4a | Cr | 91 (129) | Iso | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4b | Cr | 78 (68) | Colh | 88 (16) | Iso | 80 | 36.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4c | Cr | 71 (20) | Colh | 83 (27) | Iso | 80 | 37.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 | ||
| Fig. 2 DSC thermograms of 1c (a) and 2c (b) on the second heating. | ||
 | ||
| Fig. 3 A polarised optical photomicrograph of 2c at 75 °C (Colh). | ||
The hierarchical tuning of molecular chirality is possible for the oligo(glutamic acid) derivatives. Oligo(glutamic acid) derivative 3, the diastereomer of 2a, was prepared. Compound 3 shows a monotropic liquid–crystalline columnar phase, whilst the enantiotropic columnar phase is seen for 2a. Upon heating, compound 3 melts to the isotropic liquid at 90 °C. The melting temperature of 3 is 39 °C higher than that of 2a. Upon cooling, 2a shows the columnar phase from 37 to −25 °C. For 3, the monotropic hexagonal columnar phase is seen from 45 to 34 °C. The isotropic to columnar transition temperature of 3 is 8 °C higher than that of 2a. The change of the molecular chirality at the inner glutamic acid parts for 2a and 3 (Fig. 1) induces the stabilisation of the crystalline structures for 3.
The oligo(glutamic acid) derivatives of 2 can be further functionalised by introducing various functional moieties at the free amino groups. In the present study, a benzoyl group (4a) and alkanoyl moieties (4b,4c) have been introduced. The functionalisation of the oligo(glutamic acid)s at the focal position has affected their liquid–crystalline properties. Compound 4a melts at 91 °C to the isotropic liquid state. The introduction of the benzoyl group to 2b results in the disappearance of the liquid crystallinity. Compounds 4b and 4c, bearing flexible alkyl chains at the core, show thermotropic columnar phases, as in the case of 2b, and the thermal stability of the columnar phases of 4b and 4c is almost the same as that of 2b.
The FT–IR spectra for the oligo(glutamic acid) derivatives were measured to examine the formation of the hydrogen bonds, as shown in Fig. 4 and Table 2. The curve fitting was performed for the IR bands of the amide groups (Amide I) of 2b upon heating. The absorption band of the carbonyl groups of the oligo(glutamic acid) parts is seen around 1642 cm−1 in the crystalline state at 60 °C, which indicates that they form hydrogen bonds. After the transition to the columnar liquid–crystalline phase, the intensities of the absorption bands at 1678 and 1655 cm−1 increase (80 °C, Fig. 4). The absorption band centred at 1678 cm−1 is ascribed to the free amide groups. The band at 1655 cm−1 is due to the formation of weaker hydrogen bonds. In the columnar liquid–crystalline state at 80 °C, about 80% of the amide groups form hydrogen bonds (Table 2). In the isotropic state at 100 °C, the IR band around 1640 cm−1 is not seen. More than half of the amide groups do not form hydrogen bonds in the isotropic state. The formation of the hydrogen bonds at the oligo(glutamic acid) moieties should contribute to the induction of the columnar liquid–crystalline properties. Hydrogen bonding has been known to play a critical role in the induction of liquid–crystalline phases.2a,b,16 However, the compounds reported here are a new series of hydrogen-bonded liquid crystals.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands of
O bands of 
The X-ray diffraction profile for the columnar phase of oligo(glutamic acid) derivative 2b is shown in Fig. 5. In the wide angle region, the diffused halo of around 4.5 Å is due to the disorder of the terminal alkyl chains. In the small angle region, the sharp reflection of 37.1 Å (100) and the smaller peaks of 21.6 Å (110), 18.4 Å (200) and 13.9 Å (210) are characteristic of the hexagonal structure. The intercolumnar distance is 42.8 Å. We suggest that the columnar structures of 2b are composed of disk-like aggregates of the compounds. A molecular model of a dimer of 2b arranged side by side is shown in Fig. 6. The size of the molecular model agrees with the X-ray results. Nanoscale segregation between the hydrogen-bonded oligo(glutamic acid) parts and lipophilic alkyl chains leads to the formation of the self-assembled columnar aggregates. The diameters of the columns for the columnar liquid–crystalline phases of 1–4 are listed in Table 1. The increase in the number of amino acids in compound 1b results in a decrease in the intercolumnar spacing, from 46.8 Å for 1b to 42.8 Å for 2b. The diameters of the columns of 4b and 4c are 36.7 and 37.7 Å, respectively, which are shorter than that of 2b. One possible explanation is that the introduction of the bulky groups at the focal point disturbs a dimerisation in the column, which leads to the decrease of the column diameter. However the FT–IR spectra of 2b and 4b show that hydrogen-bonded properties of these compounds are almost the same, which may result in the similar liquid–crystalline behaviour. The column diameters increase with increasing length of the terminal alkyl chains for 2a–c.
 | ||
| Fig. 5 X-Ray diffraction profile of 2b at 80 °C. | ||
 | ||
| Fig. 6 Molecular model of a dimer of 2b. | ||
We reported on the self-assembled behaviour of folic acid derivatives 54 and 63 containing the oligo(glutamic acid)s of 1b and 2b, respectively (Fig. 7). The folic acid derivatives of 5 and 6 show thermally stable liquid–crystalline properties through the combination of the cyclic hydrogen bonds of the pterin rings and the amide hydrogen bonds of the oligo(glutamic acid) parts. For example, the isotropisation temperature of the liquid–crystalline phase of folic acid 6 is 99 °C higher than that of the oligo(glutamic acid) of 2b. The oligo(glutamic acid) derivatives shown in the present study are considered to have potential as the building blocks for various molecular self-assembled materials.3,4,17
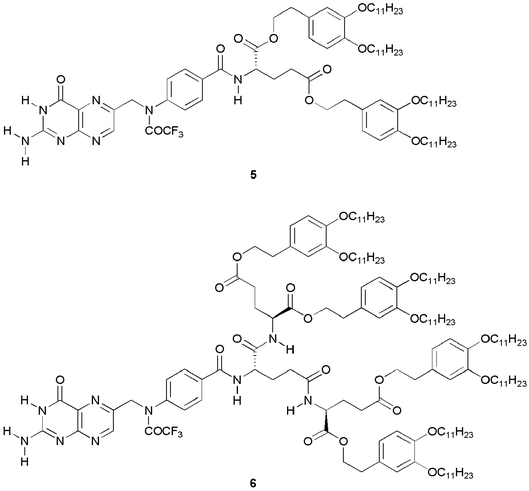 | ||
| Fig. 7 Structures of folic acid derivatives.3,4 | ||
Conclusion
The oligo(glutamic acid)s of 1–4 are the first examples of liquid crystals in which the components of amino acids substantially function to induce thermotropic liquid–crystalline properties. These taper shaped molecules show thermotropic columnar phases through the formation of hydrogen bonds and nanoscale segregation. The liquid–crystalline properties and self-assembled structures of these materials have been tuned by the molecular structures of the compounds, such as the number of amino acids, chirality and focal functionality. Such liquid crystals based on amino acids may be useful for new functional liquid–crystalline materials in bio-related fields.Experimental
General methods and materials
All reagents and solvents were purchased from Aldrich Chemical, Tokyo Kasei, and Wako Pure Chemicals and were used as received. Analytical thin layer chromatography (TLC) was performed on silica gel plates from E. Merck (silica gel F254). Silica gel column chromatography was carried out with silica gel 60 from Kanto Chemicals (silica gel 60, spherical 40–50 µm). Elemental analyses were carried out on a Perkin-Elmer CHNS/O 2400 apparatus. 1H and 13C NMR spectra were recorded on a JEOL JNM-LA400. Chemical shifts of 1H and 13C NMR signals are expressed in parts per million (δ) using internal standards Me4Si (δ = 0.00) and CDCl3 (δ = 77.00), respectively. Mass spectra (MALDI) were recorded on a PerSeptive Biosystems Voyager-DE STR spectrometer. Synthesis and spectroscopic data of compounds 1–3 have been reported in our previous papers.3,4DSC measurements were performed on a Mettler DSC 30 (scanning rate: 5 °C min−1). Transition temperatures were taken at the maximum of transition peaks. A polarising optical microscope Olympus BH-2 equipped with a Mettler FP82HT hot stage was used for visual observation. IR measurements were conducted on a JASCO FT/IR-660 Plus on KBr plates. X-Ray diffraction measurements were carried out on a Rigaku RINT 2100 diffractometer with a heating stage using Ni-filtered CuKα radiation.
Synthesis
Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research on Priority Areas of “Dynamic Control of Strongly Correlated Soft Materials” (No. 413/13031009) and a Grant-in-Aid for The 21st Century COE Program for Frontiers in Fundamental Chemistry from the Ministry of Education, Culture, Sports, Science and Technology. Partial financial support of the Sasakawa Scientific Research Grant from the Japan Science Society (MN) is also acknowledged.References
- (a) Molecular Self-Assembly, Organic Versus Inorganic Approaches , ed. M. Fujita, Springer, Berlin, 2000, Structure & Bonding, vol. 96 Search PubMed; (b) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS; (c) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS; (d) T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS; (e) G. ten Brinke and O. Ikkala, Chem. Rec., 2004, 4, 219–230 Search PubMed; (f) G. Gottarelli and G. P. Spada, Chem. Rec., 2004, 4, 39–49 Search PubMed.
- (a) T. Kato, Struct. Bonding, 2000, 96, 95–146 CAS; (b) T. Kato, N. Mizoshita and K. Kanie, Macromol. Rapid Commun., 2001, 22, 797–814 CrossRef CAS; (c) C. Tschierske, J. Mater. Chem., 2001, 11, 2647–2671 RSC.
- (a) T. Kato, T. Matsuoka, M. Nishii, Y. Kamikawa, K. Kanie, T. Nishimura, E. Yashima and S. Ujiie, Angew. Chem., Int. Ed., 2004, 43, 1969–1972 CrossRef CAS; (b) Y. Kamikawa, M. Nishii and T. Kato, Chem. Eur. J., 2004, 10, 5942–5951 CrossRef CAS.
- (a) K. Kanie, T. Yasuda, S. Ujiie and T. Kato, Chem. Commun., 2000, 1899–1900 RSC; (b) K. Kanie, M. Nishii, T. Yasuda, T. Taki, S. Ujiie and T. Kato, J. Mater. Chem., 2001, 11, 2875–2886 RSC; (c) K. Kanie, T. Yasuda, M. Nishii, S. Ujiie and T. Kato, Chem. Lett., 2001, 480–481 CrossRef CAS; (d) T. Kato and N. Mizoshita, Curr. Opin. Solid State Mater. Sci., 2002, 6, 579–587 CrossRef CAS.
- S. M. Yu, V. P. Conticello, G. Zhang, C. Kayser, M. J. Fournier, T. L. Mason and D. A. Tirrell, Nature, 1997, 389, 167–170 CrossRef CAS.
- (a) J. Watanabe, Y. Fukuda, R. Gehani and I. Uematsu, Macromolecules, 1984, 17, 1004–1009 CrossRef CAS; (b) J. Watanabe, H. Ono, I. Uematsu and A. Abe, Macromolecules, 1985, 18, 2141–2148 CrossRef CAS; (c) J. Watanabe and Y. Takashina, Macromolecules, 1991, 24, 3423–3426 CrossRef CAS.
- (a) P. A. G. Cormack, B. D. Moore and D. C. Sherrington, Chem. Commun., 1996, 353–354 RSC; (b) C. Guillermain and B. Gallot, Macromol. Chem. Phys., 2002, 203, 1346–1356 CrossRef CAS.
- W. Zhou, W. Gu, Y. Xu, C. S. Pecinovsky and D. L. Gin, Langmuir, 2003, 19, 6346–6348 CrossRef CAS.
- (a) A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS; (b) K. Hanabusa, J. Tange, Y. Taguchi, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1993, 390–392 RSC; (c) M. Suzuki, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Chem. Eur. J., 2003, 9, 348–354 CrossRef CAS; (d) N. Mizoshita, Y. Suzuki, K. Kishimoto, K. Hanabusa and T. Kato, J. Mater. Chem., 2002, 12, 2197–2201 RSC; (e) N. Yamada, K. Ariga, M. Naito, K. Matsubara and E. Koyama, J. Am. Chem. Soc., 1998, 120, 12192–12199 CrossRef CAS; (f) T. Niwa, H. Yokoi, T. Kinoshita and S. Zhang, Polym. J., 2004, 36, 665–673 CAS.
- (a) H.-A. Klok, J. J. Hwang, J. D. Hartgerink and S. I. Stupp, Macromolecules, 2002, 35, 6101–6111 CrossRef CAS; (b) R. C. Claussen, B. M. Rabatic and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 12680–12681 CrossRef CAS.
- (a) M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS; (b) N. Nakashima, S. Asakuma and T. Kunitake, J. Am. Chem. Soc., 1985, 107, 509–510 CrossRef CAS; (c) N. Nakashima, S. Asakuma, J.-M. Kim and T. Kunitake, Chem. Lett., 1984, 1709–1712 CAS; (d) T. Kunitake, Angew. Chem., Int. Ed. Engl., 1992, 31, 709–726 CrossRef; (e) T. Shimizu, Polym. J., 2003, 35, 1–22 CAS; (f) N. Sakai and S. Matile, Chem. Commun., 2003, 2514–2523 RSC.
- (a) C. R. Noller and W. C. Rockwell, J. Am. Chem. Soc., 1938, 60, 2076–2077 CrossRef CAS; (b) J. W. Goodby, Mol. Cryst. Liq. Cryst., 1984, 110, 205–219 CrossRef CAS; (c) G. A. Jeffrey, Acc. Chem. Res., 1986, 19, 168–173 CrossRef CAS; (d) F. Dumoulin, D. Lafont, P. Boullanger, G. Mackenzie, G. H. Mehl and J. W. Goodby, J. Am. Chem. Soc., 2002, 124, 13737–13748 CrossRef CAS; (e) V. Vill and R. Hashim, Curr. Opin. Colloid Interface Sci., 2002, 7, 395–409 Search PubMed.
- (a) K. Borisch, S. Diele, P. Göring and C. Tschierske, Chem. Commun., 1996, 237–238 RSC; (b) K. Borisch, S. Diele, P. Göring, H. Kresse and C. Tschierske, J. Mater. Chem., 1998, 8, 529–543 RSC; (c) C. Tschierske, J. Mater. Chem., 1998, 8, 1485–1508 RSC; (d) V. Percec, W.-D. Cho and G. Ungar, J. Am. Chem. Soc., 2000, 122, 10273–10281 CrossRef CAS; (e) V. Percec, W.-D. Cho, G. Ungar and D. J. P. Yeardley, J. Am. Chem. Soc., 2001, 123, 1302–1315 CrossRef CAS; (f) S. Lecommandoux, H.-A. Klok, M. Sayar and S. I. Stupp, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3501–3518 CrossRef CAS; (g) J. H. Cameron, A. Facher, G. Lattermann and S. Diele, Adv. Mater., 1997, 9, 398–403 CrossRef CAS; (h) M. Yoshio, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2004, 126, 994–995 CrossRef CAS; (i) J. H. K. Ky Hirschberg, R. A. Koevoets, R. P. Sijbesma and E. W. Meijer, Chem. Eur. J., 2003, 9, 4222–4231 CrossRef; (j) Y. Takaguchi, T. Tajima, Y. Yanagimoto, S. Tsuboi, K. Ohta, J. Motoyoshiya and H. Aoyama, Org. Lett., 2003, 5, 1677–1679 CrossRef CAS.
- (a) J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3713–3725 CrossRef CAS; (b) D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- (a) B. Dardel, D. Guillon, B. Heinrich and R. Deschenaux, J. Mater. Chem., 2001, 11, 2814–2831 RSC; (b) D. Guillon and R. Deschenaux, Curr. Opin. Solid State Mater. Sci., 2002, 6, 515–525 CrossRef CAS; (c) J.-M. Rueff, J. Barberá, B. Donnio, D. Guillon, M. Marcos and J.-L. Serrano, Macromolecules, 2003, 36, 8368–8375 CrossRef CAS; (d) I. M. Saez and J. W. Goodby, Chem. Eur. J., 2003, 9, 4869–4877 CrossRef CAS; (e) P. H. J. Kouwer and G. H. Mehl, Angew. Chem., Int. Ed., 2003, 42, 6015–6018 CrossRef CAS; (f) M. W. P. L. Baars, S. H. M. Söntjens, H. M. Fischer, H. W. I. Peerlings and E. W. Meijer, Chem. Eur. J., 1998, 4, 2456–2466 CrossRef CAS.
- (a) T. Kato and J. M. J. Fréchet, J. Am. Chem. Soc., 1989, 111, 8533–8534 CrossRef CAS; (b) T. Kato, M. Nakano, T. Moteki, T. Uryu and S. Ujiie, Macromolecules, 1995, 28, 8875–8876 CrossRef CAS; (c) H. Kihara, T. Kato, T. Uryu and J. M. J. Fréchet, Chem. Mater., 1996, 8, 961–968 CrossRef CAS.
- Y. Kamikawa, T. Kato, H. Onouchi, D. Kashiwagi, K. Maeda and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4580–4586 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |