Antioxidant activity of olive phenols: mechanistic investigation and characterization of oxidation products by mass spectrometry
Marjolaine
Roche
,
Claire
Dufour
*,
Nathalie
Mora
and
Olivier
Dangles
UMR A 408 INRA – University of Avignon, Safety and Quality of Plant Products, Site Agroparc, 84914 Avignon cedex 9, France. E-mail: cdufour@avignon.inra.fr; Fax: +33 4 32 72 24 92; Tel: +33 4 32 72 25 15
First published on 21st December 2004
Abstract
In this work, the antioxidant activity of olive phenols is first characterized by their stoichiometries ntot (number of radicals trapped per antioxidant molecule) and their rate constants for the first H-atom abstraction k1 by the stable radical DPPH. It appears that oleuropein, hydroxytyrosol and caffeic acid have the largest k1 values, whereas dihydrocaffeic acid, an intestinal metabolite of caffeic acid, is the best antioxidant in terms of ntot. For phenols with a catechol moiety ntot is higher than two, implying an antioxidant effect of their primarily formed oxidation products. A HPLC–MS analysis of the main products formed in the AAPH-induced oxidation of olive phenols reveals the presence of dimers and trimers. With hydroxytyrosol and dihydrocaffeic acid, oligomerization can take place with the addition of water molecules.
The antioxidant activity of olive phenols is then evaluated by their ability to inhibit the AAPH-induced peroxidation of linoleic acid in SDS micelles. It is shown that olive phenols and quercetin act as retardants rather than chain breakers like α-tocopherol. From a detailed mechanistic investigation, it appears that the inhibition of lipid peroxidation by olive phenols can be satisfactorily interpreted by assuming that they essentially reduce the AAPH-derived initiating radicals. Overall, olive phenols prove to be efficient scavengers of hydrophilic peroxyl radicals with a long lasting antioxidant effect owing to the residual activity of some of their oxidation products.
Introduction
Olive phenols belong to the broad class of naturally occurring phenolic antioxidants that may have beneficial effects on human health via a diet rich in plant products such as the mediterranean diet.1,2 These health effects mainly exert themselves through the prevention of degenerative pathologies such as cardiovascular diseases and cancers.3,4 Although low molecular weight phenols are found in virgin olive oil, a high percentage is discarded in olive mill wastewaters (OMW), a by-product of the extraction process of olive oil.5 Most OMW are accumulated over a short period (typically November–December) then spread over fields or dumped into rivers, thus causing pollution of soils and aquatic area. Olive phenols are partially responsible for this pollution due to their toxicity to plants, bacteria and aquatic organisms.6–8 Hence, chemical and biotechnological processes (e.g., Fenton reaction or oxidation catalyzed by fungal enzymes) have been developed to degrade OMW phenols.9,10 An interesting alternative could be the extraction of phenols from OMW and their valorization as antioxidants in the food and pharmaceutical industries.The antioxidant efficiency of olive phenols has been assessed in various tests such as the inhibition of low density lipoprotein oxidation.11 In addition, bioavailability studies have shown that olive phenols can be absorbed from the intestine and enter the blood circulation as conjugates.12,13 The aim of this study is to obtain a deeper insight in the mechanism of their antioxidant action by combining three distinct approaches: a quantitative DPPH radical scavenging test, a partial elucidation of the oxidation products formed upon peroxyl radical scavenging and a detailed analysis of the inhibition by olive phenols of lipid peroxidation in SDS micelles.
Results and discussion
The olive phenols selected for this study are: oleuropein, hydroxytyrosol, tyrosol, caffeic acid and p-coumaric acid (Fig. 1).14 Dihydrocaffeic and ferulic acids are incorporated because they are typical caffeic acid metabolites in the intestine and plasma respectively.15,16 Chlorogenic acid, the main dietary source of caffeic acid, the flavonol quercetin, α-tocopherol, ascorbic acid and n-hexadecylcaffeate, a chemically synthesized amphiphilic analog of caffeic acid, are also investigated for comparison. | ||
| Fig. 1 Chemical structures of compounds used as antioxidants. | ||
Scavenging of the DPPH radical in methanol
DPPH is a stable nitrogen-centered free radical. A quantitative analysis of the H-atom transfer reaction from a given phenol to DPPH provides a very simple and efficient way to characterize the phenol by a set of parameters (i.e., rate constants and stoichiometries) tightly related to its intrinsic antioxidant activity.17 The H-transfer reactions are monitored by UV/VIS spectroscopy by recording the decay of the DPPH visible absorption band (λmax = 515 nm in MeOH) that reflects the conversion of the DPPH radical into the corresponding colorless hydrazine (DPPH–H) by the antioxidant. The experiments are run at a DPPH–antioxidant molar ratio of four in order to exhaust the H-donating ability of the antioxidant. With potent antioxidants, the visible absorbance quickly decays over 1–3 min as a result of the transfer of the most labile H-atoms of the antioxidant (fast step, monitored over 250 s, Fig. 2). This step may be followed by a much slower decrease of the visible absorbance featuring the residual H-donating ability of the antioxidant degradation products (slow step) as already observed with dihydrocaffeic acid.18 Only the fast step is kinetically analyzed. Experiments extending over 10 min were used for the determination of the total stoichiometry ntot of the antioxidant, according to: ntot = (A0 − Af)/(εC) (Af: final absorbance, A0: initial absorbance, C: initial antioxidant concentration) (Table 1).| Antioxidant | Δt/s![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
k/M−1 s−1 | n | k 1 = k × n/M−1 s−1 | n tot at 600 s |
|---|---|---|---|---|---|
| a Tyrosol and p-coumaric acid: very slow reaction with DPPH. Values are means (SD) n = 6. b Data interval used for the calculation of k and n. | |||||
| Hydroxytyrosol | 60 | 491 (15) | 2.24 (0.04) | 1100 | 2.48 (0.04) |
| Oleuropein | 100 | 478 (10) | 1.97 (0.02) | 942 | 2.09 (0.07) |
| Caffeic acid | 50 | 414 (20) | 1.71 (0.10) | 708 | 2.29 (0.06) |
| Dihydrocaffeic acid | 100 | 97 (2) | 2.35 (0.07) | 228 | 3.01 (0.11) |
| Chlorogenic acid | 300 | 96 (7) | 2.06 (0.06) | 198 | 2.06 (0.12) |
| Ferulic acid | 150 | 153 (7) | 0.99 (0.02) | 151 | 1.36 (0.01) |
 | ||
| Fig. 2 Decay of the visible absorbance at 515 nm of a 0.2 mM DPPH solution in MeOH following addition of a phenol (final concentration = 50 µM). ■ = ferulic acid, △ = chlorogenic acid, ▲ = dihydrocaffeic acid, □ = caffeic acid, ◆ = oleuropein, ◇ = hydroxytyrosol. | ||
The general kinetic model used for analyzing the H-atom transfer reaction between DPPH and a given antioxidant during the fast step (50–300 s) makes no hypothesis about the mechanism of antioxidant degradation. An antioxidant of stoichiometry n is simply regarded as n independent antioxidant subunits (AH) which all transfer a single H-atom to DPPH with the same second-order rate constant k. Hence, the curve fitting of the absorbance vs time plots can be carried out using simple second-order kinetics, the initial AH concentration being set at nC.17 Moreover, rate constant k can be identified with k1/n, k1 being the rate constant for the first (most labile) H-atom abstraction from the antioxidant.
Ortho-diphenols noted AH2 (caffeic acid, dihydrocaffeic acid, chlorogenic acid, oleuropein and hydroxytyrosol) typically give n values close to two (Table 1) in agreement with the stepwise formation of semiquinone radicals and quinones during the fast step: AH2 + 2DPPH → A + 2DPPH–H. Monophenols noted AH (e.g., ferulic acid) must be primarily converted into dimers upon recombination of the corresponding aryloxyl radicals. In this case, the partial stoichiometry is close to 1 : 2AH + 2DPPH → A2 + 2DPPH–H.
The antioxidants can be ranked according to their k1 value (Table 1): hydroxytyrosol > oleuropein > caffeic acid ≫ dihydrocaffeic acid > chlorogenic acid > ferulic acid. The ortho-diphenols caffeic acid, oleuropein and hydroxytyrosol are strong hydrogen donors with k1 values ranging from 700 to 1100 M−1 s−1 in agreement with the formation of semiquinone radicals that are strongly stabilized by a combination of electronic and intramolecular H-bond effects. However, ortho-diphenols react more slowly with DPPH than the flavonol quercetin.19 Comparing caffeic acid and chlorogenic acid on the one hand and oleuropein and hydroxytyrosol on the other hand, it can be concluded that the quinic acid moiety of chlorogenic acid hampers DPPH scavenging whereas the elenolic acid moiety of oleuropein does not. The k1 value of caffeic acid is higher by a factor of ca. three than that of dihydrocaffeic acid, and is likely to be a consequence of a larger electron delocalization and/or stronger intramolecular H-bond in the caffeoyl radical (H-atom abstraction from the 4′-OH group). These differences are correctly reflected in the values of the phenolic bond dissociation energies (BDE) deduced from semi-empirical quantum mechanic calculations after optimization of hydrogen bonding in both the parent phenol and the corresponding aryloxyl radical (Table 2).
| Phenol | Hydrogen bond | E, BDEa/kcal−1 mol−1 |
|---|---|---|
| a BDE (bond dissociation energy) = E(phenol) − E(aryloxyl radical). | ||
| Caffeic acid | O4–H⋯O3–H | −2335.72 |
| Caffeoyl radical | O4–H⋯O3˙ | −2263.15, 72.6a |
| Caffeic acid | O3–H⋯O4–H | −2335.72 |
| Caffeoyl radical | O3–H⋯O4˙ | −2264.96, 70.8a |
| Ferulic acid | O4–H⋯O3–Me | −2603.56 |
| Feruloyl radical | — | −2530.49, 73.1a |
| Dihydrocaffeic acid | O4–H⋯O3–H | −2463.15 |
| Dihydrocaffeoyl radical | O4–H⋯O3˙ | −2391.47, 71.7a |
| Dihydrocaffeic acid | O3–H⋯O4–H | −2463.41 |
| Dihydrocaffeoyl radical | O3–H⋯O4˙ | −2391.93, 71.5a |
| p-Coumaric acid | — | −2232.50 |
| p-Coumaroyl radical | — | −2157.32, 75.2a |
The total stoichiometry ntot provides a second opportunity to compare antioxidants. Ranking according to decreasing ntot values gives: dihydrocaffeic acid > hydroxytyrosol > caffeic acid > oleuropein = chlorogenic acid > ferulic acid. With ntot values higher than two, dihydrocaffeic acid, hydroxytyrosol and caffeic acid can be proposed to form o-quinones that evolve towards products endowed with an additional H-atom donating activity. This residual activity seems to be quenched by steric hindrance with chlorogenic acid and oleuropein whose ntot value is only two. With a ntot value higher than one, ferulic acid probably forms dimers that are still able to transfer labile H-atoms to DPPH.20 The rankings of the antioxidants according to both k1 and ntot highlight the singular behavior of dihydrocaffeic acid, which, although reacting with DPPH more slowly than the other o-diphenols, emerges as the best antioxidant in terms of the number of radicals trapped.
Product characterization in the AAPH-induced oxidation of olive phenols
The thermal decomposition of the hydrophilic diazo compound AAPH (2,2′-azo-bis(2-amidinopropane) dihydrochloride, noted R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R) in the presence of dioxygen delivers peroxyl radicals (ROO˙) at a constant rate. This is a common way to apply an oxidative stress in aqueous solutions containing biological targets for antioxidant testing. The ROO˙ radical is a strong electron/H-atom abstracting agent and is expected to rapidly react with the olive phenols to form oxidation products that must be quite similar to those formed in the DPPH test, with the additional advantage of more biologically relevant conditions. In addition, product characterization in the AAPH-induced oxidation of olive phenols should be an important point for the interpretation of the antioxidant effects observed in AAPH-induced lipid peroxidation (see below). The oxidation reactions are conducted in a pH 7.4 phosphate buffer at 37 °C and analyzed by HPLC–MS (Table 3). The experiments were especially conclusive for caffeic acid, hydroxytyrosol, dihydrocaffeic acid, ferulic acid and p-coumaric acid, with each of these antioxidants showing a strong propensity for oligomerization and covalent dimers and trimers were systematically detected in agreement with literature.21–23 According to their fragmentation, two types of caffeic acid dimers can be proposed, one having unbreakable C–C linkages (‘C–C dimers’, e.g., biphenyl type) and the other having breakable C–O linkages (‘C–O dimers’, e.g., biaryl ether type). Indeed, in addition to the fragmentation pattern common to both types of dimers (decarboxylation), the C–O dimers give monomeric fragments whereas the C–C dimers do not. Moreover, the C–O dimers display one less OH group than the C–C dimers and are consistently eluted later on the C18 silica chromatography column. Remarkably, in the case of o-diphenols with unconjugated chains (hydroxytyrosol and dihydrocaffeic acid), dimerization can occur with incorporation of a water molecule. Hence, it can be proposed that o-quinones derived from hydroxytyrosol and dihydrocaffeic acid, or more probably tautomeric p-quinone methides, undergo water addition before oxidative coupling with a second o-diphenol molecule (Scheme 1). For comparison, oxidation of caffeic acid was also carried out by potassium nitrosodisulfonate, a one-electron oxidant, and sodium periodate, a two-electron oxidant that is expected to directly yield the o-quinone (data no shown).24 No significant differences in product distribution could be observed in agreement with a dimerization primarily occurring via addition of a caffeic acid molecule onto the corresponding o-quinone.
N–R) in the presence of dioxygen delivers peroxyl radicals (ROO˙) at a constant rate. This is a common way to apply an oxidative stress in aqueous solutions containing biological targets for antioxidant testing. The ROO˙ radical is a strong electron/H-atom abstracting agent and is expected to rapidly react with the olive phenols to form oxidation products that must be quite similar to those formed in the DPPH test, with the additional advantage of more biologically relevant conditions. In addition, product characterization in the AAPH-induced oxidation of olive phenols should be an important point for the interpretation of the antioxidant effects observed in AAPH-induced lipid peroxidation (see below). The oxidation reactions are conducted in a pH 7.4 phosphate buffer at 37 °C and analyzed by HPLC–MS (Table 3). The experiments were especially conclusive for caffeic acid, hydroxytyrosol, dihydrocaffeic acid, ferulic acid and p-coumaric acid, with each of these antioxidants showing a strong propensity for oligomerization and covalent dimers and trimers were systematically detected in agreement with literature.21–23 According to their fragmentation, two types of caffeic acid dimers can be proposed, one having unbreakable C–C linkages (‘C–C dimers’, e.g., biphenyl type) and the other having breakable C–O linkages (‘C–O dimers’, e.g., biaryl ether type). Indeed, in addition to the fragmentation pattern common to both types of dimers (decarboxylation), the C–O dimers give monomeric fragments whereas the C–C dimers do not. Moreover, the C–O dimers display one less OH group than the C–C dimers and are consistently eluted later on the C18 silica chromatography column. Remarkably, in the case of o-diphenols with unconjugated chains (hydroxytyrosol and dihydrocaffeic acid), dimerization can occur with incorporation of a water molecule. Hence, it can be proposed that o-quinones derived from hydroxytyrosol and dihydrocaffeic acid, or more probably tautomeric p-quinone methides, undergo water addition before oxidative coupling with a second o-diphenol molecule (Scheme 1). For comparison, oxidation of caffeic acid was also carried out by potassium nitrosodisulfonate, a one-electron oxidant, and sodium periodate, a two-electron oxidant that is expected to directly yield the o-quinone (data no shown).24 No significant differences in product distribution could be observed in agreement with a dimerization primarily occurring via addition of a caffeic acid molecule onto the corresponding o-quinone.
| Phenol, na | Retention time/min | λmax/nm | m/z | Proposed structure for product |
|---|---|---|---|---|
| a Stoichiometry for peroxyl radical scavenging using Ra = Ri/n. A n value of 2.8 was estimated for oleuropein. | ||||
| Caffeic acid, 2.2 | 8.4 | 295, 326 | 179, 135 | AH2 |
| 9.6 | 324 | 313, 269, 147, 121 | (AH)2, C–C type | |
| 11.1, 11.9, 12.2, 12.8 | 290, 320 | 313, 269, 179, 177, 135 | (AH)2, C–O type | |
| 9.3, 10.8 | 320 | 489, 445 | (2AH + A) − 2H | |
| Hydroxytyrosol, 3.6 | 7.3 | 272, 442 | 303, 267, 239, 219, 183 | (AH)2 − 2H |
| 8.2 | 280 | 153, 123 | AH2 | |
| 8.3 | 272, 414 | 319, 289, 241 | (AH)2 + H2O − 4H | |
| 8.7 | 268, 388 | 167, 149, 137 | A + H2O − 2H | |
| 16.5 | 322, 380 | 455, 303, 273 | (2AH + A) − 2H | |
| Dihydrocaffeic acid, 8.2 | 6.9 | 282, 392 | 589, 475, 361, 209 | (2AH + A) + 3H2O − 6H |
| 7.6 | 266, 390 | 375, 331, 287, 269, 259, 151, 123 | (AH)2 + H2O − 4H | |
| 7.9 | 282 | 181, 137, 121, 109 | AH2 | |
| 8.3 | 294, 486 | 495, 179, 135 | (2AH + A) − 2H | |
| 8.5 | 295, 324 | 361, 179, 135 | (AH)2 | |
| 10.3 | 280, 325, 416 | 495, 315 | (2AH + A) − 2H | |
| Ferulic acid, 1.7 | 10.3 | 295, 324 | 193, 149, 135 | AH |
| 12.8 | 336 | 385, 341, 297, 283,173, 159, 123 | 2AH − 2H | |
| 13.6 | 326 | 385, 341, 297, 283, 173, 159, 123 | 2AH − 2H | |
| 15 | 290, 336 | 577, 533, 489, 445 | 3AH − 4H | |
| 18.2 | 295, 322 | 489, 339, 295, 193 | 3AH − 4H | |
| p-Coumaric acid | 9.2 | 298, 310 | 163, 119 | AH |
| 10.7 | 304 | 325, 281, 237, 219 | 2AH − 2H | |
| 11.4 | 320 | 281, 237, 143, 93 | 2AH − 2H | |
| 11.9 | 302 | 487, 443 | 3AH − 4H | |
| 12.9 | 298, 316 | 325, 281, 237 | 2AH − 2H | |
| 18.6 | 298, 315 | 443, 399, 355, 279, 235 | 3AH − 4H | |
 | ||
| Scheme 1 Proposed mechanism for hydroxytyrosol oxidation (R = CH2OH). | ||
The rate of antioxidant consumption Ra (determined by HPLC) allows an estimate of the stoichiometry (n) of peroxyl radical scavenging by using the following relationship: Ra = Ri/n, Ri being the constant flow of AAPH-derived peroxyl radicals ROO˙. Ri is expressed as 2ekd(AAPH), where kd is the dissociation rate constant of the diazo compound and e the molar fraction of AAPH-derived peroxyl radicals that escape recombination in the solvent cage and become available for reduction by the antioxidant. The relationship Ra = Ri/n assumes a steady-state for ROO˙, with each ROO˙ generated reacting with the antioxidant. It does not hold for poorly reactive antioxidants (e.g., tyrosol or p-coumaric acid) for which recombination of peroxyl radicals into non-radical products cannot be neglected. On the other hand, Ri can also be estimated from the lag phase (T) of α-tocopherol-inhibited peroxidation of linoleic acid (see below): Ri = 2(α-toc)/T (assuming a stoichiometry of two for α-tocopherol). With an AAPH concentration of 1 mM typically used in the peroxidation experiments, the Ri value is roughly 1.3 × 10−9 M s−1. Hence, in the oxidation experiments (typical AAPH concentration = 50 mM), a Ri value of 6.5 × 10−8 M s−1 can be used. The antioxidant stoichiometries n thus calculated are reported in Table 3. The order of decreasing antioxidant stoichiometry is close to that deduced from the DPPH test (only, the rankings of oleuropein and caffeic acid are exchanged): dihydrocaffeic acid >> hydroxytyrosol > oleuropein > caffeic acid > ferulic acid. It confirms that the antioxidants with a catechol nucleus substituted by a saturated carbon chain experience the most extensive oxidation, thereby delivering a large number of H atoms to the radicals (DPPH, ROO˙).
The antioxidant stoichiometry for peroxyl radical scavenging can be interpreted in more detail by using the information deduced from product analysis by HPLC–MS. For example, starting with o-diphenol AH2, dimerization to form (AH)2 requires the scavenging of one peroxyl radical per AH2 molecule (n = 1). Additional scavenging of two peroxyl radicals to form oxidized dimers A2 raises the stoichiometry to two. This seems to be the main fate of caffeic acid in its reaction with ROO˙. With o-diphenols having a saturated carbon chain (hydroxytyrosol, dihydrocaffeic acid), dimerization can also take place with water incorporation to form (AH)2 + H2O − 2H, with a stoichiometry of two. With such antioxidants, it can be speculated that o-quinone–p-quinone methide tautomerism allows water addition on the exo-cyclic carbon atom of the latter tautomer (Scheme 1). Interestingly, oxidation can proceed further to yield (AH)2 + H2O − 4H dimers with an overall stoichiometry of three. Oxidized dimers and trimers typically display UV/VIS absorption bands with λmax > 380 nm, in agreement with the presence of p-quinone methide chromophores (Table 3).
Inhibition of AAPH-induced linoleic acid peroxidation in SDS micelles
This popular test is aimed at comparing the ability of the olive oil antioxidants to scavenge peroxyl radicals derived from AAPH (ROO˙, inhibition of initiation) and/or from the polyunsaturated fatty acid (LOO˙, inhibition of the propagation step of radical-chain peroxidation) (Scheme 2). Although the antioxidant hierarchy is expected to primarily reflect the intrinsic ability of the antioxidant to transfer H-atoms to peroxyl radicals, it may be critically influenced by the partitioning of the antioxidant between the aqueous phase, where the ROO˙ radicals are thought to be generated, and the micellar phase, where the LOO˙ radicals reside.25L-Ascorbic acid, quercetin and α-tocopherol, the main component of vitamin E and a typical amphiphilic chain breaking antioxidant, are included as reference compounds. The experiments are monitored by UV/VIS spectroscopy by recording the accumulation of the lipid hydroperoxides LOOH (λmax = 234 nm) in the absence of antioxidant (constant peroxidation rate Rp0) and in the presence of the antioxidant (initial concentration C, initial peroxidation rate Rp) (Fig. 3). For a first evaluation of the antioxidant capacity, the Rp/Rp0 ratio was plotted as a function of C. Hence, IC50 parameters (antioxidant concentration corresponding to 50% inhibition, i.e.Rp/Rp0 = 0.5) could be estimated (Table 4). Remarkably, oleuropein has almost the same IC50 value as α-tocopherol and quercetin, the other phenols being less efficient. However, the antioxidant hierarchy is concentration dependent. For example, at a high antioxidant concentration of 3 µM, α-tocopherol completely inhibits peroxidation over the whole period of monitoring and thus appears as the best antioxidant, followed by oleuropein = quercetin = n-hexadecylcaffeate > hydroxytyrosol (Fig. 3, A). These results are in agreement with the order determined for IC50 values. At a low, and more biologically relevant, antioxidant concentration of 0.2 µM, α-tocopherol is still the best antioxidant as far as the first period following the addition of the antioxidant (lag phase of α-tocopherol-inhibited peroxidation) is considered. However, the rapid consumption of α-tocopherol causes the peroxidation to sharply resume after the lag-phase (Fig. 3, B). In contrast, quercetin and the olive o-diphenols inhibit lipid peroxidation without a lag-phase but exert a more persistent protection, probably owing to their higher stoichiometry in peroxyl radical scavenging. At the 0.2 µM concentration, the antioxidant activity appears to be similar for all the phenolic compounds, except for n-hexadecylcaffeate which is essentially inactive. Finally, the endogenous antioxidant ascorbic acid shows a weak antioxidant effect at high concentrations and no effect at the lowest concentration.| Antioxidant | IC50/µM | AE 1 | AE 2 | r 2 |
|---|---|---|---|---|
| a NA: no applicable treatment because of an underlying pro-oxidant effect. b Mean Rp0 values of 9.0 × 10−9 and 6.5 × 10−9 used in the calculations (in M s−1) for, respectively, α-tocopherol and quercetin. Values are means (SD) from n = 3. | ||||
| α-Tocopherol | 0.18 (0.02) | 6200 | 1100b | 0.994 |
| Oleuropein | 0.49 (0.20) | 6139 (661) | — | 0.998 |
| Quercetin | 0.59 (0.11) | 7498 (689) | 9.1 (4.5)b | 0.997 |
| n-Hexadecylcaffeate | 1.09 (0.04) | 2927 (481) | 2.6 (2.0)b | 0.996 |
| Hydroxytyrosol | 2.53 (0.40) | 1619 (222) | — | 0.995 |
| Chlorogenic acid | 5.70 (1.59) | NAa | NAa | |
| Caffeic acid | 11.32 (2.96) | NAa | NAa | |
| Ascorbic acid | 13.53 (1.61) | 348 (52) | — | 0.992 |
| Dihydrocaffeic acid | 30.00 (0.00) | NAa | NAa | |
![Relative accumulation at 234 nm of hydroperoxides issued from lipid peroxidation of 2.55 mM linoleic acid in 0.1 M SDS micelles (initiated by 1 mM AAPH). (A)
[antioxidant]
= 3 µM, (B)
[antioxidant]
= 0.2 µM. △= dihydrocaffeic acid, ◆
=
l-ascorbic acid, □
= chlorogenic acid, ▲
= caffeic acid, ●
= hydroxytyrosol, ○
= quercetin, ■
= oleuropein, ◇
=
α-tocopherol,
=
n-hexadecylcaffeate, +
= no antioxidant.](/image/article/2005/OB/b416101g/b416101g-f3.gif) | ||
Fig. 3 Relative accumulation at 234 nm of hydroperoxides issued from lipid peroxidation of 2.55 mM linoleic acid in 0.1 M SDS micelles (initiated by 1 mM AAPH). (A)
[antioxidant]
= 3 µM, (B)
[antioxidant]
= 0.2 µM. △= dihydrocaffeic acid, ◆
=
L-ascorbic acid, □
= chlorogenic acid, ▲
= caffeic acid, ●
= hydroxytyrosol, ○
= quercetin, ■
= oleuropein, ◇
=
α-tocopherol, ![[Cross with vertical bar through centre]](https://www.rsc.org/images/entities/char_e113.gif) =
n-hexadecylcaffeate, +
= no antioxidant.
=
n-hexadecylcaffeate, +
= no antioxidant. | ||
 | ||
| Scheme 2 Mechanism of inhibited lipid peroxidation. | ||
Radical-chain mechanism
Assuming a steady-state for the initiating radicals ROO˙, one can write: Ri = k1(ROO˙)(LH) + ka1(ROO˙)(AH). Thus, the initiation rate R1 = k1(ROO˙)(LH) can be expressed as f1Ri, where f1 is the fraction of freely diffusing ROO˙ radicals that escape reduction by the antioxidant and initiate the peroxidation: f1 = 1/[1 + AE1(AH)/(LH)] with AE1 = ka1/k1 (antioxidant efficiency for inhibition of initiation). Assuming a steady-state for the lipid peroxyl radicals LOO˙, one gets:| k1(ROO˙)(LH) = ka2(LOO˙)(AH) + 2kt(LOO˙)2 | (1) |
The rate of lipid hydroperoxide formation is: Rp = d(LOOH)/dt = k2(LOO˙)(LH) + ka2(LOO˙)(AH). Solving eqn. 1 for (LOO˙) gives:
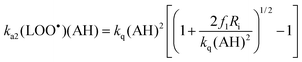 | (2) |
 | (3) |
The following parameters have been introduced: antioxidant efficiency for inhibition of propagation AE2 = ka2/k2, kq = ka22/(4kt) = (r2AE2)2/2, with r2 = k2/(2kt)1/2 (lipid oxidizability).
In the absence of antioxidant, eqn. 3 becomes eqn. 4:
| Rp0 = k2(LOO˙)(LH) = r2(LH)Ri1/2 | (4) |
Combining eqn. 2–4 readily gives eqn. 5:
 | (5) |
Parameter (2Ri/kq)1/2 = (8Rikt)1/2/ka2, which is in concentration units, can be noted CA to give eqn. 6 that is used in the curve fitting and simulation procedures (C: initial antioxidant concentration, C0: lipid concentration, n: antioxidant stoichiometry, f1 = 1/(1 + AE1nC/C0)).
 | (6) |
Moreover, parameters AE2 and CA are bound through the relationship: AE2CA = (8Rikt)1/2/k2 = 2Ri1/2/r2 = 2C0Ri/Rp0. Taking Ri = 1.3 × 10−9 M s−1 and the mean Rp0 value (typically in the range 6–10 × 10−9 M s−1) for each experiment, it becomes possible to eliminate CA from the set of adjustable parameters. Finally, the antioxidant stoichiometry is set at two.
At high antioxidant concentrations, and assuming no significant inhibition of initiation (f1 = 1, AE1 = 0), the Rp/Rp0 ratio tends to a constant non-zero value of AE2CA/(2C0) = Ri/Rp0. Indeed, even at high antioxidant concentrations, although chain propagation is totally quenched, lipid hydroperoxides still accumulate at the rate of initiation (Rp = Ri) via the reduction of the LOO˙ radicals by the antioxidant. The Rp/Rp0 lower limit can be estimated as 0.13–0.22, i.e. 13–22% of peroxidation should persist at high antioxidant concentrations if inhibition of initiation does not take place. On the other hand, if inhibition of initiation is important, f1 and consequently the Rp/Rp0 ratio should drop to zero. Simulations of the Rp/Rp0vs.C curves using eqn. 4 (Fig. 4) show that pure inhibition of initiation (AE2 = 0) leads to a relatively smooth decrease to zero of the Rp/Rp0 ratio. In the case of pure inhibition of propagation (AE1 = 0), the decay is sharper with saturation at the Ri/Rp0 limit. Clearly, inhibition of both initiation and propagation is needed to account for α-tocopherol inhibited peroxidation, whereas the more hydrophilic antioxidants hydroxytyrosol, oleuropein and quercetin seem to inhibit peroxidation essentially via scavenging of the AAPH-derived peroxyl radicals. Accordingly, the corresponding AE1 values (Table 4) can be estimated from the curve-fittings of the Rp/Rp0vs.C plots against a greatly simplified version of eqn. 6 (AE2 = 0, CA = ∞): Rp/Rp0 = √f1. Finally, except for n-hexadecylcaffeate, the behavior of hydroxycinnamic acids as peroxidation inhibitors cannot be described by eqn. 6. Indeed, after a rather sharp decrease of the Rp/Rp0 ratio for low C values, a saturation well above the Rp/Rp0 limit for a pure inhibition of propagation is observed, thus suggesting that an underlying pro-oxidant effect is operating. Thus, it may be proposed that, in this system, the aryloxyl radicals derived from the hydroxycinnamic acids are reactive enough to abstract one of the labile bis-allylic H-atoms of linoleic acid, thereby reinitiating peroxidation. This process would take place in competition with radical dimerization and/or disproportionation. Interestingly, n-hexadecylcaffeate does not display this pro-oxidant effect as if its positioning in the SDS micelles with respect to the linoleic acid molecule did not allow reinitiation to take place. Despite its amphiphilic character, n-hexadecylcaffeate seems to essentially inhibit initiation. This suggests that its polar (more likely partially anionic) head protrudes in the aqueous phase and is not available for reaction with the lipid peroxyl radicals. Overall, the o-diphenols that are typical of olive phenols, especially oleuropein, emerge as the stronger olive antioxidants in our model in agreement with the ranking provided by the DPPH scavenging test.
 | ||
| Fig. 4 Inhibition of initiation vs. inhibition of propagation in linoleic acid peroxidation. (AE1, AE2) couples used in the simulations (solid lines) are: 1 (1500, 0), 2 (5000, 0), 3 (0, 1000), 4 (1000, 1000) and 5 (5000, 1000). Mean Rp0 value used in the simulations = 7.7 × 10−9 M s−1. Experimental plots: ■ = α-tocopherol, ● = hydroxytyrosol. | ||
Olive phenols, which are found in high concentrations in olive mill waste waters, display potent antioxidant activities. It should be worth extracting them for industrial applications as naturally occurring antioxidants.
Experimental
Chemicals
Caffeic acid, chlorogenic acid, dihydrocaffeic acid, p-coumaric acid, (±) α-tocopherol, quercetin, L-ascorbic acid, 3,4-dihydroxyphenylacetic acid, DPPH, AAPH, potassium nitrosodisulfonate, linoleic acid, SDS, lithium aluminium hydride, dimethylaminopyridine (DMAP) and 1-hexadecanol were purchased from Sigma-Aldrich (L′Isle d'Abeau, France). All reagents were of the highest purity available (95–99%) and were used without further purification. Ferulic acid, p-tyrosol and oleuropein were purchased from Extrasynthèse (Genay, France). Sodium periodate was obtained from Prolabo (Paris, France). Silica gel was obtained from Merck (Darmstadt, Germany). All solvents used were analytical grade. Phosphate buffers (pH 7.4, 50 mM NaH2PO4 with and without 100 mM NaCl) were prepared with Millipore Q-Plus water and eluted on a chelating resin (Chelex 100, 0.4 mequiv. per mL, Bio-Rad) to remove contaminating metal traces.Analyses
UV/visible spectra were recorded on a Hewlett-Packard 8453 diode array spectrometer equipped with a magnetically stirred cell (optical pathlength 1 cm). The temperature in the cell was kept constant by means of a water thermostated bath. 1H- and 13C-NMR spectra were recorded on a 300 MHz Bruker Advance DPX-300 spectrometer at 27 °C. Chemical shifts (δ) are given in ppm relative to Me4Si. 1H–1H coupling constants (J) are given in Hz. High resolution mass analysis was carried out on a JEOL SX102 spectrometer. HPLC–MS analyses were carried out on a Hewlett Packard 1050 apparatus coupled to a UV/visible diode array detector and to a Micromass platform LCZ 4000 mass spectrometer. Mass analyses were performed in the negative electrospray ionization mode with a capillary voltage of 25 and 50 V and a desolvation temperature of 250 °C. An Alltima C18 column (150 × 4.6 mm, precolumn of 4.6 × 7.5 mm,) was used for the chromatographic separations at 35 °C. The solvent system was a gradient of A (0.05% aqueous HCOOH) and B (acetonitrile) with 5% B at 0 min and 100% B at 30 min with a flow rate of 1 mL min−1. TLC analysis was performed on aluminium sheets coated with silica gel 60 F 254. Detection was achieved by exposure to UV light (254 nm) and by heating after exposure to a 10% H2SO4 solution in EtOH. Purifications were performed by column chromatography on silica gel Si 60 (40–63 µm).Chemical synthesis
Synthesis of n-hexadecylcaffeate
CH).
CH). HRMS (FAB, positive mode): m/z 405.3005 ([M + H]+)
(405.30049, calcd for C25H41O4).
Antioxidant tests
240 M−1 cm−1 assuming a purity of 95%) placed in the spectrometer cell was added 20 µL of a freshly prepared 2.5 mM solution of antioxidant in MeOH. The reaction was monitored at 25 °C over 250–600 s. Each experiment was repeated six times. Standard deviations were lower than 5%.
100 M−1 cm−1)28 was below 2% in all experiments. The uninhibited and inhibited peroxidation rates were calculated from the slope of the absorbance at 234 nm vs time lines before and after antioxidant addition using fixed time intervals. Each experiment was run in triplicate. Standard deviations were lower than 10%.
Acknowledgements
M. Roche is grateful to the General Council of the PACA region and to INRA for financial support.References
- A. Bendini, M. Bonoli, L. Cerretani, B. Biguzzi, G. Lercker and T. G. Toschi, J. Chromatogr., A, 2003, 425–433 CrossRef CAS.
- K. L. Tuck and P. J. Hayball, J. Nutr. Biochem., 2002, 13, 636–644 CrossRef CAS.
- R. W. Owen, A. Giacosa, W. E. Hull, R. Haubner, B. Spiegelhalder and H. Bartsch, Eur. J. Cancer, 2000, 36, 1235–1247 CrossRef CAS.
- E. Cartron, M. A. Carbonneau, G. Fouret, B. Descomps and C. L. Léger, J. Nat. Prod., 2001, 64, 480–486 CrossRef CAS.
- L. Lesage-Meessen, D. Navarro, S. Maunier, J.-C. Sigoillot, J. Lorquin, M. Delattre, J.-L. Simon, M. Asther and M. Labat, Food Chem., 2001, 75, 501–507 CrossRef CAS.
- R. Capasso, G. Cristinzio, A. Evidente and F. Scognamiglio, Phytochemistry, 1992, 31, 4125–4128 CrossRef CAS.
- J. Pérez, T. de la Rubia, J. Moreno and J. Martínez, Environ. Toxicol. Chem., 1992, 11, 489–495 CrossRef CAS.
- A. Fiorentino, A. Gentili, M. Isidori, P. Monaco, A. Nardelli, A. Parrella and F. Temussi, J. Agric. Food Chem., 2003, 51, 1005–1009 CrossRef CAS.
- A. Jaouani, S. Sayadi, M. Vanthournhout and M. J. Penninckx, Enzyme Microb. Technol., 2003, 33, 802–809 CrossRef CAS.
- F. J. Rivas, F. J. Beltrán, O. Gimeno and J. Frades, J. Agric. Food Chem., 2001, 49, 1873–1880 CrossRef CAS.
- R. Leenen, A. J. C. R. Roodenburg, M. N. Vissers, J. A. E. Schurbiers, K. P. A. M. van Putte, S. A. Wiseman and F. H. M. M. van de Put, J. Agric. Food Chem., 2002, 50, 1290–1297 CrossRef CAS.
- C. Manna, P. Galletti, G. Maisto, V. Cucciolla, S. D'Angelo and V. Zappia, FEBS Lett., 2000, 470, 341–344 CrossRef CAS.
- E. Miro-Casas, M.-I. Covas, M. Farre, M. Fito, J. Ortuño, T. Weinbrenner, P. Roset and R. de la Torre, Clin. Chem., 2003, 49, 945–952 CrossRef CAS.
- R. Limiroli, R. Consonni, A. Ranalli, G. Bianchi and L. Zetta, J. Agric. Food Chem., 1996, 44, 2040–2048 CrossRef CAS.
- A. Chesson, G. J. Provan, W. R. Russel, L. Scobbie, A. J. Richardson and C. Stewart, J. Sci. Food Agric., 1999, 79, 373–378 CrossRef CAS.
- K. Azuma, K. Ippoushi, M. Nakayama, H. Ito, H. Higashio and J. Terao, J. Agric. Food Chem., 2000, 48, 5496–5500 CrossRef CAS.
- C. Dufour, E. da Silva, P. Potier, Y. Queneau and O. Dangles, J. Agric. Food Chem., 2002, 50, 3425–3430 CrossRef CAS.
- F. A. M. Silva, F. Borges, C. Guimarães, J. L. F. C. Lima, C. Matos and S. Reis, J. Agric. Food Chem., 2000, 48, 2122–2126 CrossRef CAS.
- P. Goupy, C. Dufour, M. Loonis and O. Dangles, J. Agric. Food Chem., 2003, 51, 615–622 CrossRef CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Lebensm.-Wiss. Technol., 1995, 28, 25–30 CAS.
- H. Fulcrand, A. Cheminat, R. Brouillard and V. Cheynier, Phytochemistry, 1994, 35, 499–505 CrossRef CAS.
- D. Vogna, A. Pezzella, L. Panzella, A. Napolitano and M. d'Ischia, Tetrahedron Lett., 2003, 44, 8289–8292 CrossRef CAS.
- R. Arakawa, M. Yamaguchi, H. Hotta, T. Osakai and T. Kimoto, J. Am. Soc. Mass Spectrom., 2004, 15, 1228–1236 CrossRef CAS.
- M. Antolovich, D. R. J. Bedgood, A. G. Bishop, D. Jardine, P. D. Prenzler and K. Robards, J. Agric. Food Chem., 2004, 52, 962–971 CrossRef CAS.
- E. Niki, Methods Enzymol., 1990, 186, 100–108 CrossRef CAS.
- P. G. Baraldi, D. Simoni, S. Manfredini and E. Menziani, Liebigs Ann. Chem., 1983, 24, 684–686 CrossRef.
- S. Gibbons, K. T. Mathew and A. I. Gray, Phytochemistry, 1999, 51, 465–467 CrossRef CAS.
- W. A. Pryor, J. A. Cornicelli, L. J. Devall, B. Tait, B. K. Trivedi, D. T. Witiak and M. Wu, J. Org. Chem., 1993, 58, 3521–3532 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |