(+)-Echinobetaine B: isolation, structure elucidation, synthesis and preliminary SAR studies on a new nematocidal betaine from a southern Australian marine sponge, Echinodictyum sp.
Robert J.
Capon
*a,
Dat
Vuong
a,
Michelle
McNally
a,
Torsten
Peterle
a,
Nicholas
Trotter
a,
Ernest
Lacey
b and
Jennifer H.
Gill
b
aCentre for Molecular Biodiversity, Institute for Molecular Biosciences, University of Queensland, St. Lucia, Queensland, 4072, Australia. E-mail: r.capon@imb.uq.edu.au; Fax: +61 7 334 62101; Tel: +61 7 334 62979
bMicrobial Screening Technologies Pty. Ltd., Kemps Creek, New South Wales, 2171, Australia
First published on 17th November 2004
Abstract
The principle nematocidal agent present in a southern Australian marine sponge of the genus Echinodictyum has been isolated and identified as the novel betaine (+)-echinobetaine B (6), and the structure assigned by spectroscopic analysis has been confirmed by total synthesis. Preliminary SAR conclusions are drawn from analysis of synthetic intermediates and the known marine metabolites zooanemonin (12) and norzooanemonin (13), and the new sponge metabolite norzooanemonin methyl ester (14). The latter compound is reported for the first time from a selection of Australian sponges, including an Axinyssa sp., a Niphates sp., an Axinella sp. and a Ptilocaulis sp.
Introduction
During earlier investigations into new agrochemical agents from the Australian marine sponge Echinodictyum sp. we reported a series of novel antibacterial metabolites, echinosulfonic acids A–C (1–3) and echinosulfone (4),1 along with the nematocidal betaine (−)-echinobetaine A (5).2 Although initial interest in this Echinodictyum sp. was prompted by promising nematocidal bioassay data against the endo parasite Haemonchus contortus, it is noteworthy that 1–4 displayed no nematocidal activity while the nematocidal properties of (−)-echinobetaine A (5) were insufficient (LD99 83 µg mL−1) to account for the level of activity observed in the crude EtOH extract (LD99 22 µg mL−1). The identity of the more potent Echinodictyum sp. nematocidal agent can now be revealed as the novel betaine (+)-echinobetaine B (6) (LD99 8.3 µg mL−1).
Results and discussion
The EtOH extract of the Echinodictyum specimen was decanted, concentrated in vacuo and triturated with DCM after which the residual solid was partitioned between n-BuOH and H2O. Whereas the n-BuOH solubles displayed antibacterial activity and ultimately led to the isolation and identification of the echinosulfonic acids A–C (1–3) and echinosulfone (4),1 the crude H2O solubles displayed significant nematocidal activity. The H2O solubles were initially further fractionated by elution through Sephadex G-10 (H2O) followed by isocratic C18 HPLC (0.1% TFA in H2O) to yield nematocide (−)-echinobetaine A (5).2 More extensive HPLC studies on nematocidal active fractions have since yielded the principle nematocidal agent as (+)-echinobetaine B (6).High resolution ESI(+)MS analysis of 6 revealed a highest mass ion at m/z 185 consistent with a molecular formula (C8H12N2O3, Δ 0.5 mmu) requiring four double bond equivalents. Examination of the NMR data for 6 (see Table 1) revealed resonances consistent with an OMe (1H: δ 3.22 (s); 13C: 57.1 ppm), two NMe's (1H: δ 3.78 and 3.63; 13C: 35.7 and 33.9 ppm), two sp2 methines (1H: δ 8.52 and 7.31; 13C: 138.1 and 123.6 ppm), one deshielded sp3 oxymethine (1H: δ 4.78; 13C: 73.3 ppm), one carboxylic acid or amide carbonyl (13C: δ 172.5 ppm), and a quaternary sp2 hybridized carbon (13C: 130.7 ppm). The presence of the carbonyl functionality was further supported by a characteristic IR absorbance (1689 cm−1), while a measurable [α]D (+30) confirmed the presence of a chiral centre. With a total of four sp2 carbons, one attributed to a carboxylic acid moiety, 6 was determined to contain an N-heterocycle. Analysis of the gHMBC NMR data (Table 1) confirmed that the chiral tertiary carbon (C-2) was substituted by both OMe and carbonyl moieties. Further gHMBC NMR correlations from N5-Me to C-4 and C-6, from N7-Me to C-3 and C-6, and from H-2 to C-3 and C-4, suggested that (+)-echinobetaine B was the substituted imidazole betaine as shown. To confirm the assigned structure a total synthesis of (±)-echinobetaine B (11) was undertaken following the synthetic pathway outlined in Scheme 1.
| Number | 13C (ppm) | 1H δ (multiplicity) | gHMBC (1H–13C) |
|---|---|---|---|
| 1 | 172.5 | ||
| 2 | 73.3 | 4.78 (s) | C-1, C-3, C-4, 2-OMe |
| 2-OMe | 57.1 | 3.22 (s) | C-2 |
| 3 | 130.7 | ||
| 4 | 123.6 | 7.31 (s) | N5–Me, C-3, C-6, |
| N5-Me | 35.7 | 3.78 (s) | C-6, C-4 |
| 6 | 138.1 | 8.52 (s) | C-3, C-4 |
| N7-Me | 33.9 | 3.63 (s) | C-6, C-3 |
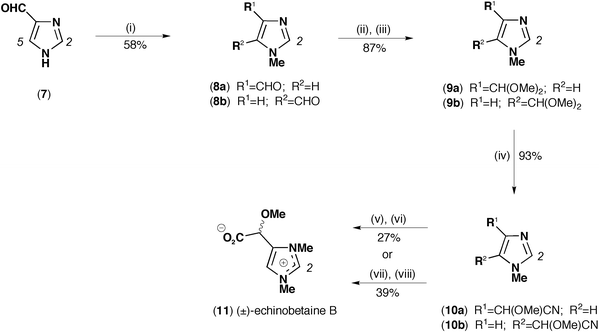 | ||
| Scheme 1 Synthesis of (±)-echinobetaine B (11). Reagents and conditions: (i) NaH, MeI, DMF, N2, 0 °C to RT, 3.5 h; (ii) 10% HCl(aq), RT; (iii) p-TsOH, MeOH, N2, 64 °C, 12 h; (iv) TMSCN, SnCl2, CH3CN, N2, RT, 96 h; (v) MeI, acetone, N2, 56 °C, 15 h; (vi) 10 M HCl(aq), 100 °C, 36 h; (vii) 10 M HCl(aq), 100 °C, 15 h; (viii) MeI, DMF, N2, 70 °C, 14 h. | ||
Commercially available imidazole carboxaldehyde (7) was viewed as a suitable starting material for the synthesis of (±)-echinobetaine B (11). As initially conceived, treatment of 7 with cyanide might be expected to return the cyanohydrin, which on per-methylation about oxygen (2-OMe) and nitrogen (N5-Me and N7-Me) could yield a suitable organonitrile precursor. This precursor would in turn be a potential substrate for commercially available stereoselective nitrilases3,4 leading to an asymmetric synthetic outcome. In practice, the proposed cyanohydrin derived from treatment of 7 with either cyanide salts or HCN proved unstable and difficult to handle, necessitating a modification (see Scheme 1) to the strategy as detailed above. Monomethylation of 7 yielded the mixture 8a and 8b, which was readily converted in good yield to the mixed acetals 9a and 9b. Subsequent treatment with trimethylsilylcyanide and the Lewis acid SnCl25,6 successfully converted the acetals in high yield to the unstable organonitrile mixture 10a and 10b. The noted instability of cyanohydrin and organonitrile precursors precluded stereoselective enzymatic hydrolysis, and necessitated a more direct chemical approach. Acid hydrolysis of the organonitrile moiety followed by N-methylation proceeded in low yield (27%), with the alternative strategy of N-methylation followed by acid hydrolysis returning a slightly improved yield (39%). The product obtained by either route, (±)-echinobetaine B (11), was identical by NMR, MS, UV and HPLC retention time comparison to the natural product (+)-echinobetaine B (6), differing only in the lack of a measurable optical rotation, as befits a racemate. Attempts to resolve the racemate by chiral HPLC proved unsuccessful.
Of particular note, the nematocidal properties for (±)-echinobetaine B (11) (LD99 65 µg mL−1) were considerably lower than those for the natural product (+)-echinobetaine B (6) (LD99 8.3 µg mL−1), implicating stereochemistry as a critical factor in the echinobetaine B pharmacophore. A similar observation had been made regarding the pharmacophore of the nematocidal co-metabolite (−)-echinobetaine A (5).2
(+)-Echinobetaine B (6) is a new example of a rare class of naturally occurring imidazole betaine. The first example of this structure class was zooanemonin (12) reported by Ackermann and Menssen in 1960 from the “horse sponge”, Hippospongia equina.7 Although the structure of 12 was revised shortly thereafter (following communication on its synthesis by Woodward and List),8 few subsequent references have appeared in the scientific literature. The most recent reference to 12 was in 2001 by Hattori et al., in which 12 was assessed as an antifouling constituent of the sponge Protophilaspongia aga.9 As with zooanemonin (12), the closely related homologue norzooanemonin (13) is known but remains equally elusive in the scientific literature. First reported in 1973 by Weinheimer et al. from the gorgonian Pseudopterogorgia americana,1013 was also described in 1977 by Gupta et al. from the hydroid Tubularia larynx.11

During our earlier investigation into Australian marine sponges we isolated norzooanemonin (13) from an Axinyssa and a Niphates sp. The latter sponge, together with an Axinella sp. and a Ptilocaulis sp., also yielded the previously undescribed norzooanemonin methyl ester 14. The molecular formula assigned to 14 by high resolution mass spectrometry (C7H11N2O2, Δmmu −0.3) was consistent with a salt possessing 4 double bond equivalents. Analysis of the 1H NMR (400 MHz, D2O) data for 14 revealed resonances diagnostic for a deshielded sp2 methine (Hδ 8.27, H-5) that proved to be D2O exchangeable (see below), as well as an isolated deshielded sp2 methine (Hδ 7.07, H-3), a CO2Me (Hδ 3.46) and two quaternary ammonium methyl moieties (Hδ 3.56 and 3.66, N6-Me and N4-Me respectively). The data presented above supported a doubly N-methylated imidazole heterocycle substituted by a methyl ester moiety (as shown). Both 13C and 2D NMR correlations supported this determination and facilitated assignment of the NMR data. In this way 14 was confirmed to be the methyl ester of norzooanemonin as indicated. As all sponge samples were extracted with EtOH, and were stored for several years in EtOH prior to investigation, we are confident that the methyl ester 14 is not an esterification artifact. Curiously, during acquisition of 1H NMR data in neutral deuterated protic solvents (CD3OD or D2O) the imidazole methine H-5 in both 13 and 14 underwent slow but complete deuterium exchange. While such exchange processes have been described for a range of heterocycles, they are typically pH or metal-ion activated.12,13 Such an exchange was not observed for 6 or 12 suggesting that the conjugated carbonyl present in 13 and 14 may play a pivotal role in this process, and revealing a measure of specificity not previously recorded.
It is worthwhile noting that the nematocidal activity displayed against Haemonchus contortus by the natural product (+)-echinobetaine B (6) (LD99 8.3 µg mL−1) in our assay system is comparable to that of the two commercial synthetic anthelmintics closantel and levamisole (LD99 5–10 µg mL−1). By contrast, none of 12–14 displayed nematocidal activity, and the synthetic racemate 11 was considerably less active (LD99 65 µg mL−1). Thus it would appear that the 2-OMe and stereochemistry are critical components in the echinobetaine B nematocidal pharmacophore. Ongoing studies are directed at confirming enantiopurity and assignment of absolute stereochemistry for 6, and towards further SAR investigations.
Experimental
General procedures
Solvents were dried according to standard techniques. Anhydrous SnCl2 was prepared by heating commercial SnCl2 dihydrate to 200 °C under an atmosphere of N2 for 1 hour. Column chromatography (flash) was performed using Merck silica gel 60. Thin layer chromatography (TLC) was performed on Merck Silica 60 F254 sheets. Compounds were visualised with 254 nm UV light and/or heating of the TLC plate that had been dipped in p-anisaldehyde, phosphomolybdic acid or potassium permanganate reagent solutions. High performance liquid chromatography (HPLC) was performed using either a Waters 2790 Separations Module equipped with a Waters 996 Photodiode Array Detector, Alltech 500 Evaporative Light Scattering Detector and a Waters Fraction Collector II, running Waters Millennium software or an Agilent 1100 Series Separations Module equipped with a six column switching capability, Agilent 1100 Series Diode Array and/or Multiple Wavelength Detectors, Polymer Laboratories PL-ELS1000 Evaporative Light Scattering Detector (ELSD) and an Agilent 1100 Series Fraction Collector and running ChemStation Rev.9.03A and Purify version A.1.2 software. Unless otherwise specified, a constant level of 0.01% TFA was used in all HPLC separations. Chiroptical measurements ([α]D), given in 10−1 deg cm2 g−1, were obtained on either a Jasco Dip-1000 digital polarimeter or a Jasco P-1010 Intelligent Remote Module type polarimeter, at ambient temperature, in a 100 × 3 mm cell. Ultraviolet (UV) absorption spectra were obtained using either a Shimadzu UV-1650PC or a CARY3 UV-visible Spectrophotometer. Infrared (IR) spectra were acquired using either a Shimadzu FTIR-8400 or a Jasco FT/IR-460Plus Spectrometer, with samples examined as films on NaCl discs. 1H and 13C NMR spectra were acquired on a Varian Inova 400, a Varian Unity 400 plus, a Bruker Avance 500 or a Bruker Avance 600 Spectrometer, in the solvents indicated and referenced to residual 1H and 13C signals in the deuterated solvents. Electrospray Ionisation Mass Spectra (ESI-MS) and Atmospheric Pressure Chemical Ionisation Mass Spectra (APCI-MS) were acquired using either a Waters 2790 Separations Module equipped with a Micromass ZMD mass detector or an Agilent 1100 Series Separations Module equipped with an Agilent 1100 Series LC/MSD mass detector. High resolution (HR) ESI-MS measurements were obtained on either a Bruker BioApex 47E FT mass spectrometer at a cone voltage of 100 kV or a Finnigan MAT 900 XL-Trap instrument with a Finnigan API III source, while EI-MS measurements were obtained on a Kratos MS25RFA mass spectrometer at 70 eV.Collection, extraction and isolation
The marine sponge Echinodictyum sp. (Museum of Victoria Registry Number MVF88741) was collected by beam trawl from the Great Australian Bight at a depth of 85 m at position 32°58′92″S : 128°00′84″E. A taxonomic description has been reported.1 The sponge was diced, steeped in EtOH and kept at −20 °C prior to extraction. The EtOH extract was decanted and concentrated in vacuo to yield a bright orange solid (4.08 g). Trituration with DCM returned insoluble soluble material that was partitioned between n-BuOH and H2O, with the nematocidal activity of the crude extract concentrated into the H2O solubles (2.96 g, 73%). The n-BuOH solubles yielded (in prior studies) echinosulfonic acids A-C (1–3) and echinosulfone (4).1 This nematocidal H2O soluble material was concentrated in vacuo and fractionated by Sephadex G-10 (H2O) chromatography, followed by isocratic C18 HPLC (2 mL min−1 H2O + 0.1% TFA, Zorbax C18 10 µm 250 × 10 mm column), to yield the nematocidal agent (−)-echinobetaine A (5) (as documented in prior studies).2 Further bioassay directed HPLC fractionation, with careful peak shaving, has yielded the dominant nematocidal agent as (+)-echinobetaine B (6) (25 mg, 0.044%) (H. contortus, LD99 8.3 µg mL−1) as the trifluoroacetate salt. Note that percentage yields of natural products are calculated against the total dry weight mass of the crude EtOH extract.The marine sponge Axinyssa sp. (Museum of Victoria Registry Number MVF83453) was collected by hand (SCUBA) off Beware Reef, Cape Conran, Victoria, 38°12′S : 148°47′E. The sponge was diced, steeped in EtOH and kept at −20 °C prior to extraction. The crude EtOH extract was processed as described above for the Echinodictyum sp. to return an H2O soluble fraction that was fractionated by Sephadex G-10 (H2O) chromatography, followed by isocratic C18 HPLC (2 mL min−1 H2O + 0.1% TFA, Zorbax C18 10 µm 250 × 10 mm column), to yield trigonelline14 (0.4 mg, 0.037%) together with norzooanemonin (13) (1.1 mg, 0.10%). Norzooanemonin possessed MS and 1H NMR data consistent with those reported in the literature.15
The marine sponge Axinella sp. (Northern Territory Museum Registry Number NTMZ3725) was collected by trawling in the Great Australian Bight. The sponge was diced, steeped in EtOH and kept at −20 °C prior to extraction. The crude EtOH extract was processed as described above for the Axinyssa sp. to return an H2O soluble fraction that was fractionated by Sephadex G-10 (H2O) chromatography, followed by isocratic C18 HPLC (1 mL min−1 H2O + 0.1% TFA, Phenomenex Aqua C18 5 µm 250 × 10 mm column), to yield norzooanemonin methyl ester (14) (1.1 mg, 1.45%). λmax (MeOH)/nm 264; δH (400 MHz; D2O) 8.27 (1 H, s), 7.07 (1 H, s), 3.66 (3 H, s), 3.56 (3 H, s) and 3.46 (3 H, s); δC (100 MHz; D2O) 175.0 (C-1), 131.0 (C-2), 35 (N4-Me), 33.0 (N6-Me) and 32.0 (CO2Me); ESI(+)MS m/z 177 (M − H + Na), 155 (M); HRESI(+)MS m/z 177.0639 (C7H10N2O2Na requires 177.0640) and 155.0817 (C7H11N2O2 requires 155.0820).
The marine sponge Niphates sp. (Museum of Victoria Registry Number MVF83541) was collected by trawling off the east coast of Tasmania. The sponge was diced, steeped in EtOH and kept at −20 °C prior to extraction. The crude EtOH extract was processed as described above for the Axinyssa sp. to return norzooanemonin (13) (1.6 mg, 0.36%) and the methyl ester 14 (27.0 mg, 6.14%).
The marine sponge Ptilocaulis sp. (Museum of Victoria Registry Number MVF83529) was collected by scientific trawling in the Great Australian Bight, 38°02′S : 128°28′E. The sponge was diced, steeped in EtOH and kept at −20 °C prior to extraction. The crude EtOH extract was processed as described above for the Axinyssa sp. to return norzooanemonin methyl ester (14) (1.7 mg, 0.7%).
Acknowledgements
We acknowledge the assistance of L. Goudie in identifying sponge material, D. Howse for database support, and T. Friedel and K. Heiland for facilitating access to bioassays. This research was partially funded by the Australian Research Council, together with Novartis Animal Health Australasia Pty Ltd.References
- S. P. B. Ovenden and R. J. Capon, J. Nat. Prod., 1999, 62(9), 1246–1249 CrossRef CAS.
- R. J. Capon, D. Vuong, E. Lacey and J. H. Gill, J. Nat. Prod., 2004 Search PubMed (accepted).
- C. O'Reilly and P. D. Turner, J. Appl. Microbiol., 2003, 95(6), 1161–1174 CrossRef CAS.
- L. Martinkova and V. Mylerova, Curr. Org. Chem., 2003, 7(13), 1279–1295 CAS.
- K. Utimoto, Y. Wakabayashi, Y. Shishiyama, M. Inoue and H. Nozaki, Tetrahedron Lett., 1981, 22(43), 4279–4280 CrossRef CAS.
- K. Utimoto, Y. Wakabayashi, T. Horiie, M. Inoue, Y. Shishiyama, M. Obayashi and H. Nozaki, Tetrahedron, 1983, 39(6), 967–973 CrossRef CAS.
- D. Ackermann and H. G. Menssen, Hoppe-Seyler’s Z. Physiol. Chem., 1960, 322, 198–207 CAS.
- D. Ackermann and P. H. List, Hoppe-Seyler’s Z. Physiol. Chem., 1960, 318(3/6), 281 CAS.
- T. Hattori, S. Matsuo, K. Adachi and Y. Shizuri, Fish. Sci., 2001, 67(4), 690–693 Search PubMed.
- A. J. Weinheimer, E. K. Metzner and M. L. Mole Jr., Tetrahedron, 1973, 29(20), 3125–3126 CrossRef CAS.
- K. C. Gupta, R. L. Miller, J. R. Williams and J. F. Blount, Experientia, 1977, 33(12), 1556 CAS.
- E. Buncel, O. Clement and I. Onyido, Acc. Chem. Res., 2000, 33(10), 672–678 CrossRef CAS.
- E. Buncel and I. Onyido, J. Labelled Compd. Radiopharm., 2002, 45(4), 291–306 CrossRef CAS.
- E. Schulze and G. Trier, Hoppe-Seyler’s Z. Physiol. Chem., 1911, 67, 46–58.
- T. Jahn, G. M. Konig, A. D. Wright, G. Worheide and J. Reitner, Tetrahedron Lett., 1997, 38(22), 3883–3884 CrossRef CAS.
- M. Kodera, N. Terasako, T. Kita, Y. Tachi, K. Kano, M. Yamazaki, M. Koikawa and T. Tokii, Inorg. Chem., 1997, 36(18), 3861–3868 CrossRef.
| This journal is © The Royal Society of Chemistry 2005 |