7-N,7′-N′-(1″,2″-Dithianyl-3″,6″-dimethylenyl)bismitomycin C: synthesis and nucleophilic activation of a dimeric mitomycin†
Sang Hyup Lee and Harold Kohn*
Division of Medicinal Chemistry and Natural Products, School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7360, USA. E-mail: harold_kohn@unc.edu; Fax: (919) 843-7835; Tel: (919) 966-2680
First published on 6th January 2005
Abstract
Dimeric alkylating agents that modify complementary DNA strands have engendered significant interest. We have prepared the novel dimeric mitomycin, 7-N,7′-N′-(1″,2″-dithianyl-3″,6″-dimethylenyl)bismitomycin C (9), in which the mitomycins are bridged by a dithiane unit. Dimer 9, like the clinically tested acyclic disulfides KW-2149 (3) and BMS-181174 (4), was designed to activate under nucleophilic and reductive conditions. Successive nucleophile-mediated disulfide cleavage transformations of 9 are expected to generate thiol species ideally positioned to render the two mitomycin systems vulnerable to nucleophilic attack and permit DNA interstrand cross-link formation. The dithiane linker, strategically positioned between the two mitomycins, distinguished 9 from 3 and 4. Nucleophilic activation of this cyclic disulfide permitted both activated mitomycins to remain tethered to one another. We report the synthesis of 9, and show that the nucleophile Et3P markedly enhances the activation and consumption of 9, compared with the reference compound 7-N, 7′-N′-(cyclohexanyl-trans-1″,4″-dimethylenyl)bismitomycin C (27). We further demonstrated that 9 provides higher levels of DNA interstrand cross-links than either the dimeric reference compounds, 27 and 7-N,7-N′-(2″,5″-dihydroxy-1″,6″-hexanediyl)bismitomycin C (28), or the monomeric mitomycins, 1 and 3, when Et3P is added to solutions containing EcoRI-linearized pBR322 DNA.
Introduction
Mitomycin C (1) is a clinical anticancer agent of significant importance.1 Bioreduction of 1 leads to the sequential activation of the C(1) and C(10) sites necessary for the generation of mono- and bis-(cross-linked) alkylated DNA adducts.2 While both DNA lesions contribute to the antitumor activity of 1, it is the DNA interstrand cross-link adducts (DNA ISC) that have attracted the most interest. The cross-linking of complementary DNA strands by 1 (1–DNA) is expected to inhibit DNA replication and subsequent cell proliferation.2 Consistent with this notion 1–DNA ISC have been shown to be ca. 60 times more lethal than the corresponding 1–DNA monoadducts.3
There has been an increased interest in the design and evaluation of novel dimeric agents that target DNA.4,5 Among these compounds are intercalating and irreversible alkylating agents that cross-link complementary strands of DNA. We have prepared a series of dimeric mitomycins (2) designed to take advantage of the enhanced reactivity of the C(1) site in 1, compared with the C(10) position.6

The clinical successes and limitations of 1 has led to the syntheses and evaluation of more than 1000 analogues.10 In the 1980s, two mitomycins were disclosed, 3 (KW-2149)11 and 4 (BMS-181174),12 where each contained a C(7) aminoethylene disulfide moiety in place of the C(7) amino unit in 1.

Compounds 3 and 4 are members of an emerging class of anticancer agents that contain a multi-sulfide linkage. Cytotoxicity in these agents is associated, in part, with a nucleophile-assisted, sulfur–sulfur cleavage transformation.11,12,17–22 These reports led us to prepare C(8) iminoporfiromycin 6 that contained a cyclic disulfide unit and its hydrocarbon counterpart 7.23

In this study, we report the synthesis of the dimeric mitomycin 9, which contains a cyclic disulfide unit. Dimer 9 was designed to undergo facile DNA ISC by a nucleophile-assisted disulfide cleavage process permitting successive modification of the two mitomycin distal C(1) sites that remain tethered together. We demonstrated that phosphines dramatically accelerate the activation and utilisation of 9 compared with 1 and provide enhanced levels of DNA ISC adducts.

Results and discussion
Selection of dimeric mitomycin 9
Compound 9 retained the key structural features found for 1 and for many semi-synthetic mitomycins. Thus, we expected that this dimeric mitomycin would likely be activated by both reductases and acids, pathways previously documented for 1.2 Four structural elements are, however, unique to 9 and were incorporated into our design of this mitomycin to promote nucleophile-assisted DNA ISC. First, 9 had two mitomycin units to permit DNA adduction at the reactive C(1) site.6 Second, the disulfide unit was strategically placed three atoms away from the C(7) position in both mitomycins. This arrangement has been important for intramolecular thiol-mediated mitomycin activation processes.16 Third, the dithiane linker in 9 ensured that the two mitomycin units would remain together after activation and DNA adduction. This feature differentiated 9 from 3 and 4 where disulfide cleavage leads to a monomeric species. Fourth, the dithiane linker is of sufficient size to permit modification of the two distal mitomycin C(1) sites in 9 by guanine residues on complementary DNA strands. We estimate with computational programs (Sybyl 6.0, HyperChem 7.1) that the distance for 9 and its expected intermediates to be between 7–25 Å depending upon their conformation. This distance can cross-link guanines on different DNA strands separated by as many as five base pairs.The envisaged nucleophile-mediated route unique to 9 is shown in Scheme 1. Activation of 9 begins with intracellular thiol- or serum albumin (CSH)-mediated disulfide cleavage14–16,24–26 to give 10, which is then converted to 11 through intramolecular cyclization. In Scheme 1, we show that intramolecular cyclization occurs at the C(8); however, we recognize that alternative cyclization sites [C(7), C(6)]14,16,23 may initiate mitomycin activation. A second round of disulfide cleavage beginning with 11 provides 12, which is then converted to 13 by intramolecular cyclization. Formation of 11 and 13 disrupts the N(4)–C(4a)–C(8a)–C(8)–O conjugated system and is expected to lead to the rapid loss of methanol from C(9) and C(9a) and mitosene formation, permitting the successive DNA adduction at the two distal C(1) sites in the dimer. If correct, 9 nucleophile-mediated activation in the presence of DNA will lead to DNA ISC.
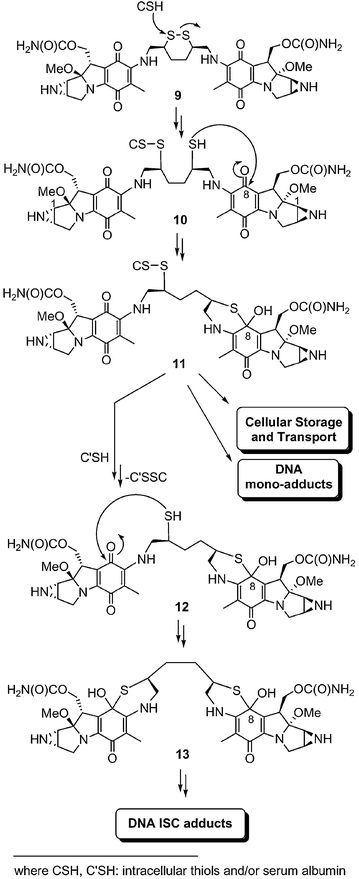 | ||
| Scheme 1 Proposed nucleophile-mediated activation pathway for 9. | ||
Thus, three objectives were established for this study: the synthesis and characterization of 9, the documentation of the nucleophile-mediated activation pathway for 9, and the assessment of the 9 DNA ISC efficiencies under nucleophile-mediated conditions using linearized pBR322 DNA. We also determined the in vitro cellular cytotoxicities of 9 against human lung adenocarcinoma cell line A549. While we found it interesting, the cytotoxicity information for 9 was not used in our assessment of the structural requirements for nucleophile activation of this mitomycin since neither the effective concentration of 9 within the A549 cells nor the factors that contributed to the mitomycin inhibition of cell replication were determined.
Synthesis of the cyclic disulfide bridge: (3R,6S)-meso-3,6-bis(aminomethyl)-1,2-dithiane di-trifluoroacetate salt (14)
Synthesis of 9 required the preparation of (3R,6S)-meso-3,6-bis(aminomethyl)-1,2-dithiane di-trifluoroacetate salt (14) (Scheme 2). We began by converting 1,5-hexadiene (15) to diepoxide 16 using m-chloroperbenzoic acid.27,28 Epoxide cleavage with phthalimide in DMF gave 17.28 Three stereoisomers of 17 exist: meso-(R,S) and the enantiomeric pair, threo-(R,R), and threo-(S,S). We found that the isolated product existed as a mixture of diastereomers (2.7 : 1, 13C NMR analysis).28 Recrystallization (twice) of 17 in DMF (80 °C) provided a sample enriched in one isomer (9.4 : 1, 13C NMR analysis) in 64% yield.28 X-Ray crystallographic analysis identified this adduct as meso-17,28 indicating that the major product in the initially isolated 17 mixture (2.7 : 1 diastereomeric mixture) was the meso-adduct. Hydrazine deprotection of the phthalimide units in 17 (meso : dl = 2.7 : 1) gave 18![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 29 (meso : dl = 2.7 : 1, 13C NMR analysis) in near quantitative yields after acid work-up. Subsequent protection of the amino groups in 18 with BOC2O and Et3N afforded 19 in moderate yield (66%) as a mixture of diastereomers (meso : dl = 3.5 : 1, 13C NMR analysis). Recrystallization of 19 from ethyl acetate provided a single isomer (≥10 : 1, 13C NMR analysis) (75% yield). The product yields and the ratio of diastereomers obtained for the conversion of 17 to 19 indicated that the major 19 diastereomer in the 3.5 : 1 mixture was the meso-isomer. Several methods were explored to replace the hydroxyl groups in 19 by thioacetate units. Mitsunobu reaction30 of 19 with thioacetic acid produced no reaction (data not shown). Next, we treated 19 (meso : dl = 3.5 : 1) with MsCl in pyridine to give 20 in a moderate yield (67%). 13C NMR analysis showed that 20 was isolated as a single diastereomer. Once again, the observed yield for this transformation indicated that this compound was the meso-isomer. We suspect that conditions employed in the isolation work-up of this reaction (addition of H2O) led to the selective precipitation of the meso-20. Displacing mesylate units in 20 with KSAc in EtOH gave 21 in a 60% yield. 13C NMR analysis showed only a single isomer, thus providing evidence that substitution proceeded with stereochemical control by an SN2 pathway. We have, therefore, assigned 21 as the meso-stereoisomer. Hydrolysis of the thioacetate groups in 21 with aqueous methanolic K2CO3 gave the dithio derivative 22, which was directly oxidized in basic methanol with O2 to yield cyclic 23 (55% yield). Cyclic disulfide 23 was also obtained in 89% yield from 21 by sequential hydrolysis and oxidation without isolation of 22. The BOC groups in 23 were removed in near quantitative yield by trifluoroacetic acid (TFA) to give cyclic disulfide 14. The overall yield for 14 from 15 was 11% (9 steps) and our reaction sequence provided 14 as a single diastereomer (meso).
29 (meso : dl = 2.7 : 1, 13C NMR analysis) in near quantitative yields after acid work-up. Subsequent protection of the amino groups in 18 with BOC2O and Et3N afforded 19 in moderate yield (66%) as a mixture of diastereomers (meso : dl = 3.5 : 1, 13C NMR analysis). Recrystallization of 19 from ethyl acetate provided a single isomer (≥10 : 1, 13C NMR analysis) (75% yield). The product yields and the ratio of diastereomers obtained for the conversion of 17 to 19 indicated that the major 19 diastereomer in the 3.5 : 1 mixture was the meso-isomer. Several methods were explored to replace the hydroxyl groups in 19 by thioacetate units. Mitsunobu reaction30 of 19 with thioacetic acid produced no reaction (data not shown). Next, we treated 19 (meso : dl = 3.5 : 1) with MsCl in pyridine to give 20 in a moderate yield (67%). 13C NMR analysis showed that 20 was isolated as a single diastereomer. Once again, the observed yield for this transformation indicated that this compound was the meso-isomer. We suspect that conditions employed in the isolation work-up of this reaction (addition of H2O) led to the selective precipitation of the meso-20. Displacing mesylate units in 20 with KSAc in EtOH gave 21 in a 60% yield. 13C NMR analysis showed only a single isomer, thus providing evidence that substitution proceeded with stereochemical control by an SN2 pathway. We have, therefore, assigned 21 as the meso-stereoisomer. Hydrolysis of the thioacetate groups in 21 with aqueous methanolic K2CO3 gave the dithio derivative 22, which was directly oxidized in basic methanol with O2 to yield cyclic 23 (55% yield). Cyclic disulfide 23 was also obtained in 89% yield from 21 by sequential hydrolysis and oxidation without isolation of 22. The BOC groups in 23 were removed in near quantitative yield by trifluoroacetic acid (TFA) to give cyclic disulfide 14. The overall yield for 14 from 15 was 11% (9 steps) and our reaction sequence provided 14 as a single diastereomer (meso).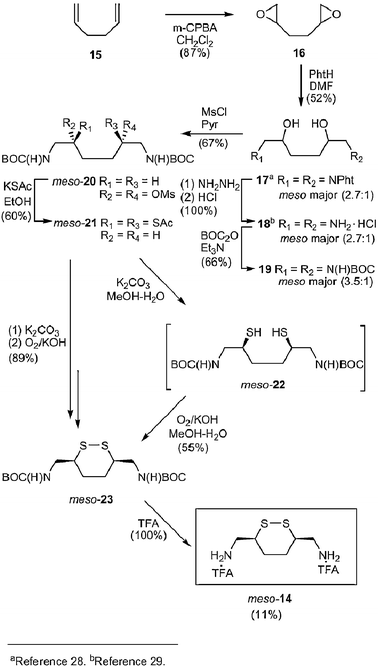 | ||
| Scheme 2 Synthesis of cyclic disulfide meso-14. | ||
Synthesis of mitomycin dimers 9, 27, and 28
Treatment of mitomycin A31 (MMA, 24) with 14 in the presence of Et3N gave 9 (53% yield) along with the mono-substituted products 25a and 25b (21% yield). Several reaction conditions and solvents (i.e. dichloromethane, ethanol, methanol, chloroform) were examined for this transformation, and we found that methanol (room temperature, 1 d) gave the best results. In the synthesis of 9, the solution color changed from violet to dark blue as the reaction proceeded (1 d), with monomers 25a and 25b being formed initially and then converted to dimer 9 (TLC and HPLC analyses). On TLC, 25a and 25b exhibited lower Rf values (0.15) than dimer 9 (0.47). After 1 day the product ratio of dimer 9 to monomers 25 was ca. 2.5. Using HPLC, we observed two peaks of equal intensity for the monomers (tR 21.8, 22.3 min) that were consistent with the formation of two diastereomers (25a, 25b); only one HPLC peak (tR 29.8 min) was observed for 9.

Using similar reaction conditions, we prepared the two dimeric mitomycins, 27 (70% yield) and 28 (59% yield), upon treatment of 24 with 1,4-bis(aminomethyl)cyclohexane (26) and 18, respectively. Compound 27 was chosen as the reference compound for the chemical studies. In 27, we replaced the dithiane moiety in 9 with a cyclohexyl moiety to determine the importance of the disulfide cleavage processes for nucleophile-mediated mitomycin activation. We recognized that 27 existed as the trans-isomer while 9 was the cis-isomer. Our decision to prepare 27 was based on the commercial availability of trans-26.

Chemical reactivity of dimeric mitomycins
We determined the rate of methanolysis of 9 and 27 in the absence of nucleophiles and in the presence of a phosphine and thiols to learn if this mitomycin is activated by nucleophiles (see later in Tables 1 and 2). The kinetic measurements were monitored by UV-vis spectroscopy (200–600 nm) or HPLC using UV-vis detection for greater than two half-lives, when possible, and then the absorbance of the starting mitomycin (374 nm) was plotted against time. We used non-linear regression analysis to fit the observed exponential decay of 9 using the SigmaPlot Program 2001 to provide pseudo-first-order rate constants. The reactions were conducted in duplicate, and the results averaged. The products were verified, when possible, by HPLC and TLC analyses with authentic samples.The methanolysis of 9 was first monitored by HPLC using a MeOH–CHCl3 (1 : 1) solution (effective ‘pH’ 3, room temperature, 3 d). The HPLC chromatograms for 9, 29 and 30

 | ||
| Fig. 1 HPLC profiles of 9 (a), 29 (b) and 30 (c). The reactions were monitored at 374 nm for 9 (tR 29.8 min) and 316 nm for 29 (tR 29.6, 30.0, 30.3 and 30.8 min) and 30 (tR 30.6, 31.3 and 32.1 min). | ||
Purification of the product mixture by preparative thin layer chromatography (PTLC) afforded an authentic sample of the C(1) methoxymitosenes 30 as a mixture of diastereomers. Solvolytic products 30 were identified by their HPLC, UV-vis, 1H NMR and mass spectrometric properties. In the 1H NMR spectra for 30 we observed the expected resonance (δ 3.51) for the C(1) methoxy units and the downfield resonances (δ 5.73 and 5.78) for the C(10) methylene unit.33 The expected molecular ion peak (m/z 813 [M + 1]+) for the C(1) methoxymitosenes 30 was observed in the low-resolution mass spectrum.
We next determined the rate of methanolysis for mitomycin dimer 9 in buffered methanolic solutions at effective ‘pH’ 7.4, 5.5 and 4.0 in the absence of nucleophiles at 25 °C (Table 1). We observed that reduction of pH led to enhanced solvolysis rates. The observed rate increases were not proportional to the pH. We observed only a modest increase in the conversion of 9 to 29 and to 30 as we reduced the pH from 7.4 to 5.5. The observed half-life (t1/2) for 9 at pH 5.5 was comparable with that reported for 1 (t1/2 = 13.7 d).34 These results suggested that 9 underwent acid-catalyzed activation along a pathway similar to that of 1.32,35 A more substantial rate increase was observed as the pH was lowered from 5.5 to 4.0. We observed a clear isosbestic point at 337 nm (data not shown). Analysis of the rate data was consistent with our assumption that the methanolysis followed pseudo-first-order kinetics and that the two mitomycin units underwent change independent of one another. Using HPLC we observed the steady depletion of 9, the appearance of 29 followed by appearance of 30, and the concomitant disappearance of 29 over a 75 h period (data not shown). When we examined the solvolysis of 27 at pH 7.4 by UV-vis spectroscopy we observed little solvolysis after 10 d.
a
| ‘pH’ | kobs/d−1 | t1/2/d |
|---|---|---|
| a Reactions were run in buffered methanolic solution (0.1 M Tris·HCl, pH 7.4; 0.1 M bis-Tris·HCl, pH 5.5; 0.1 M bis-Tris·HCl, pH 4.0) at 25 °C. All reactions were run in duplicate and the values averaged. The concentration of 9 was 0.03 mM and the data were obtained using a Cary 3Bio Varian UV-visible spectrophotometer and the reactions monitored at 374 ± 2 nm unless otherwise indicated.b The data were obtained using HPLC. | ||
| 7.4 | 0.0347 | 20b |
| 5.5 | 0.0533 | 13 |
| 4.0 | 1.65 | 0.42 |
Since 9 and 27 underwent little change at pH 7.4, we determined if the solvolysis rates of these dimers were increased in the presence of thiols [L-dithiothreitol (L-DTT), and glutathione (GSH)] at pH 7.4 (Table 2). Adding L-DTT (10, 20 equiv.) gave no significant increases (≤5%) in the consumption of either 9 or 27 after 6–10 d, compared with solutions without L-DTT. Likewise, when GSH was used as the nucleophile (20 equiv.) the activation rates of 9 and 27 were not affected (<5%) after 10 d. The similarity of the 9 and 27 rate data and the lack of detectable rate enhancements as the thiol and the thiol concentration were varied led us to conclude that L-DTT and GSH did not contribute to the methanolysis of mitomycins 9 and 27 at pH 7.4. This finding mirrored the low activation level observed for C(8) iminoporfiromycin 6 when L-DTT was added.23 For 6, we suggested that the thiol-cleaved C(8) iminoporfiromycin product 31, if formed, can revert back to 6 due to the ease of formation of the 6-membered ring.23 A similar pathway may have occurred with the L-DTT–9 cleaved product 32.
 a
a
| Reagents | 9 | 27 | |||
|---|---|---|---|---|---|
| Nu− | Equiv. | kobs/d−1 | t1/2/d | kobs/d−1 | t1/2/d |
| a Reactions were run in buffered methanolic solution (0.1 M Tris·HCl, pH 7.4) at 25 °C. The reactions were run in duplicate and the values averaged. The data were obtained using a Cary 3Bio Varian UV-visible spectrophotometer and the reactions monitored at 374 ± 3 nm unless otherwise indicated. The concentration of the mitomycin was 0.03 mM.b The data were obtained using HPLC.c No appreciable change in 10 d (less than 5% of original amount). The unreacted mitomycin was identified by HPLC and TLC.d No appreciable change in 6 d (less than 5% of original amount). The unreacted mitomycin was identified by HPLC and TLC.e The data were obtained by UV spectroscopy and are in agreement with the HPLC data (t1/2 = 0.13 d). | |||||
| None | — | 0.0347b | 20 | c | c |
| L-DTT | 10 | d | d | c | c |
| 20 | d | d | c | c | |
| GSH | 20 | c | c | c | c |
| 2 | c | c | — | — | |
| 5 | d | d | — | — | |
| Et3P | 10 | 2.67 | 0.26 | cb | c |
| 20 | 5.33e | 0.13 | cb | c | |
| 50 | 12.0 | 0.058 | cb | c | |
We next asked if phosphines affected the activation of 9 and 27 (Table 2). Previously, Gates and co-workers showed that Ph3P triggered the activation of leinamycin.22 We subsequently showed that Et3P increased the activation of C(8) iminoporfiromycin 6.23 We found that Et3P (10–50 equiv.) markedly enhanced 9 activation. Rate increases for 9 were only observed upon addition of ≥10 equiv. of Et3P; a similar finding was observed for 6.23 We suspect that this represented the threshold level of Et3P needed for mitomycin nucleophilic activation under our conditions, and that sub-threshold levels of Et3P are consumed by the trace amounts of O2 not purged by Ar. For 10, 20 and 50 equiv. of Et3P we observed a 77-, 154- and 345-fold increase, respectively, in the rate of mitomycin consumption and the formation of mitosene products (Table 2), documenting that the activation rate was linear with Et3P concentration. In contrast to 9, no appreciable rate enhancements (<5%) were observed for 27 upon addition of Et3P (10–50 equiv.) (Table 2). Our findings that Et3P affected only the consumption of 9 and not of 27 led us to conclude that the disulfide group in 9 underwent cleavage to thiol 33 thereby activating the appended mitomycin units.

When the reactions of 9 with excess Et3P were monitored by UV-vis spectroscopy we observed the progressive increase in the 314 nm absorption with time, which was consistent with the formation of mitosene products. Careful inspection of the HPLC chromatograms for the 9 plus Et3P (10 equiv.) reaction revealed minor amounts of the C(1) substituted products 29 and 30 during the initial stages (0–45 min) of the reaction. The products disappeared with time with the concomitant appearance of a new, unidentified peak (tR 24.0 min) that showed UV-vis absorption maxima at 233 and 314 nm (data not shown). When 9 was treated with 20 equiv. of Et3P we observed only 29 in the HPLC at the initial stages of the reaction followed by the appearance of the unidentified 24 min peak. Several attempts to identify this new peak by LC–MS and after chemical treatment (e.g. hydrolysis, oxidation) gave inconclusive results (data not shown).
Our finding that 9 consumption was nearly linear with Et3P concentration and that Et3P mediated the conversion of 9 to mitosenes 29 and then to 30 led us to conclude that phosphine cleavage of the disulfide unit in 9 provided thiol(s) (i.e.33) capable of activating the quinone ring in 33. The kinetic data further suggest that the disulfide cleavage is the rate-limiting step. In Scheme 3, we propose a phosphine-mediated activation pathway for 9 that is consistent with these findings. This mechanism is similar to that suggested for C(8) iminoporfiromycin 6.23 The generation of thiol 33 is key to this pathway. We expect that 33 undergoes intramolecular cyclization to give hemi-thioketal 34 leading to the disruption of the N(4)–C(4a)–C(8a)–C(8)–O conjugated system. Mitosene formation and aziridine ring-opening then occur to give the C(1)-activated product 35. A second round of activation is initiated after the decomposition of the thiophosphonium species 35 by H2O (or MeOH)36,37 to provide thiol 36. Similar to the conversion of 33 to 35, intramolecular cyclization of thiol 36 leads to 37 and then 38, in which both C(1) sites in the two distal mitomycin units have been modified. The reaction is completed when 38 converts to 40. Our pathway derives support from recent studies showing that Et3P rapidly promotes L-DTT disulfide cleavage,37 and comparable activation results obtained for C(8) iminoporfiromycin 6 with phosphines.23
 | ||
| Scheme 3 Proposed phosphine-mediated activation pathway for 9. | ||
DNA bonding profiles for mitomycin dimers 9, 27, and 28
The ability of dimeric mitomycins 9, 27 and 28 to cross-link complementary EcoRI-linearized pBR322 DNA was determined using denaturing alkaline agarose gel electrophoresis as reported by Cech38a and Tepe and Williams.38b The size of the DNA product(s) was estimated using λ DNA digested with HindIII as a molecular weight marker. The extent of DNA ISC formation for 9, 27 and 28 was determined in the absence and the presence of Et3P and L-DTT. We also included 1 and 3 in this study.We first examined the extent of 9 DNA ISC induced by Et3P for varying concentrations of 9 (0.1–0.01 mM) and Et3P (5, 10 equiv.). The experiments were run at room temperature (2 h) in aqueous buffered solutions (pH 7.4). We found that treatment of 0.1 mM concentrations of 9 with Et3P provided appreciable levels of DNA ISC adducts, and we used this mitomycin concentration in our subsequent studies. Next, we added Et3P (5 equiv.) and compared the extent of DNA ISC for 9 with those obtained for 1, 27 and 28 (Fig. 2). We observed that 9 efficiently generated DNA ISC (83%) while 27 and 28 generated 14 and 21%, respectively. Under these conditions, 1 gave 5% of DNA ISC. These findings differentiate dimeric mitomycins 9, 27, and 28 from their monomeric counterpart 1, and distinguish dimeric mitomycins that contain a disulfide-containing linker (9) from those that do not (27, 28). Dimers 9, 27, and 28 all showed higher levels (14–83%) of DNA ISC than 1 (5%). Since 1 can only generate DNA ISC by sequential adduction at C(1) and C(10) but 9, 27, and 28 can cross-link complementary strands by DNA adduction processes that occur at the two mitomycins, we have tentatively attributed the increased levels of DNA ISC for the dimers, compared with 1, in part, to C(1) modification processes. This finding is in agreement with our earlier studies that 2 gave enhanced levels of DNA ISC compared with 1 under reductive (Na2S2O4) conditions6 and was supported by the recent study of Tomasz and co-workers.7 Significantly, when we compared the extent of DNA ISC for 9, 27, and 28 we observed a 4.0–5.9-fold increase in DNA ISC upon inclusion of a disulfide unit in the linker. We have attributed the enhanced level of DNA ISC for 9, in part, to the functional role provided by the disulfide unit in the Et3P-mediated mitomycin activation process (Scheme 3). Significantly, the extent of DNA ISC for 9 (0.1 mM) under Et3P-mediated conditions exceeded those reported for dimeric mitomycins 2 (0.2 mM) under reductive conditions (Na2S2O4).6
 | ||
Fig. 2 Denaturing 1.2% alkaline agarose gel for 1, 9, 27 and 28 (0.1 mM) using Et3P (5 equiv.). DNA cross-linking experiments using 0.1 mM concentrations and EcoRI-linearized pBR322 plasmid DNA with Et3P (5 equiv.). All reactions were incubated at room temperature (2 h). Lane 1: λ HindIII DNA molecular weight marker. Lane 2: control (only linearized pBR322). Lane 3: 1 + Et3P (5 equiv.). Lane 4: 28 + Et3P (5 equiv.). Lane 5: 27 + Et3P (5 equiv.). Lane 6: 9 + Et3P (5 equiv.). Lane 7: only Et3P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (5 equiv.). (5 equiv.). | ||
We also examined the effect of L-DTT on mitomycin 9–DNA ISC and compared these findings with 1, 27 and 28. Using the conditions established for Et3P, we treated aqueous buffered solutions (pH 7.4) containing 1, 9, 27 and 28 (0.1 mM) and DNA with L-DTT (5 equiv.) at room temperature (2 h) (Fig. 3). We found that L-DTT (5 equiv.) led to detectably higher levels of DNA ISC for 9 (77%) than 27 (24%) or 28 (31%). Significantly, we observed noticeable amounts of DNA ISC for dimers 27 (24%) and 28 (31%) using L-DTT, and these values exceeded those seen with Et3P (Fig. 2). Dimers 27 and 28 cannot undergo disulfide activation yet DNA ISC adducts were observed. We are uncertain if the observed differences in the DNA ISC levels for 9 and 27 (28) are due to L-DTT-mediated cleavage of the disulfide unit in 9. The kinetic experiments showed that the consumption of 9 and 27 were not affected by L-DTT (10 equiv.) (Table 2). Thus, we have attributed the 27 and 1 DNA ISC products, in part, to trace levels of L-DTT activation pathways other than the one outlined for 9, which proceeded through disulfide cleavage (Scheme 3). Among these, the most likely routes are quinone reductive and base-mediated C(9) proton abstraction pathways. Our finding that L-DTT treatment of 9 led to substantial levels of DNA ISC (Fig. 3) yet provided only trace levels of mitosene products (Table 2) documented the sensitivity of the denaturing agarose gel electrophoresis assay. Generation of only a single DNA ISC within the 4361 bp EcoRI pBR322 DNA leads to a marked change in the electrophoretic mobility of the DNA adduct.
 | ||
| Fig. 3 Denaturing 1.2% alkaline agarose gel for 1, 9, 27 and 28 (0.1 mM) using L-DTT (5 equiv.). DNA cross-linking experiments using 0.1 mM mitomycins and EcoRI-linearized pBR322 plasmid DNA with L-DTT (5 equiv.). All reactions were incubated at room temperature (2 h). Lane 1: λ HindIII DNA molecular weight marker. Lane 2: control (only linearized pBR322). Lane 3: 1 + L-DTT (5 equiv.). Lane 4: 28 + L-DTT (5 equiv.). Lane 5: 27 + L-DTT (5 equiv.). Lane 6: 9 + L-DTT (5 equiv.). Lane 7: only L-DTT (5 equiv.). | ||
Similar DNA-bonding profiles were obtained when the reaction time was reduced from 2 h to 6 min and the amount of nucleophile was increased from 5 to 10 equiv. For Et3P, we observed that 9 gave moderate levels of DNA ISC (58%), and the extent of DNA ISC for 27, 28, and 1 was 8, 12%, and 3%, respectively (data not shown). For L-DTT (10 equiv.), we found that 9 gave moderate levels of DNA ISC (30%), and 27, 28, and 1 gave reduced levels, 13, 16, and 3%, respectively (data not shown).
Mitomycin 9 was more efficient than 1 in generating DNA ISC upon treatment with Et3P (Fig. 2). But 9 is a dimeric mitomycin with an appended disulfide unit and 1 is a monomeric mitomycin without an attached disulfide moiety. Thus, we compared 1 with 3 to determine the importance of the C(7) aminoethylene disulfide unit for DNA ISC. Significantly, McAdam et al. showed that 3 upon treatment with thiols provided DNA ISC.25b Treatment (6 min) of 0.1 mM solutions of 1 and 3 with Et3P (10 equiv.) gave levels of DNA ISC corresponding to 5 and 14%, respectively (data not shown). Since the effective monomeric mitomycin unit concentration of a 0.1 mM solution of dimeric 9 is twice that of a 0.1 mM solution of 3, the observed 14% DNA ISC for 3 will likely increase with increasing 3 concentration. Nonetheless, the 58% DNA ISC found for 9 indicates that the two mitomycin units and the cyclic disulfide bridge contributed to the DNA ISC efficiency of this agent.
The effect of Et3P on 9 and 27 DNA ISC transformations paralleled the observed consumption rates for these mitomycins (Table 2). However, the effect of Et3P on the kinetic data appeared greater than its effect on the extent of DNA ISC formation. The DNA ISC experiments did not permit us to measure the number of ISC adducts within EcoRI DNA (4631 bp) for 9, 27 or 28. Thus, the true effect of Et3P in these experiments remains unknown. Moreover, we have not determined the site(s) of mitomycin covalent adduction [i.e. C(1), C(10)]. Dimeric mitomycin activation may have given tri- and tetra-functionalized adducts,7 and DNA products where linkages exist between duplexes. These questions await further study. Nonetheless, we concluded that the enhanced levels of DNA ISC for 9 compared with 27, 28, and 3 stemmed, in part, from nucleophile-mediated mitomycin activation processes leading to DNA adduction (Scheme 3).
Cytotoxicities
Using an antiproliferative activity assay we tested whether mitomycin dimer 9 inhibited tumor cell line replication.39 As discussed earlier, we did not use the IC50 values as a measure of the extent and efficiency of DNA ISC for the mitomycins in this test group since we did not determine the relative levels of the mitomycins within the cells and the factors that contributed to the inhibition of cell replication. The in vitro antiproliferative activity tests were conducted at the Kyowa Hakko Pharmaceutical Company (Shizuoka, Japan) using the human tumor cell line A549 (lung adenocarcinoma). This cell line was chosen because of its sensitivity to KW-2149 (3). Since compound 9 was designed to undergo non-reductive, acidic, and nucleophile-mediated activation, we were also interested in evaluating the antiproliferative activities of this compound under both aerobic and hypoxic conditions and determining the activity ratio [IC50 (hypoxic)/IC50 (aerobic)] of 9 compared with 1, 3, 27 and 28.The results for in vitro antiproliferative activity tests for mitomycin dimer 9 and reference compounds 1, 3, 27 and 28 are listed in Table 3. We found that under aerobic and hypoxic conditions, 9 was 17-fold and >5-fold, respectively, less potent than its monomer prototype, mitomycin C (1). Similar differences were previously observed for dimeric mitomycins 2 tethered by alkyl linkers with a central heteroatom [N(H), O] against the A549 tumor cell line.6 The activity ratio (hypoxic/aerobic) for 9 was >2.4 while for 1 and 3 it was 8.2 and 3.4, respectively. When 9 was compared with 27 and 28, we found small differences under aerobic and hypoxic conditions. The activity ratios (hypoxic/aerobic) for 9, 27 and 28 appeared to be similar (>1.6 to >2.4) and these values did not permit us to differentiate the compounds. Consequently, we have concluded that for the A549 cell line incorporating the disulfide unit within the dimeric assembly did not measurably contribute to cell death and that the advantages of this unit observed for nucleophile-mediated mitomycin activation and DNA adduction processes did not extend to cell cytotoxicity.
a
| Compound | IC50/μmol L−1 | IC50 (hypoxic)/IC50 (aerobic) | |
|---|---|---|---|
| Aerobic | Hypoxic | ||
| a A549 cell lines were used. The IC50 (μmol/L) value is the concentration that inhibits cell replication by 50% under the assay condition. | |||
| 9 | 41 | >100 | >2.4 |
| 27 | 51 | >100 | >2.0 |
| 28 | 63 | >100 | >1.6 |
| 1 | 2.4 | 20 | 8.2 |
| 3 | 0.12 | 0.41 | 3.4 |
Conclusions
In this study, the synthesis and evaluation of the novel mitomycin dimer 9 is reported. This mitomycin contained four key structural elements that were expected to improve drug activation and DNA adduction upon nucleophilic activation. First, there was the inclusion of two mitomycin units in 9. This feature permits DNA adduction processes to occur at the two reactive mitomycin C(1) sites. Second, there was the disulfide moiety. Cleavage of this unit in 3 and 4 is likely to be responsible for the activities of these mitomycins in 1-resistant and non-hypoxic cells.14–16 Similarly, nucleophilic cleavage of the disulfide unit in 9 generates a thiol species ideally positioned to render the ring system vulnerable to nucleophilic attack (Scheme 1, 10→11, 12→13). Third, the disulfide unit was incorporated within a dithiane permitting the two mitomcyin moieties to remain tethered upon disulfide cleavage. This feature differentiated 9 from 3 and 4, in which disulfide cleavage leads to monomeric mitomycins. Fourth, the dithiane linker allowed for guanines on complementary DNA strands to modify the two distal mitomycin C(1) sites to give DNA ISC adducts.We compared the rate of methanolysis of 9 with the cyclohexyl-linked dimeric mitomycin 27 in the presence and absence of nucleophiles. Et3P markedly increased the rate of 9 consumption and mitosene product formation but not for 27. The kinetic studies indicated that Et3P-mediated 9 activation proceeded by phosphine attack at the disulfide unit to give 33 (Scheme 3) and was supported by a previous investigation using DTT and Et3P.37
The efficiency of mitomycin nucleophile-mediated activation processes for DNA ISC was determined using pBR322 DNA and a denaturing alkaline agarose gel electrophoresis assay.38 We found that 9 generated higher levels of DNA ISC adducts upon Et3P addition than did the reference compounds 1, 3, 27, and 28. The extent of DNA ISC for 9 with Et3P exceeded that of dimeric mitomycins 2 under reductive conditions.6 The higher levels of DNA ISC adducts for 9 compared with dimeric mitomycin 27 paralleled the rates of mitomycin consumption observed in the kinetic studies. Taken together, these findings indicated that the proposed nucleophile-mediated mechanism for disulfide cleavage (Scheme 3) contributed to the increased levels of DNA ISC. This pathway complements the reductive and acid-catalyzed DNA ISC pathways previously established for the mitomycins.2,40 The DNA ISC efficiency of this non-reductive activation route for 9 warrants the further investigation of this transformation where the sequence specificity, site of mitomycin adduction [i.e. C(1), C(10)], and the extent of inter-duplex cross-linking products are determined.
Experimental
General
Mitomycins A (24) and C (1) used in this study were generously provided by Kyowa Hakko Co., Ltd. (Shizuoka, Japan). 1H and 13C NMR spectra were recorded on a General Electric QE-300 spectrometer. Mass spectral (MS) data were obtained by Dr Mehdi Moini at the University of Texas at Austin. The low-resolution MS studies were run on a Finnegan TSQ-70 triple quadrupole mass spectrometer, and the high-resolution MS studies were conducted on a Micromass ZAB-E mass spectrometer. FT-IR spectra were run on a Mattson Galaxy Series FT-IR 5000 spectrometer. Melting points were determined in open capillary tubes using a Thomas-Hoover melting point apparatus and are uncorrected. pH Measurements of aqueous solutions were determined on a Radiometer pHM26 meter using a Radiometer pHC4000 glass electrode. The effective ‘pH’ of the buffered methanolic solutions was similarly determined and the meter and electrode was standardized against aqueous buffer solutions.LC–MS analyses were conducted with Agilent 1100 LC/MSD by Dr Voyksner (LCMS Limited, Raleigh, NC). The products were analyzed with a Zorbak C18 SB column (2.1 × 50 mm, 3.5 μm particles) using the following linear gradient condition: 80% A [0.025 M ammonium acetate in H2O–CH3CN (95 : 5), pH 6.5], 20% B [0.025 M ammonium acetate in H2O–CH3CN (5 : 95), pH 6.5] isocratic for 1 min, and then from 80% A, 20% B to 20% A, 80% B for 30 min. The flow rate was 0.3 mL min−1, and the eluent was monitored at 365 and 313 nm. The mass spectral mode of operation was positive ion electrospray (+ESI) and scan range was 300–1900 Da with 45 psi of nebulization pressure.
7-N,7′-N′-(1″,2″-Dithianyl-3″,6″-dimethylenyl)bismitomycin C (9). Yield, 2.2 mg (53%); HPLC tR 29.8 min; Rf 0.47 (20% MeOH–CHCl3); λmax (CHCN–H2O)/nm 222, and 374; δH (300 MHz; pyridine-d5) 1.83–2.04 [m, 4 H, C(3′)H2], 2.14 [s, 6 H, C(6)CH3], 2.75 [d, J = 3.9 Hz, 2 H, C(2)H], 3.15 [d, J = 3.9 Hz, 2 H, C(1)H], 3.22 [s, 6 H, C(9a)OCH3], 3.62 [br d, J = 12.6 Hz, 2 H, C(3)HH′], 3.83–3.95 [m, 4 H, C(1′)H2], 4.02 [dd, J = 10.8, 3.9 Hz, 2 H, C(9)H], 4.56 [d, J = 12.6 Hz, 2 H, C(3)HH′], 5.06 [dd, J = 10.8, 10.2 Hz, 2 H, C(10)HH′], and 5.41 [dd, J = 10.2, 3.9 Hz, 2 H, C(10)HH′], the signals for the C(2′)H, C(7)NH, C(10)OC(O)NH2 and N(1a)H protons were not detected and are believed to overlap with the observed peaks, the 1H NMR data were in agreement with the COSY spectrum; δC (75 MHz; pyridine-d5) 11.7 [C(6)CH3], 30.2 [C(3′)], 34.4 [C(2)], 38.4 [C(1)], 46.0 [C(9)], 46.7 [C(2′)], 48.1 [C(1′)], 51.3 [C(9a)OCH3], 52.3 [C(3)], 64.1 [C(10)], 106.3 [C(6)], 108.6 [C(9a)], 112.6 [C(8a)], 148.7 [C(7)], 157.4 [C(4a)], 159.8 [C(10a)], 178.5 [C(8)], and 180.9 [C(5)]; m/z (+FAB) 813 ([M + 1]+, 100%); HRMS (+FAB) C36H45N8O10S2 [M + 1]+ requires 813.27001, found 813.26906.
7-N-(6′-Aminomethyl-1′,2′-dithianyl-3′-methylenyl)mitomycin C (25a and 25b). Yield, 0.5 mg (21%); HPLC tR 21.8, 22.3 min (two peaks, 1 : 1); Rf 0.15 (20% MeOH–CHCl3); λmax (CH3CN–H2O)/nm 222 and 374; δH (300 MHz; pyridine-d5) 1.75–2.06 [m, 4 H, C(3′)H2, C(4′)H2], 2.13 [s, 3 H, C(6)CH3], 3.14 [d, J = 4.8 Hz, 1 H, C(1)H], 3.21 [s, 3 H, C(9a)OCH3], 3.22–3.38 [m, 3 H, C(2′)H, C(6′)H2], 3.62 [br d, J = 12.9 Hz, 2 H, C(3)HH′], 3.78–3.95 [m, 2 H, C(1′)H2], 3.99 [dd, J = 10.8, 3.9 Hz, 1 H, C(9)H], 4.56 [d, J = 12.9 Hz, 1 H, C(3)HH′], 5.06 [dd, J = 10.8, 10.2 Hz, 1 H, C(10)HH′], and 5.41 [dd, J = 10.2, 3.9 Hz, 1 H, C(10)HH′], the signals for the C(5′)H, C(7)NH, C(10)OC(O)NH2 and N(1a)H protons were not detected and are believed to overlap with the observed peaks, the signal for the C(2)H proton overlapped with the (CH3CH2)3N peak; m/z (+FAB) 496 ([M + 1]+, 100%); HRMS (+FAB) C21H30N5O5S2 [M + 1]+ requires 496.16884, found 496.17002.
General procedure for the solvolysis of mitomycins (kinetic studies)
To a buffered methanolic solution (0.1 M Tris·HCl, pH 7.4; 0.1 M bis-Tris·HCl, pH 5.5 and 4.0) (final volume 1.5 mL) maintained at 25 °C containing the mitomycins (10–60 μL of 4 mM methanolic solution, final concentration 0.015–0.03 mM) was added a methanolic solution (5–50 μL) of the nucleophile of choice (stock solution: 4–20 mM, final nucleophile concentration 0.03–0.6 mM). The reaction was monitored by UV-visible spectroscopy (200–600 nm), and typically followed for greater than two half-lives. The effective pH of the solution was determined at the conclusion of the reaction and found to be within ± 0.1 pH units of the original solution. The reaction products were identified by coinjection of authentic samples where possible in the HPLC and cospot of authentic samples where possible in the TLC. The absorbance for the λmax of mitomycin (ca. 374 nm) was plotted versus time and found to decrease in a first-order decay (exponential decay) process. The non-linear regression analysis to fit the observed exponential decay by SigmaPlot Program (SigmaPlot, 2001) yielded pseudo-first-order rate constants (kobs) and half-lives (t1/2). The reactions were done in duplicate and the results averaged.General procedure for alkaline agarose gel electrophoresis23,38
The agarose gels were prepared by adding 1.2 g of agarose to 100 mL of an aqueous 100 mM NaCl and 2 mM EDTA solution (pH 8.0). The suspension was heated in a microwave oven until all of the agarose was dissolved (1 min). The gel was poured and was allowed to cool and solidify at room temperature (1 h). The gel was soaked in an aqueous alkaline running buffer solution (50 mL) containing 40 mM NaOH and 1 mM EDTA (1 h) and then the comb was removed. The buffer solution was refreshed prior to electrophoresis. To an aqueous solution of ca. 85 μL of H2O (sterile) and 2.5 μL of 1 M Tris·HCl (pH 7.4) was added a solution of EcoRI-linearized pBR322 (5 μL, 5 μg) in 10 mM Tris solution containing 1 mM EDTA (pH 8.0). After deaeration with Ar (15 min), the mitomycin (1–5 μL of 1–2 mM DMSO solution, final concentration 0.01–0.1 mM) and a nucleophile of choice (1–5 μL of 1–20 mM DMSO solution, final concentration 0.05–1.0 mM) were added and the resulting solution (final volume 100 μL) was incubated at room temperature (2 h). The solution was washed with 1 : 1 PhOH/CHCl3 (100 μL) and CHCl3 (2 × 100 μL), and precipitated [12.1 μL of 3 M NaOAc and 250 μL of EtOH, −70 °C (10 min)]. The mixture was centrifuged at 0 °C (15 min), and the EtOH was decanted off and evaporated in vacuo. The remaining DNA was dissolved in 25 μL of 10 mM Tris solution containing 1 mM EDTA (pH 8.0). Agarose loading dye (5 μL) was added to the sample (5 μL) and the samples were loaded onto the wells. The gel was run at 75 mA/25 V (30 min) and then at 145 mA/38 V (3–4 h). The gel was then neutralized for 45 min in an aqueous 100 mM Tris pH 7.0 buffer solution containing 150 mM NaCl, which was refreshed every 15 min. The gel was stained with an aqueous 100 mM Tris pH 7.5 buffer solution (100 mL) containing ethidium bromide [20 μL of an aqueous ethidium bromide stock solution (10 mg in 10 mL)] and 150 mM NaCl for 20 min. The background staining was then removed by soaking the gel in an aqueous 50 mM NH4OAc and 10 mM β-mercaptoethanol solution (3 h). The gel was then analyzed by two methods. In one method, the gel was visualized by UV and photographed using Polaroid film 667. In the second method, the gel was analyzed with a StormTM 860 phosphorimager operating in the blue fluorescence mode and ImageQuant 5.0 software (Molecular Dynamics, Sunnyvale, CA).General procedure for antiproliferative activity test39
In vitro antiproliferative tests were conducted using human tumor cell line A549 (lung adenocarcinoma) by Dr Hitoshi Arai (Kyowa Hakko Kogyo Co., Shizuoka, Japan). The cells (2 × 103 cells per well) were precultured at 37 °C (24 h) in 96-well microtiterplates containing the culture medium [RPMI-1640 supplemented with 10% (v/v) fetal bovine serum, 100 U mL−1 of penicillin, and 100 μg mL−1 of streptomycin] under either aerobic (5% of CO2 and 95% of air) or hypoxic (5% of CO2 and <2% of O2) conditions. The cells were then treated with the drug candidates (1 h), washed twice with the medium and further incubated (71 h) in the drug-free medium. The antiproliferative activity of drugs against tumor cells was measured by MTT assay. Cell growth (%) was calculated by the equation, {[A − Ao]/[Ac − Ao]} × 100 (A: absorbance, Ao: blank absorbance, Ac: control absorbance), and the activity was expressed by IC50 values (concentration required for 50% inhibition).Acknowledgements
The authors gratefully acknowledge the NIH (CA29756) for support of these studies. We thank Dr Junji Kanazawa and Ms Yoshino Yamada (Kyowa Hakko Kogyo Co., Shizuoka, Japan) for conducting the in vitro antiproliferative test and Dr Masaji Kasai and Dr Hitoshi Arai (Kyowa Hakko Co., Ltd., Shizuoka, Japan) for generously supplying mitomycins A (24) and C (1), and KW-2149 (3).References
- Mitomycin C: Current Status and New Developments, S. K. Carter and S. T. Crooke, ed., Academic Press, New York, 1979 Search PubMed; W. T. Bradner, Antitumor Treat., 2001, 27, 35–50 Search PubMed.
- (a) V. N. Iyer and W. Szybalski, Science, 1964, 145, 55–58 CrossRef CAS; (b) W. Szybalski and V. N. Iyer, in Antibiotics: Mechanism of Action, D. Gottlieb and P. D. Shaw ed., Springer-Verlag, New York, 1967, vol. 1, pp. 211–245 Search PubMed; (c) H. W. Moore and R. Czerniak, Med. Res. Rev., 1981, 1, 249–280 CrossRef CAS.
- S. R. Keyes, R. Loomis, M. P. DiGiovanna, C. A. Pritsos, S. Rockwee and A. C. Sartorelli, Cancer Commun., 1991, 3, 351–356 CAS; L. A. Ramos, R. Lipman, M. Tomasz and A. K. Basu, Proc. Am. Assoc. Cancer Res., 1997, 38, 182 Search PubMed; M. Tomasz and Y. Palom, Pharmacol. Ther., 1997, 76, 73–87 CrossRef CAS.
- S. R. Rajaski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2795 CrossRef CAS.
- For representative examples, see: D. S. Bose, A. S. Thompson, J. A. Ching, J. A. Hartley, M. D. Berardine, T. C. Jenkins, S. Neidle, L. H. Hurley and D. E. Thurston, J. Am. Chem. Soc., 1992, 114, 4939–4941 Search PubMed; J. A. Hartley, A. Hazrati, T. J. Hodgkinson, L. R. Kelland, R. Khanim, M. Shipman and F. Suzenet, Chem. Commun., 2000, 2325–2326 CrossRef CAS; S. A. Gamage, J. A. Spicer, G. J. Atwell, F. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 1999, 42, 2383–2393 RSC; S. Blanchard, I. Rodriguez, C. Tardy, B. Baldeyrou, C. Bailly, P. Colson, C. Houssier, S. Léonce, L. Krause-Berthier, B. Pfeiffer, P. Renard, A. Pierré, P. Caubère and G. Guillaumet, J. Med. Chem., 2004, 47, 978–987 CrossRef CAS; S. J. Gregson, P. W. Howard, D. R. Gullick, A. Hamaguchi, K. E. Corcoran, N. A. Brooks, J. A. Hartley, T. C. Jenkins, S. Patel, M. J. Guille and D. E. Thurston, J. Med. Chem., 2004, 47, 1161–1174 CrossRef CAS; J. P. Jeyadevan, P. G. Bray, J. Chadwick, A. E. Mercer, A. Byrne, S. A. Ward, B. D. Park, D. P. Williams, R. Cosstick, J. Davies, A. P. Higson, E. Irving, G. H. Posner and P. M. O'Neill, J. Med. Chem., 2004, 47, 1290–1298 CrossRef CAS; G. H. Posner, A. J. McRiner, I.-H. Paik, S. Sur, K. Borstnik, S. Xie, T. A. Shapiro, A. Alagbala and B. Foster, J. Med. Chem., 2004, 47, 1299–1301 CrossRef CAS; J. B. Chaires, F. Leng, T. Przewloka, I. Fokt, Y.-H. Ling, R. Perez-Soler and W. Priebe, J. Med. Chem., 1997, 40, 261–266 CrossRef CAS.
- Y. Na, V.-S. Li, Y. Nakanishi, K. F. Bastow and H. Kohn, J. Med. Chem., 2001, 44, 3453–3462 CrossRef CAS.
- M. M. Paz, G. S. Kumar, M. Glover, M. J. Waring and M. Tomasz, J. Med. Chem., 2004, 47, 3308–3319 CrossRef CAS.
- M. Tomasz, R. Lipman, D. Chowdary, J. Pawlak, G. L. Verdine and K. Nakanishi, Science, 1987, 235, 1204–1208 CAS; M. Tomasz, in DNA Adducts: Identification and Biological Significance, K. Hemminki, A. Dipple, D. E. G. Shuker, D. Segerback, F. F. Kadlubar and H. Bartsch, ed., IARC, Lyon, France, 1994, pp. 349–357 Search PubMed.
- V.-S. Li and H. Kohn, J. Am. Chem. Soc., 1991, 113, 275–283 CrossRef; H. Kohn, V.-S. Li and M.-s. Tang, J. Am. Chem. Soc., 1992, 114, 5501–5509 CrossRef CAS.
- H. Arai, Y. Kanda, T. Ashizawa, M. Morimoto, K. Gomi, M. Kono and M. Kasai, J. Med. Chem., 1994, 37, 1794–1804 CrossRef CAS; W. T. Bradner, W. A. Remers and D. M. Vyas, Anticancer Res., 1989, 9, 1095–1099 CAS.
- M. Kono, Y. Saitoh, M. Kasai, A. Sato, K. Shirahata, M. Morimoto and T. Ashizawa, Chem. Pharm. Bull., 1989, 37, 1128–1130 CAS.
- D. M. Vyas, Y. Chiang, D. Benigni, W. C. Rose and W. T. Brander, in Recent Advances in Chemotherapy. Anticancer Section, J. Tshigami, ed., University of Tokyo Press, Tokyo, 1985, pp. 485–486 Search PubMed.
- E. Kobayashi, M. Okabe, M. Kono, H. Arai, M. Kasai, K. Gomi, J.-H. Lee, M. Inaba and T. Tsuruo, Cancer Chemother. Pharmacol., 1993, 32, 20–24 CrossRef CAS.
- Q.-Y. He, H. Maruenda and M. Tomasz, J. Am. Chem. Soc., 1994, 116, 9349–9350 CrossRef CAS.
- S. Wang and H. Kohn, J. Med. Chem., 1999, 42, 788–790 CrossRef CAS.
- Y. Na, S. Wang and H. Kohn, J. Am. Chem. Soc., 2002, 124, 4666–4677 CrossRef CAS.
- R. A. Norton, A. J. Finlayson and G. H. N. Towers, Phytochemistry, 1985, 24, 356–357 CrossRef CAS; C. P. Constabel, F. Blaza and G. H. N. Towers, Phytochemistry, 1988, 27, 3533–3535 CrossRef CAS; Y. Wang, M. Koreeda, T. Chatterji and K. S. Gates, J. Org. Chem., 1998, 63, 8644–8645 CrossRef CAS; R. Furumai, A. Matsuyama, N. Kobashi, K.-H. Lee, M. Nishiyama, H. Nakajima, A. Tanaka, Y. Komatsu, N. Nishino, M. Yoshida and S. Horinouchi, Cancer Res., 2002, 62, 4916–4921 CAS.
- M. Hara, I. Takahashi, M. Yoshida, K. Asano, I. Kawamoto, M. Morimoto and H. Nakano, J. Antibiot., 1989, 42, 333–335 CAS; M. Hara, K. Asano, I. Kawamoto, T. Takiguchi, S. Katsumata, K. Takahashi and H. Nakano, J. Antibiot., 1989, 42, 1786–1774; S. J. Behroozi, W. Kim and K. S. Gates, J. Org. Chem., 1995, 60, 3964–3966 CrossRef CAS.
- M. D. Lee, G. A. Ellestad and D. B. Borders, Acc. Chem. Res., 1991, 24, 235–243 CrossRef CAS; G. A. Ellestad, P. R. Hamann, N. Zein, G. O. Morton, M. M. Siegel, M. Pastel, D. B. Borders and W. J. McGahren, Tetrahedron Lett., 1989, 30, 3033–3036 CrossRef CAS; A. G. Myers, S. B. Cohen and B. M. Kwon, J. Am. Chem. Soc., 1994, 116, 1255–1271 CrossRef CAS.
- T. N. Makarieva, V. A. Stonik, A. S. Dmitrenok, B. B. Grebnev, V. V. Isakov, N. M. Rebachyk and Y. W. Rashkes, J. Nat. Prod., 1995, 58, 254–258 CrossRef CAS.
- B. S. Davidson, T. F. Molinski, L. R. Barrows and C. M. Ireland, J. Am. Chem. Soc., 1991, 113, 4709–4710 CrossRef CAS; P. A. Searle and T. F. Molinski, J. Org. Chem., 1994, 59, 6600–6605 CrossRef CAS.
- H. Zang, L. Breydo, K. Mitra, J. Dannaldson and K. S. Gates, Bioorg. Med. Chem. Lett., 2001, 11, 1511–1515 CrossRef CAS.
- S. H. Lee and H. Kohn, J. Am. Chem. Soc., 2004, 126, 4281–4292 CrossRef CAS.
- S. Kobayashi, J. Ushiki, K. Takai, S. Okumura, M. Kono, M. Kasai, K. Gomi, M. Morimoto, H. Ueno and T. Hirata, Cancer Chemother. Pharmacol., 1993, 32, 143–150 CrossRef CAS.
- (a) J. R. W. Masters, R. J. Know, J. A. Hartley, L. R. Kelland, H. R. Hendricks and T. Conners, Biochem. Pharmacol., 1997, 53, 279–285 CrossRef CAS; (b) S. R. McAdam, R. J. Knox, J. A. Hartley and J. R. W. Masters, Biochem. Pharmacol., 1998, 55, 1777–1783 CrossRef CAS.
- T. Yasuzawa and K. B. Tomer, Bioconjugate Chem., 1997, 8, 391–399 CrossRef CAS.
- C. Baylon, M.-P. Heck and C. Mioskowski, J. Org. Chem., 1999, 64, 3354–3360 CrossRef CAS; J. L. Everett and G. A. R. Kon, J. Chem. Soc., 1950, 3131–3135 RSC.
- S. H. Lee and H. Kohn, J. Org. Chem., 2002, 67, 1692–1695 CrossRef CAS.
- For the free amine, see: I. S. Matveev and N. N. Politun, US Pat., 170,520, April 23, 1965 (Chem. Abstr., 1965, 63, 9811h); I. S. Matveev and N. N. Politun, Chem. Heterocycl. Compd., 1966, 2, 375–377 Search PubMed.
- O. Mitsunobu, Synthesis, 1981, 1, 1–28 CrossRef.
- T. Hata, T. Hoshi, K. Kanamori, A. Matsumae, Y. Sano, T. Shima and R. Sugawara, J. Antibiot., 1956, 9, 141–146.
- M. Tomasz and R. Lipman, J. Am. Chem. Soc., 1979, 101, 6063–6067 CrossRef CAS; M. Tomasz and R. Lipman, Biochemistry, 1981, 20, 5056–5061 CrossRef CAS.
- Y. P. Hong and H. Kohn, J. Am. Chem. Soc., 1991, 113, 4634–4644 CrossRef CAS; I. Han and H. Kohn, J. Org. Chem., 1991, 56, 4648–4653 CrossRef CAS.
- S. Wang and H. Kohn, J. Org. Chem., 1997, 62, 5404–5412 CrossRef CAS.
- C. L. Stevens, K. G. Taylor, M. E. Munk, W. S. Marshall, K. Noll, G. D. Shah, L. G. Shah and K. Uzu, J. Med. Chem., 1964, 8, 1–10; B. S. Iyengar and W. A. Remers, J. Med. Chem., 1985, 28, 963–967 CrossRef CAS; U. Hornemann, P. J. Keller and K. Takeda, J. Med. Chem., 1985, 28, 31–36 CrossRef CAS; J. Rebek, S. H. Shaber, Y.-K. Shue, J.-C. Gehret and S. Zimmerman, J. Org. Chem., 1984, 49, 5164–5174 CrossRef; R. A. McClelland and K. Lam, J. Am. Chem. Soc., 1985, 107, 5182–5186 CrossRef CAS; R. C. Boruah and E. B. Skibo, J. Org. Chem., 1995, 60, 2232–2243 CrossRef.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- S. H. Lee and H. Kohn, Heterocycles, 2003, 60, 47–56 CrossRef CAS.
- (a) T. R. Cech, Biochemistry, 1981, 20, 1431–1437 CrossRef CAS; (b) J. J. Tepe and R. M. Williams, J. Am. Chem. Soc., 1999, 121, 2951–2955 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Viotica, J. T. Warren, H. Bokesch, S. Kenny and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- W. A. Remers, The Chemistry of Antitumor Antibiotics, Wiley, New York, 1979, vol. 1, pp. 271–276 Search PubMed; M. Tomasz, in Topics in Molecular and Structural Biology: Molecular Aspects of Anticancer Drug-DNA Interactions, S. Neidle and M. Waring, ed., Macmillan, New York, 1994, vol. 2, pp. 312–347 Search PubMed.
Footnote |
| † Dedicated to Laurence H. Hurley on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2005 |