Solid-phase synthesis of peptide-platinum complexes using platinum-chelating building blocks derived from amino acids
Marc S. Robillard, Sophie van Alphen, Nico J. Meeuwenoord, Bart A. J. Jansen, Gijs A. van der Marel, Jacques H. van Boom† and Jan Reedijk*
Leiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands. E-mail: reedijk@chem.leidenuniv.nl; Fax: +31-71-527-4671; Tel: +31-71-527-4459
First published on 8th December 2004
Abstract
Amino acids have been employed as precursors in the synthesis of platinum-chelating solid-phase building blocks. These chelating molecules were subsequently successfully used in the solid-phase synthesis of peptide-platinum complexes. The newly introduced functionality in the chelating part, as well as the nature of the pendant peptide, was shown to have an important influence on the anticancer activity of the complexes.
Introduction
The use of combinatorial chemistry complemented by high-throughput screening has developed into a well-established protocol in drug discovery. The development of an efficient synthetic procedure for platinum complexes would significantly facilitate the implementation of such a protocol in the search for novel anticancer platinum drugs. In this respect, we have demonstrated the compatibility of solid-phase synthetic strategies with platinum chemistry by the synthesis of a variety of cisplatin-like complexes and even dinuclear platinum complexes.1 Intrigued by the finding that amino acid residues and peptides can be successfully employed as site-specific DNA-interacting elements conjugated to metal complexes,2 we recently constructed a library of dichloroplatinum(II) tripeptide complexes, 1, by an automated parallel solid-phase synthesis protocol (Fig. 1).1c | ||
| Fig. 1 Peptide-platinum complexes 1 and 2 and platinum-chelating building blocks 3 and 4. | ||
Dichloroplatinum(II) tripeptide complexes 1 were designed on the basis of the assumption that the use of dipeptides B conjugated to a dichloroplatinum moiety A (from building block 3) would not only improve targeting to the DNA but also provide specific, possibly favorable additional interactions with the platinated DNA.2–4 The modular nature of structure 1 allowed for the facile introduction of a high degree of diversity by varying the amino acids in the glycine-tethered dipeptide (B).
Inspection of the structure of complexes 1 indicates that introduction of additional functionalities in the platinum chelating building block (A) itself would give complexes 2, thereby further increasing the diversity of accessible platinum structures. An appealing strategy therefore involves the construction of analogs of the platinum-chelating ethylenediamine derivative 3 (Fig. 1) from amino acids as versatile key building blocks to give ligands 4. Incorporation of 4 to give 2 would lead to the introduction of chiral centers near to the platinum complex and potential DNA-interacting moieties close to the DNA platination site. In this paper we report the preparation of platinum-chelating building blocks 4 (see Fig. 1) and their application in the solid-phase assembly of tripeptide-platinum complexes 2. The effect of the functionalities close to the platinum core, as well as the appended functionalities (e.g. arginine), on the anticancer properties of the complexes was investigated.
Results and discussion
The solid-phase assembly of the projected platinum complexes 2 commences with the solution-phase synthesis of platinum-chelating building block 4, using amino acids as starting compounds. One approach to the synthesis of target compounds 4 (Scheme 1) comprises the reductive amination of Boc–aminoaldehyde (6) with amino acids 5,5 and subsequent protective group manipulation, thereby yielding target compounds 4. Reductive amination of Boc–aminoethanal 6 with tert-butyl protected alanine (5a) or phenylalanine (5b), using NaBH3CN as reducing agent, gave tert-butyl esters 7a,b, respectively. The secondary nitrogen in 7a,b was protected with a Fmoc group, affording compounds 8a,b. Subsequent acidolysis of the Boc and tert-butyl groups, followed by Fmoc protection of the primary amine in 9a,b, resulted in bis-Fmoc protected building blocks 4a and 4b.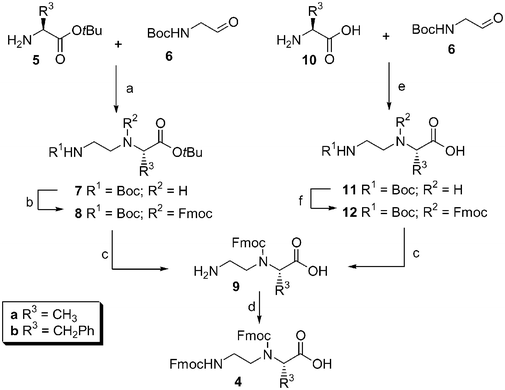 | ||
| Scheme 1 Synthesis of alanine- and phenylalanine-derived building blocks 4a and 4b. Reagents and conditions: (a) 1.3 equiv. NaBH3CN, 3% AcOH in MeOH; (b) 2 equiv. DiPEA, 1.1 equiv. Fmoc–OSu, DCM; (c) DCM–TFA (1∶1); (d) 3 equiv. DiPEA, 1.1 equiv. Fmoc–OSu, dioxane; (e) 1.1 equiv. NaBH3CN, MeOH; (f) 1 equiv. DiPEA, 1.75 equiv. BTSA, DCM, 1.1 equiv. Fmoc–Osu. | ||
An alternative approach6 uses unprotected amino acids (Scheme 1). Reductive amination of a suspension of phenylalanine (10b) in methanol afforded insoluble 11b. Dissolution of 11b in dichloromethane was mediated by in situ silylation of the carboxylic acid and the secondary amine by bis(trimethylsilyl)acetamide.7 Subsequently, the secondary amine was Fmoc-protected, giving 12b. Acidolysis of the Boc group, providing 9b, and Fmoc-protection of the resulting primary amine, furnished the phenylalanine-based building block 4b in an improved overall yield compared to the approach starting from 5.
To investigate the effect on anticancer activity of an appended potential DNA-binding guanidinium function, as well as methyl and benzyl moieties near to the platinum core, the obtained building blocks 4a and 4b and unfunctionalized 3 (R3 = H) were applied in the solid phase construction of peptide-platinum complexes 18a–f (Scheme 2). To this end, immobilized dipeptides glycine–glycine and glycine–arginine (14), prepared via a standard peptide synthesis protocol, were elongated with building blocks 3 and 4a,b to furnish the six immobilized tripeptides 15a–f (Scheme 2). The incorporation of the two new building blocks 4a and 4b proceeded successfully, as ascertained by determining the loading of 15d and 15f. Unmasking of the ethylenediamine moiety in 15, followed by treatment with K2PtCl4 (∼6 eq.) in DMF–H2O (9∶1) for 48 h in the dark, provided immobilized dichloroplatinum complexes 17a–f. Treatment of 17a–f with TFA–H2O (95∶5) resulted in removal of the arginine-protecting Pbf group and concomitant cleavage from the solid support, yielding crude 18a–f. Purification of these complexes with gel permeation chromatography (HW-40, 1% aq. AcOH) was troublesome, due to unwanted adsorption onto the column material. In response to this, the elution solvent was changed to 1% HCl in MeOH–H2O, which countered this effect and furnished homogeneous complexes 18a–f in 14–58% yield. The identity of tripeptide-platinum complexes 18a–f was fully ascertained by 195Pt NMR, 1H NMR and mass spectroscopy. In this respect it is of interest that 18b–f occur as diastereomers, due to the chiral coordinating nitrogen. The diastereomers of complexes 18c–f could be observed separately with LCMS (18c–e), 1H NMR (18c and 18d) and 195Pt NMR (18d and 18f). However, 195Pt and 1H NMR failed to detect separate diastereomers of complex 18b and insufficient column retention precluded further investigation of 18b with LCMS. The marked difference between 18b and 18c–f can be explained by the lack of the additional chiral center in the platinum chelating part of 18b. Thus, the chiral coordinating nitrogen in 18b is too far removed from the next chiral center in the molecule, the αCH of arginine, to significantly impose diastereomeric characteristics.
 | ||
| Scheme 2 Solid-phase synthesis of peptide-platinum complexes 18a–f. Reagents and conditions: (a) piperidine; (b) Fmoc–AA–OH, BOP, HOBt, DiPEA, NMP; (c) BOP, HOBt, DiPEA, NMP; (d) K2PtCl4, DMF–H2O (9∶1); e) TFA–H2O (19∶1). | ||
Having complexes 18a–f in hand, the cytotoxicity of each of them was evaluated in the human ovarian carcinoma A2780 (cisplatin sensitive) and A2780cisR (cisplatin resistant) cell lines. IC50 is defined as the concentration of the drug that results in 50% growth inhibition. The cytotoxicity results are summarized in Table 1.
| Complex | A2780 | A2780cisR |
|---|---|---|
| 18a | 24.1 | 203 |
| 18b | 131 | ≫152 |
| 18c | 123 | ≫195 |
| 18d | 136 | ≫154 |
| 18e | 186 | ≫170 |
| 18f | 31.8 | 105 |
| Cisplatin | 0.37 | 2.40 |
These results reveal that the peptide-platinum complexes 18a–f have a reduced activity compared to cisplatin and are also cross-resistant with cisplatin. Unfunctionalized 18a displayed the highest activity, closely followed by phenyalanine-derived and arginine-tethered 18f. Complexes 18b–d have similar activities and 18e was shown to be the least potent. Even though the pendant dipeptide, as well as the incorporated amino acid in the chelate backbone, seem to have a tremendous effect on the anticancer activity of these platinum complexes, the determined sequence of activity of this small set of peptide-platinum complexes does not yet allow to draw a clear structure-activity relationship.
Concluding remarks
The results described in this paper present a straightforward method for the synthesis of platinum-chelating building blocks from amino acids. The synthetic strategy is likely to be amendable to the incorporation of a wide range of amino acid residues in various positions on the backbone of the platinum chelating unit. Such modifications in the platinum complex impart structural diversity, which is required to expand the collection of distinct types of platinum structures available through a solid-phase combinatorial or parallel synthesis approach. Chelating ligands 4a and 4b were successfully used in the solid-phase synthesis of a range of peptide-platinum complexes. The newly introduced functionality in the chelating part, as well as the nature of the pendant peptide, was shown to influence the anticancer activity of the complexes. Future studies will deal with larger groups of molecules, to allow the determination of a more quantitative structure-activity relationship.Experimental
Methods and materials
1H NMR and 13C NMR spectra were recorded using a Bruker DPX 300 spectrometer. Chemical shifts (δ) are given in ppm relative to TMS as an internal standard. 195Pt spectra were taken on a Bruker DPX 300 spectrometer and were calibrated using K2PtCl4 as an external reference at δ = −1614 ppm. Electrospray mass spectra were recorded on a Finnigan MAT TSQ-70 equipped with a custom-made electrospray ionization (ESI) interface. LCMS analysis was performed on a Jasco HPLC system (detection simultaneously at 214 and 254 nm) coupled to a Perkin Elmer Sciex API 165 mass instrument. An analytical Econosphere® C18 column (AllTech, 4.6 mm × 250 mm, 5 µm particle size) was used. The applied eluents were (A) H2O; (B) CH3CN and (C) 0.5% aq. TFA. Gel permeation chromatography was performed using a Fractogel HW-40 column (26 mm × 60 cm). The flow speed was 1.5 ml min−1. The applied eluents were 1% aq. AcOH or 0.01 M HCl in H2O–MeOH (1∶1 v/v). Detection was performed at 214 nm.All solvents and reagents were obtained from commercial sources and were used as received unless stated otherwise. DCM (Baker, p.a.) and DiPEA (Biosolve) were dried by refluxing over CaH2 (5 g l−1) for 5 h, then distilled and stored over molecular sieves (4 Å). MeOH (Rathburn, HPLC grade) was stored over 3 Å molecular sieves.
TLC analysis was conducted on DC Fertigfolien (Schleicher & Schuell, F 1500, LS 254). Compounds were visualized by UV light and by spraying with a ninhydrin solution followed by charring. Eluents for column chromatography were of technical grade and distilled before use. Column chromatography was performed on Baker silica gel 60 (0.063–0.200 mm). All reactions were performed at room temperature unless stated otherwise.
General procedure for solid-phase synthesis of peptides
Peptides were synthesized on an ABI 433A (Applied Biosystems, division of Perkin Elmer) peptide synthesizer employing a FastMoc® peptide synthesis protocol. Generally, after cleavage of the Fmoc group from the resin with piperidine, a five-fold excess of amino acid was dissolved in NMP and activated by sequential addition of 1 equiv. BOP–HOBt (0.5 M BOP and 0.5 M HOBt in DMF–NMP 1∶1 v/v), and 2 equiv. DiPEA (1.25 M in NMP). The resulting solution was transferred to the reaction vessel, which was then shaken for 1 h. The coupling procedure of the first amino acid was performed twice to ensure a high initial loading. All solvents used in the automated peptide synthesis were of peptide synthesis grade and were purchased from Biosolve. Piperidine, DiPEA and TFA were from Biosolve. BOP and HOBt were from Neosystem Laboratoire (France). Rink Amide MBHA resin [4-(2′,4′-dimethoxyphenyl–Fmoc–aminomethyl) phenoxyacetamidonorleucyl–MBHA; 0.5 mmol g−1] was purchased from NovaBiochem (Switzerland). The standard Fmoc–amino acids used in the synthesis were Fmoc–Arg(Pbf)–OH and Fmoc–Gly–OH.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −tBu]+.
−tBu]+.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 145.2 (Cq arom., 4 × Fmoc), 142.6 (Cq arom., 4 × Fmoc), 128.8, 128.1, 126.0, 120.9 (CH arom., 16 × Fmoc), 68.6 (CH2, Fmoc), 67.5 (CH2, Fmoc), 57.6 (CH, αAla), 56.6 (CH, 2 × Fmoc), 47.1 (CH2, NC2H4N), 40.8 (CH2, NC2H4N), 15.6 (CH3, βAla); ESI-MS: m/z 575 [M−H]−.O), 157.3 (Cq, CO), 145.2, 145.2, 145.2, 145.1, 142.9, 142.6 (Cq arom., Fmoc), 139.6 (Cq arom., Phe), 129.9-123.7 (CH arom., 5 × Phe + 12 × Fmoc), 121.0 (CH arom., 4 × Fmoc), 68.5 (CH2, Fmoc), 67.6 (CH2, Fmoc), 64.7 (CH, αPhe), 57.1 (CH, 2 × Fmoc), 40.2 (CH2, NC2H4N), 38.7 (CH2, NC2H4N), 35.9 (CH2, βPhe); ESI-MS: m/z 651 [M−H]−.
O), 145.2 (Cq arom., 4 × Fmoc), 142.6 (Cq arom., 4 × Fmoc), 128.8, 128.1, 126.0, 120.9 (CH arom., 16 × Fmoc), 68.6 (CH2, Fmoc), 67.5 (CH2, Fmoc), 57.6 (CH, αAla), 56.6 (CH, 2 × Fmoc), 47.1 (CH2, NC2H4N), 40.8 (CH2, NC2H4N), 15.6 (CH3, βAla); ESI-MS: m/z 575 [M−H]−.O), 157.3 (Cq, CO), 145.2, 145.2, 145.2, 145.1, 142.9, 142.6 (Cq arom., Fmoc), 139.6 (Cq arom., Phe), 129.9-123.7 (CH arom., 5 × Phe + 12 × Fmoc), 121.0 (CH arom., 4 × Fmoc), 68.5 (CH2, Fmoc), 67.6 (CH2, Fmoc), 64.7 (CH, αPhe), 57.1 (CH, 2 × Fmoc), 40.2 (CH2, NC2H4N), 38.7 (CH2, NC2H4N), 35.9 (CH2, βPhe); ESI-MS: m/z 651 [M−H]−.Cytotoxicity studies
A2780 (human ovarian carcinoma) and A2780cisR (cisplatin-resistant) cell lines were grown as monolayers in DMEM (Gibco BRL™, Invitrogen Corporation, Netherlands) supplemented with 10% fetal calf serum (Gibco, Paisley, Scotland), penicillin (100 units ml−1; Duchefa, Netherlands) and streptomycin (100 µg ml−1; Duchefa, Netherlands) in a humidified 6% CO2, 94% air atmosphere, 37 °C. Cells were passed after trypsinization when the plates were 80–90% full.Cell growth inhibition by the platinum compounds was determined using the MTT assay.8 After trypsinization, cells were divided into 96-wells plates at concentrations of 3–5 × 103 cells per well in 100 µl growth medium. The cells were incubated for 24 h prior to drug testing to allow cell adhesion. Stock solutions (1 mg ml−1 complete millipore) of the compounds were freshly prepared. The dilutions (5 step dilutions) were prepared in complete medium. The concentrations used were 0.2, 0.1, 0.02, 0.01, 0.002 mg ml−1. Each concentration was tested in quadruplicate, using 100 µl per well added to the 100 µl of complete medium in the well. In the control group, 100 µl of complete medium was added. The plates were incubated for 72 h, after which MTT (5 mg ml−1, 50 µl) was added to each well, followed by incubation for 2 h. The medium was discarded and the blue formazan crystals were dissolved in DMSO (100 µl). Optical density of the wells was measured at 590 nm using a Biorad 550 microplate reader. The IC50 values (drug concentration that results in 50% reduction of cell growth with respect to the untreated control) were determined graphically using GraphPad Prism® software (version 3.05, 2000).
Acknowledgements
This research was supported by the Council for Chemical Sciences of The Netherlands Organization for Scientific Research (CW–NWO) and by The Netherlands Foundation for Technical Sciences (STW). Support and sponsorship by COST Action D8/00097 (Biocoordination Chemistry) and D20/0003 (Metal Compounds in the Treatment of Cancer) is kindly acknowledged. The authors wish to thank Johnson & Matthey (Reading, UK) for their generous loan of K2PtCl4.References
- (a) M. S. Robillard, A. R. P. M. Valentijn, N. J. Meeuwenoord, G. A. van der Marel, J. H. van Boom and J. Reedijk, Angew. Chem., Int. Ed., 2000, 39, 3096 CrossRef CAS; (b) S. van Zutphen, M. S. Robillard, G. A. van der Marel, H. S. Overkleeft, H. den Dulk, J. Brouwer and J. Reedijk, Chem. Commun., 2003, 634 RSC; (c) M. S. Robillard, M. Bacac, H. van den Elst, A. Flamigni, G. A. van der Marel, J. H. van Boom and J. Reedijk, J. Comb. Chem., 2003, 5, 821 CrossRef CAS.
- (a) N. Y. Sardesai, K. Zimmermann and J. K. Barton, J. Am. Chem. Soc., 1994, 116, 7502 CrossRef CAS; (b) N. Y. Sardesai, S. C. Lin, K. Zimmermann and J. K. Barton, Bioconjugate Chem., 1995, 6, 302 CrossRef CAS; (c) M. Lee, J. E. Simpson, Jr., A. J. Burns, S. Kupchinsky, N. Brooks, J. A. Hartley and L. R. Kelland, Med. Chem. Res., 1996, 6, 365 Search PubMed; (d) R. H. Terbrueggen, T. W. Johann and J. K. Barton, Inorg. Chem., 1998, 37, 6874 CrossRef CAS and references therein; (e) J. W. Trauger, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1998, 120, 3534 CrossRef CAS; (f) X. Huang, M. E. Pieczko and E. C. Long, Biochemistry, 1999, 38, 2160 CrossRef CAS; (g) C. A. Hastings and J. K. Barton, Biochemistry, 1999, 38, 10042 CrossRef CAS; (h) P. G. Baraldi, R. Romagnoli, A. Duatti, C. Bolzati, A. Piffanelli, N. Bianchi, C. Mischiati and R. Gambari, Bioorg. Med. Chem. Lett., 2000, 10, 1397 CrossRef CAS.
- J. Luo and T. C. Bruice, J. Am. Chem. Soc., 1997, 119, 6693 CrossRef CAS.
- M. S. Robillard, N. P. Davies, G. A. van der Marel, J. H. van Boom, J. Reedijk and V. Murray, J. Inorg. Biochem., 2003, 96, 331 CrossRef CAS.
- (a) K. L. Dueholm, K. H. Petersen, D. K. Jensen, M. Egholm, P. E. Nielsen and O. Buchardt, Bioorg. Med. Chem. Lett., 1994, 4, 1077 CrossRef CAS; (b) G. Haaima, A. Lohse, O. Buchardt and P. E. Nielsen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1939 CrossRef CAS; (c) A. Farèse, N. Patino, R. Condom, S. Dalleu and R. Guedj, Tetrahedron Lett., 1996, 37, 1413 CrossRef CAS; (d) A. Püschl, S. Sforza, G. Haaima, O. Dahl and P. E. Nielsen, Tetrahedron Lett., 1998, 39, 4707 CrossRef CAS; (e) B. Falkiewicz, K. Kowalska, A. S. Kolodziejczyk, K. Wisniewski and L. Lankiewics, Nucleosides Nucleotides, 1999, 18, 353 CAS; (f) Y. Wu and J.-C. Xu, Tetrahedron, 2001, 57, 8107 CrossRef CAS; (g) B. Falkiewicz, W. Wisniowski, A. S. Kolodziejczyk, K. Wisniewski and L. Lankiewics, Nucleosides Nucleotides, 2001, 20, 1393 CrossRef CAS; (h) B. Falkiewicz, A. S. Kolodziejczyk, B. Liberek and K. Wisniewski, Tetrahedron, 2001, 57, 7909 CrossRef CAS.
- G. Bitan, D. Muller, R. Kasher, E. V. Gluhov and C. Gilon, J. Chem. Soc., Perkin Trans. 1, 1997, 1501 RSC.
- D. R. Bolin, I.-I. Sytwu, F. Humiec and J. Meienhofer, Int. J. Peptide Protein Res., 1989, 33, 353 CAS.
- H. Tada, O. Shiho, K. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157 CrossRef CAS.
Footnote |
| † Deceased July 31, 2004. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |