Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton†
Fang C.
Mei
and
Xiaodong
Cheng
*
Department of Pharmacology and Toxicology, The University of Texas Medical Branch, Galveston, Texas 77555, USA. E-mail: xcheng@utmb.edu; Tel: 409-772-9656; Fax: 409-772-9642
First published on 26th September 2005
Abstract
“Exchange protein directly activated by cAMP” (Epac) is a newly discovered cAMP receptor that mediates the intracellular cAMP actions in addition to the classic cAMP-dependent protein kinase system. In this study, we show that Epac interacts directly with tubulin, co-purifies with cellular microtubules, and co-localizes with the mitotic spindle assembly. Association with microtubules suppresses Epac-mediated Rap1 activation, while the binding of Epac promotes microtubule formation. These results demonstrate that Epac plays an important role in connecting the microtubule cytoskeleton network and intracellular cAMP-signalling.
Introduction
Cyclic AMP, one of the most common intracellular second messengers, mediates the effect of extracellular signals associated with G-protein coupled receptors. cAMP regulates a wide range of important biological processes including metabolism, growth and differentiation, secretion, neoplastic transformation, cell division, and cytoskeleton dynamics. Microtubules are critical components of the cellular cytoskeleton network that is essential for maintaining normal cell structure and coordinating intracellular organelles. Microtubules also play a crucial role in cell division. Upon entry into mitosis, the interphase microtubule networks rapidly disintegrate and reassemble into the mitotic spindle.1 Although purified α/β tubulin heterodimers can undergo dynamic assembly/disassembly to form microtubules in vitro, microtubules in living cells are tightly modulated by a variety of accessory proteins. Microtubule-associated proteins (MAPs) facilitate microtubule formation by enhancing microtubule assembly and suppressing disassembly,2 while other proteins promote the depolymerization of microtubule either by increasing disassembly of microtubules at their ends or by generating internal breaks within microtubules.3–5 Regulation of microtubules by these accessory proteins is vital for in vivo microtubule functions.Ties between second messenger cAMP and the microtubule cytoskeleton network have long been established. cAMP regulates microtubule-associated cellular functions such as cell morphology and shape,6–10 secretion/organelle translocation,11–13 and cell division/growth.14,15 Cyclic AMP induced morphological changes in 3T3, L929, and CHO cells correlate with altered intracellular distribution of microfilaments and microtubules.6,16 This close connection between cAMP and microtubule-related cell functions suggests cAMP may play an important role in regulating the microtubule network in vivo, or vice versa. At present the mechanism and functional consequence of cyclic AMP and microtubule interaction is not clear. Two major intracellular cAMP receptors are known to mediate cAMP effects in vivo:17–19 the classic cAMP-dependent protein kinase (PKA) and the newly discovered exchange protein directly activated by cAMP (Epac). Both the catalytic and regulatory subunits of PKA have been shown to associate with microtubules. PKA can also phosphorylate MAPs such as tau20,21 and MAP222 although exactly how PKA regulates the microtubule network in vivo is not clear. The discovery of Epac opens up a new dimension in studying cAMP-mediated signalling. It is most likely that the cAMP-mediated signalling mechanism is much more complex than believed earlier, and many cAMP-mediated effects that were previously thought to act through PKA may also be transduced through Epac. The finding that Epac and PKA can exert opposing effects on cross talking with the PI3K/AKT pathways also suggests that the net outcome of cAMP signalling depends on the dynamic abundance and distribution of intracellular Epac and PKA.23 Although Epac was cloned as an upstream regulator for the small G protein Rap1, the cellular functions of Epac are not clearly understood. The finding that the subcellular localization of Epac is cell cycle dependent suggests that Epac is a dynamic and multifunctional protein.24 In the present study, we demonstrate that Epac is associated with microtubule/tubulin in vivo and in vitro. While interaction between Epac and microtubule can modulate the activation of Epac, purified recombinant Epac in turn, is capable of promoting the polymerization of microtubules via direct interaction with microtubule/tubulin in vitro. These results demonstrate that Epac's cellular functions may be closely related to the microtubule-associated cytoskeleton network.
Results
Association of Epac and α/β tubulin dimer in cells revealed by tandem immunoaffinity purification and mass spectrometry (MS)
To identify potential Epac interaction partners in vivo, we performed tandem immunoaffinity purification with immobilized anti-FLAG and anti-HA antibodies to purify cellular Epac-containing complexes from 12 litres of Hela cells that stably express Epac with FLAG and HA epitopes tagged at its C-terminus. The immunoprecipitated protein complexes were sequentially eluted from anti-FLAG and anti-HA matrixes using purified FLAG and HA peptides, respectively. Proteins that associated with Epac were analyzed by 10% Tricine–SDS gel. As shown in Fig. 1a, lane 2, several protein bands were clearly presented after tandem immunoaffinity purification, while few proteins were detected in the control mock purification using non-transfected Hela cells (Fig. 1a, lane 1). Using tryptic in-gel digestion and peptide mass fingerprinting (see Fig. S1, in the electronic supplementary information), we identified the major Epac-interacting protein band around 55 kD as α/β tubulin subunits (Fig. 1a), which was confirmed by Western blot analysis using anti-tubulin antibodies. To ensure that we mapped the interactions between Epac and its cellular partners under the most physiologically relevant conditions, we used a retroviral system that expresses moderate amounts of epitope-tagged Epac similar to the endogenous Epac level (refer to the supplementary information, Fig. S2). However, it is still possible that the apparent interaction between Epac and tubulin revealed by tandem immunoaffinity purification is non-specific, since tubulin is one of the most abundant proteins inside the cell. To further validate our finding, affinity pull-down experiments were carried out by passing an S100 fraction of COS-7 cell lysate through a column immobilized with purified recombinant Δ(1-148)Epac. Δ(1-148)Epac was used in our study as the soluble full-length Epac protein cannot be expressed and purified.25 Furthermore, published literatures so far indicate that the function of the N-terminal 148 residues is mainly associated with cellular targeting of Epac in vivo.26,24 Extensive biophysical and biochemical characterization by Wittinghofer and colleagues has shown that Δ(1-148)Epac retains all measurable biochemical functional properties such as cAMP binding and Rap1 activation. Therefore, Δ(1-148)Epac can be used as a proper model for studying Epac in vitro where membrane targeting of Epac is not necessary for its function.25 After extensive washing with high salt to remove non-specific proteins, the remaining proteins in association with the Epac affinity column were analyzed by SDS-PAGE. One major band with an apparent molecular weight of 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was observed to associate with the Epac affinity resin, but not with the beads alone (Fig. 1b). When this predominant protein band was subjected to automatic N-terminal sequencing, an amino acid sequence of MREIVHIQAG was obtained. This specific sequence matched the N-terminal sequence of β tubulin.27 After the identification of tubulin as a major component of cellular Epac complexes, we further examined if Epac could interact directly with α/β tubulin dimer. As shown in Fig. 1c, immobilized GST-Δ(1-148)Epac was capable of pulling down purified tubulin protein. This observation indicates that Epac can directly interact with tubulin.
000 was observed to associate with the Epac affinity resin, but not with the beads alone (Fig. 1b). When this predominant protein band was subjected to automatic N-terminal sequencing, an amino acid sequence of MREIVHIQAG was obtained. This specific sequence matched the N-terminal sequence of β tubulin.27 After the identification of tubulin as a major component of cellular Epac complexes, we further examined if Epac could interact directly with α/β tubulin dimer. As shown in Fig. 1c, immobilized GST-Δ(1-148)Epac was capable of pulling down purified tubulin protein. This observation indicates that Epac can directly interact with tubulin.
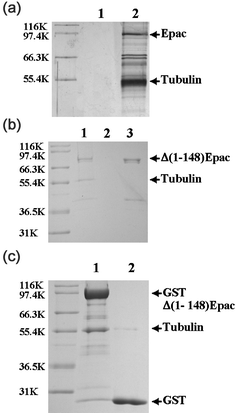 | ||
| Fig. 1 Association of cellular and purified tubulins with Epac. (a) Cellular Epac-containing complexes from Hela S cells expressing Epac-FLAG-HA fusion protein purified by tandem immunoprecipitation with antibodies specific for FLAG and HA epitopes (line 2) and control mock purification using non-transfected Hela cells (lane 1). (b) Affinity pull-down of cellular tubulin from COS-7 cell lysate by Δ(1-148)Epac affinity resin. Lanes 1 and 2, AminoLink gel with and without Δ(1-148)Epac protein coupled, respectively; Lane 3, Δ(1-148)Epac protein before coupling to AminoLink resin. (c) Affinity pull-down of purified tubulin by GST-Δ(1-148)Epac affinity resin. Lane 1, pull-down with immobilized GST-Δ(1-148)Epac affinity GSH resin; Lane 2, control pull-down with GST protein immobilized to GSH resin. Similar results were obtained with three independent experiments. | ||
Co-purification of Epac with microtubules
To test if Epac interacts with microtubule in vivo, we isolated microtubules from COS-7 cell lysate using the classic assembly–disassembly preparation procedure based on temperature-dependent, spontaneous polymerization–deploymerization of tubulin/microtubule. Microtubule-associated proteins (MAPs) can be co-purified by differential centrifugation under conditions that favor microtubule assembly.28 Microtubule preparations from cell lysates of COS-7 cells were separated on SDS-PAGE after two cycles of assembly/disassembly to remove soluble and particular cytoplasmic contaminants. Immunoblotting analysis of the isolated microtubule protein preparation using affinity-purified, anti-Epac antibody revealed that endogenous Epac co-purified with the microtubule preparation, along with other known microtubule associated proteins such as heat-shock protein 70 (Hsp70) and pyruvate kinase identified by peptide mass fingerprinting (refer to supplementary Fig. S3) and N-terminal automatic protein sequencing, respectively (Fig. 2a). Similar results were obtained when cell lysate from Hela S3 cells expressing FLAG-HA epitope-tagged Epac were used (Fig. 2a). Moreover, when purified tubulin and Δ(1-148)Epac proteins were mixed and subjected to polymerization–deploymerization cycles, Epac was observed to co-sediment with the microtubule preparation (Fig. 2b), while incubation of Epac protein alone under the same conditions did not lead to significant precipitation of Epac (date not shown). These observations suggest that Epac is a bona fide microtubule binding protein and may interact with the microtubule filaments directly in vivo.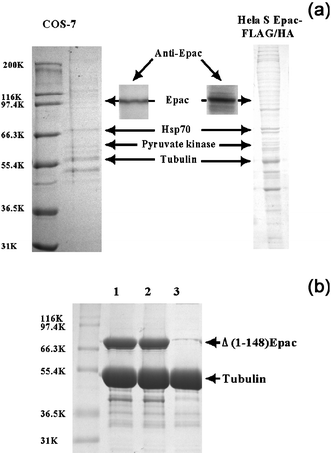 | ||
| Fig. 2 Association of Epac with cellular microtubules and purified tubulin. (a) Cellular microtubule binding proteins from COS-7 and Hela S Epac-FLAG-HA cell extracts after two cycles of warm–cold polymerization–deploymerization analyzed by Coomassie-blue staining and Western blot analysis using affinity-purified Epac antibody. The presence of Epac in the cellular microtubule preparations was demonstrated by immunoblotting analysis using affinity purified Epac antibodies (inserts). Microtubule-associated proteins, pyruvate kinase and Hsp70, were identified by N-terminal automatic protein sequencing or peptide mass fingerprinting, respectively. (b) Association of Δ(1-148)Epac with in vitro assembled microtubules. Lanes 1 and 2, tubulin with Δ(1-148)Epac in the presence or absence of cAMP, respectively, Lane 3, tubulin alone. Three experiments were performed with similar results. | ||
Epac interacts with mitotic spindle and regulates cell morphology in vivo
To further confirm the interaction between Epac and microtubule in cells, the endogenous Epac of COS-7 cells was probed by an affinity-purified Epac antibody. Immuno-fluorescent staining of Epac was observed to associate with the β-tubulin staining in centrosome/ester structures during interphase, and colocalize with the mitotic spindle and centrosomes in mitotic cells (Fig. 3a). Similar results were obtained when Epac fusion protein with green fluorescent protein tagged at the C-terminus (Epac-GFP) was used as a localization marker. Epac-GFP was observed to localize to centrosome and mitotic spindle in dividing cells (Fig. 3b). These observations were consistent with our earlier subcellular localization study of Epac.24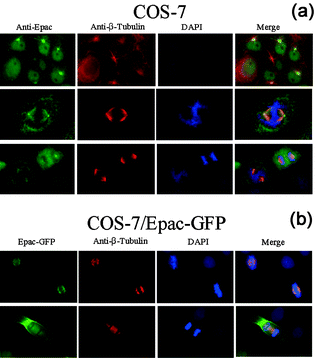 | ||
| Fig. 3 Colocalization of Epac and microtubules. (a) Co-localization of endogenous Epac with β-tubulin of microtubules in mitotic spindles as monitored by immuno-fluorescence microscopy. (b) Co-localization of Epac-GFP with microtubules in mitotic spindles as monitored by fluorescence microscopy. | ||
Association of microtubule suppresses Epac-mediated Rap activation
To determine the cellular function of Epac and microtubule interaction, we tested the effect of this interaction on Epac cellular activity by monitoring the Epac-mediated Rap1 activation in the presence of microtubule disrupting agent nocodazole. As shown in Fig. 4, pretreatment of COS-7 cells by nocodazole led to an increase of Rap1 activation. In addition, nocodazole exerted a synergistic effect on Rap1 activation when used in combination with Epac-specific agonist, 8-CPT-2′-O-Me-cAMP. This apparent effect of nocodazole on Epac-mediated Rap1 activation suggests that depolymerization of microtubule leads to Rap1 activation.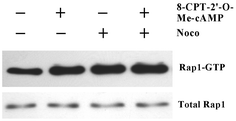 | ||
| Fig. 4 Synergistic activation of Rap1 by 8-CPT-2’-O-Me-cAMP and nocodazole. COS-7 cells, starved in serum-free medium for 24 h and pre-incubated with 5–10 µM of Nocodazole or vehicle for 45 min, were treated with or without 100 µM 8-CPT-2′-O-Me-cAMP for 15 min. GTP-bound Rap1 (Rap1GTP), probed by a GST pull-down assay and total cellular Rap1 were detected by immunoblotting with Rap1 specific antibody. Three experiments were performed with similar results. | ||
Epac promotes microtubule formation in vitro
To test if Epac interaction with tubulin affects the dynamics of microtubule polymerization–depolymerization, in vitro microtubule assembly with purified tubulin in the presence or absence of purified Δ(1-148)Epac and cAMP was monitored. When MAP-free tubulin was incubated at 37 °C in the presence of Epac, a modest increase in tubulin proteins were observed in the microtubule pellet fraction than that of tubulin only. Addition of cAMP to the reaction mixture did not lead to a further significant change in microtubule polymerization when compared to that of Epac addition alone (Lanes 1–3, Fig. 5a). The ability of Epac to promote microtubule polymerization was much more apparent when GDP was present in the reaction mixture (Lanes 4–7, Fig. 5a). To test if Epac formed specific complex with microtubule, the tubulin polymerization reaction mixture was incubated at 4 °C for 30 min before centrifugation so that the microtubule, but not the irreversible tubulin aggregates, would de-polymerize. As shown in Fig. 5a, most Epac and tubulin proteins in the pellet disappeared after the sample was incubated at 4 °C (Lane 8, Fig. 5a), suggesting that Epac in the pellet indeed bound to microtubules, which undergo reversible temperature-induced polymerization–depolymerization. These results thus demonstrate that Epac can modulate microtubule assembly. To further confirm our observations, we determined the critical concentration for microtubule assembly in the presence or absence of Epac by the classic turbidity measurement. As shown in Fig. 5b, incubation of Epac with phosphocellulose-purified tubulin led to an increase in microtubule formation as monitored by increases in light scattering at 350 nm. A protein concentration-dependence study of microtubule formation in the presence and absence of Epac showed that Epac decreased the critical concentration of tubulin needed for polymerization from 0.8 mg ml−1 (without Epac) to 0.5 mg ml−1 (with Epac); (Fig. 5b insert), further supporting the fact that Epac promotes microtubule formation in vitro.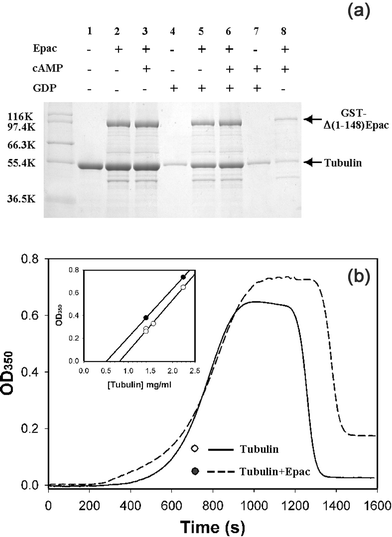 | ||
| Fig. 5 Effects of Epac on microtubule assembly. (a) Microtubule formation of purified tubulin (1 mg ml−1) with 0.2 mM GTP in the absence or presence of Epac (1 mg ml−1), cAMP (0.5 mM), and GDP (1 mM). Reaction mixtures were incubated at 37 °C for 30 min to induce the polymerization of tubulin. Microtubules were spun down at 100000 g at 37 °C for 30 min. Protein pellets were re-suspended in SDS sample buffer and analyzed on SDS-PAGE. Lane 8 was identical to lane 3 except the sample was incubated at 4 °C for 30 min to induce the depolymerization of microtubules before centrifugation. (b) In vitro microtubule assembly of purified tubulin (2.3 mg ml−1) monitored by turbidity measurement in the presence (dashed line) and absence (solid line) of Epac. Insert, microtubule assembly as a function of tubulin concentrations in the presence (closed circles) and absence (open circles) of Epac. Similar results were obtained with two independent experiments. | ||
Discussion
Since the initial discovery of Epac as a cAMP-regulated guanine nucleotide exchange factor (GEF) for Rap1, several studies have shown that Epac is a multifunctional protein that regulates an array of diverse cellular functions, such as excytosis,29 integrin-mediated cell–cell adhesion,30 cell division,24 PI3K/Akt pathways23 and secretion of the non-amyloidogenic soluble form of amyloid precusor protein.31 In this study, we have used several independent approaches to demonstrate the interaction between Epac and microtubule/tubulin. Using a tandem immunoaffinity purification approach, we isolated native Epac protein complexes from Hela S cells and further identified the α/β-tubulin dimer as a major intracellular Epac interaction partner. We have shown that cytosolic Epac co-purifies with the microtubule/tubulin polymerization–depolymerization cycles and Epac co-localizes with tubulin in the mitotic spindle in a cell cycle-specific fashion by immunofluorescence analysis. Taken together, these results suggest that tubulin/microtubule is a physiological interaction partner for Epac, and that Epac is a microtubule-associated protein that interacts directly with microtubule/tubulin.Further examination of the biochemical nature of the interaction between Epac and tubulin/microtubule is therefore of critical importance in understanding the cellular function of Epac. It has been recently reported that microtubule polymerization–depolymerization modulates the activity of nucleotide exchange factor GEF-H1.32 Could tubulin/microtubule potentially act as an upstream regulator of Epac by modulating its guanine nucleotide exchange activity? Answers to these questions are important not only for understanding the cellular functions of Epac, but also for elucidating the general mechanism of guanine nucleotide exchange factors as many GEFs have been shown to associate with the microtubule cytoskeleton. Our study demonstrates that disruption of microtubule networks by nocodazole and increase of intracellular cAMP levels synergistically activates Rap1, an Epac downstream effector. These results suggest that interaction with microtubule is indeed capable of suppressing the Epac-mediated Rap1 activation in vivo. Microtubule binding can modulate Epac activation either directly by inhibiting the catalytic activity of Epac, or indirectly by sequestering Epac in the cytoskeleton fraction to prevent the access of its substrate, Rap1. When we measured the nucleotide exchange activity of Epac on Rap1 using an in vitro fluorescence-based assay,33,25 no significant difference was observed in the presence and absence of tubulin/microtubule. Therefore, it is unlikely that tubulin/microtubule directly regulates Epac/Rap1 exchange activity in cells, and the inhibitory effect of microtubule on Epac/Rap1 activation most likely occurs through the latter mechanism—by sequestering Epac in the cytoskeleton fraction to prevent the access of its substrate, Rap1.
The ability of Epac to enhance microtubule assembly coupled with the fact that Epac associates with the microtubules/mitotic spindle imply that Epac may also play a functional role in regulating the mitotic cell division where the microtubule network undergoes rapid changes in the dynamics of polymerization–deploymerization. This is consistent with our prior observation that over-expression of Epac proteins leads to an increase in multi-nuclear cell population.24 Although our data suggest that Epac interacts directly with microtubules and promotes microtubule formation, this effect of Epac on tubulin polymerization is not regulated by cAMP in vitro when only purified tubulin and Epac were used. On the other hand, we have shown in an earlier study that interaction between Epac and microtubule network is most likely modulated by intracellular cAMP concentrations as wild-type Epac only binds to the mitotic spindle in M-phase when cAMP concentration is low, while mutant Epac defective in cAMP binding can associate with microtubules in interphase.24 Taken together, these data imply that the interaction between Epac and microtubules in vivo is probably more complicated and most likely regulated by additional cellular components. The mechanism of this cell cycle-specific colocalization of Epac with microtubules is currently under investigation.
In summary, we have provided several lines of evidence that Epac interacts with microtubule/tubulin and promotes the formation of microtubules. Our current study opens up a new dimension in our thinking about the connection between cAMP signalling and microtubule organization. The intimate relationship between Epac and microtubules suggests that Epac may be associated with biological functions such as cell division, intracellular transport, cell motility, and cell morphology, in which dynamic assembly/disassembly of microtubules is essential. Epac, in addition to PKA, may play an important role in coordinating these biological events by connecting the second messenger, cAMP, with the microtubule cytoskeleton network.
Experimental
Cell culture and transfection
HEK 293 and COS-7 cells were maintained in Dulbecco's Modified Eagle Medium with 10% fetal bovine serum at 37 °C in a humidified chamber supplemented with 5% CO2. Transfection was carried out with 50–75% confluent cells in 6-well plates and purified plasmid DNA at 1 µg per well using Lipofectamine Plus™ Reagent (GibcoBRL). Stable transfectants were selected using G418.A stable Hela S3 cell line expressing C-terminal double epitope-tagged Epac protein was generated by retrovirus infection. Briefly, amphotropic retroviral packaging Phoenix cells were transfected with pOZ-FH-C-Epac or vector alone by the calcium phosphate method, and the resulting supernatants containing retroviral particles were used to infect the Hela S3 cells. Infected cells were selected by two to three rounds of magnetic affinity sorting with anti-IL2Rα antibody-conjugated magnetic beads to achieve pure population of stable Epac-FLAG-HA expressing cells. Expression of the epitope-tagged Epac protein were confirmed by Western blot analysis using anti-FLAG and anti-HA antibodies, while the expression levels of epitope-tagged and endogenous Epac were compared using affinity purified anti-Epac antibody. Hela S3 cells were maintained in tissue culture flasks and grown in spinner bottles in S-MEM (GibcoBRL) with 10% fetal bovine serum and 0.584 mg ml−1L-glutamine at 37 °C and 5% CO2 for large-scale culture.
Tandem affinity purification of cellular Epac complexes
Cellular Epac complexes were purified from cytoplasmic S100 fraction using 12 litres of HeLa S3 cells stably expressing Epac fused with a C-terminal FLAG/HA tandem epitope tag by immunoprecipitation on anti-FLAG antibody-conjugated agarose. The bound protein complexes were eluted with purified FLAG peptide and were further affinity purified by anti-HA antibody-conjugated agarose as described previously.34Peptide mass fingerprinting
Individual protein bands of interest were excised from gel and cut into pieces smaller than 1 mm. The gel slices were sequentially washed with 250 µl of 100 mM ammonium bicarbonate, 250 µl of 50 mM ammonium bicarbonate in 50% acetonitrile, and 50 µl of 100% acetonitrile each for 30 min, and dried under a Speedvac. Depending on the band intensity and gel volume, 8–26 µl of 30 ng µl−1 sequencing grade modified trypsin (Promega) in 20 mM ammonium bicarbonate were added to cover the gel slices. After incubation at 37 °C for 6–8 h, peptides were extracted twice with 100 µl of 60% acetonitrile–0.1% TFA, dried under a Speedvac, and resuspended in 10 µl of 60% acetonitrile–0.1% TFA. One microlitre of this peptide sample was mixed with equal volume of matix (α-cyano-4-hydroxy-trans-cinnamic acid) and analyzed by MALDI-TOF mass spectrometry (Voyager-DE STR, Perkin Elmer) in the Biomedical Resource Facility to obtain peptide mass. Data were summed over 150 acquisitions in delayed extraction mode, with sensitivity ca. 10 fmol (20 kV accelerating voltage, 10 V guide wire voltage, 100 ns delay). Internal calibrations were performed using tryptic autolysis peaks at 842.5090 and 2211.1064 Da. Peptide mass data were analyzed by searching against the National Center for Biotechnology Information (NCBI) nonredundant database using search engines Protein Prospector (http://prospector.ucsf.edu/, University of California, San Francisco) and Profound (http://prowl.rockefeller.edu/cgi-bin/ProFound, Rockefeller University), with a mass tolerance of 0.10 Da and allowing 1 missed cleavage and peptide modifications by acrylamide adducts with cysteine and methionine oxidation.Protein expression and purification
GST-Δ(1-148)Epac was expressed and purified as described.25 Δ(1-148)Epac was obtained by thrombin cleavage of GST-Δ(1-148)Epac and further purified on MonoQ and Superdex 200 FPLC columns. Porcine brain tubulin (PC-tubulin, >99% pure) free of MAPs was purchased from Cytoskeleton Inc.Affinity pull-down of cellular Epac-binding proteins by immobilized Δ(1-148)Epac column
Δ(1-148)Epac affinity column was prepared by coupling 0.3 mg of purified recombinant Δ(1-148)Epac with 0.2 ml of AminoLink Coupling Gel (Pierce). Unreacted sites were fully blocked by 1.0 M Tris (pH 7.4). Ten plates of 150-mm 80% confluent COS-7 cells were harvested and lysed via French Press in buffer B (20 mM Hepes, pH 7.9, 10 mM KCl, 4 mM MgCl2, 2 mM CaCl2, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and complete protease inhibitors from Roche). S100 fraction was added to the Δ(1-148)Epac affinity resin and incubated at 4 °C overnight with gentle mixing. The resin was washed with 50 bed volumes of buffer B plus 1.0 M NaCl. Proteins that remained bound to the resin were then eluted by directly boiling in SDS sample buffer and separated on SDS-PAGE. A control experiment was done in parallel using uncoupled AminoLink Gel with all reactive sites blocked.Automatic N-terminal protein sequence analysis
Protein sample was separated on SDS-PAGE and transferred to PVDF membrane by electroblotting. Protein bands of interest were excised from the membrane and submitted to the Biomedical Resource Facility for sequencing analysis. N-terminal protein sequencing was done using an Applied Biosystem, Inc. (ABI) 475A gas phase protein/peptide microsequencer online with an ABI 120A PTH amino acid analyzer. Data were analyzed with an ABI 900A data analysis unit.Assembly/disassembly of cellular tubulin
Eight 150-mm plates of 90% confluent COS-7 or Hela S/Epac-FLAG/HA cells were harvested and re-suspended in buffer A (100 mM PIPES, pH 6.94, 2 mM MgSO4, 1 mM EGTA, 1 mM DTT). Cytoplasmic S100 fraction was generated by centrifugation of the crude cell extract at 100000 g for 60 min at 4 °C. GTP was added to the preparations at a final concentration of 2 mM and two cycles of microtubule assembly/disassembly were performed as described.35 The resulting samples were separated on SDS-PAGE. Microtubule associated proteins were identified by N-terminal automatic protein sequencing and peptide mass fingerprinting. The presence of Epac and Epac-FLAG/HA was monitored by Western blot analysis using anti-Epac and anti-HA antibodies, respectively.
Immunostaining and fluorescence microscopy
For visualization of the endogenously expressed Epac protein, COS-7 cells were grown on cover slips in 6-well plates overnight, fixed in 2% formaldehyde and 0.2% glutaraldehyde in PBS and permeabilized with 0.1% Triton X-100. After incubation with affinity-purified primary Epac antibody (2.5 µg ml−1) for 1 h at room temperature, the cells were incubated with Alexa Fluro 488 labeled goat anti-rabbit IgG secondary antibodies (5 µg ml−1). To examine the subcellular localization of Epac-EGFP, cells expressing Epac-EGFP fusion protein were subcultured in 6-well plates with a poly-L-lysine-coated cover slip in each well and grown for 16–24 h. Then the cells were either observed live or fixed in 2% paraformaldehyde in PBS at room temperature for 10 min and then in methanol at −20 °C for 1 min. For immunolabelling of microtubules, cells were fixed with methanol at −20 °C for 10 min, rinsed twice with cold acetone for 10 s, rehydrated in PBS for 1 h, and followed by immunochemical staining using Cy3 conjugated monoclonal anti-β-tubulin antibody and DAPI solution. The samples were then rinsed with PBS, mounted on glass slides and sealed in 70% glycerol. Fluorescent signals were revealed under the fluorescence microscope (Olympus BX51), using appropriate filters. Digital images were recorded using a Hamamatsu CCD camera (C4742-95).Rap1 activation assay
The GTP loading status of Rap1 was assessed using a glutathione S-transferase (GST) fusion of the Rap1 binding domain (RBD) of RalGDS as described earlier (27). Briefly, cells were grown to 75% confluence in a 100 mm Corning culture dish, starved in serum-free medium for 48 h, and treated with 10 µM forskolin or 100 µM 8-CPT-2’-O-Me-cAMP for 15 min. Following three washes in PBS, the cells were lysed in a buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 20 mM MgCl2, 5 mM EGTA, 1% Triton X100, and Roche EDTA-free protease inhibitors. The cell lysate was mixed with 40 µl of glutathione–Sepharose beads with 30 µg of GST-RalGDS-RBD bound and incubated at 4°C for 2 h with gentle agitation. Following three washes in lysis buffer, the beads were suspended in 40 µl of SDS sample buffer. 15 µl of protein samples were loaded onto a 15% SDS poly(acrylamide) gel and further analyzed with Western blot using Rap1 specific antibodies.In vitro assembly of tubulin
Microtubule assembly with purified tubulin in the presence or absence of Δ(1-148)Epac or GST-Δ(1-148)Epac was monitored by sedimentation or light scattering assay following the development of turbidity at 350 nm in a temperature controlled Hitachi 3010 spectrophotometer as described previously.36 Briefly, PC-purified tubulin at various concentrations was incubated in buffer A with additional 14 mM MgCl2 and 2.5 mM GTP in the presence and absence of 1 : 5 molar ratio of Epac at 10 °C in a thermostatted cell holder. After collecting a two-minute baseline, the sample temperature was shifted to 37 °C and the formation of microtubules was monitored continuously as a function of time until a plateau was reached, at which point the temperature was again switched back to 10 °C to induce the depolymerization of microtubules.Acknowledgements
This work is supported by NIH grant GM66170. We are indebted to Dr James C. Lee and Dr John Lew (University of California, Santa Barbara) for continuous support and insightful discussions. We thank Dr Chunming Liu for support and advice on large scale immunoaffinity purification of human Epac complexes, Dr Alex Kurosky and Dr Tony Haag in the Biomedical Resource Facility (supported by NCI grant R24CA88317) for mass spectrometry analysis, and Dr Istvan Boldogh and Dr Denial Liebenthal in the Cell Biology Service Core Laboratory of the NIEHS Center (supported by NIEHS Center Grant ES06676) for large-scale cell culture.References
- Y. Zhai, P. J. Kronebusch, P. M. Simon and G. G. Borisy, J. Cell Biol., 1996, 135, 201–214 CrossRef CAS.
- D. N. Drechsel, A. A. Hyman, M. H. Cobb and M. W. Kirschner, Mol. Biol. Cell, 1992, 3, 1141–1154 CAS.
- L. D. Belmont and T. J. Mitchison, Cell, 1996, 84, 623–631 CrossRef CAS.
- R. D. Vale, Cell, 1991, 64, 827–839 CrossRef CAS.
- N. Shiina, Y. Gotoh and E. Nishida, EMBO J., 1992, 11, 4723–4731 CAS.
- M. C. Willingham and I. Pastan, J. Cell Biol., 1975, 67, 146–159 CrossRef CAS.
- L. S. Borman, J. N. Dumont and A. W. Hsie, Exp. Cell Res., 1975, 91, 422–428 CrossRef CAS.
- K. Nath, J. W. Shay and A. P. Bollon, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 319–323 CAS.
- S. R. Heidemann, H. C. Joshi, A. Schechter, J. R. Fletcher and M. Bothwell, J. Cell Biol., 1985, 100, 916–927 CrossRef CAS.
- T. T. Puck, P. Webb and R. Johnson, Ann. N. Y. Acad. Sci., 2002, 968, 122–138 CrossRef CAS.
- M. W. Bitensky, C. N. Keirns and J. J. Keirns, Ann. N. Y. Acad. Sci., 1975, 253, 685–691 CrossRef CAS.
- G. Weissmann, I. Goldstein, S. Hoffstein and P. K. Tsung, Ann. N. Y. Acad. Sci., 1975, 253, 750–762 CrossRef CAS.
- P. J. Sammak, S. R. Adams, A. T. Harootunian, M. Schliwa and R. Y. Tsien, J. Cell Biol., 1992, 117, 57–72 CrossRef CAS.
- T. T. Puck, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4491–4495 CAS.
- Z. W. Wang and E. Rozengurt, J. Cell Biol., 1983, 96, 1743–1750 CrossRef CAS.
- K. R. Porter, T. T. Puck, A. W. Hsie and D. Kelley, Cell, 1974, 2, 145–162 CrossRef CAS.
- S. S. Taylor, J. A. Buechler and W. Yonemoto, Annu. Rev. Biochem., 1990, 59, 971–1005 CrossRef CAS.
- J. de Rooij, F. J. Zwartkruis, M. H. Verheijen, R. H. Cool, S. M. Nijman, A. Wittinghofer and J. L. Bos, Nature, 1998, 396, 474–477 CrossRef CAS.
- H. Kawasaki, G. M. Springett, N. Mochizuki, S. Toki, M. Nakaya, M. Matsuda, D. E. Housman and A. M. Graybiel, Science, 1998, 282, 2275–2279 CrossRef CAS.
- C. W. Scott, R. C. Spreen, J. L. Herman, F. P. Chow, M. D. Davison, J. Young and C. B. Caputo, J. Biol. Chem., 1993, 268, 1166–1173 CAS.
- H. C. Tseng, Q. Lu, E. Henderson and D. J. Graves, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9503–9508 CrossRef CAS.
- W. E. Theurkauf and R. B. Vallee, J. Biol. Chem., 1982, 257, 3284–3290 CAS.
- F. C. Mei, J. Qiao, O. M. Tsygankova, J. L. Meinkoth, L. A. Quilliam and X. Cheng, J. Biol. Chem., 2002, 277, 11497–11504 CrossRef CAS.
- J. Qiao, F. C. Mei, V. L. Popov, L. A. Vergara and X. Cheng, J. Biol. Chem., 2002, 277, 26581–26586 CrossRef CAS.
- A. Kraemer, H. R. Rehmann, R. H. Cool, C. Theiss, J. de Rooij, J. L. Bos and A. Wittinghofer, J. Mol. Biol., 2001, 306, 1167–1177 CrossRef CAS.
- J. de Rooij, H. Rehmann, M. van Triest, R. H. Cool, A. Wittinghofer and J. L. Bos, J. Biol. Chem., 2000, 275, 20829–20836 CrossRef CAS.
- C. D. Wilde, C. E. Crowther, T. P. Cripe, L. M. Gwo-Shu and N. J. Cowan, Nature, 1982, 297, 83–84 CrossRef CAS.
- D. B. Murphy, Methods Cell Biol., 1982, 24, 31–49 Search PubMed.
- N. Ozaki, T. Shibasaki, Y. Kashima, T. Miki, K. Takahashi, H. Ueno, Y. Sunaga, H. Yano, Y. Matsuura, T. Iwanaga, Y. Takai and S. Seino, Nat. Cell Biol., 2000, 2, 805–811 CrossRef CAS.
- S. Rangarajan, J. M. Enserink, H. B. Kuiperij, J. de Rooij, L. S. Price, F. Schwede and J. L. Bos, J. Cell Biol., 2003, 160, 487–493 CrossRef CAS.
- M. Maillet, S. J. Robert, M. Cacquevel, M. Gastineau, D. Vivien, J. Bertoglio, J. L. Zugaza, R. Fischmeister and F. Lezoualc'h, Nat. Cell Biol., 2003, 5, 633–639 CrossRef CAS.
- M. Krendel, F. T. Zenke and G. M. Bokoch, Nat. Cell Biol., 2002, 4, 294–301 CrossRef CAS.
- B. N. Van den, R. H. Cool, G. Horn and A. Wittinghofer, Oncogene, 1997, 15, 845–850 CrossRef.
- Y. Nakatani and V. Ogryzko, Methods Enzymol., 2003, 370, 430–444 CAS.
- B. W. Nagle, K. H. Doenges and J. Bryan, Cell, 1977, 12, 573–586 CrossRef CAS.
- F. Gaskin, C. R. Cantor and M. L. Shelanski, J. Mol. Biol., 1974, 89, 737–755 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3 including Western blot analysis of ectopic and endogenous Epac expression levels, mass spectrograms of tryptic in-gel digestions for tubulin and heat-shock protein 70. See DOI: 10.1039/b511267b |
| This journal is © The Royal Society of Chemistry 2005 |