Synthesis of an IGD peptidomimetic with motogenic activity†‡
Natalia
Shpiro
a,
Ian R.
Ellis
b,
Trevor J.
Dines
c,
Ana M.
Schor
b,
Seth L.
Schor
b,
David G.
Norman
d and
Rodolfo
Marquez
*a
aDivision of Biological Chemistry and Mol. Microbiology, School of Life Sciences, University of Dundee, Dundee, Scotland, UK. E-mail: r.marquez@dundee.ac.uk; Fax: +44 (0)1382 345517; Tel: +44 (0)1382 348969
bUnit of Cell and Molecular Biology, The Dental School, University of Dundee, Dundee, Scotland, UK
cDivision of Physical and Inorganic Chemistry, School of Life Sciences, University of Dundee, Dundee, Scotland, UK
dCancer Research UK, Nucleic Acid Structure Research Group, School of Life Sciences, University of Dundee, Dundee, Scotland, UK
First published on 26th August 2005
Abstract
Rational design and synthesis of an IGD peptidomimetic substrate with significant motogenic activity.
Introduction
Migration Stimulating Factor (MSF) is a novel stress-response molecule expressed by epithelial and stromal cells in fetal skin and common human tumours.1 MSF is not normally present in healthy adult skin, but is transiently re-expressed during wound healing. Human recombinant MSF displays a number of potent bioactivities relevant to wound healing and cancer progression, including the stimulation of cell migration (target: carcinoma cells, keratinocytes, dermal fibroblasts, endothelial cells), stimulation of hyaluronan synthesis by fibroblasts, and angiogenesis in vivo and in vitro.1MSF is a truncated isoform of fibronectin produced from the primary fibronectin gene transcript by a bypass of normal alternative splicing involving read-through of the intron separating exons III-1a and -1b. Intron retention results in the inclusion of a unique 30 bp coding sequence. MSF protein is consequently identical to the 70 kDa N-terminus of fibronectin, including nine type I and two type II modules, and terminates with the sequence coded by module III-1a and a unique decamer not present in any previously described “full-length” fibronectin isoform.
The IGD (isoleucine, glycine, aspartate) tripeptide motif, a highly conserved feature of the fibronectin type I module, is present within the third, fifth, seventh and ninth constituent type I modules of MSF (Fig. 1).2 Interestingly, synthetic trimer and tetramer peptides containing the IGD motif exhibit the same range of biological activities as those displayed by MSF.3 Furthermore, in vitro mutagenesis and analysis of IGD-recombinant constructs has demonstrated that the motogenic activity of MSF on target fibroblasts is mediated by the IGD sequences.1
On the basis of this information, our objective has been to synthesise an IGD peptidomimetic framework which expresses MSF/IGD bioactivities and shows improved stability compared to the IGD tripeptide. Such a peptidomimetic would be a starting point for the development of a new family of therapeutic agents for the management of patients with impaired wound healing and other pathologies requiring the stimulation of cell migration and angiogenesis.
 | ||
| Fig. 1 Primary sequence of the fibronectin/MSF type I modules–the IGD sequences are shown in bold, and the conserved cysteine positions are indicated by light-grey highlight. | ||
The feasibility of generating biologically active small molecule peptidomimetics has been previously demonstrated by the synthesis of RGD peptide mimetics as possible inhibitors of angiogenesis, metastasis and other processes dependent upon cell adhesion and migration.
Careful examination of the NMR structural information available for the fnI-5 and fnI-7 modules revealed that the desired IGD motif is present as a tightly structured peptide turn (Fig. 2). We believe that it is this highly conserved structure which is then recognised by the relevant cell receptors.4,5
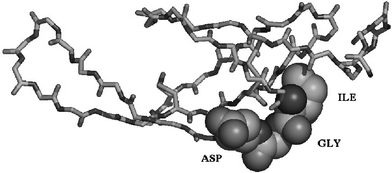 | ||
| Fig. 2 Three dimensional structure of fnI-7 backbone with space filling representation of the IGD motif. | ||
The observation that the IGD containing loops in fnI-5 and fn1-7 are tightly defined (unlike most of the known RGD containing loops which are largely unstructured and flexible), led us to predict that small molecule IGD peptidomimetics bearing similar geometrical and stereo-electronic arrangements to those observed in the fnI structure might be recognised by the MSF (IGD) receptors. Successful receptor recognition should translate into a similar dose–response curve to that of an IGD peptide and, by extension, to that of intact MSF.
Results and discussion
Rational IGD mimetic design
Our initial step into the IGD peptidomimetic design involved the determination and measurement of the backbone dihedral angles observed in the IGD sequences of two independently obtained NMR structures of fnI-5 and fnI-7 (Table 1). Furthermore, the relative closeness of the measured dihedral angles across both structures made us confident that a suitable mimetic could be designed.| PHI fnI-7 | PHI fnI-5 | PSI fnI-7 | PSI fnI-5 | |
|---|---|---|---|---|
| I | −52 | −83 | 124 | 132 |
| G | 104 | 82 | −2 | 9 |
| D | −92 | −89 | 162 | −177 |
After careful consideration of the dihedral angles present within the two loops, a number of small molecular weight entities that could potentially mimic the geometrical features of the IGD motif, within the protein framework, were selected. The selected structures were then optimised and their energy minimised using GAUSSIAN ab initio level calculations using the hybrid SCF-DFT method B3-LYP6 with the Dunning–Huzinaga double-zeta (DZ) basis set, incorporating polarization functions on C, N and O atoms.
The results of these modelling studies clearly indicated that a benzodiazepine (BDP) bicyclic ring system should closely resemble the IGD backbone conformation (Fig. 3).
 | ||
| Fig. 3 (A) Highlight of the backbone bonds of the IGD turn of fnI-7. (B) Highlight of the bonds of the benzodiazepine (BDP) unit that fit to the IGD turn. | ||
Having determined that the benzodiazepine ring system should closely resemble the IGD backbone, we then focused on the benzodiazepine substitution pattern that could then mimic the steric and electronic properties of the IGD unit within fnI.
GAUSSIAN calculations, as previously described, were used to test spatial and electronic arrangements which would mimic the isoleucine and aspartic acid residue mimics with a view to maintaining the correct conformation of the BDP core. The glycine residue was envisioned as being closely mimicked by the BDP core itself.
In the first instance, our modelling results indicated that the aspartic acid mimic as the carboxymethyl (ester or free acid) should be located at the C2 position of the BDP skeleton, with the corresponding S configuration at the newly designed stereo-centre. A number of alternatives for the isoleucine side chain mimic were modelled before eventually settling for a 3-(methyl) butyl unit connected by an ether linkage at the C9 position. A rather less flexible amide linkage was contraindicated by our modelling studies, which predicted an unfavourable intramolecular hydrogen bond which would cause an undesired conformational change.
The combined results of this modelling approach generated the benzodiazepine unit 1. Not surprisingly, our proposed unit 1 shares a degree of similarity with other γ-turn peptidomimetics such as benzodiazepine 2, which has been previously used as an RGD mimetic (Fig. 4).7–10
 | ||
| Fig. 4 Proposed first generation IGD peptidomimetic 1 and RGD peptidomimetic unit 2. | ||
Synthesis of the first generation IGD peptidomimetic unit 1
Once our modelling studies provided us with a suitable candidate for development, our efforts turned to the design and implementation of a flexible synthesis of benzodiazepine 1. Thus, our synthesis of the prototype IGD peptidomimetic unit 1 began with commercially available nitro-cresol 3, which was cleanly alkylated and then brominated under radical conditions to generate the benzylic bromide 4 together with a small amount of the undesired dibromide 5 as an inseparable mixture of compounds.11,12An efficient SN2 nucleophilic substitution of methylamine on the benzylic bromides 4 and 5 then proceeded to cleanly afford the desired benzyl amines 6 and 7 respectively.13 BOC protection of the newly introduced benzylic amine then generated the nitrobenzene units 8 and 9, which under palladium hydrogenation conditions reduced both the nitro and the tertiary bromide functionalities to produce the free amine unit 10 as a single compound (Scheme 1).14
 | ||
| Scheme 1 Synthesis of aniline 10. | ||
Condensation of aniline 10 with dimethyl acetylenedicarboxylate proceeded cleanly to produce the expected dimethyl diester 11.15 Palladium mediated hydrogenation of the alkene double bond reduction then afforded the desired diester 12 as a racemic mixture.
Finally, efficient removal of the BOC protecting group afforded the free benzylic methyl amine 13 in excellent yield, which was then selectively cyclised with the adjacent methyl ester to generate the first IGD mimetic 1 ever reported thus far (Scheme 2).16
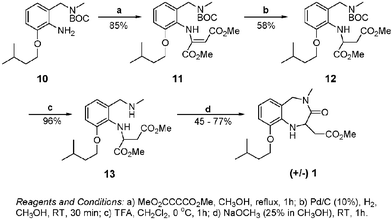 | ||
| Scheme 2 Synthesis of IGD peptidomimetic 1. | ||
Biological assessment
The potency and efficacy of the IGD mimetic 1 was tested on the migration of human skin fibroblasts in the 3D collagen gel assay, as previously used to demonstrate the motogenic activity of IGD synthetic peptides.3Significantly, the IGD peptidomimetic 1 stimulated target cell migration with a bell-shaped dose–response similar in potency and profile to that of IGDS under the same assay conditions. Crucially, the control reverse peptide (SDGI) was completely devoid of bioactivity in this assay (Fig. 5).
 | ||
| Fig. 5 The motogenic activity of IGD peptidomimetic 1. The effects of peptidomimetic 1 (BDP 1) and synthetic peptides (IGDS and SDGI) on fibroblast migration were assessed in the collagen gel assay. Data are expressed as fold-stimulation of migration relative to control fibroblasts in the absence of the test compound. The range of baseline (control) migration is indicated by the horizontal dotted lines. | ||
Conclusion
We have successfully designed and synthesised the first IGD peptidomimetic, with significant biological activity, reported to date. Recent animal model studies indicating that IGDS synthetic peptides are potent stimulators of new blood vessel formation (angiogenesis) and significantly accelerate dermal wound healing in genetically diabetic mice (unpublished data) provide a rational platform for the development of clinically relevant IGD peptidomimetics. We are currently working on the development of non-racemic versions of our IGD mimetic as well as on new structural derivatives, with an aim to increasing their biological potency and/or altering their biological action.Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council and the Scottish Enterprise Proof of Concept Fund for financial support.Notes and references
- (a) S. L. Schor, I. R. Ellis, S. J. Jones, R. Baillie, K. Seneviratne, J. Clausen, K. Montegi, B. Vojtesek, K. Kankova, E. Furrie, M. J. Sales, A. M. Schor and R. A. Kay, Cancer Res., 2003, 63, 8827 CAS; (b) S. L. Schor, A. M. Schor, R. P. Keatch and J. J. F. Belch, The Wound Healing Manual, McGraw Hill, New York, 2005, pp. 109–121 Search PubMed; (c) S. L. Schor and A. M. Schor, unpublished results Search PubMed.
- R. Hynes, Fibronectins, Springer-Verlag, New York, 1990 Search PubMed.
- S. L. Schor, I. Ellis, J. Banyard and A. M. Schor, J. Cell Sci., 1999, 112, 3879 Search PubMed.
- M. Baron, D. Norman, A. Willis and I. D. Campbell, Nature, 1990, 345, 642 CrossRef CAS; M. J. Williams, I. Phan, T. S. Harvey, A. Rostagno, L. I. Gold and I. D. Campbell, J. Mol. Biol., 1994, 235, 1302 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- R. M. Keenan, J. F. Callahan, J. M. Samanen, W. E. Bondinell, R. R. Calvo, L. C. Chen, C. DeBrosse, D. S. Eggleston, R. C. Haltiwanger, S. M. Hwang, D. R. Jakas, T. W. Ku, W. H. Miller, K. A. Newlander, A. Nichols, M. F. Parker, L. S. Southhall, I. Uzinskas, J. A. Vasko-Moser, J. W. Venslavsky, A. S. Wong and W. F. Huffman, J. Med. Chem., 1999, 42, 545 CrossRef CAS.
- W. H. Miller, D. P. Alberts, P. K. Bhatnagar, W. E. Bondinell, J. F. Callahan, R. R. Calvo, R. D. Cousins, K. F. Erhard, D. A. Heerding, R. M. Keenan, C. Kwon, P. J. Manley, K. A. Newlander, S. T. Ross, J. M. Samanen, I. N. Uzinskas, J. W. Venslavsky, C. C. K. Yuan, R. C. Haltiwanger, M. Gowen, S. M. Hwang, I. E. James, M. W. Lark, D. J. Rieman, G. B. Stroup, L. M. Azzarano, K. L. Salyers, B. R. Smith, K. W. Ward, K. O. Johanson and W. F. Huffman, J. Med. Chem., 2000, 43, 22 CrossRef CAS.
- T. W. Ku, F. E. Ali, L. S. Barton, J. W. Bean, W. E. Bondinell, J. L. Burgess, J. F. Callahan, R. R. Calvo, L. C. Chen, D. S. Eggleston, J. G. Gleason, W. F. Huffman, S. M. Hwang, D. R. Jakas, C. B. Karash, R. M. Keenan, K. D. Kopple, W. H. Miller, K. A. Newlander, A. Nichols, M. F. Parker, C. E. Peishoff, J. M. Samanen, I. Uzinskas and J. W. Venslavsky, J. Am. Chem. Soc., 1993, 115, 8861 CrossRef CAS.
- E. Addicks, R. Mazitschek and A. Giannis, ChemBioChem, 2002, 3, 1078 CrossRef CAS.
- T. Kimura, N. Watanabe, M. Matsui, K. Hayashi, H. Tanaka, I. Ohtsuka, T. Saeki, M. Kogushi, H. Kabayashi, K. Akasaka, Y. Yamagishi, I. Saitou and I. Yamatsu, J. Med. Chem., 1993, 36, 1641 CrossRef CAS.
- D. D. Tanner, C. P. Meintzer, E. C. Tsai and H. Oumarmahamat, J. Am. Chem. Soc., 1990, 112, 7369 CrossRef CAS.
- M. E. Krolski, A. F. Renaldo, D. E. Rudisill and J. K. Stille, J. Org. Chem., 1988, 53, 1170 CrossRef CAS.
- B. C. Soderberg, S. R. Rector and S. N. O'Neil, Tetrahedron Lett., 1999, 40, 3657 CrossRef CAS.
- W. H. Miller, K. A. Newlander, D. S. Eggleston and R. C. Haltiwanger, Tetrahedron Lett., 1995, 36, 373 CrossRef CAS.
- W. E. Bondinell, W. H. Miller, F. E. Ali, A. C. Allen, C. W. DeBrosse, D. S. Eggleston, K. F. Erhard, R. C. Hatiwanger, W. F. Huffman, S.-M. Hwang, D. R. Jakas, P. F. Koster, T. W. Ku, C. P. Lee, A. J. Nichols, S. T. Ross, J. M. Samanen, R. E. Valocik, J. A. Vasko-Moser, J. W. Venlavsky, A. S. Wong and C.-K. Yuan., Bioorg. Med. Chem., 1994, 2, 897 CrossRef CAS.
- 1H NMR ( 300 MHz, CDCl3) δH 6.64 (1H, dd, J = 7.0, 2.4 Hz), 6.54–6.46 (2H, m), 5.41 (1H, d, J = 16.3 Hz), 5.03–4.97 (1H, m), 4.34 (1H, d, J = 4.2 Hz), 3.94–3.86 (2H, m), 3.66 (3H, s), 3.64 (1H, d, J = 16.2 Hz), 2.99 (3H, s), 2.98 (1H, dd, J = 15.8, 7.4 Hz), 2.61 (1H, dd, J = 15.8, 6.3 Hz), 1.76–1.69 (1H, m), 1.61 (2H, q, J = 6.6 Hz), 0.89 (6H, d, J = 6.5 Hz). 13C NMR (75 MHz, CDCl3) δC 172.1, 169.9, 147.0, 135.3, 121.7, 119.6, 117.1, 111.0, 67.2, 53.4, 52.3, 51.9, 38.3, 36.6, 35.0, 25.6, 23.0.
Footnotes |
| † Dedicated to Professor Steve Ley on his 60th Birthday. |
| ‡ Electronic supplementary information (ESI) available: Migration Assay and Spectral Data for IGD mimetic 1. See http://dx.doi.org/10.1039/b509023g |
| This journal is © The Royal Society of Chemistry 2005 |