Xenopus as a model organism in developmental chemical genetic screens†
Matthew L.
Tomlinson
a,
Robert A.
Field
b and
Grant N.
Wheeler
*a
aSchool of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: grant.wheeler@uea.ac.uk; Fax: +44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (0)01603592250; Tel: +44(0)01603593988
(0)01603592250; Tel: +44(0)01603593988
bSchool of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, NR4 7TJ, UK. Fax: +44(0)1603592003; Tel: +44(0)1603593983
First published on 5th August 2005
Abstract
Chemical genetics is a potentially powerful tool for studying developmental processes in vertebrate systems. We present data showing Xenopus laevis as a model organism in which systematic chemical genetic screens can be carried out. Previous forward chemical genetic screens, including those with developing zebrafish embryos, have demonstrated the nature and value of biological information gained with this approach. We show how amenable Xenopus is to chemical genetics by investigating a series of compounds either with known biochemical effects, or previously identified to give developmental phenotypes, on a range of biological functions, including the development of pigmentation, the heart and the central nervous system in zebrafish. We have found that the compounds give comparable phenotypes when applied to developing Xenopus embryos. We have also studied the penetrance and expressivity of these chemical genetic phenotypes in relation to genetic variation and the developmental window during which the compound is present. Finally, we assess the feasibility and the potential throughput of a screen in this vertebrate species.
Introduction
Chemical genetics has been a term used since the 1930s to describe studies of the difference in chemical makeup between mutant strains of various organisms.1 Over the past 10–15 years it has altered such that the chemicals studied are not generated by the organism, but rather are generated by synthetic chemists often using combinatorial chemistry techniques.2 Chemical genetics has recently received renewed interest from the biological community (reviewed in ref. 3–9). The premise of modern chemical genetics10–13 is that, with the aid of appropriate high throughput synthesis (chemical and/or enzymatic) and biological screening strategies (typically whole cell phenotype or isolated enzyme assays), it may be feasible to identify specific small molecule inhibitors (either synthetic compounds or natural products) of any biochemical event. In parallel to this renewed interest from the academic community, there has also been growing interest from the drug discovery community (see ref. 14 for a review) in using phenotype-based hit-to-lead type screens, with either cell based assays15 or model organisms.16Chemical genetics offer a complimentary approach to loss-of-function mutations in the analysis of complex, multi-component biological processes, such as development or signal transduction. This approach provides fundamental challenges for synthetic and analytical chemistry on one hand, and novel opportunities to manipulate biological systems on the other.4,17,18 The drive to use small molecules as general tools to explore biological questions has led to the development of combinatorial chemistry approaches to generate libraries of structurally and functionally diverse molecules.19–21 These new libraries, along with high throughput screening methods, have been applied in a number of labs to look at questions in bacteriology,22 cancer biology,23 neurobiology24 and stem cell biology.13
Whole animal high-throughput screens have been and are being developed with the model organisms Drosophila and C. elegans (see ref. 25 for a review). These screens have concentrated on elucidating proteins in pathways for human disease models and thus are more concentrated on physiology than development. The divergence of their protein structure and function from vertebrates (Fig. 1) has meant that, although a great deal of physiological information has been discovered, their role in developmental biology, especially with respect to organogenesis, may be limited in scope. The use of genetic model systems has made large-scale mutagenesis screens in early development feasible.26 However, these screens do not allow for the finer temporal control of protein function. During embryonic development, many patterning and signalling systems are used multiple times.27–29 This means that mutagenesis screens will disguise later developmental events, in particular during organogenesis. The ability to have temporal control over compound addition, and thus the modulation of protein function, provides a more focused approach to phenotypic assays. With respect to vertebrate development, the number of compounds so far identified as having a specific developmental effect is small and such compounds often come from natural sources, as in the case of cyclopamine which blocks the hedgehog signalling pathway.30 The present exploitation of zebrafish in chemical genetic screening has opened up new opportunities. To date, chemical genetic screens have identified compounds that modulate embryonic development31 as well as enabling drug-target discovery,32 target validation, and toxicological studies.33,34
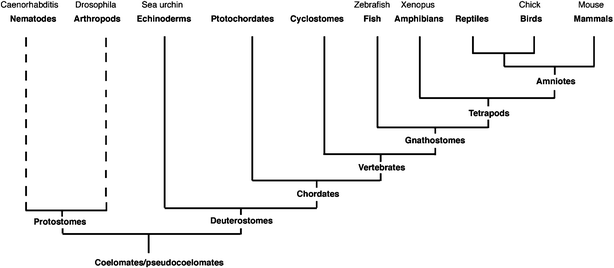 | ||
| Fig. 1 Phylogenetic tree, showing the main model organisms commonly used in research and their evolutionary relationship. | ||
The use of chemical inhibitors as tools for functional analysis in Xenopus development has been demonstrated using cyclopamine and FGF receptor inhibitors such as SU5402.35,36Xenopus laevis has been at the forefront of research in developmental biology for much of the second half of the last century. Its ease of use with respect to the generation of embryos, surgical manipulation and molecular intervention including transgenic methods is widely recognised. Much current understanding of the development of the central nervous system, gastrulation and early events in organogenesis of the eye, heart, kidney has come from studies in Xenopus.37–41 However, as a genetic system it has not been useful due to the tetraploid genome and long maturation time for breeding. In this paper we show that Xenopus laevis can be used as a model organism for chemical genetic screens and is amenable to high throughput screening techniques. Xenopus laevis should appeal to the chemical geneticist looking to begin investigation with a higher vertebrate system (Fig. 1) for developmental and/or hit to lead studies in drug discovery. In addition, chemical genetics approaches will expand the tool kit for this already versatile and highly useful model organism.
Results
Compounds assayed in developing Xenopus embryos
In order to assess the amenability of Xenopus laevis to chemical genetic studies we identified the most applicable and relevant set of compounds from a developmental chemical genetic screen previously performed using zebrafish.31Table 1 shows the chemical structures of the assayed compounds. Fig. 2a–f and Table 2 show that 5 out of 7 of the small molecules described in the previous screen in zebrafish give similar phenotypes in Xenopus laevis: compound 31B4 resulted in a total loss of pigment in both the retina and melanocytes (Fig. 2b), as did 33M20 (Fig. 2d). Treatment with 32N8 showed an enlargement of the developing hindbrain (Fig. 2c) and 33M20 shows a buckled notochord caused by over elongation (arrow Fig. 2d). 32P6 and 31J6 both resulted in heart phenotypes with an enlarged heart sac (Fig. 2e–f). 32N5 and 33N14 were expected to give hindbrain or pigment phenotypes; neither were observed in Xenopus laevis. Table 2 summarizes these results and also shows that for all these small molecules the developmental time at which the compound was added had no effect on the phenotype, with identical results after application at either 8–16 cell stage or stage 15 neurula (post-gastrulation). We also looked at the result of doubling the concentration for each of these compounds. An enhanced effect was observed for 32P6, and 32N8 which caused either death or showed a blistering phenotype when added at the 8–16 cell stage or stage 15 respectively (data not shown).![Chemical genetic mutations previously identified in zebrafish and observed in Xenopus; (a) stage 41 embryo with only the addition of DMSO at the 8–16 cell stage, showing the wild type state for comparison and also a higher magnification of normal heart development [(b–f) show repeats of the phenotypes seen by Peterson et al 2000 and (g–i) the effects of compounds with a defined biochemical activity applied to developing Xenopus embryos]; (b) retinal and melanocyte pigment loss phenotype observed with compound 31B4; (c) enlarged hindbrain phenotype caused by compound 32N8, with a closer view of the deformed hindbrain; (d) compound 33M20 showing both a loss of pigmentation and also a clearly observed distortion of the notochord (arrow), caused by over elongation; (e) heart sac enlargement seen with compound 32P6, also causes abnormal heart development, shown in higher magnification; (f) another compound causing the enlarged heart sac; 31J6; (g) castanospermine causes an enlarged heart sac and head area; (h) the compound PP2 caused an arrest of both growth and development as indicated by a greatly reduced trunk and lack of eye development; (i) a greatly enlarged hindbrain was observed with the addition of compound Ro-31-8220. All compounds were used at 2µg ml−1 and applied at the 8–16 cell stage. All embryos are shown in lateral view, with anterior to the left.](/image/article/2005/MB/b506103b/b506103b-f2.gif) | ||
| Fig. 2 Chemical genetic mutations previously identified in zebrafish and observed in Xenopus; (a) stage 41 embryo with only the addition of DMSO at the 8–16 cell stage, showing the wild type state for comparison and also a higher magnification of normal heart development [(b–f) show repeats of the phenotypes seen by Peterson et al 2000 and (g–i) the effects of compounds with a defined biochemical activity applied to developing Xenopus embryos]; (b) retinal and melanocyte pigment loss phenotype observed with compound 31B4; (c) enlarged hindbrain phenotype caused by compound 32N8, with a closer view of the deformed hindbrain; (d) compound 33M20 showing both a loss of pigmentation and also a clearly observed distortion of the notochord (arrow), caused by over elongation; (e) heart sac enlargement seen with compound 32P6, also causes abnormal heart development, shown in higher magnification; (f) another compound causing the enlarged heart sac; 31J6; (g) castanospermine causes an enlarged heart sac and head area; (h) the compound PP2 caused an arrest of both growth and development as indicated by a greatly reduced trunk and lack of eye development; (i) a greatly enlarged hindbrain was observed with the addition of compound Ro-31-8220. All compounds were used at 2µg ml−1 and applied at the 8–16 cell stage. All embryos are shown in lateral view, with anterior to the left. | ||
| Supplier’s name | Name given by Peterson et al. 2000 | Structure |
|---|---|---|
| 5101231 | 31B4 |
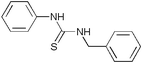
|
| 5266239 | 31J6 |
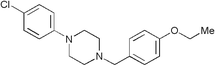
|
| 5175083 | 32N8 |
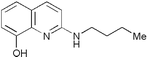
|
| 5243294 | 32P6 |
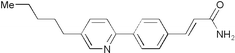
|
| 5234221 | 33M20 |
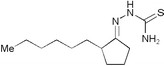
|
| Castanospermine | — |
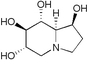
|
| PP2 | — |
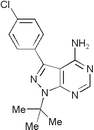
|
| Ro-31-8220 | — |
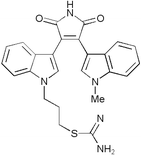
|
| Stage compound added | Compound concentration/µg ml−1 | 31B4 | 32N5 | 32N8 | 33M20 | 32P6 | 31J6 | 33N14 |
|---|---|---|---|---|---|---|---|---|
| 8–16 Cell | 2 | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ |
| 8–16 Cell | 4 | ✓ | ✗ | ✠ | ✓ | ✓✓ | ✓ | ✗ |
| Stage 15 | 2 | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Stage 15 | 4 | ✓ | ✗ | ? | ✓ | ✓ | ✓ | ✗ |
Compounds with previously identified biochemical targets
We next performed a targeted chemical assay, taking a selection of known protein kinase inhibitors and a general array of molecular inhibitors to determine if they had an effect on development. Thirteen inhibitors were tested; 3 caused death and some of the others showed varying developmental phenotypes such as major morphological defects in the head and trunk (Fig. 2g and h) or strong defects in the head (Fig. 2i). A summary of all the compounds tested is shown in both Table 1 and in the electronic supplementary information (Table S4).Conditional chemical genetic phenotype
As shown, compound 31B4 (Fig. 2b) has an effect on pigment production. Peterson et al.31 showed that, when 31B4 was removed from the embryo buffer, pigmentation returns to both the retina and the melanocytes, restoring the wild type state. Fig. 3a shows a gradual, but clear recovery of pigmentation over time as would be expected with the release of a reversible enzyme inhibitor. Interestingly neither 31B4 nor 33M20 affect colour of the cement gland (Fig. 2b and d and Fig. 3a).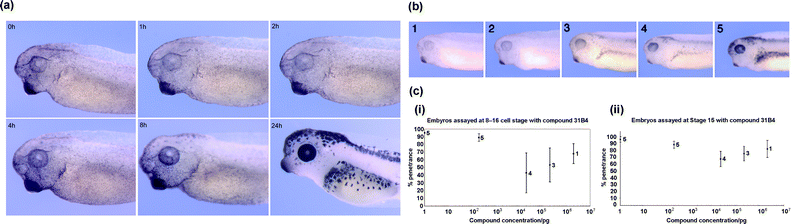 | ||
| Fig. 3 Temporal release of compound 31B4 in Xenopus embryos. (a) Buffer exchanged and compound removed at time = 0 h, no effect was observed until 4 h, where some retinal pigmentation could be seen to return. At 8 h after release of the compound, both the retinal and melanocyte pigmentation were observed to return and at 24 h the wild type state had returned. (b) Graded phenotypic index for embryos exposed to decreasing amounts (left to right) of 31B4. (c) Graphs (i) and (ii) illustrating the phenotypic response to a serial dilution of compound 31B4. All embryos are shown in lateral view, with anterior to the left. | ||
Phenotypic variation to a compound
It is common to observe variations in the quality, pigmentation and survival rates of different batches of eggs from different frogs. We therefore tested compound 31B4 at different concentrations and with eggs from different frogs to assess variation of phenotype (Fig. 3b). Firstly it was observed that batches of embryos showed two slightly different amounts of pigmentation when exposed to the same concentration of small molecule. These results were used to generate a graded index of the varying pigmentation (shown in Fig. 3b). A score of 1 corresponds to a total loss of retinal and melanocyte pigment and a score of 5 to normal pigmentation (Fig. 3b). As the concentration of 31B4 decreased there was a corresponding trend for pigmentation to return to the wild type state (refer to the supplementary information, Tables S5–6). The embryonic stage at which the compound was added (8–16 cell or stage 15) did not affect the degree of phenotype observed as both these stages precede pigment production. However, the graphs in Fig. 3c (i and ii) show that much greater phenotypic variation was observed in embryos treated at the 8–16 cell stage versus stage 15. In addition, lower penetrance was seen at certain concentrations (i.e. 200 ng ml−1).Screening throughput
Xenopus laevis embryos are slightly larger than zebrafish (1–1.3 mm versus 0.7 mm) so we determined if screens could be routinely done in 96 well plates, compared with 48 well plates. In the zebrafish screens 3 embryos were added to each well.31 Five Xenopus embryos per well were investigated, to increase the numbers of embryos being screened and strengthen the statistical significance of the assay. Table 3 shows that both embryo survival rate and the penetrance of chemical genetic phenotypes are similar in 96 well plates and 48 well plates with 5 embryos in each.| Plate | Negative control | 31B4 | 32N8 | 33M20 | 32P6 | 31J6 | |
|---|---|---|---|---|---|---|---|
| a Assay performed with 8–16 cell stage embryos. b Assay repeated with embryos from stage 15. | |||||||
| Dead/deformed (%)a | 48 | 3 | 20 | 6 | 3 | 0 | 6 |
| 96 | 0 | 0 | 3 | 0 | 0 | 6 | |
| Chemical genetic mutation (%)a | 48 | 0 | 80 | 94 | 97 | 97 | 94 |
| 96 | 0 | 100 | 97 | 100 | 94 | 94 | |
| Normal development (%)a | 48 | 97 | 0 | 0 | 0 | 3 | 0 |
| 96 | 100 | 0 | 0 | 0 | 6 | 0 | |
| Dead/deformed (%)b | 48 | 0 | 0 | 0 | 0 | 0 | 0 |
| 96 | 3 | 0 | 3 | 0 | 0 | 3 | |
| Chemical genetic mutation (%)b | 48 | 0 | 100 | 100 | 97 | 97 | 94 |
| 96 | 0 | 100 | 94 | 100 | 97 | 97 | |
| Normal development (%)b | 48 | 100 | 0 | 0 | 3 | 3 | 6 |
| 96 | 97 | 0 | 3 | 0 | 3 | 0 | |
Discussion
Our results show that large-scale screens to identify small molecules that disrupt developmental events are clearly feasible using Xenopus laevis. To date, most such screens on vertebrates have been done using zebrafish.42 We have shown that small compounds known to cause certain phenotypes in developing zebrafish embryos also give similar phenotypes in Xenopus (Fig. 2b–f). We have also shown that known biochemical inhibitors of protein kinases and glucosidases can lead to profound developmental problems (Fig. 2g–i). Xenopus thus offers an alternative model system for chemical genetic screens, in particular screens targeting early events in cell differentiation and organogenesis.Using chemical genetics to identify molecules that can perturb known or unknown elements of signalling pathways is cost effective, rapid and requires no long-term breeding of animals.42 With the use of defined screens it is also possible to target any gene product or signalling pathway in the developing embryo. This is often referred to as forward chemical genetics and can rival forward genetic screens carried out using mutagenesis or RNAi with the important additional aspect of temporal control over protein function. Another approach for large scale screens is to use reverse genetics. Here, a protein of interest is overexpressed and an assay is carried out for ligands that bind to or modulate its function. Potential ligands can then be tested on Xenopus and assayed for a phenotypic outcome. Thus, testing the ligand for uptake, specificity and potential non-specific toxic effects becomes feasible. At the moment advances in chemical library synthesis and the availability of commercial libraries allow for the potential identification of many small molecules, each generating specific phenotypes. The drawback to this is currently the identification of the cellular target of these molecules. There have been recent advances in tagging the compounds to allow affinity purification of the target protein.43–45 These technologies, although rapidly advancing, are still very much in development. Phenotype-based forward-screens have the benefit of allowing protein function, in a disease or developmental pathway, to be modulated even if the molecular targets of the pathway are unknown.
Chemical genetics will enable powerful large-scale phenotypic screens to be carried out in Xenopus laevis which, as a non-genetic organism, has been limited in this respect. The potential use of morpholinos for large screens in Xenopus embryos is powerful, but has the problem that the phenotypes observed are confined to effects on early development, as the morpholinos are injected into cleavage stage embryos and so act at the earliest occurrence of the protein’s function in development.46 If the protein concerned is additionally required later in development, this will not be uncovered. Also if the protein is not activated until later in development, its function may not be affected by the morpholino due to the dilution of the morpholino during cell division and growth. With chemical genetics, small compounds can be applied to the embryo at any time during development at defined concentrations, permitting screens to identify effects on organogenesis (i.e. heart, kidney and even limb development).
In Xenopus research the use of whole mount in situ hybridisation to analyse gene activity is a standard procedure. Large scale screens of gene expression patterns have resulted in the identification of many interesting genes and synexpression groups.47 Automated in situ machines, which are increasingly being used, make feasible the analysis of large screens at the level of the gene rather than morphological phenotype, thus allowing for more focused screens targeting known proteins or signalling pathways.
In recent years, Xenopus tropicalis has begun to be developed as a genetic organism and pilot mutagenesis screens are being carried out successfully (Lyle Zimmerman, personal communication and ref. 48). Using mutants from tropicalis and transgenic systems such as the GAL4/UAS system49 it will be possible to use enhancer and suppressor screens to look for small molecules that regulate known signalling pathways. Using this method, potential new avenues of research may open up. Xenopus has been used for many years in developmental and environmental toxicology studies.50–52 Our results suggest Xenopus embryos and tadpoles can also be used in toxicology studies of new compounds being developed for biomedical purposes and would help flag effects on development at an early stage before clinical trials.
Materials and methods
Small molecules used
Synthetic small molecules were obtained from the DiverSet E from the Chembridge Corporation and prepared as 5 mg ml−1 stock solutions in dimethyl sulfoxide. Chemical names and structures are shown in Table 1, chemicals where the phenotype is not shown are available in the electronic supplementary information (Table S4).Screening
All experiments were performed in compliance with the relevant laws and institutional guidelines at UEA. The research has been approved by the local ethical review committee according to UK Home Office regulations. Xenopus laevis (Daudin) embryos were generated53 and staged according to Nieuwkoop and Faber.54 The embryos were arrayed by Pasteur pipette, 5 embryos per well in 48 or 96 well plates containing 1000 or 200 µl of 0.1XMMR (Marc's modified ringers,53 respectively, supplemented with 50 µg ml−1 of gentamycin sulfate). Embryos were grown at 18 °C and examined visually with a dissecting microscope at 1,2 and 3 d post fertilisation. Embryos were photographed as described.55 To determine the throughput of a potential screen (Table 3) the numbers of surviving embryos and those showing naturally-occurring gross morphological abnormalities were counted as well as those showing the expected chemical genetic phenotype.Genetic variation and temporal release of small molecule 31B4
For the temporal release assay of small molecule 31B4, embryos were arrayed as described previously and the compound added at either 8–16 cell stage or stage 15. The compound was removed from stage 37 (0 h), then the embryos were scored at 1, 2, 4, 8 and 24 h later. The compounds were removed by a complete buffer exchange for 0.1XMMR (50 µg ml−1 gentamycin sulfate). The phenotypic assay to investigate genetic variation to the compound 31B4, was performed in 9 cm Petri dishes with the compound added to 30 ml of 0.1XMMR (50 µg ml−1 gentamycin sulfate) to the appropriate concentration and 100 embryos were added per dish at the 8–16 cell stage or stage 15. Embryos were then transferred to 5 cm Petri dishes at stage 37, 20 embryos per dish to facilitate phenotypic scoring. The percentage of each phenotypic category was determined by the number of embryos showing the phenotype divided by the number of surviving embryos. Fig. 3c (i) and (ii) was constructed by taking the average for the highest percentage score in a concentration category. Average deviation around the mean was calculated by using the highest percentage scores in a concentration category.Acknowledgements
We would like to thank Andrea Munsterberg, Dylan Sweetman, Lyle Zimmerman, Jelena Gavrilovic and Mark Fidock for helpful discussions and comments on the manuscript. This work has been supported by a MRC Discipline Hopper award (RAF and GNW), a BBSRC New investigator grant to GNW and a Pfizer BBSRC CASE studentship to MLT.References
- H. von Euler, H. Hellstrom and N. Lofgren, Z. Physiol. Chem., 1935, 234, 151–164 Search PubMed.
- H. E. Blackwell and Y. Zhao, Plant Physiol., 2003, 133, 448–455 CrossRef CAS.
- T. U. Mayer, Trends Cell Biol, 2003, 13, 270–277 CrossRef CAS.
- J. Yeh and C. Crews, Dev. Cell, 2003, 5, 11–19 Search PubMed.
- S. M. Khersonsky and Y. T. Chang, Comb. Chem. High Throughput Screening, 2004, 7, 645–652 Search PubMed.
- N. J. Westwood, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2761–2774 CrossRef CAS.
- S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64–66 Search PubMed.
- R. S. Lokey, Curr. Opin. Chem. Biol., 2003, 7, 91–96 CrossRef CAS.
- D. R. Spring, Chem. Soc. Rev., 2005, 34, 472–482 RSC.
- B. R. Stockwell, Trends Biotechnol., 2000, 18, 449–455 CrossRef CAS.
- C. M. Crews and U. Splittgerber, Trends Biochem. Sci., 1999, 24, 317–320 CrossRef CAS.
- R. L. Strausberg and S. L. Schreiber, Science, 2003, 300, 294–295 CrossRef CAS.
- S. Ding and P. G. Schultz, Nat. Biotechnol., 2004, 22, 833–840 CrossRef.
- T. N. Doan, C. D. Eilertson and A. L. Rubinstein, Drug Discovery Today: TARGETS, 2004, 3, 191–197 Search PubMed.
- J. Huang, H. Zhu, S. J. Haggarty, D. R. Spring, H. Hwang, F. Jin, M. Snyder and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16594–16599 CrossRef CAS.
- R. G. Pendleton, F. Parvez, M. Sayed and R. Hillman, J. Pharmacol. Exp. Ther., 2002, 300, 91–96 CrossRef CAS.
- T. J. Mitchison, Chem. Biol., 1994, 1, 3–6 CAS.
- S. L. Schreiber, Bioorg. Med. Chem., 1998, 6, 1127–1152 CrossRef CAS.
- H. E. Blackwell, L. Perez, R. A. Stavenger, J. A. Tallarico, E. Cope Eatough, M. A. Foley and S. L. Schreiber, Chem. Biol., 2001, 8, 1167–1182 CrossRef CAS.
- S. Ding, N. S. Gray, X. Wu, Q. Ding and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 1594–1596 CrossRef CAS.
- M. D. Burke, E. M. Berger and S. L. Schreiber, Science, 2003, 302, 613–618 CrossRef CAS.
- U. S. Eggert, N. Ruiz, B. V. Falcone, A. A. Branstrom, R. C. Goldman, T. J. Silhavy and D. Kahne, Science, 2001, 294, 361–364 CrossRef CAS.
- C. J. Torrance, V. Agrawal, B. Vogelstein and K. W. Kinzler, Nat. Biotechnol., 2001, 19, 940–945 CrossRef CAS.
- B. R. Stockwell, Neuron, 2002, 36, 559–562 CrossRef CAS.
- P. M. Carroll, B. Dougherty, P. Ross-Macdonald, K. Browman and K. FitzGerald, Pharmacol. Ther., 2003, 99, 183–220 Search PubMed.
- P. Haffter and C. Nusslein-Volhard, Int. J. Dev. Biol., 1996, 40, 221–227 Search PubMed.
- T. Reya and H. Clevers, Nature, 2005, 434, 843–850 CrossRef CAS.
- T. J. Van Raay and M. L. Vetter, Dev. Neurosci., 2004, 26, 352–358 Search PubMed.
- E. H. Davidson, J. P. Rast, P. Oliveri, A. Ransick, C. Calestani, C. H. Yuh, T. Minokawa, G. Amore, V. Hinman, C. Arenas-Mena, O. Otim, C. T. Brown, C. B. Livi, P. Y. Lee, R. Revilla, A. G. Rust, Z. Pan, M. J. Schilstra, P. J. Clarke, M. I. Arnone, L. Rowen, R. A. Cameron, D. R. McClay, L. Hood and H. Bolouri, Science, 2002, 295, 1669–1678 CrossRef CAS.
- M. K. Cooper, J. A. Porter, K. E. Young and P. A. Beachy, Science, 1998, 280, 1603–1607 CrossRef CAS.
- R. T. Peterson, B. A. Link, J. E. Dowling and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12965–12969 CrossRef CAS.
- U. Langheinrich, E. Hennen, G. Stott and G. Vacun, Curr. Biol., 2002, 12, 2023–2028 CrossRef CAS.
- U. Langheinrich, Bioessays, 2003, 25, 904–912 CrossRef CAS.
- R. T. Peterson, J. D. Mably, J. N. Chen and M. C. Fishman, Curr. Biol., 2001, 11, 1481–1491 CrossRef CAS.
- H. A. Chung, J. Hyodo-Miura, A. Kitayama, C. Terasaka, T. Nagamune and N. Ueno, Genes Cells, 2004, 9, 749–761 CrossRef CAS.
- K. Koebernick, T. Hollemann and T. Pieler, Dev. Biol., 2003, 260, 325–338 CrossRef CAS.
- E. M. De Robertis and H. Kuroda, Annu. Rev. Cell. Dev. Biol., 2004, 20, 285–308 CrossRef.
- R. Keller, L. A. Davidson and D. R. Shook, Differentiation, 2003, 71, 171–205 CrossRef.
- C. A. Zygar, T. L. Cook and R. M. Grainger, Jr., Development, 1998, 125, 3509–3519 Search PubMed.
- T. Mohun, R. Orford and C. Shang, Trends Cardiovasc. Med., 2003, 13, 244–248 CrossRef.
- E. A. Jones, J. Am. Soc. Nephrol., 2005, 16, 313–321 Search PubMed.
- L. I. Zon and R. T. Peterson, Nat. Rev. Drug. Discovery, 2005, 4, 35–44 CrossRef CAS.
- L. Burdine and T. Kodadek, Chem. Biol., 2004, 11, 593–597 CrossRef CAS.
- G. Mitsopoulos, D. P. Walsh and Y. T. Chang, Curr. Opin. Chem. Biol., 2004, 8, 26–32 CrossRef CAS.
- D. Jung, D. Williams, S. M. Kershonsky, T. Kang, N. Heidary, Y. T. Chang and S. J. Orlow, Mol. BioSyst., 2005, 1, 85–92 RSC.
- S. Kenwrick, E. Amaya and N. Papalopulu, Dev. Dyn., 2004, 229, 289–299 CrossRef CAS.
- N. Pollet, N. Muncke, B. Verbeek, Y. Li, U. Fenger, H. Delius and C. Niehrs, Mech. Dev., 2005, 122(3), 365–439 CrossRef CAS.
- N. Hirsch, L. B. Zimmerman and R. M. Grainger, Dev. Dyn., 2002, 225, 422–433 CrossRef CAS.
- K. O. Hartley, S. L. Nutt and E. Amaya, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1377–1382 CrossRef CAS.
- J. R. Snape, S. J. Maund, D. B. Pickford and T. H. Hutchinson, Aquat. Toxicol., 2004, 67, 143–154 CrossRef CAS.
- D. J. Fort, E. L. Stover, T. L. Propst, B. C. Faulkner, T. A. Vollmuth and F. J. Murray, Food Chem. Toxicol., 1998, 36, 591–600 CrossRef CAS.
- T. B. Hayes, A. Collins, M. Lee, M. Mendoza, N. Noriega, A. A. Stuart and A. Vonk, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5476–5480 CrossRef CAS.
- H. L. Sive, R. M. Grainger and R. M. Harland, Early Development of Xenopus laevis, Cold Spring Harbor Laboratory Press, New York, 2000 Search PubMed.
- Normal table of Xenopus laevis (Daudin), ed. P. D. Nieuwkoop and J. Faber, Garlan Publishing, New York, 1994 Search PubMed.
- M. Harrison, M. Abu-Elmagd, T. Grocott, C. Yates, J. Gavrilovic and G. N. Wheeler, Dev. Dyn., 2004, 231, 214–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Assays of compound 31B4 at cell stages 8–16 or stage 15 and a table of all assayed compounds showing chemical information. See http://dx.doi.org/10.1039/b506103b |
| This journal is © The Royal Society of Chemistry 2005 |