Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections
Zuben E.
Sauna
,
Suneet
Shukla
and
Suresh V.
Ambudkar
*
Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, DHHS, Bethesda, Maryland 20892-4256. E-mail: ambudkar@helix.nih.gov; Fax: (301) 435-8188; Tel: (301) 402-4178
First published on 26th May 2005
Abstract
Disulfiram, a drug used to treat alcoholism, has recently been indicated to play a primary as well as an adjuvant role in the treatment of many cancers and in the reversal of fungal drug-resistance. This review discusses the molecular mechanism of action of disulfiram and its potential use in the treatment of human cancers and fungal infections.
Introduction
Disulfiram, or tetraethylthiuram disulfide, has been used for over half a century for alcohol aversion therapy; its pharmacokinetics have been extensively studied and it has an excellent safety record. Recent reports indicate that disulfiram and other dithiocarbamates may have a significant potential in the treatment of human cancers. Disulfiram has been reported to induce apoptosis, shows metal ion-dependent antineoplastic activity, and arrests angiogenesis. Disulfiram has also been shown to inhibit the activating transcription factor/cyclic-AMP-responsive element binding protein, which is implicated in the growth and progression of melanomas. Moreover, recent studies show that disulfiram inhibits the activity of ABC drug transport proteins, which are responsible for the development of multiple drug resistance in cancer and fungal cells. Thus, disulfiram may have an important role as an adjuvant in the chemotherapy of human cancers and in the treatment of drug-resistant fungal infections. In addition to empirical evidence, the biochemical mechanisms and cellular pathways that underlie the action of disulfiram have also begun to emerge. Cancer is a disease involving complex networked systems that would benefit from treatments that intervene in multiple pathways. Drugs such as disulfiram with limited toxicity and manageable side effects that perturb several biomolecules and pathways may provide useful strategies in the treatment of complex diseases.Circumventing multidrug resistance: lessons learned from generations of failure
Systemic chemotherapy is extensively used as the treatment of choice in cancers that are metastatic, i.e. approximately 50% of all cancers.1 However, no more than 10% of patients are cured by chemotherapy. The phenomenon of multidrug resistance (MDR) is recognized as frustrating efforts in the clinic to formulate effective chemotherapy against several blood cancers,2 as well as the solid tumors associated with breast,3 ovarian4 and lower gastrointestinal tract cancers.5 This resistance of cells to chemically diverse drugs with multiple mechanisms of action is defined as MDR. The expression of P-glycoprotein (Pgp), a product of the MDR1 (ABCB1) gene, was correlated with the degree of drug resistance in several cell lines.6,7 A large body of evidence has since accumulated to strongly implicate Pgp and other energy-dependent pumps, MDR-associated protein 1 (MRP1, ABCC1) and the mitoxantrone resistance-associated protein (MXR, ABCG2) that extrude chemotherapeutic agents from cells (for a recent review see 8). Pgp, MRP1 and ABCG2 are all members of the ATP-binding cassette (ABC) superfamily of transport proteins. The ABC family of transport proteins represents one of the largest families of proteins in living organisms.9 These membrane proteins utilize the energy derived from the hydrolysis of ATP for the vectorial transport of a variety of substrates, which include ions, sugars, lipids, peptides and natural product hydrophobic anticancer drugs, across the cell membrane.10 The functional unit of an ABC transporter is a nucleotide binding domain (NBD) and one transmembrane domain comprised of six putative transmembrane α-helices.11 The NBD consists of the highly conserved Walker A and B motifs and the ABC signature motif. Pgp consists of two putative transmembrane domains, each consisting of six hydrophobic transmembrane helices and one NBD or ATP-binding domain.12 While Pgp has two membrane spanning domains, MRP1 has three.13,14 Unlike Pgp and MRP1, ABCG2 has only one NBD domain and a single membrane spanning domain.15,16 The functional unit of Pgp appears to be a monomer and both its halves are necessary for function,10 whereas the minimal functional unit of ABCG2 seems to be a dimer.17,18Besides being a significant clinical problem in the treatment of human cancers, MDR-mediated by ABC transport proteins is rapidly gaining clinical importance in the treatment of fungal infections. Clinical antifungal resistance is not a major problem in normal patients, but immunocompromised individuals frequently develop systemic infections with a high incidence of antifungal resistance which develops during clinical therapy.19,20 The clinical therapy of systemic candidiasis, caused by the fungal pathogen Candida albicans, involves the use of antifungal azoles such as fluconazole, ketoconazole, itraconazole or voriconazole but the fungal pathogen develops resistance to these drugs, particularly after prolonged treatment. The active extrusion of these antifungal agents by ABC transporters like Candida drug resistance protein-P (Cdr1p and Cdr2p) is one of the major mechanisms which has been implicated for the development of MDR in yeast.21
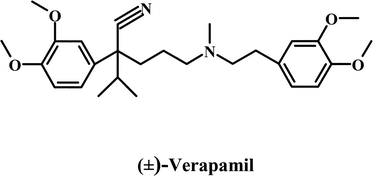
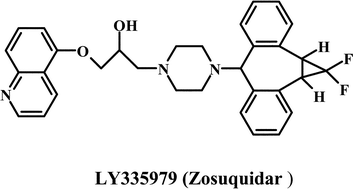
Most strategies for reversing MDR have focused on the modulation of Pgp activity by using compounds, which are referred to as chemosensitizers or MDR modulators. The strategy involves co-administrating the chemosensitizer with an anticancer drug.22 This impairs the Pgp function, resulting in enhanced intracellular anticancer drug accumulation. Compounds belonging to chemical classes as diverse as calcium channel blockers, calmodulin inhibitors, coronary vasodilators, alkaloids, quinolines, hormones, cyclosporines, surfactants and antibiotics have been shown to act as chemosensitizers.10,22,23 The first generation of Pgp chemosensitizers followed the discovery by Tsuruo and coworkers24 that the calcium channel blocker, verapamil, enhances the intracellular accumulation of many anticancer drugs. Many other calcium channel blockers tested for their ability to reverse the MDR phenotype showed comparable results in vitro.25 Besides the calcium channel blockers, the immunosuppressant, cyclosporin A, remains one of the most effective first generation MDR modulators. The major limitation to the clinical use of most first generation MDR modulators was that they typically reversed MDR at concentrations that made them toxic.22,23,25,26 The development of second-generation MDR modulators focused on more potent and considerably less toxic analogs of the first generation agents. The in vitro and preclinical studies suggested that PSC 833 (Valspodar), an analog of cyclosporin A, was promising.1 However, though several Phase III protocols are in progress, overall the results have not been very encouraging23 and Novartis has discontinued development of this drug. In recent years, several MDR modulators have been developed specifically for Pgp unlike the first and second-generation chemosensitizers, which were initially developed for other disease conditions. These third generation modulators exhibit effective MDR reversal in the 100–200 nM range and include the cyclopropyldibenzosuberane LY 335979,27,28 the acridonecarboxamide GF 120918,29,30 the diketopiperazine XR9576,31 the diarylimidazole OC144-09332 and the amido-keto-pipecolinate derivative, VX-71033,34 (see Table 1 for chemical structures of selected first, second, and third generation modulators). These agents are currently used in various clinical trials. Nonetheless, despite sustained efforts for over 20 years, there is still no effective MDR modulator in clinical use. The most important reason that MDR modulators fail in clinical trials is the unmanageable toxicity and adverse effects of doses required to reverse drug resistance.
| First generation modulators | |

|
|
| Second generation modulators | |

|

|
| Third generation modulators | |
|

|
Using the dithiocarbamate disulfiram to sensitize drug resistant tumors to chemotherapy
Disulfiram, first synthesized in 1881, was used to accelerate the vulcanization of rubber.35 It was only in the 1930s that disulfiram found a medicinal use as a scabiescide and, subsequently, as a vermicide36 because it was toxic to lower animal forms due to its ability to chelate copper; an essential component of the respiratory chain of these organisms. In 1948 it was proposed that disulfiram be used in the treatment of chronic alcoholism as alcohol aversion therapy.37 This has proved to be immensely successful and disulfiram, under the trade name Antabuse, continues to be used clinically to this day.38 Moreover, disulfiram is finding increasing use in cocaine addiction.39–41 There are several excellent reviews on the use of disulfiram in narcotic addiction and due to space constraints this aspect will not be addressed here.There has been renewed interest in disulfiram in the last few years, particularly in elucidating the mechanistic basis for its action in reversal of MDR.42,43 We have seen above that several generations of MDR modulators that have been extremely successful in vitro have failed in the clinic. Disulfiram, as a reversal agent for MDR, comes with a huge advantage. Chick has reviewed a vast literature on the hepatotoxicity, CNS adverse effects, neuropathology and drug interactions of disulfiram in the clinic.44 Though monitoring of patients on high doses of disulfiram (over 250 mg per day) is recommended,38 there are few adverse effects associated with long term treatment. Moreover, acute disulfiram overdose is uncommon. In adults, clinical manifestations after acute overdose are rare with doses less than 3 g.44 In fact, when disulfiram was first introduced, doses of 1–3 g per day were employed38 and some practitioners have argued that the current doses are not sufficient for all patients and have routinely used 300–500 mg per day.45 Hepatotoxicity is the most common and serious cause for concern during treatment with disulfiram. Reports from Denmark, where the prescription of disulfiram is the highest in the world, indicate that over a 22 year period fatalities associated with disulfiram induced hepatotoxicity were 1 per 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients.46 Moreover, hepatotoxicity is generally reversible if disulfiram is stopped prior to clinical manifestation,47 which can be managed using liver function tests. The toxicity data thus suggest that, if necessary, disulfiram could be used in cancer chemotherapy at doses considerably higher than those used in alcohol aversion therapy. The pharmacokinetics of disulfiram have also been extensively studied and provide some clues as to its success in clinical practice. There is >80% bioavailability after an oral dose and the elimination of disulfiram and its metabolites is a very slow process. Approximately 20% of the drug remains in the body for 1–2 weeks post-ingestion.48 Considering the relatively non-discriminate biochemical actions of disulfiram in chelating metal ions and modifying cysteine residues,49 the excellent tolerance and absence of serious adverse events and side effects may appear surprising. However, these are well established and undisputed clinical facts.38,44,46
000 patients.46 Moreover, hepatotoxicity is generally reversible if disulfiram is stopped prior to clinical manifestation,47 which can be managed using liver function tests. The toxicity data thus suggest that, if necessary, disulfiram could be used in cancer chemotherapy at doses considerably higher than those used in alcohol aversion therapy. The pharmacokinetics of disulfiram have also been extensively studied and provide some clues as to its success in clinical practice. There is >80% bioavailability after an oral dose and the elimination of disulfiram and its metabolites is a very slow process. Approximately 20% of the drug remains in the body for 1–2 weeks post-ingestion.48 Considering the relatively non-discriminate biochemical actions of disulfiram in chelating metal ions and modifying cysteine residues,49 the excellent tolerance and absence of serious adverse events and side effects may appear surprising. However, these are well established and undisputed clinical facts.38,44,46
The effect of disulfiram on Pgp-mediated MDR was first reported by Loo and Clarke.42 The most common approach taken to disabling the molecular pumps associated with MDR has been to develop competitive or non-competitive inhibitors that prevent the pumps from effluxing the chemotherapeutic agent.1,22,26 However, Loo and Clark found that disulfiram inhibited Pgp through a novel mechanism, by inhibiting the maturation of the transporter.42 Disulfiram did not inhibit the synthesis of Pgp as the total amount of immunoreactive Pgp was the same at all concentrations of disulfiram. However, the mature, glycosylated form of the protein at the cell surface was drastically reduced. This had a physiological consequence. Drug-resistant cells treated with disulfiram showed sensitivity to colchicine and vinblastine comparable to control cells not transfected with Pgp. Moreover, disulfiram, in addition to reducing the cell surface expression, also inhibited Pgp-mediated ATP hydrolysis. Several groups have demonstrated that disulfiram inactivates alcohol dehydrogenase by chemically modifying cysteines in the active site of the enzyme49,50 and it appears that disulfiram may interact with the two cysteines in the Walker A domains of both ATP sites of Pgp.42 However, we observed a very interesting phenomenon when we measured ATP hydrolysis in the presence of disulfiram after first protecting the ATP sites with excess ATP. Instead of inhibition, we observed stimulation of ATP hydrolysis.43 This suggested an interaction between the drug–substrate binding site of Pgp and disulfiram, which was confirmed by monitoring the displacement of the photoaffinity substrate analog [125I]-iodoarylazidoprazosin (IAAP) and [3H]-azidopine. In addition to Pgp, disulfiram has also been shown to interact with two other ABC transporters, MRP1 and MRP4.43 The interactions of disulfiram with these ABC transporters are complex and are outlined in Fig. 1A. When the ATP sites are accessible, disulfiram interacts with cysteines at or near the active site, inhibits ATP hydrolysis and inactivates the protein. It must be noted that unlike compounds such as N-ethylmaleimide (NEM) that covalently modify a single sulfhydryl, disulfiram can form disulfide bridges across two cysteines that lie within a distance of approximately 8 Å (Fig. 1B). When the ATP sites are protected, disulfiram stimulates ATP hydrolysis by Pgp (and to a limited extent by MRP1) and there is direct evidence that disulfiram also competes for the substrate-binding site of Pgp. The treatment of Pgp with NEM has no effect on drug–substrate binding, while both disulfiram and NEM inhibit the binding of [α-32P]-8-azidoATP to Pgp. Thus, inhibition of drug–substrate binding occurs by a mechanism distinct from the effects of cysteine modification by disulfiram. The evidence suggests that disulfiram is unique in that as a single chemical moiety it neutralizes Pgp-mediated MDR by three independent mechanisms at two different levels. It (i) arrests Pgp maturation, (ii) inhibits the transport function and (iii) obstructs the ATP hydrolysis that powers drug transport.
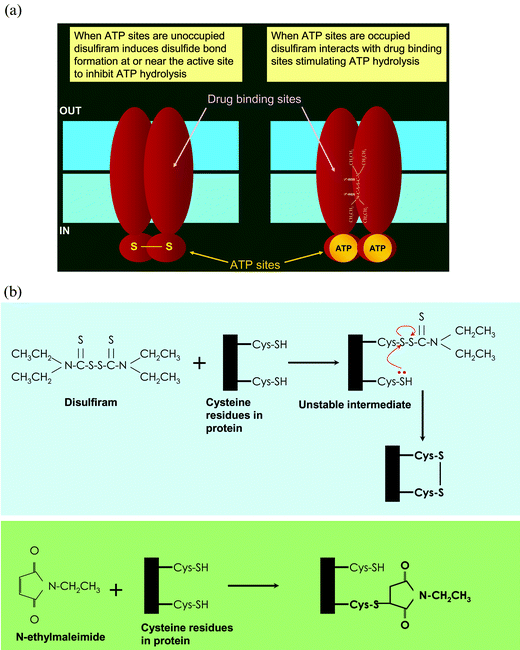 | ||
| Fig. 1 The biochemical basis of the action of disulfiram on the ABC drug transporters Pgp and Cdr1p. (A) Disulfiram interacts with both the transport-substrate and ATP-binding sites of Pgp. When the ATP sites are unoccupied, disulfiram modifies cysteine residues (disulfide bond formation) at or near the ATP sites (see B below), resulting in inhibition of ATP hydrolysis. When the ATP sites are protected with excess ATP (although ATP bound to both sites is shown, experimental evidence suggests binding of only one ATP molecule possibly at the inter-phase of two ATP sites), disulfiram (depicted in its chemical structure) interacts with the transport-substrate sites (in the membrane domains) and stimulates ATP hydrolysis. (B) Mechanism of modification of cysteines at or near the active site by disulfiram and NEM. Disulfiram first interacts with a cysteine residue to form an unstable mixed disulfide. If there are additional cysteine moieties in the vicinity (6–8 Å) of the unstable intermediate, it reacts with the thiol group to form an intramolecular disulfide bond (upper panel). On the other hand, NEM covalently modifies cysteine residue (lower panel). | ||
Though ATP-dependent molecular pumps that extrude anticancer agents are the principal routes by which cancer cells evade chemotherapy, there are several other more specific mechanisms of drug resistance. 5-Fluorouracil (5-FU) is a significant component in the treatment of colorectal cancer51 and other solid tumors. Cancer cells with high nuclear activity of the transcription factor NF-κB are extremely resistant to 5-FU.52,53 5-FU treatment activates NF-κB, which makes the cells resistant to apoptosis.53,54 It was demonstrated by Fernandez et al. that several dithiocarbamates, including disulfiram, inhibit NF-κB and induce apoptosis in T-cells.55 Recently, Wang et al. demonstrated that for the 5-FU resistant colorectal cell line H6305-FU treatment with a combination of disulfiram and 5-FU resulted in an IC50 (5-FU) 83-fold lower than treatment with 5-FU alone.56 At the molecular level, disulfiram strongly inhibited NF-κB nuclear translocation and the binding of NF-κB to DNA,56 activities that are correlated with the antiapoptotic properties of NF-κB.57–59 All members of the NF-κB family have an N-terminal Rel homology (RH) domain that plays a central role in NF-κB dimerization, nuclear translocation and DNA binding.60 The RH domain contains several sulfhydryl groups that are part of the DNA binding domain58 and disulfiram and its metabolite diethyldithio-carbamate target these residues.56 The resistance of colorectal cancers to 5-FU is a very significant clinical problem54 and an approach that involves induction of the inhibitor of NF-κB (IκB) via gene therapy has had some success.61,62 However, the obstacles to the clinical fruition of this approach are considerable and a small molecule approach using disulfiram is considered the more viable strategy.56
Disulfiram as an antifungal agent
Diethyldithiocarbamate and the two different substituted analogs of this compound were evaluated for their anticandidial effect.63 These compounds were tested for their in vitro inhibitory effect on the growth of Candida strains and it was observed that sodium diethyldithio-carbamte (DDTC) and sodium dimethylthiocarbamate (DMDTC) produced inhibitory effects comparable to amphotericin-B, a drug clinically used to treat candidiasis. The in vivo effects of these dithiocarbamates were also encouraging with N-methyl-D-glucamine dithiocarbamate (NMGDTC) being the most effective. Furthermore, the effectiveness of the dithiocarbamates was considerably enhanced if the infected mice were immunosuppressed with cisplatin. Finally, combinatorial therapy using dithiocarbamates with amphotericin-B had advantages over the use of the either agent alone. A systematic study testing six chemically synthesized dithiocarbamate derivatives on 40 clinical isolates of Candida64 demonstrated that DDTC, DMDT, and sodium pyrrolidene dithiocarbamate (PYRDTC) showed significant inhibitory effects. Based on early experience with the use of disulfiram as a scabiescide and vermicide, it was suggested that the dithiocarbamates exert their antifungal action by chelating metals that are indispensable components of the respiratory chains of lower organisms. To test this hypothesis, excess metal ions were added to chelate the dithiocarbamates. This resulted in only partial reversal of their antifungal activity, which suggests that additional inhibitory mechanisms for dithiocarbamates may exist. These could include interaction with the ABC multidrug transporters like Cdr1p and Cdr2p, which have been implicated in the development of fungal drug resistance.65,66 In a screen to determine the drug resistance profile of the ABC transporters of the yeast pleiotropic drug resistance network in S. cerevisiae, it was found that the ABC-MDR transporters mediate resistance to classes of clinically and agriculturally important fungicides and dithiocarbamates, which were identified as the compounds to which the Pdr5p, Snq2p or Yor1p, the yeast multidrug transporters, confer resistance.66 This suggests that the ABC multidrug transporters from S. cerevisiae interact with dithiocarbamates and a direct interaction of disulfiram with the known ABC transporter in C. albicans, Cdr1p, was recently demonstrated.67Cdr1p has been shown to be involved in the energy-dependent efflux of antifungal compounds like fluconazole, miconazole, itraconazole and nystatin. We showed that disulfiram can be used as an inhibitor of Cdr1p-mediated drug resistance.67 Disulfiram inhibited the binding of both [125I]-IAAP and [3H]-azidopine to Cdr1p, suggesting a direct interaction with substrate binding sites of the transporter. Moreover, disulfiram inhibited Cdr1p-mediated ATP hydrolysis, which was reversed by dithiothreiotol. These studies suggest that fungal and mammalian ABC transport proteins are inhibited by disulfiram via comparable mechanisms. The inhibition of the Cdr1p activity by disulfiram would suggest that it could be used in combination with antifungal agents to reverse the drug resistance mediated by Cdr1p. Therefore, we checked the ability of disulfiram to modulate the drug resistance mediated by Cdr1p in S. cerevisiae. We found that the presence of the non-toxic concentration of disulfiram with the other antifungal agents made the cells more susceptible to the drugs, which indicates the potential usefulness of disulfiram in antifungal therapy to sensitize drug resistant cells.
Use of disulfiram as an anticancer agent
Reviewing the status of cancer research over the past 25 years, Hanahan and Weinberg 68 describe the scientific literature as “complex beyond measure” but argue that new progress will now come from being able to understand cancer in terms of a small number of underlying principles. They devote their essay to sketching a broad outline of such principles and identify six traits that most, if not all, cancers have acquired: sustained angiogenesis, evading apoptosis, tissue invasion and metastasis, limitless replicative potential, self sufficiency in growth signals and insensitivity to antigrowth signals. It is interesting that disulfiram has been shown to directly impact at least three of these traits—inducing apoptosis, acting as an antiangiogenesis agent and preventing tissue invasiveness and metastasis (Fig. 2).69–74 This of course is in addition to a very significant role in synergistically enhancing the potency of anticancer drugs by disabling MDR pumps.42,43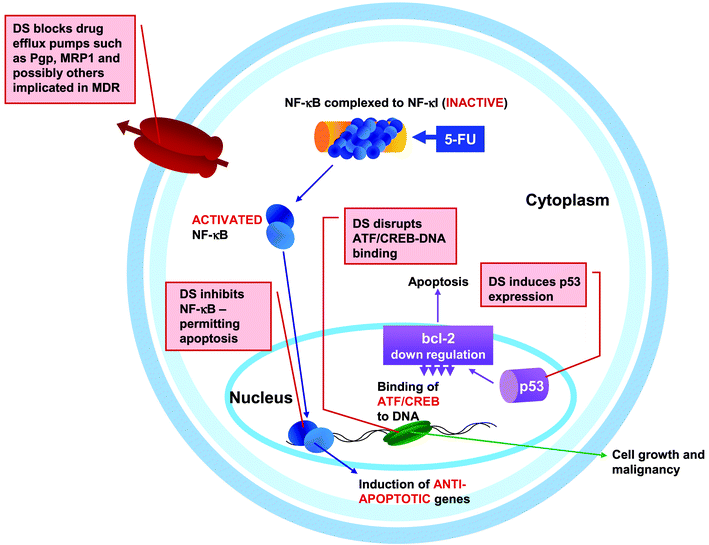 | ||
| Fig. 2 Potential therapeutic targets for disulfiram in a cancer cell. Illustration showing some of the interactions demonstrated between disulfiram and cellular mechanisms implicated in human cancers. See text for details of the mechanisms. DS, disulfiram; 5-FU, 5-fluorouracil. | ||
Disulfiram-mediated apoptosis has been shown to occur in thymocytes,75 smooth muscle cells76 and human hepatoma cells.77 There is evidence that disulfiram induces p53.78,79 The modulation of p53 occurs at an early stage in the apoptotic pathway, resulting in inhibition of bcl-279 and subsequent apoptosis. It has also been suggested that cell death induced by disulfiram is the result of transport of the Cu2+ ion into the cell.75 The apoptotic effects of disulfiram have, however, generated some controversy as several groups have found that dicarbamates can be inhibitors of apoptosis,74,80–82 as they inhibit TNF-κ, itself a potent inducer of apoptosis.73 A recent report, however, claims that the two effects occur at widely separated time scales,74 suggesting that the metabolites of disulfiram may have a more significant role in these processes than disulfiram alone.
In addition to inducing apoptosis, disulfiram has been shown to directly inhibit the growth of cancer cells both in vitro and in vivo70,83 and divalent metal ions (especially Cu2+) have been shown to enhance its antineoplastic activity.69 Cysteine residues are located at critical DNA binding regions of transcription factors84–86 and DNA transcription factor binding is severely disrupted when these cysteines are modified by the formation of mixed disulfides. It has been directly demonstrated that disulfiram mediates the inhibition of the activating transcription factor/cyclic-AMP-responsive element binding protein (ATF/CREB), which is potentiated by Cu2+ ions.70 This had the effect of arresting the growth of malignant melanoma, which are notoriously resistant to chemotherapeutic agents.87 A recent report illustrates the potential for this strategy, where the combination of oral zinc gluconate and disulfiram induced reduction in hepatic metastases and clinical remission in a patient with stage IV metastatic ocular melanoma.70 This study also clearly shows that disulfiram is a safe drug, as continuous therapy for 53 months had negligible side effects.
Finally, disulfiram has been suggested to show antiangiogenic activity.71,72,88 The mechanisms underlying disulfiram-mediated inhibition of angiogenesis have recently been elucidated by Shiah et al.71 Degradation of the extracellular matrix precedes angiogenesis and cell invasion, which in turn are prerequisites for cancer metastasis.89,90 This degradation of the extracellular matrix is affected by the matrix metalloproteinases.91,92 Disulfiram was shown to be an effective inhibitor of the matrix metalloproteinases and blocked angiogenesis in vivo in ten day old chicken embryos.71 While it has been well established that metalloproteinases-inhibitors prevent angiogenesis, tumor dissemination and metastasis transition to the clinic has been fraught with difficulties. Two favored candidates were TIMPs (tissue inhibitors of metalloproteinases), which are naturally occurring proteins,93 and BB-94 (batimastat), a synthetic low molecular weight inhibitor.94 However, TIMPs have an extremely short half-life in vivo and are not promising for clinical applications while BB-94 has very poor solubility and has to be injected as a detergent emulsion.94
Conclusions and future outlook
Investigating the role of disulfiram in the treatment of human cancers may at first glance seem an unrealistic proposition. In an era of molecular modeling and rational drug design, we would be investigating a 125 year old compound developed for the vulcanization of rubber. Disulfiram has the added disadvantage that its effects on biomolecules appear to be non-discriminatory. On the other hand, disulfiram has a 75 year history in medicine, is a commercially available product (as Antabuse™) with a known pharmacology profile and over 50 years of clinical experience. The normal adult dose of disulfiram in the treatment of alcoholism is 500 mg per day, which translates to an approximate concentration of 20 µM in a 100 kg individual. This is a concentration that is sufficient to elicit the in vitro responses of disulfiram. The last decade has seen numerous reports suggesting disulfiram may play a very useful role in the treatment of human cancers. Disulfiram does have very significant advantages: it is a drug with a very good safety record and has excellent bioavailability. Both these factors are of critical importance. Several MDR modulators have failed in the clinic due to unmanageable side effects.8,22,26,95 Similarly, the lack of bioavailability has limited the use of drugs that inhibit angiogenesis and tumor progression.93,94 Also, sulfhydryl-reacting agents, despite their apparently unspecific mode of action, provide useful drugs against human diseases96 and disulfiram shows remarkably few side-effects and is not toxic, even with prolonged use.44,45,70,97 Recent investigations have applied a range of biochemical techniques to understand the mechanistic basis of the specific actions of disulfiram. However, as this review enumerates, the actions of disulfiram are extremely diverse and this drug may have multiple therapeutic targets. This ties in with the emerging paradigm in systems biology: human cells and tissues are a complex networked system. Thus, a drug discovery approach (especially for complex diseases such as cancer) necessitates identification of combinations of small molecules that perturb cellular networks in a desired fashion.98–100 While combinations of synergistic compounds have been exploited for decades, these are generally limited to agents known to be effective in a therapeutic area or to agents with a clear rationale for the combination. Systematic discovery based on systems biology, however, advocates a much wider combination space. Borisy et al.101 have designed a robotic screening and informatics system to explore such combinatorial spaces. They implemented this system to test the inhibition of fluconazole resistant C. albicans with 30 drugs in pair-wise combinations. It is interesting that of the 435 possible two-component combinations, only six effective combinations emerged, of which two had disulfiram as one of the agents.101 Empirical studies such as these are complementary to the mechanistic studies that elucidate the interaction of disulfiram with specific cellular and molecular pathways. Taken together, these studies argue for more animal research and clinical trials to achieve the full therapeutic promise of this fascinating compound.References
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS.
- C. P. Leith, K. J. Kopecky, I. M. Chen, L. Eijdems, M. L. Slovak, T. S. McConnell, D. R. Head, J. Weick, M. R. Grever, F. R. Appelbaum and C. L. Willman, Blood, 1999, 94, 1086–1099 CAS.
- B. J. Trock, F. Leonessa and R. Clarke, J. Natl. Cancer Inst., 1997, 89, 917–931 CrossRef CAS.
- A. G. J. Vanderzee, H. Hollema, A. J. H. Suurmeijer, M. Krans, W. J. Sluiter, P. H. B. Willemse, J. G. Aalders and E. G. E. Devries, J. Clin. Oncol., 1995, 13, 70–78 CAS.
- M. Kitazono, T. Sumizawa, Y. Takebayashi, Z.-S. Chen, T. Furukawa, S. Nagayama, A. Tani, S. Takao, T. Aikou and S.-i. Akiyama, J. Natl. Cancer Inst., 1999, 91, 1647–1653 CrossRef CAS.
- M. M. Gottesman and I. Pastan, Annu. Rev. Biochem., 1993, 62, 385–427 CrossRef CAS.
- M. M. Gottesman, I. Pastan and S. V. Ambudkar, Curr. Opin. Genet. Dev., 1996, 6, 610–617 CrossRef CAS.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS.
- M. M. Gottesman and S. V. Ambudkar, J. Bioenerg. Biomembr., 2001, 33, 453–458 CrossRef CAS.
- S. V. Ambudkar, S. Dey, C. A. Hrycyna, M. Ramachandra, I. Pastan and M. M. Gottesman, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 361–398 CrossRef CAS.
- M. Dean, Y. Hamon and G. Chimini, J. Lipid Res., 2001, 42, 1007–1017 CAS.
- C. J. Chen, J. E. Chin, K. Ueda, D. P. Clark, I. Pastan, M. M. Gottesman and I. B. Roninson, Cell, 1986, 47, 381–389 CrossRef CAS.
- S. P. Cole, G. Bhardwaj, J. H. Gerlach, J. E. Mackie, C. E. Grant, K. C. Almquist, A. J. Stewart, E. U. Kurz, A. M. Duncan and R. G. Deeley, Science, 1992, 258, 1650–1654 CAS.
- A. Haimeur, G. Conseil, R. G. Deeley and S. P. C. Cole, Curr. Drug Metab., 2004, 5, 21–53 Search PubMed.
- L. A. Doyle, W. Yang, L. V. Abruzzo, T. Krogmann, Y. Gao, A. K. Rishi and D. D. Ross, Proc. Natl. Acad. Sci. USA, 1998, 95, 15665–15670 CrossRef CAS.
- K. Miyake, L. Mickley, T. Litman, Z. Zhan, R. Robey, B. Cristensen, M. Brangi, L. Greenberger, M. Dean, T. Fojo and S. E. Bates, Cancer Res., 1999, 59, 8–13 CAS.
- K. Kage, S. Tsukahara, T. Sugiyama, S. Asada, E. Ishikawa, T. Tsuruo and Y. Sugimoto, Int. J. Cancer, 2002, 97, 626–630 CrossRef CAS.
- U. Henriksen, U. Gether and T. Litman, J. Cell. Sci., 2005, 118, 1417–1426 Search PubMed.
- D. P. Kontoyiannis and R. E. Lewis, Lancet, 2002, 359, 1135–1144 CrossRef CAS.
- D. Sanglard and F. C. Odds, Lancet Infect. Dis., 2002, 2, 73–85 Search PubMed.
- H. Wolfger, Y. M. Mamnun and K. Kuchler, Res. Microbiol., 2001, 152, 375–389 CrossRef CAS.
- B. Tan, D. Piwnica-Worms and L. Ratner, Curr. Opin. Oncol., 2000, 12, 450–458 CrossRef CAS.
- G. D. Leonard, O. Polgar and S. E. Bates, Curr. Opin. Investig. Drugs, 2002, 3, 1652–1659 Search PubMed.
- T. Tsuruo, H. Iida, S. Tsukagoshi and Y. Sakurai, Cancer Res., 1981, 41, 1967–1972 CAS.
- T. Tsuruo, H. Iida, S. Tsukagoshi and Y. Sakurai, Cancer Res., 1982, 42, 4730–4733 CAS.
- R. Krishna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265–283 CrossRef CAS.
- A. H. Dantzig, R. L. Shepard, J. Cao, K. L. Law, W. J. Ehlhardt, T. M. Baughman, T. F. Bumol and J. J. Starling, Cancer Res., 1996, 56, 4171–4179 CAS.
- J. J. Starling, R. L. Shepard, J. Cao, K. L. Law, B. H. Norman, J. S. Kroin, W. J. Ehlhardt, T. M. Baughman, M. A. Winter, M. G. Bell, C. Shih, J. Gruber, W. F. Elmquist and A. H. Dantzig, Adv. Enzyme Regul., 1997, 37, 335–347 CrossRef CAS.
- A. Sparreboom, A. S. T. Planting, R. C. Jewell, M. E. L. van der Burg, A. van der Gaast, P. de Bruijn, W. J. Loos, K. Nooter, L. H. Chandler, E. M. Paul, P. S. Wissel and J. Verweij, Anti-Cancer Drugs, 1999, 10, 719–728 CAS.
- A. S. T. Planting, P. Sonneveld, A. van der Gaast, A. Sparreboom, M. E. L. van der Burg, G. P. M. Luyten, K. de Leeuw, M. de Boer-Dennert, P. S. Wissel, R. C. Jewell, E. M. Paul, N. B. Purvis and J. Verweij, Cancer Chemoth. Pharm., 2005, 55, 91–99 Search PubMed.
- P. Mistry, A. J. Stewart, W. Dangerfield, S. Okiji, C. Liddle, D. Bootle, J. A. Plumb, D. Templeton and P. Charlton, Cancer Res., 2001, 61, 749–758 CAS.
- M. J. Newman, J. C. Rodarte, K. D. Benbatoul, S. J. Romano, C. Z. Zhang, S. Krane, E. J. Moran, R. T. Uyeda, R. Dixon, E. S. Guns and L. D. Mayer, Cancer Res., 2000, 60, 2964–2972 CAS.
- U. A. Germann, D. Shlyakhter, V. S. Mason, R. E. Zelle, J. P. Duffy, V. Galullo, D. M. Armistead, J. O. Saunders, J. Boger and M. W. Harding, Anti-Cancer Drugs, 1997, 8, 125–140 CAS.
- U. A. Germann, P. J. Ford, D. Shlyakhter, V. S. Mason and M. W. Harding, Anti-Cancer Drugs, 1997, 8, 141–155 CAS.
- D. F. Twiss, S. A. Bazier and F. Thomas, J. Soc. Chem. Ind., 1922, 41, 81–88 Search PubMed.
- D. I. Eneanya, J. R. Bianchine, D. O. Duran and B. D. Andersen, Annu. Rev. Pharmacol. Toxicol., 1981, 21, 575–596 CrossRef CAS.
- J. Hald and E. Jacobsen, Lancet, 1948, 2, 1001–1004 CrossRef.
- R. K. Fuller and E. Gordis, Addiction, 2004, 99, 21–24 CrossRef.
- J. F. Cubells, M. C. Chawarski, T. George and R. S. Schottenfled, Am. J. Med. Genet., 2004, 130B, 164–165.
- D. A. Gorelick, E. L. Gardner and Z. X. Xi, Drugs, 2004, 64, 1547–1573 CAS.
- M. Jofre-Bonet, J. L. Sindelar, I. L. Petrakis, C. Nich, T. Frankforter, B. J. Rounsaville and K. M. Carroll, J. Subst. Abuse Treat., 2004, 26, 225–232 CrossRef.
- T. W. Loo and D. M. Clarke, J. Natl. Cancer Inst., 2000, 92, 898–902 CrossRef CAS.
- Z. E. Sauna, X. H. Peng, K. Nandigama, S. Tekle and S. V. Ambudkar, Mol. Pharmacol., 2004, 65, 675–684 CrossRef CAS.
- J. Chick, Drug Safety, 1999, 20, 427–435 Search PubMed.
- C. Brewer, Brit. J. Psychiat., 1984, 144, 200–202 Search PubMed.
- H. Friis and P. B. Andreasen, J. Intern. Med., 1992, 232, 133–138 CrossRef CAS.
- C. Wright, J. A. Vafier and C. R. Lake, J. Clin. Psychiat., 1988, 49, 430–434 Search PubMed.
- M. J. Ellenhorn, in Ellenhorn's Medical Toxicology: Diagnosis and treatment of human poisoning, ed. S. Schonwald, G. Ordogand and J. Wasserberger, Lippincott, Williams and Wilkins, Baltimore, 1997, vol. 2, pp. 340–349 Search PubMed.
- M. L. Shen, K. L. Johnson, D. C. Mays, J. J. Lipsky and S. Naylor, Biochem. Pharmacol., 2001, 61, 537–545 CrossRef CAS.
- M. L. Shen, J. J. Lipsky and S. Naylor, Biochem. Pharmacol., 2000, 60, 947–953 CrossRef CAS.
- S. A. Doggrell, Expert Opin. Pharmacother., 2004, 5, 2621–2624 Search PubMed.
- R. Voboril, S. N. Hochwald, J. Li, A. Brank, J. Weberova, F. Wessels, L. L. Moldawer, E. Ramsay Camp and S. L. D. MacKay, J. Surg. Res., 2004, 120, 178–188 CrossRef CAS.
- R. M. Mader, M. Muller and G. G. Steger, Gen. Pharmacol., 1998, 31, 661–666 CrossRef CAS.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer., 2003, 3, 330–338 CrossRef CAS.
- P. C. Fernandez, J. Machado, V. T. Heussler, C. Botteron, G. H. Palmer and D. A. E. Dobbelaere, Biol. Chem., 1999, 380, 1383–1394 CrossRef CAS.
- W. G. Wang, H. L. McLeod and J. Cassidy, Int. J. Cancer, 2003, 104, 504–511 CrossRef CAS.
- B. B. Aggarwal, Cancer Cell, 2004, 6, 203–208 Search PubMed.
- K. Natarajan, S. Singh, T. R. Burke, D. Grunberger and B. B. Aggarwal, Proc. Natl. Acad. Sci. USA, 1996, 93, 9090–9095 CrossRef CAS.
- M. B. Toledano and W. J. Leonard, Proc. Natl. Acad. Sci. USA, 1991, 88, 4328–4332 CAS.
- N. D. Perkins, Trends Biochem. Sci., 2000, 25, 434–440 CrossRef CAS.
- C. Y. Wang, M. W. Mayo and A. S. Baldwin, Science, 1996, 274, 784–787 CrossRef.
- C. Y. Wang, J. C. Cusack, R. Liu and A. S. Baldwin, Nat. Med., 1999, 5, 412–417 CrossRef CAS.
- E. M. Walker, Jr., D. J. Cannon, M. E. Reifsteck, K. A. Hobbs, H. F. Hardin, M. M. Jones and J. K. Skeeles, Res. Commun. Chem. Pathol. Pharmacol., 1987, 56, 253–263 Search PubMed.
- D. T. Shah, E. M. Walker, Jr., M. M. Jones, P. K. Singh and B. Larsen, Ann. Clin. Lab. Sci., 1997, 27, 282–286 Search PubMed.
- A. Decottignies and A. Goffeau, Nat. Genet., 1997, 15, 137–145 CrossRef CAS.
- M. Kolaczkowski, A. Kolaczowska, J. Luczynski, S. Witek and A. Goffeau, Microb. Drug Resist., 1998, 4, 143–158 CrossRef CAS.
- S. Shukla, Z. E. Sauna, R. Prasad and S. V. Ambudkar, Biochem. Biophys. Res. Commun., 2004, 322, 520–525 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CAS.
- D. Z. Cen, D. Brayton, B. Shahandeh, F. L. Meyskens and P. J. Farmer, J. Med. Chem., 2004, 47, 6914–6920 CrossRef CAS.
- S. S. Brar, C. Grigg, K. S. Wilson, W. D. Holder, D. Dreau, C. Austin, M. Foster, A. J. Ghio, A. R. Whorton, G. W. Stowell, L. B. Whittall, R. R. Whittle, D. P. White and T. P. Kennedy, Mol. Cancer Ther., 2004, 3, 1049–1060 Search PubMed.
- S. G. Shiah, Y. R. Kao, F. Y. H. Wu and C. W. Wu, Mol. Pharmacol., 2003, 64, 1076–1084 CrossRef CAS.
- M. Marikovsky, N. Nevo, E. Vadai and C. Harris-Cerruti, Int. J. Cancer, 2002, 97, 34–41 CrossRef CAS.
- A. P. Zhao, Z. Q. Wu, M. Pollack, F. M. Rollwagen, P. Hirszel and X. M. Zhou, Cytokine, 2000, 12, 1356–1367 CrossRef CAS.
- C. Stefan, I. Nobel, D. H. Burgess, B. Zhivotovsky, M. J. Burkitt, S. Orrenius and A. F. G. Slater, Chem. Res. Toxicol., 1997, 10, 636–643 CrossRef.
- C. S. I. Nobel, M. Kimland, B. Lind, S. Orrenius and A. F. G. Slater, J. Biol. Chem., 1995, 270, 26202–26208 CrossRef CAS.
- A. Iseki, F. Kambe, K. Okumura, S. Niwata, R. Yamamoto, T. Hayakawa and H. Seo, Biochem. Biophys. Res. Commun., 2000, 276, 88–92 CrossRef CAS.
- D. W. Pyatt, Y. Z. Yang, A. Le, W. S. Stillman and R. D. Irons, Biochem. Biophys. Res. Commun., 2000, 274, 513–518 CrossRef CAS.
- J.-C. Tsai, M. Jain, C.-M. Hsieh, W.-S. Lee, M. Yoshizumi, C. Patterson, M. A. Perrella, C. Cooke, H. Wang, E. Haber, R. Schlegel and M.-E. Lee, J. Biol. Chem., 1996, 271, 3667–3670 CrossRef CAS.
- G. Y. Liu, N. Frank, H. Bartsch and J. K. Lin, Mol. Carcinog., 1998, 22, 235–246 CrossRef CAS.
- S. Verhaegen, A. J. McGowan, A. R. Brophy, R. S. Fernandes and T. G. Cotter, Biochem. Pharmacol., 1995, 50, 1021–1029 CrossRef CAS.
- R. Bessho, K. Matsubara, M. Kubota, K. Kuwakado, H. Hirota, Y. Wakazono, Y. W. Lin, A. Okuda, M. Kawai, R. Nishikomori and T. Heike, Biochem. Pharmacol., 1994, 48, 1883–1889 CrossRef CAS.
- J. T. Wolfe, D. Ross and G. M. Cohen, FEBS Lett., 1994, 352, 58–62 CrossRef CAS.
- D. Z. Cen, R. I. Gonzalez, J. A. Buckmeier, R. S. Kahlon, N. B. Tohidian and F. L. Meyskens, Mol. Cancer Ther., 2002, 1, 197–204 Search PubMed.
- P. Costantini, A. S. Belzacq, H. La Vieira, N. Larochette, M. A. de Pablo, N. Zamzami, S. A. Susin, C. Brenner and G. Kroemer, Oncogene, 2000, 19, 307–314 CrossRef CAS.
- P. Klatt, E. P. Molina and S. Lamas, J. Biol. Chem., 1999, 274, 15857–15864 CrossRef CAS.
- P. Brennan and L. A. J. O'Neill, Biochem. Pharmacol., 1998, 55, 965–973 CrossRef CAS.
- L. Chin, Nat. Rev. Cancer, 2003, 3, 559–570 CrossRef CAS.
- M. Marikovsky, V. Ziv, N. Nevo, C. Harris-Cerruti and O. Mahler, J. Immunol., 2003, 170, 2993–3001 CAS.
- M. J. Duffy, Clin. Exp. Metastasis, 1992, 10, 145–155 CrossRef CAS.
- W. G. Stetlerstevenson, S. Aznavoorian and L. A. Liotta, Annu. Rev. Cell Biol., 1993, 9, 541–573 Search PubMed.
- M. Johnsen, L. R. Lund, J. Romer, K. Almholt and K. Dano, Curr. Opin. Cell Biol., 1998, 10, 667–671 CrossRef CAS.
- V. M. Kahari and U. Saarialho-Kere, Ann. Med., 1999, 31, 34–45 Search PubMed.
- M. D. Johnson, H. R. C. Kim, L. Chesler, G. Tsaowu, N. Bouck and P. J. Polverini, J. Cell Physiol., 1994, 160, 194–202 CrossRef CAS.
- S. WojtowiczPraga, J. Low, J. Marshall, E. Ness, R. Dickson, J. Barter, M. Sale, P. McCann, J. Moore, A. Cole and M. J. Hawkins, Invest. New Drugs, 1996, 14, 193–202 CAS.
- J. A. Shabbits, R. Krishna and L. D. Mayer, Exp. Rev. Anticancer Ther., 2001, 1, 585–594 Search PubMed.
- A. Scozzafava, A. Casini and C. T. Supuran, Curr. Med. Chem., 2002, 9, 1167–1185 CAS.
- H. E. Poulsen, S. Loft, J. R. Andersen and M. Andersen, Acta Psychiat. Scand., 1992, 86, 59–66 Search PubMed.
- H. Kitano, Nature, 2002, 420, 206–210 CrossRef CAS.
- H. Kitano, Science, 2002, 295, 1662–1664 CrossRef CAS.
- R. Brent, Cell, 2000, 100, 169–183 CrossRef CAS.
- A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, J. Lehar, E. R. Price, G. Serbedzija, G. R. Zimmermann, M. A. Foley, B. R. Stockwell and C. T. Keith, Proc. Natl. Acad. Sci. USA, 2003, 100, 7977–7982 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |