One-step, non-denaturing isolation of an RNA polymerase enzyme complex using an improved multi-use affinity probe resin
M.
Uljana Mayer
*,
Liang
Shi
and
Thomas C.
Squier
Cell Biology and Biochemistry Group, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99452, USA. E-mail: Uljana.Mayer@pnl.gov; Fax: (509) 376-6767; Tel: (509) 376-1681
First published on 14th April 2005
Abstract
The rapid isolation of protein complexes is critical to the goal of establishing protein interaction networks. High-throughput methods for identifying protein binding partners in a way suitable for mass spectrometric identification and structural analysis are required and small molecule/peptide interactions provide the key. We have now shown that a redesigned resin derivatized with a bisarsenical dye can be used to isolate the Shewanella oneidensis RNA polymerase core enzyme with a tetracysteine-tagged RNA polymerase A as bait protein. A critical advantage of this method is the ability to release the intact complex using a mild, one-step procedure with a competing dithiol. In addition to the identification of the core complex, additional interaction partners, including universal stress protein, were identified. These results provide a path forward to identifying how changes in critical protein complexes over time modulate cell function.
Cells mediate their response to environmental stresses through changes in protein–protein interactions.1,2 The direct pull-down of protein complexes followed by identification through mass spectrometry has become a method of choice for finding protein interaction partners, complementing other procedures such as crosslinking and two-hybrid methods.3–6 Since protein–protein interactions depend on cooperativity, and thus size, for strength, peptide/small molecule systems are important alternatives which minimize combined tag/probe size and give access to mild, non-denaturing elution methods. Requirements for an optimal protein tag/affinity reagent pair are 1) a small genetic tag size, 2) an uncharged tag, 3) tight tag/probe binding with minimal background interactions and 4) a mild methodology orthogonal to the wash methods that allows a one-step elution. We have developed an approach that fulfills all of the above requirements based on the bisarsenic-derivatized fluorescein reagent (FlAsH) developed by Tsien and coworkers.7–9 Proteins of interest are tagged genetically with a tetracysteine motif, which allows them to bind to a redesigned FlAsH–ethylenediamine derivatized glass resin. The protein complexes are then released in a mild, one-step procedure with a competing dithiol. We have used this new solid support to isolate the RNA polymerase complex from the bacterium Shewanella oneidensis.
It had previously been shown that highly overexpressed tetracysteine-tagged proteins can be purified using FlAsH attached to a solid support.9,10 For protein pull-downs we have modified the existing resin. We opted for a longer linker length to enhance binding. We chose a carboxylated glass bead resin (CPGBiotech), both for the lower background of glass over sepharose in pull-downs and for the high density of attachment sites. We have changed the synthetic procedure10 by starting with carboxy-fluorescein instead of aminofluorescein (see Scheme 1), thereby greatly improving the yield of the first EDC coupling step. This is followed by a one-step mercuration and deprotection. Transmetallation7,8 with arsenic yields FlAsH–ethylenediamine–EDT, which is then coupled to glass beads. The binding capacity of the substituted glass beads is about 1.5 mg protein per mL solid support, based on the purification of a 17 kDa tetracysteine-tagged protein.
 | ||
| Scheme 1 Synthesis of the FlAsH–ethylenediamine resin: a) 1) N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, dry DMSO, 2) N-Boc-ethylenediamine, 25 °C, 15 h, 95%; b) mercuric acetate, trifluoroacetic acid, 25 °C, 16 h, 90%; c) 1) arsenic trichloride, palladium acetate, N-methylpyrrolidinone, diisoproyl–ethylamine, 60 °C, then room temperature over night, 2) quench with phosphate buffer, pH 7.0, 0.5 mL ethanedithiol, 36%; d) N-hydroxysuccinimide-activated glass beads, dry DMSO. | ||
The small tag size greatly contributes to the ease of cloning, as the genetic tag can be added directly through the cloning primers and thus easily incorporated into either terminus of any protein expressed in a vector of choice for any organism of interest.11 We chose RNA polymerase (Rpo) as a model system. The bacterial core enzyme is a stable complex of two alpha (A), a beta (B) and a beta prime (C) subunits.12 In addition, the holoenzyme contains omega and sigma factor subunits. The RNA polymerase is further known to transiently interact with other proteins.
With a low-background high-capacity resin in hand, we were able to proceed to protein complex identification. For the pull-down, the tetracysteine-tagged RNA polymerase A subunit was overexpressed in S. oneidensis.13 After induction of this bait protein, the cells were lysed and exposed to the affinity resin overnight. The solid support was washed with buffer14 using mild conditions to minimize the loss of low-affinity binding components. The protein complex was eluted with the same buffer and 50 mM dimercaptopropanesulfonate. The total yield of protein was 3 mg per mL of beads. Eluted protein was run on SDS–PAGE. As shown in Fig. 1a, the major protein bands (above background) are consistent with the sizes of RpoA (36 kDa), RpoB (150 kDa) and RpoC (155 kDa). These bands make up 24% of the total protein density. There is only a single protein band on the native PAGE gel, which can be visualized by FlAsH and Coomassie blue staining and using antibodies against the V5-tag on RpoA under reducing conditions, indicating that the complex is intact after purification (Fig. 1b).
 | ||
| Fig. 1 Eluted holoenzyme protein complex characterized by SDS (a) and native PAGE (b). (a) Coommassie-stained SDS–PAGE gel of the eluted protein complex associated with 4Cys-RNA polymerase A (RpoA) bait protein (lane 2), relative to control nonspecific binding to beads (lane 1). The two major non-specific binding bands correspond in size to translation elongation factor Ef-Tu (43.3 kDa) and the inosine-5-monophosphate dehydrogenase (51.7 kDa). (b) Native gel showing only one band by 1) FlAsH staining, 2) Coommassie blue staining and 3) immunoblotting against the V5-tag on RpoA. | ||
Protein identity was verified by capillary LC–MS/MS15 on the eluate, which was trypsinized and purified by standard methods.16 SEQUEST (Thermo-Finnigan 2.0) analysis using the conservative filters pioneered by the Yates group17,18 gave good sequence coverage for the core enzyme in the bait experiments (Fig. 2), with no background for the target proteins RpoB and RpoC and a low background for RpoA. The omega subunit and sigma factor 70 were found with only a couple of peptide identifications over the course of the triplicate experiments. These peptides fulfill the Yates criteria, but do not match our criteria of at least two confident peptide hits in each experiment, indicating that under the conditions used not all binding partners remain in stoichiometric amounts (there were no hits for these proteins in the background). We are currently optimizing methods for capturing weakly and DNA- or membrane-bound binding partners.
 | ||
| Fig. 2 Schematic of the sequence coverage for the RNA polymerase core proteins from three LC–MS/MS runs. The total number of identified peptides (on left) from MS/MS are shown as coloured bars on the full protein sequence in black (red: 1 or 2 peptide hits, yellow: 3–5 peptide hits, green: 6–10 and blue: >10 identified peptides). | ||
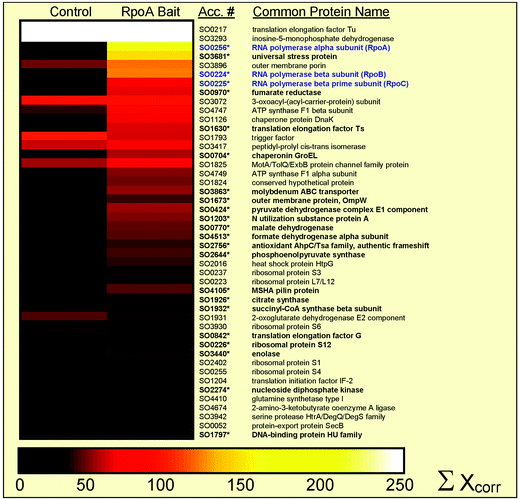 | ||
| Fig. 3 Mass spectometric identification of the protein complex. Graphical representation (Omniviz 3.6) of identified proteins associated with the RNA polymerase complex, where individual columns represent the confidence level, i.e. sum of Xcorr,17,18 for five separate experiments for eluted protein using the control beads and RpoA bait. Identified gene accession numbers and common protein names are shown to the right and core enzyme components of the RNA polymerase complex are highlighted in blue. Scale associated with colour choices for protein abundances is shown at the bottom of the figure, where white represents an Xcorr above 250. Of the 50 proteins shown, 26 are significantly above background (designated with an asterisk and in bold) as judged by 3-fold higher Xcorr values. | ||
The list of proteins which have been confidently identified with many peptide hits through mass spectrometry in the bait experiment above the background provides an interesting glimpse of additional interactors. A graphical representation (Omniviz 3.6) comparing the level of protein confidence for control and bait experiments is shown in Fig. 3. Translation elongation factor Ef-Tu and the inosine-5-monophosphate dehydrogenase are highly expressed in S. oneidensis and provide similar and higher background contamination in this and alternate pull-down methods using direct attachment, biotin- and 6His-tags. In addition to the three core RNA polymerase enzymes, a putative outer membrane protein and an analog to the E. coli universal stress protein are also found in high levels in bait over control experiments. The universal stress protein is of particular interest, since it is known to regulate stress responses in prokaryotes; however, its mechanism of action is unknown at this time and no binding partners other than a phosphorylating kinase had previously been identified.19,20
We have thus shown that the tetracysteine tag–FlAsH system is a valuable addition to existing protein pull-down methods, in that it provides a one-step, non-denaturing procedure for protein binding partner identification with yields high enough to permit structural measurements of the intact complex. In addition, in contrast to the 6His-tag–Ni-NTA system, the same tag can be used to determine the in vivo localization of the protein of interest21 and for the structural characterization of the protein and complex.22
Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under Contract DEAC06-76RLO 1830. This research was supported by the Genomics: GTL program of the Department of Energy’s Office of Biological and Environmental Research. We thank Joshua N. Adkins, Deanna L. Auberry, Joel G. Pounds, Ronald Moore and Sewite Negash for helpful discussions and the PNNL EMSL proteomics and NMR user facilities and staff.
Notes and references
- A. Bauer and B. Kuster, Eur. J. Biochem., 2003, 270, 570–578 CrossRef CAS.
- A. Dziembowski and B. Seraphin, FEBS Lett., 2004, 556, 1–6 CrossRef CAS.
- D. Figeys, Curr. Opin. Biotechnol., 2003, 14, 119–125 CrossRef CAS.
- A. C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer and G. Superti-Furga, Nature, 2002, 415, 141–147 CrossRef CAS.
- Y. Ho, A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, L. Y. Yang, C. Wolting, I. Donaldson, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. V. Hogue, D. Figeys and M. Tyers, Nature, 2002, 415, 180–183 CrossRef CAS.
- B. Seraphin, Adv. Protein Chem., 2002, 61, 99–117.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS.
- B. A. Griffin, S. R. Adams, J. Jones and R. Y. Tsien, Methods Enzymol., 2000, 327, 565–578 CrossRef CAS.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS.
- K. S. Thorn, N. Naber, M. Matuska, R. D. Vale and R. Cooke, Protein Sci., 2000, 9, 213–217 CAS.
- The gene of RNA Polymerase A (rpoA) from S. oneidensis was amplified by PCR and cloned into the pBAD-DTOPO 202 vector (Invitrogen) according the manufacturer's protocol. To add the tetracysteine tag on the C-terminus of RpoA, the reverse primer (5′-CTTGCAACAACCAGGGCAACATGCAGCTAGGTCGTCTGCTAAACTAGCTG) contained the coding sequence for AACCPGCCK (marked in bold). The forward primer (5′-CACCCAGAGTACGGTTTACAGTTACGC) was used to amplify both coding and promoter region of rpoA. pBAD-DTOPO 202 also adds the 6His and V5 tags to the recombinant RpoA for purification and identification.
- L. Snyder and W. Champness, Molecular Genetics of Bacteria, ASM Press, Washingon, DC, 2003 Search PubMed.
- L. Shi, J. T. Lin, L. M. Markillie, T. C. Squier and B. S. Hooker, BioTechniques, 2005, 38, 297–299 CrossRef CAS.
- 50 mM HEPES, 140 mM sodium chloride, 5 mM 2-mercaptoethanol, pH 7.5 was the buffer used.
- Y. Shen, N. Tolic, R. Zhao, L. Pasa-Tolic, L. Li, S. J. Berger, R. Harkewicz, G. A. Anderson, M. E. Belov and R. D. Smith, Anal. Chem., 2001, 73, 3011–3021 CrossRef CAS.
- E. F. Strittmatter, L. J. Kangas, K. Petritis, H. M. Mottaz, G. A. Anderson, Y. Shen, J. M. Jacobs, D. G. Camp and R. D. Smith, J. Proteome Res., 2004, 3, 760–769 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- D. A. Wolters, M. P. Washburn and J. R. Yates, Anal. Chem., 2001, 73, 5683–5690 CrossRef CAS.
- K. Kvint, L. Nachin, A. Diez and T. Nystrom, Curr. Opin. Microbiol., 2003, 6, 140–145 CrossRef CAS.
- M. C. Sousa and D. B. McKay, Structure, 2001, 9, 1135–1141 CrossRef CAS.
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Science, 2002, 296, 503–507 CrossRef CAS.
- B. Chen, M. U. Mayer, L.-M. Markillie, D. L. Stenoien and T. C. Squier, Biochemistry, 2005, 44, 905–914 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |