Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery†
Younsoo
Bae
a,
Woo-Dong
Jang
a,
Nobuhiro
Nishiyama
b,
Shigeto
Fukushima
a and
Kazunori
Kataoka
*ab
aDepartment of Materials Science and Engineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kataoka@bmw.t.u-tokyo.ac.jp; Fax: +81-3-5841-7139; Tel: +81-3-5841-7138
bCenter for Disease Biology and Investigative Medicine, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
First published on 30th June 2005
Abstract
A new type of multifunctional polymeric micelle drug carrier for active intracellular drug delivery was prepared and characterized in this study. The micelle is a nano-supramolecular assembly with a spherical core–shell structure, and its surface and core were modified with piloting molecules for cancer cells and pH-sensitive drug binding linkers for controlled drug release, respectively. In order to prepare such micelles, self-assembling amphiphilic block copolymers, folate–poly(ethylene glycol)–poly(aspartate hydrazone adriamycin) [Fol–PEG–P(Asp-Hyd-ADR)], were specially designed and synthesized by installing a molecular promoter to enhance intracellular transport, folate (Fol), at the end of the shell-forming PEG chain and conjugating the anticancer drug, adriamycin (ADR), to the side chain of the core-forming PAsp segment through an acid-sensitive hydrazone bond. Because folate-binding proteins (FBP) are selectively overexpressed on the cancer cell membranes, the folate-bound micelles (FMA) can be guided to the cancer cells in the body, and after the micelles enter the cells, hydrazone bonds are cleaved by the intracellular acidic environment (pH 5–6) so that the drug release profile of the micelles is controlled pH-dependently. In this regard, FBP-binding selectivity of the prepared FMA was evaluated by surface plasmon resonance (SPR) measurements. The tetrazolium dye method (MTT assay) using human pharyngeal cancer cells (KB cell) revealed that FMA significantly improved cell growth inhibitory activity in spite of a short exposure time due to the selective and strong interaction between folate molecules and their receptors. Subsequent flow cytometric analysis showed that cellular uptake of FMA significantly increased. Consequently, these findings would provide one of the most effective approaches for cancer treatment using intracellular environment-targeting supramolecular drug carriers.
Introduction
Biologically active materials for cancer chemotherapy are usually selected using cell biology and pharmacology, which provide a rationale for understanding the Absorption, Distribution, Metabolism and Excretion (ADME) of exogenous molecules in the body. A number of studies have claimed that polymer science and supramolecular chemistry can offer the most adequate and suitable means for designing functional nano-bio devices.1–5 For these reasons, growing interest is currently focused on the creation of functional polymeric nano-carriers that regulate ADME of drugs at the boundary between biochemistry and nano-technology.6,7Among such carriers, spherical supramolecular nano-assemblies from amphiphilic block copolymers, called polymeric micelles, have attracted considerable attention in the field of drug delivery systems due to their unique characteristics such as high water-solubility, high drug loading capacity and low toxicity, which are induced by the prolonged circulation in the blood and enhanced accumulation in tumor tissue.8 In particular, the intracellular environment-sensitive polymeric micelle, that can release the loaded drug, adriamycin (ADR), through sensing pH decreases in the acidic endocytic compartments such as endosomes (pH 5–6) and lysosomes (pH 4–5), has recently been reported to minimize non-specific systemic spread of toxic drugs while maximizing tumor-directed drug delivery efficiency.9,10 Indeed, animal studies have elucidated that this type of micelle significantly improves the bioavailability of existing drugs by controlling the drug release profile, inducing high antitumor activity with extremely low drug toxicity and avoiding clearance by the host defense system and uptake by normal tissue. Nevertheless, cell growth inhibitory activity still needs to be improved because the effective dose for cancer treatment using these micelles was relatively high, 4-fold compared with free drugs. However, in vitro activity of most polymer–drug conjugates is known to decrease as their biological behavior, that is involved with cellular uptake by an endocytotic pathway and relatively slow drug release, often works to the disadvantage of exerting drug efficacy compared to free drugs that rapidly move into the cell interior.11 Consequently, it is expected that if the macromolecular drug carriers were actively transported inside the cells, the bioavailability of the carriers should increase more than the drug carrier system that simply exploits passive drug delivery. In order to achieve both these goals, the micelles are also required to selectively and strongly interact with targeted cells so that intracellular transport can be controllable. On the basis of this background, we have developed a new type of multifunctional polymeric micelle drug carrier system that will actively enhance its capability to deal with intracellular drug delivery to the targeted cancer cells (Fig. 1).
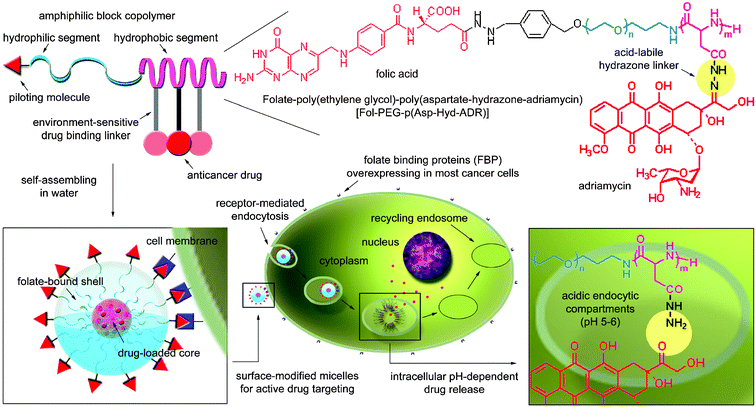 | ||
| Fig. 1 Preparation of multifunctional polymeric micelles with tumor selectivity for active drug targeting and pH-sensitivity for intracellular site-specific drug transport. Folic acid with high-tumor affinity due to the overexpression of its receptors was conjugated onto the surface of the micelle. | ||
In the case of carriers for targeting cancer cells, the fundamental problems such as intracellular transport and the release of incorporated materials are often encountered during the materials' intracellular movement. These problems are particularly crucial to the carriers in terms of the delivery of materials that should become pharmaceutically effective after entering cells. To satisfy the former requirement, the micelles we present in this study are equipped with folate-ligands, which are non-immunogenic and stable during transportation in vivo without denaturation. For the latter requirement, the pH-sensitivity that enables selective drug release in intracellular acidic compartments, endosomes (pH 5–6), is used. Folate is a vitamin with low molecular weight (MW = 441.4) and behaves as a ligand because it has high affinity for its receptors (Kd < 1 nM), folate-binding proteins (FBP), that are selectively overexpressed on the surface of cancer cells.12,13 Therefore, its conjugates with an appropriate design can be directed to the cancer cells in the body and internalized in the target cells via receptor-mediated endocytosis.14–16 In the meantime, there is notable intracellular behavior of the folate-bound materials after ligand-mediated endocytosis, which distinguishes the folate ligand from other types of ligands such as antibodies, hormones and peptides. In contrast to the fact that most ligands are internalized and transported to the lysosomes, folate-conjugates remain in ‘recycling endosomes (pH 5–6)’ or escape into the cytoplasm, and such a characteristic plays a crucial role in accelerating intracellular uptake of the materials by the cell, avoiding the possible effects of lysosomal enzyme action.17–19
Therefore, the experimental results discussed in this study will present clear information about the effects of folate ligands on biological activity of the intracellular drug delivery when using the supramolecular drug carriers that are selectively activated in the cell.
Results and discussion
Design of the micelles and synthesis of their constituent bifunctional block copolymers
To prepare the folate-bound micelles, self-assembling amphiphilic block copolymers, folate–poly(ethylene glycol)–block–poly(aspartate hydrazone adriamycin) [Fol–PEG–p(Asp-Hyd-ADR) 11], were designed and synthesized by developing a new and optimized synthetic protocol of both folate and poly(ethylene glycol) (PEG) to introduce functional groups for conjugation (Scheme 1). In general, two methods can be used to conjugate folate onto the surface of the polymeric micelles: (i) preparation of a surface-activated micelle followed by the conjugation of folate molecules by mixing them in the solution, and (ii) conjugation of folate molecules onto the constituent block copolymers followed by the preparation of the micelles with folate-bound block copolymers. The former may be a simple method to prepare the surface-modified micelles. However, it is unlikely to ensure high loading contents of folate molecules because the reaction rate is usually low and a cumbersome process of removing unreacted folate molecules is inevitably required. Moreover, the presence of unreacted ligand molecules, free folate, may hamper the binding between folate-bound micelles and folate-binding proteins (FBP) through competitive interaction. In contrast, the latter method, which was adopted in this study, requires a somewhat complex synthetic route involving the protection of functional groups to prevent side reactions, but has several advantages including quantitative reactions and characterization of the products. An impediment to the preparation of folate-conjugates, isomerization between inactive α-conjugates and active γ-conjugates, can be overcome, and this is quite crucial when preparing folate-conjugates. It has been reported that isomerization of folate-conjugated derivatives frequently occurs, in the case of DCC-coupling for instance, and affects the FBP-binding selectivity of folic acid.20 Therefore, the present synthetic route is probably one of the the best ways to conjugate folic acid as well as anticancer drugs with PEG at its γ-position to maintain FBP-binding activity and hydrazide groups at the drug-binding moiety of the side chain, respectively.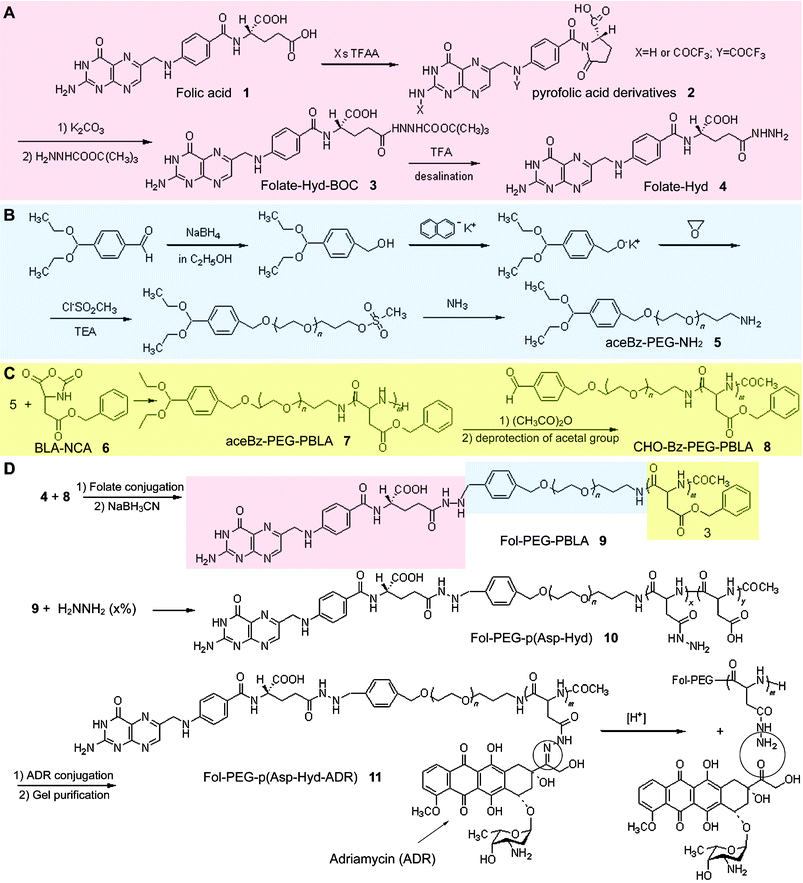 | ||
| Scheme 1 Synthetic procedure of folate–poly(ethylene glycol)–poly(aspartate hydrazone adriamycin) block copolymers (D). Folate-hydrazide (A), α-4-(diethoxymethyl)benzyl-ω-amine-poly(ethylene glycol) (B), and aldehyde-benzyl-poly(ethylene glycol)-poly(β-benzyl L-aspartate) (C) were prepared prior to synthesis of the product. | ||
Synthesis of block copolymers and preparation of the folate-bound micelles
As described above, the micelles were prepared after the synthesis of folate–polymer conjugates to maintain FBP-binding activity. This is achieved by the design of an end-group functionalized folate and PEG.Treatment of folic acid 1 (Fig. 2A) with an excess amount of trifluoroacetic anhydride in THF produces N2,10-bis(trifluoroacetyl)-pyrofolic acid 2.21 By adding water, the compound affords N10-(trifluoroacetyl)-pyrofolic acid and can be further transformed to pyrofolic acid via deacylation with K2CO3. However, both N10-(trifluoroacetyl)-pyrofolic acid and pyrofolic acid are generally known as isomers, and the two imide carbonyl groups (γ-carbonyl and pteroic carbonyl) of pyrofolic acid derivatives, N10-(trifluoroacetyl)-pyrofolic acid and pyrofolic acid, can react with nucleophiles. For these reasons, the products were purified to confirm the absence of pteroic acid derivatives by using a preparative HPLC and TLC, and pure folate–hydrazide–BOC (Fol–Hyd–BOC) 3 was successfully collected (Fig. 2B).
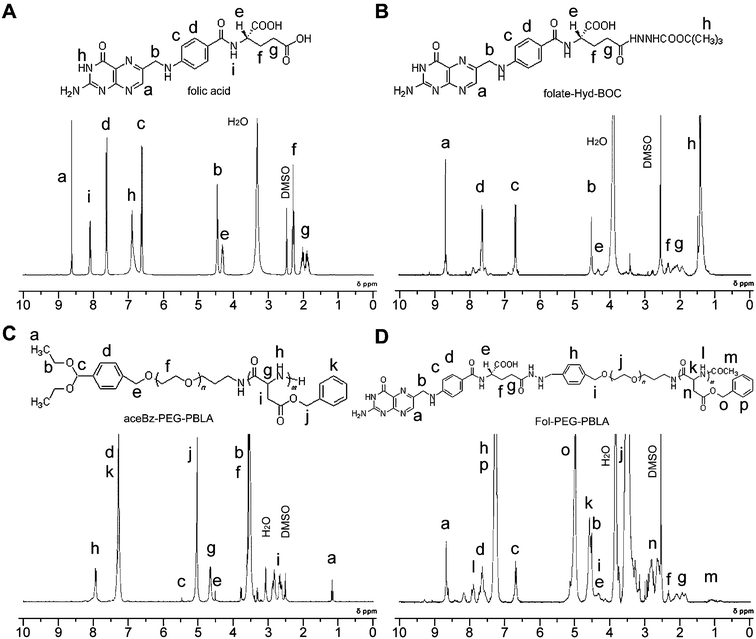 | ||
| Fig. 2 1H-NMR spectra of folate (A), folate–hydrazide–BOC (B), 4-(diethoxymethyl)benzyl–poly(ethylene glycol)–poly(β-benzyl L-aspartate) (C) and folate–poly(ethylene glycol)–poly(β-benzyl L-aspartate) (D). | ||
From the GPC analysis, the weight-average molecular weight (Mw) and the molecular weight distribution (Mw/Mn) of α-4-(diethoxymethyl)benzyl-ω-amine–poly(ethylene glycol)
[aceBz–PEG-NH25] were determined to be 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 159 and 1.02, respectively. The molecular weight was in accordance with the estimated value calculated from the ratio between monomers and initiators and its distribution was extremely narrow. These results indicate that the polymerization of EO proceeds without any side reactions by using potassium 4-(diethoxymethyl)benzylalkoxide (PDA) as an initiator.22 The end-group was then checked by 1H-NMR, comparing the peaks of PEG and 4-(diethoxymethyl)benzyl alcohol. The molecular weight was determined to be 10971 from the peak intensity ratio of PEG (-OCH2CH2-) and benzylacetal [(CH3CH2O)2CH-], which is in a good agreement with the Mw determined by GPC (10159). Fig. 2C shows the 1H-NMR peak of α-4-(diethoxymethyl)benzyl-poly(ethylene glycol)-poly(β-benzyl-L-aspartate)
[aceBz–PEG–PBLA 7] synthesized via ring-opening polymerization of β-benzyl L-aspartate–N-carboxy-anhydride (BLA–NCA) by using 5 as a macromolecular initiator.
159 and 1.02, respectively. The molecular weight was in accordance with the estimated value calculated from the ratio between monomers and initiators and its distribution was extremely narrow. These results indicate that the polymerization of EO proceeds without any side reactions by using potassium 4-(diethoxymethyl)benzylalkoxide (PDA) as an initiator.22 The end-group was then checked by 1H-NMR, comparing the peaks of PEG and 4-(diethoxymethyl)benzyl alcohol. The molecular weight was determined to be 10971 from the peak intensity ratio of PEG (-OCH2CH2-) and benzylacetal [(CH3CH2O)2CH-], which is in a good agreement with the Mw determined by GPC (10159). Fig. 2C shows the 1H-NMR peak of α-4-(diethoxymethyl)benzyl-poly(ethylene glycol)-poly(β-benzyl-L-aspartate)
[aceBz–PEG–PBLA 7] synthesized via ring-opening polymerization of β-benzyl L-aspartate–N-carboxy-anhydride (BLA–NCA) by using 5 as a macromolecular initiator.
In the case of folate–poly(ethylene glycol)–poly(β-benzyl-L-aspartate)
[Fol–PEG–PBLA 9], the molecular weight and its distribution were 22673 and 1.142 respectively which were determined by GPC equipped with a UV-VIS detector (360 nm) to confirm the conjugation between folate and PEG. 1H-NMR also supported that the conjugation of folate to PEG was successfully carried out because a peak of aldehyde groups of unreacted PEG was not observed after the reaction (Fig. 2D). The anticancer drug, adriamycin (ADR), was introduced to Fol–PEG–PBLA block copolymers through an acid-sensitive hydrazone bond, and drug-loading contents and pH-sensitive drug release profiles of the folate-bound pH-sensitive micelle with ADR (FMA) were calculated by reversed phase liquid chromatography referring to a method reported elsewhere.10 The compositions of 11 were 12–32–21, denoting that the polymer backbones consist of PEG with the molecular weight of 12000 g mol−1 for the shell-forming segment, PBLA with 32 repeating units for the core-forming segment, and 21 hydrazide groups that are substituted for benzyl groups of PBLA for drug-binding. 11 formed spherical micelles with 63.4 nm diameters in aqueous solutions, which was confirmed by DLS. In the meantime, the folate-unbound pH-sensitive micelle with ADR (MA) was prepared from α-methoxy-poly(ethylene glycol)–poly(aspartate hydrazone adriamycin)
[PEG–p(Asp-Hyd-ADR)] as control. The compositions of its constituent block copolymer PEG–p(Asp-Hyd-ADR) was 12–37–28, denoting that the polymer backbones consisted of PEG with molecular weight of 12000 g mol−1, PBLA with 37 repeating units, and 28 hydrazide groups substituted for benzyl groups of PBLA for drug-binding. The number of hydrazide groups for drug-binding was optimized by considering stability and the drug release profile of the micelles9,10
(see also supporting information).† Prepared MA had 65 nm diameters, which were also determined by DLS. The size distribution of the micelles, expressed as polydispersity index (μ/Γ2), was 0.155 and 0.168 for MA and FMA, respectively.
FBP-binding selectivity of the micelles
The FBP-binding effect of FMA was evaluated by surface plasmon resonance (SPR) measurements. For the experiments, FBP was immobilized onto a dextran-coated gold sensor chip, which was freshly prepared for each sample injection. The analysis showed that FMA not only effectively recognized but also strongly bound to the FBP (Fig. 3). Interestingly, once it binds, even though excess amounts of free folic acid (500 µg ml−1) were added, FMA was not displaced or detached. On the other hand, when both FMA and free folic acid were injected at the same time, the FBP-binding effect of FMA obviously decreased. These results can be explained by the fact that FMA can bind FBP with a strong multivalent form, and binding sites are saturated and blocked in the presence of free folate molecules. Such characteristic behavior was not shown in the case of MA used as control. Therefore, it is clearly revealed that FMA exhibits selective FBP-binding behavior, and the concept described in this research would be an effective approach for designing environment-sensitive polymeric micelle drug carriers for specific recognition of cancer cells in the body. | ||
| Fig. 3 Evaluation of FBP-binding selectivity of folate-bound micelles by using surface plasmon resonance (SPR) measurement (eluent: 100 mM phosphate buffer, pH 7.4; flow rate: 10 µl min−1; density of folate-binding protein: 3 ng mm−2 per channel; sample concentration: 200 µg ml−1). | ||
Evaluation of in vitro cytotoxic acitivity of FMA
SPR measurements showed that the FMA has strong and selective FBP-binding effects. Meanwhile, it is generally accepted that FBPs are overexpressed on the cell membranes of cancer cells.23 Folate-bound carriers, therefore, are expected not only to increase the tumor-specific accumulation but also to reduce undesired distribution in normal tissues, reducing side effects from drug toxicity. In order to verify whether the FBP-binding effect of FMA is also active against living cells, in vitro growth-inhibition tests were carried out using human pharyngeal cancer cells (KB cell).KB cells are widely used for evaluating the interaction between folate-mediated drug delivery systems and cancer cells due to their high affinity FBP-binding effect that has been studied and well elucidated by a large number of scientists over decades even though its contamination by HeLa cells is suspicious.24,25 As described in the experimental section, KB cells were incubated with FMA, MA and free ADR, with exposure times of 3 h and 24 h. Post-incubation was carried out for 24 h after drug exposure, considering the fact that the cytotoxicity of the intracellular environment-sensitive drug carriers has a tendency to increase time-dependently accompanying drug release.26–28Fig. 4 shows that FMA enhances cell-growth inhibitory activity effectively, and we hypothesized it is probably due to the accelerated intracellular transport of the micelles by folate installation on their surfaces. Although both FMA and MA showed the delayed cytotoxicity changing with exposure time, only FMA showed effective growth-inhibitory activity with a short exposure time, clearly demonstrating that each micelle undergoes a different mechanism of entering the cell. As summarized in Table 1, it is notable that cytotoxic activity of FMA was as high as 1/1.45 fold with respect to free ADR after 24 h exposure time. Interestingly, even though both FMA and MA have equivalent drug contents, cytotoxicity of FMA was 8-fold higher than that of MA in terms of IC50 after 24 h incubation. As described above, both FMA and MA are designed to release the loaded drugs only by sensing pH decrease in the cells, and therefore such a great difference in cytotoxicity indicates that a strong FBP-binding effect should be the decisive reason for the enhanced efficacy of the intracellular environment-sensitive polymeric micelles.
![Correlation between exposure time and cell-growth inhibitory activity of folate-bound pH-sensitive micelles with adriamycin (FMA), folate-unbound pH-sensitive micelles with adriamycin (MA) and free adriamycin (ADR)
[3 h exposure (A) and after 24 h incubation (B)].](/image/article/2005/MB/b500266d/b500266d-f4.gif) | ||
| Fig. 4 Correlation between exposure time and cell-growth inhibitory activity of folate-bound pH-sensitive micelles with adriamycin (FMA), folate-unbound pH-sensitive micelles with adriamycin (MA) and free adriamycin (ADR) [3 h exposure (A) and after 24 h incubation (B)]. | ||
| Sample | Exposure time/h | IC50b/µg ml−1 ± SD | Relative indexc |
|---|---|---|---|
| a KB cells were used for the eight independent experiments (n = 8). b IC50 means the inhibitory concentration of the drugs required for 50% reduction in cell population. The values of the micelles are calculated with the ADR equivalents. c Relative index means the ratio between a control and the object for comparison. Here, we evaluated the growth inhibitory effect of the micelle by converting their concentrations with respect to ADR after 24 h incubation as a control. d ND denotes ‘not determined’. | |||
| ADR | 3 | 0.103 ± 0.052 | 2.19 |
| 24 | 0.047 ± 0.013 | 1 | |
| FMA | 3 | 0.21 ± 0.104 | 4.47 |
| 24 | 0.068 ± 0.039 | 1.45 | |
| MA | 3 | NDd | NDd |
| 24 | 0.43 ± 0.065 | 9.15 | |
Enhanced cellular uptake of the micelles by folate installation
In order to confirm our hypothesis that cellular uptake of the micelles is enhanced by folate installation above, flow cytometric (FCM) analysis was carried out by changing sample exposure time to the cells. Based on the in vitro cytotoxicity results, samples (FMA, MA and ADR) were exposed to KB cells for 3 h and 24 h, and increasing fluorescence of the cells was monitored. As shown in Fig. 5, the experimental results clearly demonstrated that cellular uptake of FMA drastically increased even with short exposure time (3 h), and the extent was almost similar to that of ADR after 24 h. Although time-dependent increase in fluorescence intensity was also observed in the case of MA, its cellular uptake still remained low in comparison with FMA as well as ADR. Therefore, it is concluded that cellular uptake of the micelles can be significantly accelerated by installing folate on their surfaces, and this conclusion would be one of the most reasonable explanations for the difference between FMA and MA in their cytotoxic activity in spite of equivalent drug loading contents. | ||
| Fig. 5 Flow cytometric histogram profiles of time-dependent change in fluorescence intensity when KB cells were exposed to FMA, MA and ADR for 3 h and 24 h. | ||
Conclusion
This paper describes a novel approach by which a polymeric micelle can be modified as a multifunctional drug carrier for intracellular active drug targeting to elevate the efficacy and safety of the loaded drugs. In order to prepare such micelles, folate was conjugated to the end of shell-forming PEG of amphiphilic PEG–PBLA block copolymers, that self-assembled into a spherical nano-structure in aqueous solutions, while anticancer drugs (ADR) were conjugated through acid-sensitive hydrazone bonds to the side chains of the core-forming PBLA block.Obtained experimental results demonstrated that this micelle, with both pH sensitivity and receptor selectivity, has a capacity not only to selectively control intracellular environment-sensitive drug release but also to significantly enhance drug delivery efficiency by affecting drug efficacy with short exposure times. For the preparation of folate-bound block copolymers, Fol–PEG–p(Asp-Hyd-ADR), it must be emphasized that our synthetic route ensures the activity of folate for cancer-targeting as well as hydrazide groups for drug-binding. Folate was functionalized at its γ-position to conjugate shell-forming PEG with aldehyde groups at the end of the chain, and this method allows the active folate ligands to conjugate on the surface of the micelles efficiently. SPR analysis showed that the folate-bound micelle was selectively interacting with the immobilized FBP, whereas the folate-unbound micelle seemed unlikely to interact with FBP. Such a characteristic FBP-binding effect of the micelle was also observed against live cells. In vitro cytotoxicity assay and FCM analysis clearly indicated that cell growth inhibitory activity of the micelle was enhanced due to enhanced cellular uptake. Consequently, it is concluded that the folate-bound polymeric micelle is an excellent intelligent nano-device for actively delivering drugs inside the cell via selective protein-binding affinity, and therefore, this system would provide a safe and effective strategy for new modalities to treat cancers using macromolecular drug carriers that interact with a living body.
Experimental
Materials
β-Benzyl-L-aspartate was from Sigma and α-methoxy-ω-amino poly(ethylene glycol) (PEG; MW = 12000) was from Nippon Oil & Fats, Japan. PEG was purified using an ion-exchange gel column (CM-Sephadex C-50, Amersham Pharmacia Biotech) prior to the synthesis of the block copolymers. Adriamycin hydrochloride (ADR-HCl) was from Nippon Kayaku, Japan, and its purity was checked by reversed phase liquid chromatograpy (RPLC). Sephadex LH-20 gel was from Amersham Pharmacia Biotech, Sweden.
Tetrahydrofuran (THF), dichloromethane (CH2Cl2), N,N-dimethyl formamide (DMF), dimethylacetamide (DMAc), anhydrous methanol (MeOH), trifluoroacetic acid (TFA), trifluoroacetic anhydride (TFAA), acetic anhydride (AA), acetonitrile (CH3CN), methanesulfonyl chloride (CH3SO2Cl), triethylamine (TEA) and diethyl ether were purchased from Wako Pure Chemical Industries, Co., Ltd., Japan. THF, DMF, and CH2Cl2 were distilled twice following standard procedures. Carbazic acid tert-butyl ester (CAt-BE) and potassium carbonate (K2CO3) were purchased from Tokyo Kasei Organic Chemicals Co., Ltd., Japan. These chemicals were used without further purification. Folic acid (C19H19N7O6, MW = 441.4), 4-(diethoxymethyl)benzaldehyde and sodium cyanoborohydride (NaBH3CN) were purchased from Sigma Chemical Co. Ltd., USA. Ethylene oxide (EO) was from Sumitomo Seika Chemicals Co. Ltd., Hyogo, Japan, and dried over calcium hydride followed by the distillation.
Cell lines and animals
A human pharyngeal cancer KB cell line was purchased from Health Science Research Resources Bank, Osaka, Japan. Cells were cultured in Dulbecco's Modified Eagle cell culture Medium (DMEM, Sigma Chemical Co., Inc., USA) containing 10% FBS in a humidified atmosphere with 5% CO2 at 37 °C.Devices
Gel permeation chromatography (GPC) measurements were carried out using a TOSOH HLC−8220 equipped with TSKgel columns (G4000PWXL and G3000PWXL). Internal refractive index (RI) and ultraviolet-visible absorption (UV-Vis; 360 nm for folate) detectors were used. DMF with 10 mM LiCl was used as an eluent at a flow rate of 0.8 ml min−1 at 40 °C. 1H-NMR spectra were measured with a JEOL EX300 spectrometer (JEOL, Tokyo, Japan). Chemical shifts are reported in ppm relative to the residual protonated solvent resonance.Synthesis of folate–hydrazide (Fol–Hyd) 10
Folic acid 1 (10 g, 23 mmol) was dissolved in ice-cooled anhydrous THF (100 ml), and an excess amount of TFAA (28 ml, 197 mmol) was slowly added. The mixture gradually turned into a homogeneous solution with a brown color as the reaction proceeded (pyrofolic acid derivatives 2). After 10 h, ice (10 g) was added to the solution to prepare N10-(trifluoroacetyl)-pyrofolic acid. After precipitation in diethyl ether, the yellow powder was collected by filtration and dried under vacuum. N10-(trifluoroacetyl)pyrofolic acid (5 g, 9.6 mmol) was then dissolved in dry DMF (25 ml), and K2CO3 was slowly added at 25 °C. The solution was carefully acidified with 5% aqueous hydrochloric acid. The precipitate was washed with water and diethyl ether. Pyrofolic acid was dried under vacuum and collected. Pyrofolic acid (500 mg, 1.2 mmol) and CAt-BE (1.6 g, 10 equiv.) were dissolved in 20 ml of dry DMF, and the reaction was allowed to proceed at 40 °C in an argon atmosphere. After 24 h reaction, folate–hydrazide–BOC 3 was precipitated in ether. The precipitate was isolated by centrifugation and then purified by a recycling preparative HPLC (LC918, Japan Analytical Industry Co., Ltd., Tokyo, Japan) and a thin-layer chromatography (TLC, 50 mmol phosphate buffer, pH 7, and acetonitrile = 3 ∶ 1). Subsequently, BOC groups of folate–hydrazide–BOC were removed with TFA, giving Fol–Hyd 4.Synthesis of α-4-(diethoxymethyl) benzyl-ω-amine-poly(ethylene glycol) [aceBz-PEG-NH2] 5
4-(diethoxymethyl)benzaldehyde (15 g, 72 mmol) was dissolved in dry ethanol (300 ml) under argon and the solution was cooled on ice during the addition of sodium borohydride (NaBH4, 3.0 g, 79.3 mmol) as reported elsewhere.22 The reaction was checked by TLC, eluted with the solution from diethyl ether and hexane (1 ∶ 1). The mixture was put into water (100 ml) and extracted by CH2Cl2. The organic solution was washed with anhydrous MgSO4, evaporated, and distillated to give the products. Potassium naphthalene was prepared by adding 3.76 g (96 mmol) of potassium into 300 ml of ice-cold THF in which 11.54 g (90 mmol) of naphthalene was added. After 24 h, unreacted potassium in the solution was removed. Potassium 4-(diethoxymethyl)benzylalkoxide (PDA), that is as an initiator for the polymerization of EO, was then prepared by mixing 4-(diethoxymethyl)benzylalcohol (0.19 g, 0.90 mmol) and potassium naphthalene (0.90 mmol) into dry THF (30 ml) under Ar. After 10 min, 4.5 g (102 mmol) of EO was added to the solution, followed by the reaction at room temperature for 2 days to give aceBz–PEG–OH. Subsequently, α-4-(diethoxymethyl)benzyl-ω-amine-poly(ethylene glycol) [aceBz–PEG–NH25] was produced from the prepared aceBz–PEG–OH via aceBz–PEG–OSO2CH3. aceBz–PEG–OH was mixed with CH3SO2Cl (0.62 g, 5.4 mmol) and TEA (0.82 g, 8.1 mmol) in 25 ml of THF. After precipitation in diethyl ether, filtration and dry, aceBz–PEG–OSO2CH3 was obtained. Then, 100 ml of 25% ammonia solution was added with respect to 1 g of aceBz–PEG–OSO2CH3 and the reaction was allowed to proceed for 2 days. The volume of the solution was concentrated into 200 ml by rotary evaporation and dialyzed against 0.25% ammonia solution, followed by substitution with distilled water and lyophilization to give 5.Synthesis of folate–poly(ethylene glycol)–poly(aspartate hydrazone adriamycin) [Fol–PEG–p(Asp–Hyd–ADR) 11]
4-(diethoxymethyl)benzyl–poly(ethylene glycol)–poly(β-benzyl L-aspartate) (aceBz–PEG–PBLA 7) was synthesized via ring-opening polymerization of β-benzyl L-aspartate N-carboxy-anhydride (BLA–NCA) by using 5 as a macromolecular initiator. Acetylation of the ω-amine group of 7 was carried out with AA to prevent dimerization between aldehyde–benzyl–poly(ethylene glycol)–poly(β-benzyl L-aspartate) (CHO–Bz–PEG–PBLA 8) block copolymers, which were obtained by deprotecting acetal groups under the acidic condition with the 0.1 N hydrochloric acid aqueous solution. The end-group activated 8 (250 mg) and 4 (50 mg, 10 equiv.) were dissolved in dry DMF and conjugated through an imine formation to give folate–poly(ethylene glycol)–poly(β-benzyl L-aspartate) [Fol–PEG–PBLA 9]. Because imines are acid-labile, reduction of the bond between 8 and 4 was carried out using NaBH3CN. The extent of folate conjugation on 8 was determined and confirmed by 1H-NMR and GPC measurements, respectively. The benzyl groups of 9 were substituted with hydrazide groups for drug binding by ester–amide exchange (EAE) aminolysis reaction. 500 mg of 9 was dissolved in 10 ml of dry DMF, and anhydrous hydrazine (0.62 mg, MW = 32.05) was added to the solution. The reaction was allowed to proceed at 40 °C for 24 h, followed by the deprotection of remaining benzyl groups with 0.1 N NaOH in water at 25 °C and dialysis against 0.25% ammonia solution. After freeze-drying, 100 mg of the obtained folate–poly(ethylene glycol)–poly(aspartate hydrazide) [Fol–PEG–p(Asp-Hyd) 10] was dissolved in 50 ml of DMF, and excess amount of ADR–HCl (200 mg, MW = 580), with respect to drug binding hydrazide residues at the side chain of block copolymers, was added. The mixed solution was stirred at room temperature for 24 h while being protected from light and concentrated to a volume of 10 ml under reduced pressure, followed by gel purification using Sephadex LH-20 to completely remove unbound ADR. Applied solution was separated into two fractions and the one eluted first was collected. After precipitation from ether, the final product was evaluated by RPLC to confirm the absence of free ADR. Then, the isolated red folate–poly(ethylene glycol)–poly(aspartate hydrazone adriamycin) block copolymer [Fol–PEG–p(Asp–Hyd–ADR) 11] was freeze-dried from benzene. Drug loading content in 11 was calculated by cleaving hydrazone linkers in 0.1 N HCl solutions and measuring the released drugs, which were determined by RPLC and UV spectroscopy from the absorbance intensity at 485 nm. Purified 11 was dissolved in DMAc again to prepare micelles by a dialysis method.Evaluation of folate-binding property of the micelles using surface plasmon resonance (SPR) measurement
To study the interaction between FBP and the folate-bound intracellular pH-sensitive polymeric micelles loading ADR (FMA), surface plasmon resonance (SPR) measurements were performed (BIAcore 3000, Biacore AB, Uppsala, Sweden). The folate unbound polymeric micelles loading ADR (MA) were prepared as control. For the experiments, FBP was immobilized onto the surface of a sensor chip as follows: (1) the device was saturated with PBS buffer (100 mM PBS, pH 7.4); (2) N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) were added to the carboxylated dextran-coated sensor chip; (3) FBP dissolved in PBS buffer at a concentration of 200 µg ml−1 was injected; and (4) The sensor chip was washed with 1 M ethanolamine aqueous solution to deactivate residual NHS-esters on the surface. The immobilization protocol, which was performed at a flow rate of 10 µl min−1, allowed the binding of 3 ng mm−2 of FBP per channel. The binding effects of both FMA and MA to the sensor chip were evaluated by performing SPR measurements at a flow rate of 10 µl min−1 with a concentration of 200 µg ml−1. The micelles were allowed to interact with FBP for 30 min. All samples were tested on newly immobilized FBP.In vitro growth-inhibition assay
In order to verify the FBP-binding effect of the micelle against live cells, in vitro growth-inhibition tests were carried out using a human pharyngeal cancer KB cell line. A tetrazolium dye method, called the MTT assay, was used for the test.29 Cells were exposed to FMA, MA, and ADR for 3 h and 24 h and washed, followed by 24 h post-incubation. Live cells were then counted using a Bio-RAD Microplate Reader 550 (Bio-Rad Laboratories). Drug concentrations of the micelles were determined based on the amounts of loaded ADR so that IC50 values could be compared directly.Flow cytometric (FCM) analysis
Enhanced cellular uptake of FMA was confirmed using flow cytometry (EPICS XL Flow Cytometry Systems, Beckman Coulter, Inc). Using 12-well culture plates, exponentially growing KB cells were seeded (30000 cell per well) and pre-incubated for 24 h, followed by coincubation with 10 µg ml−1 of FMA, MA, and ADR. After exposure for 3 h and 24 h, the medium was discarded and the cells were washed three times with PBS. Cells were then detached by trypsinization, centrifuged and dispersed again in PBS for the measurements. Data were acquired and processed with the accompanying software (EXPO 32).
Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and by Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation (JST).References
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977 CAS.
- M. L. Adams, A. Lavasanifar and G. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS.
- J. Kopecek, P. Kopeckova, T. Minko, Z. R. Lu and C. M. Peterson, J. Controlled Release, 2001, 74, 147 CrossRef CAS.
- Y. Takakura and M. Hashida, Pharm. Res., 1996, 13, 820 CrossRef CAS.
- H. Maeda and Y. Matsumura, Crit. Rev. Ther. Drug, 1989, 6, 193 Search PubMed.
- R. Duncan, Nat. Rev. Drug. Discov., 2003, 2, 347 CrossRef CAS.
- D. S. Goldin, C. A. Dahl, K. L. Olsen, L. H. Ostrach and R. D. Klausner, Science, 2001, 292, 443 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640 CrossRef CAS.
- Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16, 122 CrossRef CAS.
- A. T. Jones, M. Gumbleton and R. Duncan, Adv. Drug Delivery Rev., 2003, 55, 1353 CrossRef CAS.
- S. D. Weitman, R. H. Lark, L. R. Coney, D. W. Fort, V. Frasca, V. R. Zurawski and B. A. Kamen, Cancer Res., 1992, 52, 3396 CAS.
- I. G. Campbell, T. A. Jones, W. D. Foulkes and J. Trowsdale, Cancer Res., 1991, 51, 5329 CAS.
- B. Stella, S. Arpicco, M. T. Peracchia, D. Desmaele, J. Hoebeke, M. Renoir, J. D'angelo, L. Cattel and P. Couvreur, J. Pharm. Sci., 2000, 89, 1452 CrossRef CAS.
- R. J. Lee and P. S. Low, J. Biol. Chem., 1994, 269, 3198 CAS.
- A. C. Antony, Blood, 1992, 79, 2807 CAS.
- A. Gabizon, A. T. Horowitz, D. Goren, D. Tzemach, F. Mandelbaum-Shavit, M. M. Qazen and S. Zalipsky, Bioconjugate Chem., 1999, 10, 289 CrossRef CAS.
- S. Wang and P. S. Low, J. Controlled Release, 1998, 53, 39 CrossRef CAS.
- R. J. Lee, S. Wang and P. S. Low, Biochim. Biophys. Acta, 1996, 1312, 237 CAS.
- H. Elnakat and M. Ratnam, Adv. Drug Delivery Rev., 2004, 56, 1067 CrossRef CAS.
- J. Luo, M. D. Smith, D. A. Lantrip, S. Wang and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 10004 CrossRef CAS.
- Y. Akiyama, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2004, 15, 424 CrossRef CAS.
- M. Wu, W. Gunning and M. Ratnam, Cancer Epidemiol. Biomarkers Prev., 1999, 8, 775 Search PubMed.
- H. Ogura, M. Yoshinouchi, T. Kudo, M. Imura, T. Fujiwara and Y. Yabe, Cell. Mol. Biol., 1993, 39, 463 Search PubMed.
- P. C. Elwood, J. Biol. Chem., 1989, 264, 14893 CAS.
- A. J. M. D'souza and E. M. Topp, J. Pharm. Sci., 2004, 93, 1962 CrossRef.
- K. D. Jensen, A. Nori, M. Tijerina, P. Kopeckova and J. Kopecek, J. Controlled Release, 2003, 87, 89 CrossRef CAS.
- N. Nishiyama, Y. Kato, Y. Sugiyama and K. Kataoka, Pharm. Res., 2001, 18, 1035 CrossRef CAS.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Quantitative introduction of hydrazide groups to the PBLA block and correlation between number of linkers and drug release profile of the micelle. See http://dx.doi.org/10.1039/b500266d |
| This journal is © The Royal Society of Chemistry 2005 |