Synthesis of monodispersed fcc and fct FePt/FePd nanoparticles by microwave irradiation†
H. Loc
Nguyen
a,
Luciano E. M.
Howard
a,
Sean R.
Giblin
bd,
Brian K.
Tanner
b,
Ian
Terry
b,
Andrew K.
Hughes
a,
Ian M.
Ross
c,
Arnaud
Serres
b,
Hannah
Bürckstümmer
a and
John S. O.
Evans
*a
aDepartment of Chemistry, University Science Laboratories, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: john.evans@dur.ac.uk
bDepartment of Physics, University Science Laboratories, University of Durham, South Road, Durham, UK DH1 3LE
cDepartment of Electronic and Electrical Engineering, University of Sheffield, Mappin Building, Mappin Street, Sheffield, UK S1 3JD
dPresent address: ISIS facility, Rutherford Appleton Laboratory, Chilton, Didcot, OXON.
First published on 24th October 2005
Abstract
A simple microwave heating method has been used for the stoichiometrically controlled synthesis of FePt and FePd nanoparticles using Na2Fe(CO)4 and Pt(acac)2/Pd(acac)2 as the main reactants. By varying the solvents and surfactants, the microwave assisted reactions have shown a significant advantage for the rapid production of monodisperse fcc FePt nanoparticle metal alloys which can be converted to the fct phase at low temperatures (364 °C). Microwave reactions at high pressure (closed system) have led to the direct formation of a mixture of fcc and fct phase FePt nanoparticles. Room temperature structural and magnetic properties of materials have been characterized by X-ray diffraction, HRTEM and magnetic measurements. The onset of ordering has been investigated by in situ high temperature X-ray diffraction studies.
Introduction
The preparation of nanoscale magnetic materials is an extremely active research area due to their potential uses in magnetic recording devices, biomedical applications, magnetooptical systems and in numerous other areas.1 Of the many nanoparticle alloys that have been studied for future generation magnetic storage applications, self-assembled Ll0 FePt nanoparticle arrays are promising candidates owing to their large uniaxial magnetocrystalline anisotropy [Ku ≅ 7 × 107 erg cm−3] and good chemical stability.2 Calculations indicate that particles as small as 2.8 nm have a sufficient anisotropy energy KuV (V is the magnetic grain volume) to be exploited for permanent data storage, leading to significant advances in hard disk drive areal densities over materials currently used.3Many approaches to the preparation of metal nanoparticles have been reported4 including chemical reduction,5 UV photolysis,6 thermal decomposition,7 metal vapour decomposition,8 electrochemical synthesis9 and sonochemical decomposition.10 Chemical routes11 appear to offer the best route to monodisperse FePt nanoparticles.2,7a In a typical preparation simultaneous decomposition of iron pentacarbonyl and reduction of platinum acetylacetonate by polyol reducing agents or co-reduction of iron and platinum salts in the presence of surfactants leads to formation of face centered cubic (fcc) FePt alloys.
To obtain self-assembled Ll0 FePt nanoparticle superlattices, which are required for storage applications, the as-synthesized nanoparticles typically have to be annealed at high temperature to transform the material from the fcc Fe/Pt disordered phase to the face centered tetragonal (fct) Fe/Pt ordered phase, the so called Ll0 structure (Fig. 1). During the annealing process, however, agglomeration of the particles can lead to a dramatic increase in both particle size and size dispersion.8c,12 This hinders applications as high-density recording materials. Different methods have been attempted to lower the FePt phase transition temperature (Tt) and particle sintering or to establish a direct route to fct nanoparticle formation. Introduction of a third metal into FePt alloys,13 although reported at lower Tt, has resulted in particles which retain the problems of agglomeration or decomposition on further annealing at higher temperature. Partially ordered fct FePt nanoparticles have recently been obtained by chemical routes including the simultaneous reduction of Fe(II)/Pt(II) salts and from Fe(CO)5/Pt(acac)2 using conventional heating methods.14 These preliminary results generally show a low ordering ratio, small room temperature (RT) coercivity of fct particles and frequently relatively broad particle size dispersion. Recently a multistep process involving coating particles with an inert silica coating during annealing followed by its subsequent removal in base has been described. This process can apparently lead to particle ordering without sintering.15 Disordered fcc particles have also been annealed to the fct phase with minimal sintering in a NaCl matrix.16
 | ||
| Fig. 1 Schematic representation of the FePt phase transformation from the fcc to fct structure. | ||
In a recent communication, we have presented a straightforward stoichiometrically controlled synthesis of FePt nanoparticles using Collman's reagent, Na2Fe(CO)4, as a reducing agent for platinum acetylacetonate, Pt(acac)2.17 An advantage of the method is that the electrons required to reduce Pt(II) are located on the Fe source rather than on an additional species (the reaction can be schematically written as Fe2− + Pt2+ → FePt). This process assures the ideal 1 : 1 stoichiometry is achieved which is important since the magnetically important fct phase only forms over an Fe1−xPt range of x ∼ 0.4–0.6; other workers have shown that conventional procedures lead to individual particles with a range of stoichiometries.18 Further, the reduction step that is key to nanoparticle alloy formation requires the simultaneous presence of Fe and Pt ions to occur, leading to the product alloy being intimately mixed on an atomic scale. Using this route we have shown that it is possible to produce fcc FePt nanoparticles which can be converted to the fct structure at low temperatures with minimal agglomeration without the presence of a third metal. By varying the surfactants and temperature regimes it was also possible to synthesise FePt nanoparticles with the important fct structure directly in solution, without any post-synthesis heat treatment.17
In order to improve the FePt nanoparticle preparation reported in our preliminary work, an alternative method of energy supply has been investigated for heating reactions more efficiently. Microwave dielectric heating has recently attracted the attention of chemists for, inter alia, organic reactions,19 molecular sieve preparation,20 and syntheses of inorganic complexes21 as it can lead to much higher heating rates than those achieved by conventional heating. The rapid and uniform heating provided by microwaves has potential benefits for nanoparticle synthesis. Microwave irradiation was recently reported to be a successful synthetic method for single metal nanoparticles such as Pt, Ir, Rh, Pd, Au, Ru22 and Ag.23 Application of microwave dielectric heating for binary Pt–Ru nanoparticles was also reported, in which a uniform size of 2–3 nm was obtained in the presence of a polymer as a protective layer for the particles.24 We are only aware of one previous publication on the synthesis of FePt by microwave methods in which platinum(II) chloride and iron(II) acetate were reduced in ethylene glycol; to achieve a 1 : 1 stoichiometry excess Fe was used.25 In this work the as-prepared superparamagnetic material was described as being amorphous (no peaks were present in its X-ray diffraction pattern) and crystalline FePt was only formed on heating to 600 °C. Little characterisation of the as-prepared material was given and the annealed material was reported as having a bimodal size distribution. Selected area electron diffraction (SAED) patterns reported did not show the ordering peaks one would expect for fct FePt and certain ordering peaks (e.g., the 001, 112 and 113 peaks based on a pseudo-cubic cell setting) appeared to be missing from the X-ray data presented.
In this work we have investigated the use of microwave irradiation as an energy source for the preparation of FePt using the Fe2−/Pt2+ methodology. We show that monodisperse crystalline fcc particles of controlled size (and thus suitable for self-assembly) can be readily produced. The reaction has been performed using a variety of solvents and surfactants leading to control over particle size. Under certain conditions it is also possible to prepare fct particles directly. We have also extended this chemistry to the microwave synthesis of FePd nanoparticles. The structure and properties of key materials have been characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM) and magnetic measurements.
Experimental
Materials and instruments
Platinum acetylacetonate [Pt(acac)2] was purchased from STREM, Pd(acac)2, disodium tetracarbonylferrate-dioxane complex [Na2Fe(CO)4·1.5C4H8O2], dioctyl ether, oleylamine and oleic acid from Aldrich, n-nonadecane from Lancaster. Octyl ether was degassed for 15 min before each use. All chemicals were weighed, placed into reaction flasks and sealed in a N2 filled glove box before transferring to microwave apparatus. Other AR (analytical reagents) grade organic solvents used for purification (e.g., hexane and absolute ethanol) were used as purchased. The microwave-assisted reactions were carried out in a CEM 300W Discover Focus Synthesis Microwave with a 2.45 GHz working frequency. Reactions under ambient pressure were performed in 100 ml thick glass walled vessels connected to a condenser under Ar. Reactions at elevated pressure and temperatures were performed in 10 ml sealed vials.Nanoparticle preparation in an open vessel
In a typical reaction a mixture of Pt/Pd acetylacetonate (0.3 mmol), disodium tetracarbonylferrate (0.3 mmol) with appropriate amounts of surfactants and solvents (Table 1) was placed in a 100 ml thick walled glass vessel connected to a condenser and Ar input. The mixture was sonicated at 60 °C for 1 h before transferring into the microwave apparatus. Reaction was typically carried out using control parameters of 300 W max power, 250 °C max temperature and 250 psi max pressure. A high reaction temperature (215 °C) was obtained using octyl ether as solvent whilst lower temperature (130–150 °C) was achieved for reactions carried out in nonadecane. After reaction the dark product mixture was allowed to cool to room temperature before adding 100 ml of absolute ethanol to precipitate dark particles. The product was separated by centrifugation then dispersed in hexane (20 ml) in the presence of appropriate surfactants and precipitated by adding ethanol (40 ml). After centrifugation, the material was washed one more time in a similar solvent mixture, dried in air at room temperature and stored under N2.| Reaction | System | Temperature/°C | Heating time/min | Hold time/min | Solvent | Surfactants (ratio used) | Particle size/nm |
|---|---|---|---|---|---|---|---|
| a Molecular mole ratio of surfactants in comparison with molecular mole ratio of main reactants. b Estimated standard uncertainties in parentheses, see text for definition of particle size. | |||||||
| 1 | FePt/open | 150 | 30 | 10 | Nonadecane | Oleyl amine–oleic acid (1 : 1)a | 2.66(7)b |
| 2 | FePt/open | 215 | 10 | 30 | Octyl ether | Oleyl amine–oleic acid (1 : 1) | 2.19(5) |
| 3 | FePt/closed | 150 + 250 | 1.5 + 1 | 2 + 2 | Octyl ether | Oleyl amine–oleic acid (1 : 1) | 3.26(7) |
| 4 | FePt/closed | 280 | 5 | 55 | Octyl ether | Oleyl amine–oleic acid (1 : 1) | 3.16(6) |
| 5 | FePd/open | 282 | 12 | 80 | Octyl ether | Oleic acid (3) | 7.20(2) |
Nanoparticle preparation in a closed vessel
FePt nanoparticles were synthesized from a mixture of platinum acetylacetonate (0.17 mmol), disodium tetracarbonylferrate (0.17 mmol) with appropriate amounts of surfactants and solvents for specific reactions (Table 1). Rapid heating (1 to 5 min) to the desired reaction temperature could be readily achieved. Safety warning: when using sealed systems there is a potential hazard due to rapid build up of high pressure in the reaction vessel and the release of CO during the reaction. The control system of the microwave reactor used is designed to remove power if a rapid pressure build up is encountered. To minimise such risks, sealed-system reactions were typically heated to the desired temperature in 2 stages, each taking ∼1 min. Despite these precautions rapid heating that could lead to explosions was experienced on occasions; we believe this is related to the formation of large particles which provide a self-accelerating heating mechanism. We therefore advise caution and the use of appropriate containment/shielding methods when such reactions are attempted. At the end of reactions the pressure was released and the dark mixture was allowed to cool to room temperature before adding absolute ethanol to precipitate dark particles. After centrifugation, the black product was dispersed in hexane (5 ml) in the presence of the surfactants used during the synthesis, precipitated by adding ethanol (10 ml) and centrifuged. The materials were washed one more time, dried in air at room temperature and stored under N2.Characterisation methods
XRD data used to confirm sample purity and particle size were collected on a Bruker D8 Advance diffractometer equipped with a Cu tube and a Sol-X energy dispersive detector. The sample was mounted on a zero background (511) silicon wafer. Data were typically collected from 10–90° 2θ (step size = 0.02° and time per step = 10 s) at room temperature. A variable divergence slit giving a constant area of sample illumination was used. In situ variable temperature X-ray diffraction data were collected using a Bruker AXS D8 Advance diffractometer equipped with a Cu tube, a Ge(111) incident beam monochromator (λ = 1.5406 Å) and a Vantec-1 PSD. High temperature measurements were performed using an Anton Parr HTK1200 high temperature furnace. Temperature calibration was determined using an external Al2O3–Si mixture of standards.26 The powdered sample was mounted on an amorphous silica disc. Variable temperature XRD data were collected over a temperature range of 297–924–296 K. Measurements (48 in total) were recorded over 48 h (every 25 K, 60 min each, a 0.2 K s−1 heating/cooling rate between temperatures, a 2θ range of 5–130° and a step time of 0.33 s). Data were rebinned onto a step size of 0.05° for Rietveld analysis. A slow flow of 5% H2–95% Ar gas was passed over the sample for the experiment's duration. XRD derived particle sizes quoted throughout the paper were obtained from Rietveld refinements of data sets. Peak shapes were fitted by convolution of a Scherrer-type broadening term of form (λ/size)cosθ and a strain term of the form strain × tanθ with an instrumental resolution function derived from a highly crystalline CeO2 standard recorded under equivalent conditions.Samples for transmission electron microscopy (TEM) analysis were prepared as dilute dispersions in hexane with a small amount of surfactants. A drop of particle dispersion was allowed to evaporate slowly on an amorphous carbon film supported on a standard 3 mm copper grid (200 mesh, Agar Scientific). High resolution TEM (HRTEM) was performed in a JEOL 2010F field-emission gun (FEG) TEM operating at 200 kV. This instrument is capable of forming sub-nanometre analytical electron probes facilitating high spatial resolution compositional analysis via an Oxford Instruments LINK/ISIS X-ray energy-dispersive spectrometer (EDS) (Si/Li detector, 1024 channels, 20 keV range). EDS spectra were acquired from single nanoparticles and also regions of the specimen containing clusters of ∼300 particles using a 30 s preset live time acquisition. Quantification of the data was performed using the Cliff–Lorimer thin section technique assuming an average material density of 14.6 g cm−3 and a specimen thickness equal to the average projected diameter of the particle(s) being studied.
Magnetic studies were carried out using a Quantum Design SQUID magnetometer. Magnetization curves as a function of applied field were measured with fields up to 50 kOe at temperatures of 10 K and 290 K. Zero-field cooling/field cooling (ZFC/FC) experiments were made at 100 Oe with temperatures ranging from 2 to 300 K on samples mounted in low background gelatin capsules. Data are presented per g of sample owing to the difficulty in accurately assessing the percentage of surfactant molecules in an individual sample.
Result and discussion
Initial reactions (Table 1) were performed using a 1 : 1 molar ratio of Fe and Pt sources and a molar equivalent of oleyl amine and oleic acid surfactants. Nonadecane was chosen as solvent since experiments using conventional heating have shown that it can lead to low particle agglomeration during subsequent heat treatment.17 Heating these reagents to 150–170 °C in an open system for 40 min led to the formation of a black suspension. The XRD of this material (Fig. 2a) is typical of a chemically disordered fcc structure possessing broad peaks at 41, 47, 68 and 82° 2θ which are indexed as the (111), (200), (220) and (311) reflections respectively. The fcc structure of the particles was also verified by electron diffraction where d-spacings calculated from radii of pattern rings were consistent with XRD results (see electronic supplementary information (ESI)†). Rietveld refinement of the data performed using Topas Academic27 suggested an average diameter of particles of 2.66(7) nm and cell parameter of 3.8744(9) Å, similar to fcc FePt nanoparticles synthesised under similar conditions by normal heating.17 We note that the temperature at which this reaction was performed is significantly lower than required using conventional methods (150 °C vs 330 °C). Further attempts using different conditions of temperature, time, solvent and vessel (open or closed) system have also led to production of fcc FePt nanoparticles as shown in Table 1. FePd nanoparticles could also be prepared by a similar route. Fig. 2b shows the XRD pattern of ∼7.2 nm FePd particles prepared in dioctyl ether with a 1 : 3 molar ratio of metal–oleic acid surfactant at 282 °C. The refined cell parameter was 3.8972(4) Å. | ||
| Fig. 2 Rietveld fits of X-ray patterns of (a) as-prepared fcc FePt samples (observed and calculated patterns for FePt with difference below) and (b) fcc FePd. | ||
Fig. 3 shows a TEM image of FePt particles produced by this route. These can be seen to be essentially spherical and of uniform size. Using image analysis software,28 size measurement of 188 randomly selected particles shows that the FePt particles have a narrow size distribution. Fitting with a log-normal distribution leads to a measured mean diameter of 2.58 nm and a dispersion σ of 5% (Fig. 4). The size is consistent with that indicated by XRD. The HRTEM images (Fig. 3 insert) of individual particles demonstrated that they were single crystals with lattice fringes consistent with the (200) and (220) d-spacing of ∼1.9 Å and ∼1.3 Å respectively. EDS analysis of clusters containing ∼300 particles gave an overall average Fe : Pt stoichiometry of 52(3) : 48(3). Analysis of individual particles revealed that a range of compositions are present with some particles either Fe or Pt rich. Averaging a large number of individual particles gave a stoichiometry of 48(7) : 52(7). Yu and co-workers have reported that individual particles prepared by the conventional polyol synthetic route can have a wide stoichiometry range with a significant proportion of particles being either Fe- or Pt-rich. In fact, they report that only 29% of individual particles lie in the 0.4 < x < 0.6 FexPt1−x range that would allow the transition to the L10 phase to occur.18 We note that unlike Yu et al. we find that 75% of the individual particles lie within this range. This suggests that significantly better control over individual particle stoichiometry is achieved with our Fe2−/Pt2+ synthetic route.
 | ||
| Fig. 3 HRTEM image of monodispersed fcc FePt particles, insert shows nanoparticles with a fringe spacing consistent with the (200) plane of fcc structure. | ||
 | ||
| Fig. 4 Particle diameter histogram of fcc FePt nanoparticles; the line plotted corresponds to the fit using a log-normal distribution. | ||
The fcc particles prepared by this route can be converted to the magnetically important fct L10 phase by annealing under a flow of 5% H2 in Ar. Fig. 5 shows a narrow 2θ range of a series of diffraction experiments recorded at increasing temperatures; each pattern was recorded over ∼1 h with rapid heating between data collections. Two different phenomena can be observed from these data. Firstly from a temperature of around 637 K (364 °C) extra peaks appear at 2θ values of ∼24 and 33°. These peaks can be indexed as the (001) and (110) reflections of the fct phase (using a pseudo-cubic cell of a ∼ 3.85, c ∼ 3.71 Å) and provide direct evidence of the ordering phase transition. The recorded ordering temperature is consistent with previous experiments on annealing FePt nanoparticles synthesized using this synthetic method and conventional heating, and is significantly lower than the 600 °C required to order previously reported materials.25 It is also clear from Fig. 5 that peaks sharpen on heating which is evidence of particle growth.
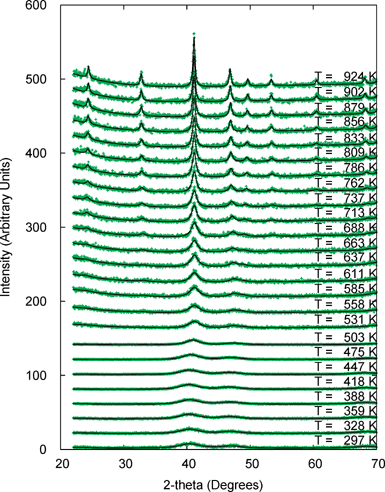 | ||
| Fig. 5 25 diffraction patterns recorded on annealing as-prepared fcc FePt as a function of temperature from 297 to 924 K. Indication of ordering is clearly visible at 637 K (364 °C) and above. Data collected on cooling are not shown but derived quantities are included in Fig. 6. | ||
Quantitative information on both these processes has been obtained by Rietveld refinement using the Topas Academic software suite. It is difficult to extract reliable quantitative information on the early stages of ordering for materials such as this as much of the information is contained in the relatively broad superlattice peaks. Due to correlations with the background (which itself has significant slowly varying contributions due to the sample mounting and furnace environment for variable temperature experiments), it is extremely hard to estimate their intensity correctly. We have therefore adopted a strategy in which a sixth order background polynomial was fitted to each of the 48 data sets in an initial round of Rietveld refinements using a fully disordered model in which 2θ ranges corresponding to ordering peaks were excluded. We believe that this produces the “least biased” estimate of the background at each temperature that can be achieved. These background polynomials were then used as fixed functions in a separate round of Rietveld refinements in which six parameters (scale factor, a and c cell parameters, overall atomic displacement parameter, particle size and order parameter) were refined at each temperature. The sample height was found to vary smoothly with temperature and was introduced to each refinement as a fixed though temperature-dependent parameter. To allow refinement through the fcc→fct phase transition a psuedo-cubic cell setting was used throughout in space group P4/mmm (Fe at 1a and 1c Wyckoff sites and Pt at 2e), and the fractional occupancy (frac) of Fe on Pt sites and Pt on Fe allowed to refine. The order parameter for the phase transition is thus given by 1–2frac, and ideally varies from 0 (fcc) to 1.0 (fct).
Fig. 6a shows the temperature dependence of the unit cell size. Below the fcc→fct ordering temperature individual values of a and c are ill-defined by Rietveld refinement (the material is cubic and peaks are broad so psuedo-cubic tetragonal values show considerable scatter) so we choose to plot (volume)1/3 on warming which provides an average measure of cell parameter over the whole temperature range. A significant reduction in this parameter is seen from around 450 K. A reduction in volume is expected for the fcc→fct transition and is well known in, for example, AuCu binary alloys. We note that the reduction in cell volume occurs before significant ordering peaks are visible in Fig. 5, and before significant particle growth. Cell volume is therefore perhaps the most accessible indication that particle ordering is beginning to occur.
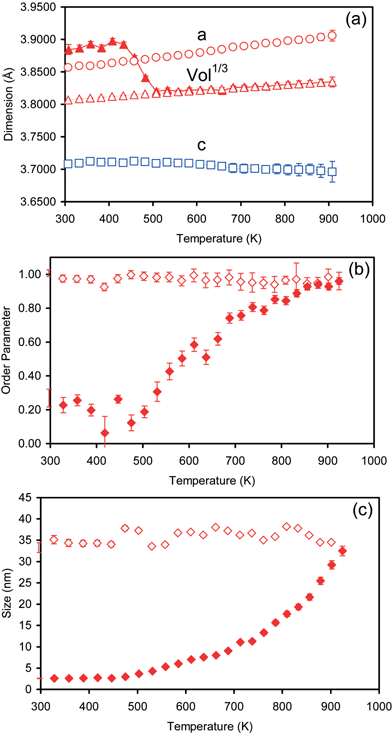 | ||
| Fig. 6 (a) Temperature dependence of the (cell volume)1/3 on warming and cooling (closed/open triangles respectively) and a (open circles) and c (open squares) cell parameters on cooling; (b) order parameter on warming (closed symbols) and cooling (open symbols); (c) particle size on warming (closed symbols) and cooling (open symbols). Error bars show ±1 standard uncertainty as derived by Rietveld refinement, and are probably an underestimate of the true uncertainty on parameters. Where error bars are not shown they are smaller than the size of the plotted symbol. | ||
On cooling the material retains the fct structure as expected. The overall cell volume reveals a positive thermal expansion coefficient throughout, though the c-axis shows a small thermal contraction (αa = +2.0 × 10−5, αc = −5.2 × 10−6 K−1 from 925–295 K) with the c/a ratio varying from 0.9464(2) at 925 K to 0.9615(3) at 295 K. The c-axis also shows a marked contraction just above 700 K which is presumably associated with the Curie temperature which is around 723 K.29 Cell parameters on cooling of a = 3.857(7), c = 3.708(1) Å at 296 K compare to literature values of a = 3.855, c = 3.71130 or a = 3.85 and c = 3.71 Å.31 A recent neutron scattering measurement on large single crystals of bulk FePt shows a similar temperature dependence of the c cell parameter.32
The order parameter and particle size dependence on temperature are shown in Figs. 6b and 6c. Below around 450 K the order parameter is approximately constant. Early in the ordering process precise values of the order parameter are hard to derive and the low temperature values plotted of ∼0.2 on warming are probably not significantly different from zero; the increase in R-factor on forcing the order parameter to be exactly 0.0 for these refinements is < 0.15% for temperatures below 558 K. A significant rise in order parameter can be seen above ∼500 K, a temperature slightly higher than that at which the cell volume decrease occurs. On cooling the material retains its fct structure with the room temperature order parameter refining to 1.01(3). The indication of perfect Fe/Pt order provides further support for the stoichiometric nature of the particles. The mean particle size also increases significantly from ∼500 K indicating particle sintering as the protective surfactants burn off. The particle size remains, as expected, essentially unchanged on cooling.
Fig. 7a shows the magnetization of sample 1 as a function of increasing temperature after cooling in a small residual field of <1 Oe (“zero” field cooling) and in a 100 Oe field. Clear evidence for superparamagnetic behaviour with a blocking temperature of T ∼ 17 K is seen. The sharp rise of the ZFC data indicates that the particle size dispersion is low, in support of the TEM conclusions. The magnetization vs field loops (Fig. 7b) of the as-synthesized sample showed small coercivity values at both 10 and 290 K, confirming the superparamagnetic properties of the particles. The data also indicate the presence of a very minor component which saturates at low field. Hysteresis loops of an annealed sample (see Fig. S4 in the ESI†) gave coercivities of 14.7 and 10.6 kOe at 10 and 290 K respectively.
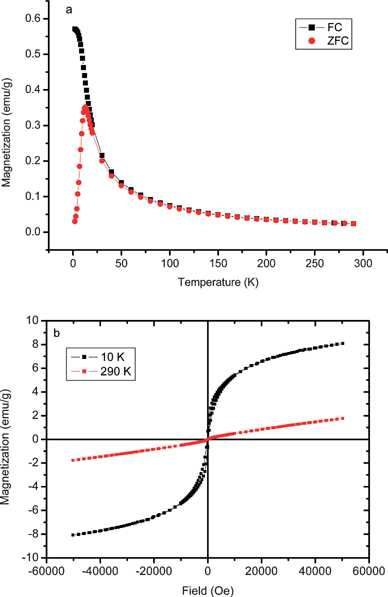 | ||
| Fig. 7 (a) ZFC and FC magnetisation curves as function of temperature from 5 to 290 K at a field 100 Oe; (b) Magnetic hysteresis loops measured at 10 and 290 K of as-prepared fcc FePt particles as prepared in reaction 1 of Table 1. | ||
We have recently reported the direct preparation of ordered fct FePt nanoparticles using the Fe2−/Pt2+ route with conventional heating at 389 °C in tetracosane. This has prompted us to attempt the preparation of ordered fct particles using microwave irradiation. A closed microwave system was chosen to access the high temperatures at which fcc FePt particles might be transformed directly to the ordered phase in solution. It proved, however, difficult to control heating rates of such reactions and pressure build-up in reaction vessels sometimes led to reaction vessel bursting. Fig. 8 shows a diffraction pattern of an FePt sample prepared with octyl ether and an oleyl amine surfactant (2 : 1 surfactant to metal ratio) at 280 °C under microwave irradiation for 10 min. Superlattice peaks are clearly present at ∼24 (001) and 33° (110) 2θ indicating FePt particles in the fct phase have formed. The overlap of sharp diffraction peaks on broader peaks at ∼40.5 (111) and 47° (200) 2θ respectively suggests the coexistence of fcc and fct FePt structural phases. Rietveld refinement confirms the existence of the ordered FePt structure giving a and c cell lattice parameters of 3.8463(2) and 3.7214(3) Å, an order parameter of 0.90(1), and an estimated particle size of 24 nm. The cell parameter of the cubic component refines to 3.872(2) Å. These results suggest that rapid heating had simultaneously caused a phase transformation from the fcc to fct phase and decomposition of surfactants leading to rapid particle size increase. Hysteresis loops measured at 10 K and 290 K (Fig. S5 in the ESI†) also suggest two phase behaviour with a kink at low field. The measured coercivities were 7.0 kOe at 290 K and 9.0 kOe at 10 K.
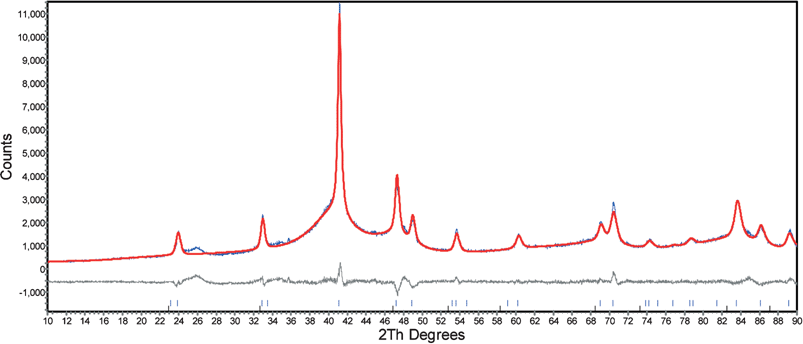 | ||
| Fig. 8 Rietveld fit of X-ray diffraction data of as-prepared FePt samples containing a mixture of fcc and fct particles. (observed and calculated patterns for FePt, and difference below). | ||
Conclusions
The general synthetic route presented here provides a straightforward and stoichiometrically controlled synthesis of FePt nanoparticles. Using microwaves for reaction heating shows significant advantages for production of monodispersed fcc FePt nanoparticle alloys which can be conveniently converted into the ordered fct phase on annealing at low temperature (364 °C). Reactions can be performed very rapidly (6 minutes or less) and at temperatures lower than using conventional heating. The Fe2−/Pt2+ route allows good control over both the overall stoichiometry and the stoichiometry of individual particles. High temperature reactions in the microwave led to the direct formation of a mixture of fcc and fct FePt nanoparticles. The fct nanoparticles were shown to have a particle size of ∼24 nm and strong coercivity indicating ferromagnetic behavior. Further extensions of FePt nanoparticles synthesis by microwave heating with varying solvent, surfactants and metals have been investigated.Acknowledgements
The authors thank Vivian Thompson for TEM images, Prof Todd Marder and Dr Patrick Steel for access to microwave facilities EPRSC and ONE-NE, via the Durham Nanotechnology Innovation Centre and Seagate Technology for financial support.References
- (a) K. J. Klabunde, Nanoscale Materials in Chemistry, John Wiley & Sons, New York, 2001 Search PubMed; (b) D. L. Peng, T. Hihara and K. Sumiyama, J. Magn. Magn. Mater., 2004, 277, 201 CrossRef CAS; (c) V. F. Puntes, K. M. Krisknan and A. P. Alivisatos, Science, 2001, 291, 2115 CrossRef CAS.
- (a) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS; (b) D. Weller and A. Moser, IEEE Trans. Magn., 1999, 35, 4423 CrossRef CAS.
- M. Plumer, J. Van Ek and D. Weller, The Physics of Ultra-High-Density Magnetic Recording, Springer, New York, 2001 Search PubMed.
- (a) C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS; (b) B. L. Cushing, V. L. Kolesnichenko and C. L. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS.
- (a) S. Sun, S. Anders, T. Thomson, J. E. E. Baglin, M. F. Toney, H. F. Hamann, C. B. Murray and B. D. Terris, J. Phys. Chem. B, 2003, 107, 5419 CrossRef CAS; (b) T. Iwaki, Y. Kakihara, T. Toda, M. Abdullah and K. Okuyama, J. Appl. Phys., 2003, 94, 6870; (c) B. Jeyadevan, A. Hobo, K. Urakawa, C. N. Chinnasamy, K. Shinoda and K. Tohji, J. Appl. Phys., 2003, 93, 7574 CrossRef CAS; (d) B. M. Leonard, N. S. P. Bhuvanesh and R. E. Schaak, J. Am. Chem. Soc., 2005, 127, 7326 CrossRef CAS.
- K. Torigoe and K. Esumi, Langmuir, 1993, 9, 1664 CrossRef CAS.
- (a) S. Sun, E. E. Fullerton, D. Weller and C. B. Murray, IEEE Trans. Magn., 2001, 37, 1239 CrossRef CAS; (b) B. Stahl, N. S. Gajbhiye, G. Wilde, D. Kramer, J. Ellrich, M. Ghafari, H. Hahn, H. Gleiter, J. Weißmüller, R. Würschum and P. Schlossmacher, Adv. Mater., 2002, 14, 24 CrossRef CAS; (c) Y. Hou, H. Kondoh, T. Kogure and T. Ohta, Chem. Mater., 2004, 16, 5149 CrossRef CAS; (d) M. Chen and D. E. Nikles, J. Appl. Phys., 2002, 91, 8477 CrossRef CAS.
- (a) K. Kakizaki, Y. Yamada, Y. Kuboki, H. Suda, K. Shibata and N. Hiratsuka, J. Magn. Magn. Mater., 2004, 272–276, 2200 CrossRef CAS; (b) P. T. L. Minh, N. P. Thuy and N. T. N. Chan, J. Magn. Magn. Mater., 2004, 277, 187 CrossRef CAS; (c) D. L. Peng, T. Hihara and K. Sumiyama, J. Magn. Magn. Mater., 2004, 277, 201 CrossRef CAS.
- M. T. Reetz and W. Helbig, J. Am. Chem. Soc., 1994, 116, 7401 CrossRef CAS.
- K. S. Suslick, M. Fang and T. Hyeon, J. Am. Chem. Soc., 1996, 118, 11960 CrossRef CAS.
- T. Hyeon, Chem. Commun., 2003, 8, 927 RSC.
- (a) T. Thomson, B. D. Terris, M. F. Toney, S. Raoux, J. E. E. Baglin, S. L. Lee and S. Sun, J. Appl. Phys., 2004, 95, 6738 CrossRef CAS; (b) H. Zeng, S. Sun, T. S. Vedantam, J. P. Liu, Z.-R. Dai and Z.-L. Wang, Appl. Phys. Lett., 2002, 80, 2583 CrossRef CAS.
- (a) S. Wang, S. S. Kang, D. E. Nikles, J. W. Harrell and X. W. Wu, J. Magn. Magn. Mater., 2003, 266, 49 CrossRef CAS; (b) Y. K. Takahashi, M. Ohnuma and K. Hono, J. Magn. Magn. Mater., 2002, 246, 259 CrossRef CAS; (c) C. L. Platt, K. W. Wierman, E. B. Svedberg, R. Van De Veerdonk, J. K. Howard, A. G. Roy and D. E. Laughlin, J. Appl. Phys., 2002, 92, 6104 CrossRef CAS.
- (a) B. Jeyadevan, K. Urakawa, A. Hobo, N. Chinnasamy, K. Shinoda, K. Tohji, D. D. J. Djayaprawira, M. Tsunoda and M. Takahashi, Jpn. J. Appl. Phys., 2003, 42, L350 CrossRef CAS; (b) S. Kang, Z. Jia, S. Shi, D. E. Nikles and J. W. Harrell, J. Appl. Phys., 2005, 97, 10J318; (c) S. Kang, Z. Jia, S. Shi, D. E. Nikles and J. W. Harrell, Appl. Phys. Lett., 2005, 86, 62503 CrossRef; (d) M. Takahashi, T. Ogawa, D. Hasegawa and B. Jeyadevan, J. Appl. Phys., 2005, 97, 10J307.
- S. Yamamoto, Y. Morimoto, T. Ono and M. Takano, Appl. Phys. Lett., 2005, 87, 32503 CrossRef.
- K. Elkins, D. Li, N. Poudyal, V. Nandwana, Z. Jin, K. Chen and J. P. Liu, J. Phys. D: Appl. Phys., 2005, 38, 2306 CrossRef CAS.
- L. E. M. Howard, H. L. Nguyen, S. R. Giblin, B. K. Tanner, I. Terry, A. K. Hughes and J. S. O. Evans, J. Am. Chem. Soc., 2005, 127, 10140 CAS.
- A. C. C. Yu, M. Mizuno, Y. Sasaki and H. Kondo, Appl. Phys. Lett., 2004, 85, 6242 CrossRef CAS.
- (a) R. N. Gedye, W. Rank and K. C. Westaway, Can. J. Chem., 1991, 69, 706 CAS; (b) M. Larhed and A. Hallberg, J. Org. Chem., 1996, 61, 9582 CrossRef CAS; (c) D. R. Baghurst and D. M. P. Mingos, J. Organomet. Chem., 1990, 384, C57 CrossRef CAS.
- C. -G. Wu and T. Bein, Chem. Commun., 1996, 8, 925 RSC.
- (a) A. R. Barron and C. C. Landry, Science, 1993, 260, 1653 CrossRef CAS; (b) A. G. Whittaker and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 2002, 3967 RSC; (c) A. G. Whittaker and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 2000, 1521 RSC; (d) A. G. Whittaker, Chem. Mater., 2005, 17, 3426 CrossRef CAS.
- (a) W. Tu and H. Liu, J. Mater. Chem., 2000, 10, 2207 RSC; (b) W. Yu, W. Tu and H. Liu, Langmuir, 1999, 15, 6 CrossRef CAS.
- K. Patel, S. Kapoor, D. P. Dave and T. Mukherjee, J. Chem. Sci., 2005, 117, 53 Search PubMed.
- F. Bensebaa, N. Patrito, Y. Le. Page, P. L'Ecuyer and D. Wang, J. Mater. Chem., 2004, 14, 3378 RSC.
- R. Harpeness and A. Gedanken, J. Mater. Chem., 2005, 15, 698 RSC.
- (a) K. G. Lyon, G. L. Salinger, C. A. Swenson and G. K. White, J. Appl. Phys., 1977, 48, 865 CrossRef CAS; (b) Y. Okada and Y. Tokumaru, J. Appl. Phys., 1984, 56, 314 CrossRef CAS; (c) D. Taylor, Br. Ceram. Trans. J., 1984, 83, 92 CAS.
- Topas Academic: http://pws.prserv.net/Alan.Coelho/.
- Image Tool: http://ddsdx.uthscsa.edu/dig/itdesc.html/.
- T. S. Vedantam, J. P. Liu, H. Zeng and S. Sun, J. Appl. Phys., 2003, 93, 7184 CrossRef CAS.
- (a) S. Saita and S. Maenosono, Chem. Mater., 2005, 17, 3705 CrossRef CAS; (b) T. J. Klemmer, N. Shukla, C. Liu, X. W. Wu, E. B. Svedberg, O. Mryasov, R. W. Chantrell, D. Weller, M. Tanase and D. E. Laughlin, Appl. Phys. Lett., 2002, 81, 2220 CrossRef CAS.
- Joint Committee for Powder Diffraction Standards, International Centre for Diffraction Data, Newton Square, PA Search PubMed.
- Y. Tsunoda and H. Kobayashi, J. Magn. Magn. Mater., 2004, 272-276, 776 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Magnetic results of sample 1, 2, 4, HRTEM and SAED results of sample 1 of Table 1. See DOI: 10.1039/b511850f |
| This journal is © The Royal Society of Chemistry 2005 |