Synthesis and characterization of highly amine functionalized mesoporous organosilicas by an “all-in-one” approach
Rebecca
Voss
a,
Arne
Thomas
a,
Markus
Antonietti
*a and
Geoffrey A.
Ozin
b
aDepartment of Colloid Chemistry, MPI of Colloids and Interfaces, Research Campus Golm, D-14424 Potsdam, Germany
bMaterials Chemistry Research Group, Lash Miller Chemical Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto M5S3H6, Canada
First published on 1st August 2005
Abstract
Mesoporous organosilicas (MOs) represent a promising class of organic–inorganic nanocomposites for a broad range of applications like catalysis, sensing, separation, or microelectronics. Their distinct feature is the presence of organic groups incorporated into the channel walls of a mesoporous structure. Here, we present a convenient “all-in-one” approach using silsesquioxane surfactant precursors for the functionalization of the channel walls with primary amine groups. The monomer is made by a hydroboration/aminolysis sequence on the base of a commercial monomer, with the template bound to the functionalization site by hydroboration and released after silica condensation and aminolysis. This combination ensures both the placement of the amine groups exclusively along the channel interface as well as optimal use of the template.
Introduction
Many efforts have been made to create mesoporous materials with ordered and uniform pores based on organic molecules, as this would allow convenient functionalization and host–guest chemistry with unrivalled performance and potential. For purely inorganic materials (with their restrictions in functionality and polarity), the discovery of the first periodic mesoporous silicas in 1992, of which MCM-41 is the most visible example,1 has opened a new landscape of possibilities. There was immediate interest afterwards in the incorporation of organic groups into this novel structure, to give them distinctive properties for a broad range of applications like catalysis, sensing or separation. This was accomplished either by grafting organic groups onto the channel walls using the reactivity of the silanol groups of the material or by co-assembling TEOS with an organosiloxane having terminal organic groups of the type RSi(OR′)3.2,3 This achievement has created materials with interesting properties, such as periodic mesoporous silicas with alkanethiol groups in the channels, which can have a large surface area for removing toxic heavy metals like lead or mercury.However, these approaches suffer from some inherent problems and limitations.4,5 First, both synthesis methods can lead to an inhomogeneous distribution of the organic groups in the pores, which essentially spoils, for instance, most chromatographic applications. In addition within the condensation approach, a significant fraction of functional groups are non-accessible, buried through condensation within the pore walls, thus only weakening the material but not contributing to its performance.
Also because of that, such precursor molecules with terminal organic groups cannot be assembled as pure compounds, as either the template process fails for reasons of incompatibility, or the structure does not possess sufficient mechanical stability. A way to bypass these problems is co-assembly with the pure inorganic precursor (usually at least 80%) to retain a stable periodic mesoporous structure, which however limits the organic content of the resulting material. Nevertheless, studies relevant for the present work have shown that the use of bridging amines6,7 and cyclam8–10 moieties generated networks able to bind and hold a variety of transition metal ions. When N,N′-(bis(3-trimethoxysilyl)propyl)ethylenediamine is co-condensed with TEOS around a suitable template, the resultant material shows an increase in affinity for Cu2+ ions, while the material’s affinity for Ni2+ and Zn2+ is comparable to that of pristine MCM-41. However the point of saturation occurs when only 5–7% of the potential binding sites are occupied. This low level of binding is typical for most ethylenediamine functionalized silica systems and quantifies the problem of non-accessible functionality and incorporation into the walls discussed above even for the mixed materials.
In 1999, three groups independently developed a novel class of organic–inorganic nanocomposites known as “periodic mesoporous organosilicas”—the PMOs.11–13 In PMOs the organic groups are located within the structural tectons bridging at least two Si tetrahedral centers. Such materials can be prepared akin to MCM-41, with a very high degree of order and uniformity of pores, using a silsesquioxane with bridging organic groups of the type (R′O)3Si–R–Si(OR′)3 as the sole precursor. As co-assembly with an inorganic precursor like TEOS is unnecessary, a homogeneous distribution of the organic groups is ensured. Thus, a much higher organic loading can be achieved than with terminal organic groups as the whole material consists entirely of SiO1.5R0.5 building blocks.
Since their invention, a large amount of work has been done on trying to make PMO type materials suitable for real world applications. With one exception,14 the pore walls of PMS and PMO materials are amorphous. To make PMO materials more functional, a variety of organic groups have been introduced into PMOs, ranging from simple alkyl chains,11–13,15 to unsaturated or aromatic groups,16–19 to very large organometallic complexes.20 In order to introduce basic groups, a methylene PMO was treated with gaseous ammonia at elevated temperatures of up to 850 °C to give a material in which Si–N bonds have replaced some of the Si–O and Si–C bonds that make up the framework, effectively incorporating a basic site within the wall of a previously non-functional PMO.21
The present paper is intended to address a new approach to generating mesoporous organosilicas with a maximal extent of amino groups, all positioned at the pore wall surface. To achieve this objective, a convenient “all-in-one” approach using silsesquioxane surfactant precursors is explored, the general concept being presented in Scheme 1.
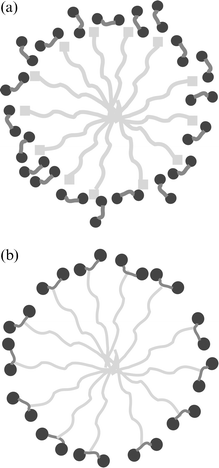 | ||
| Scheme 1 Illustration of a) the conventional nanocasting approach; b) the “all-in-one” method delineated here. Light grey: surfactant molecule or long chain hydrocarbon; dark grey: alkoxysilane motifs, mid-grey: organic functionality. | ||
A commercial PMO monomer of the type (EtO)3Si–R–Si(OEt)3 with a bridging ethylene moiety is activated by borohydride addition, and a second long chain hydrocarbon is added afterwards to generate a molecule which is an activated monomer and template at the same time (Scheme 2). Within that route, we make use of the fact that the second addition of the borohydride is sterically very demanding and does not take place a second time with bis(trimethylsilyl)ethene (thus leading to an active dimer). Addition of a linear, alkyl chain with a terminal double bond in slight excess however still allows the reaction reach practically complete conversion, as shown by 1H-NMR (data not shown).
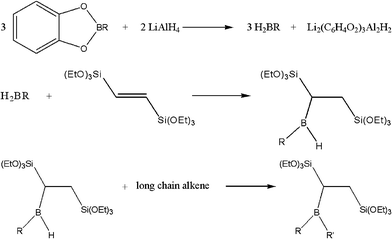 | ||
| Scheme 2 Reaction scheme for the preparation of the silsesquioxane surfactant precursor. | ||
This precursor, when hydrolyzed in the inorganic part, does form micelles and lyotropic phases by itself, with all the activated boron sites located at the interface. Cleavage of the CB bonds after condensation of the MO network, for instance by oxidation or ammonolysis, finally results in the functional MO material where the location of functionality has been “imprinted” by the template moiety.
The resulting materials are characterized by TEM, nitrogen and copper adsorption measurements, SAXS and solution and solid state NMR.
Although we restrict ourselves in the present paper to very simple micelle forming templates, we think that this approach can be adapted to more complicated imprinting/templating strategies for the generation of multifunctional cavities.
Experimental
Materials
Bis(triethoxysilyl)ethene was synthesised by a procedure described elsewhere1 or purchased from Gelest. 2-Propyl-1,2,3-benzodioxaborole, 1 M lithium aluminium hydride solution in tetrahydrofuran (THF), dry THF, hexadecene, pentene, tetraethoxysilane (TEOS), hydroxylamine-O-sulfonic acid, diglyme and bis(triethoxysilyl)ethane (BTSE) were purchased from Aldrich, ethanol and hexane from Fischer. All commercial chemicals were used without further purification.Preparation of the boron containing precursor for the “all-in-one” approach 1
In a typical synthesis 0.49 g (3 mmol) 2-propyl-1,2,3-benzodioxaborole were dissolved in 3 ml of dry THF and cooled to 0 °C. At this temperature 2 ml (3 mmol) of a 1 M lithium aluminium hydride solution were added. The solution was stirred at 0 °C for 1 hour. Then 0.83 g (2.4 mmol) of bis(triethoxysilyl)ethene were added and the solution was allowed to warm up to room temperature and kept there for at least 4 hours. Then 0.63 ml (2.4 mmol) hexadecane were mixed into the solution, which was then heated at 45 °C overnight. After the solution was cooled down to room temperature, 0.5 ml of pentene (as a security measure to react the potential remainder of B–H groups, the high excess of pentene is easily removed) were added and all was stirred for 1 hour. Addition of hexane led to precipitation of a white salt, which was filtered off. The solvent was removed to give 1.52 g (2.4 mmol) of pure precursor 1.13C NMR (CDCl3): δ 59.6 (Si(OCH2CH3)3), 32.8 (CH3CH2CH2(CH2)10CH2CH2CH2), 30.5 (CH3CH2CH2(CH2)10CH2CH2CH2), 26.5 (CH3CH2CH2(CH2)10CH2CH2CH2 and CH3CH2CH2), 23.5 (CH3CH2CH2(CH2)10CH2CH2CH2 and CH3CH2CH2), 18.3 (Si(OCH2CH3)3), 14.6 (CH3CH2CH2(CH2)10CH2CH2CH2 and CH3CH2CH2).
Preparation of the porous organosilica material based on the “all-in-one” method
1.5 g (2.4 mmol) of 1 and 9.6 mmol of silica atoms either from TEOS or from BTSE were dissolved in 5.0 g of ethanol. After homogenisation of the mixture 0.5 g of a 0.01 M aqueous HCl solution was added, and the mixture was homogenised again. The ethanol was removed from the reaction mixture in vacuum and the resulting viscous solution was aged for 3 day at 60 °C in air to give monolithic solids.Reaction to the amine MO
1 g of the freshly condensed MO was mechanically crushed and suspended in 15 g of diglyme. For every 1 mmol of boron contained in the MO, 4 mmol of hydroxylamine-O-sulfonic acid were added, and the mixture was stirred at 110 °C for a period of 3 hours. After the suspension was cooled down to room temperature it was filtered and the solid was washed with conc. HCl and stirred in it at room temperature for 2 hours. After that the solid was filtered and thoroughly washed with water. Then it was stirred in acetone, hexane and acetone for 2 hours each to remove the organic molecules formed during the reaction. The product was filtered off and dried at 60 °C to give the final amine functionalized MO.Characterization of mesoporous materials
Transmission electron microscopy (TEM) images were taken using a Zeiss EM 912Ω operated at an acceleration voltage of 120 kV. Samples were ground in a ball mill and dispersed in acetone. One droplet of the suspension was applied to a 400 mesh carbon-coated copper grid and left to dry in air.Nitrogen sorption data were obtained with a Quantachrome autosorb 1 at liquid nitrogen temperature.
Solid-state 11B and 29Si cross-polarization magic-angle spinning NMR spectra were recorded on a Bruker DSX 200 NMR spectrometer. For 11B NMR NaBEt4 was used as reference.
SAXS measurements were done using a Nonius rotating anode.
Results and discussion
The resulting materials were obtained as white powders. Characterization of the samples with TEM revealed the presence of a worm-like pore architecture (Fig. 1) | ||
| Fig. 1 Typical TEM picture of the amine functionalised MO material prepared from the silsesquioxane surfactant precursor and TEOS as a comonomer. White bar 100 nm. | ||
The occurrence of such worm-like pores is rather typical for the assembly behaviour of non-ionic surfactants (note that the monomer after hydrolysis is a T-type non-ionic surfactant) and has been described in similar fashion for pure silica.22,23
The mesoporosity is confirmed by nitrogen adsorption measurements (Fig. 2) showing a type VI isotherm without a capillary condensation step as is typical for many other non-ionic surfactants. BET evaluation of the surface area revealed a specific surface area of 974 m2 g−1, while the BJH evaluation of the desorption branch gave a pore diameter of 3.9 nm. The total pore volume was determined to be 0.65 cm3 g−1. These values are consistent with the TEM pictures and the composition of the system (note that the porogen is linked to the monomer, that is the stoichiometry is fixed). Both from the isotherms and from the surface area we can deduce the coexistence of smaller pores, presumably on the scale of 1 nm. This is explained by the assumption that the condensation traps not only micellar aggregates, but also monomeric species. This phenomenon might be due to cmc effects in the mixed solvent situation, but also to connectivity and trapping effects; the micellar self-assembly and the required sterics for complete silica condensation might in fact interfere with each other. The formation of a real wormhole pore structure is also reflected in the SAXS measurements (Fig. 3).
 | ||
| Fig. 2 Nitrogen sorption isotherm of the final amine-functionalized MO with TEOS as a comonomer. | ||
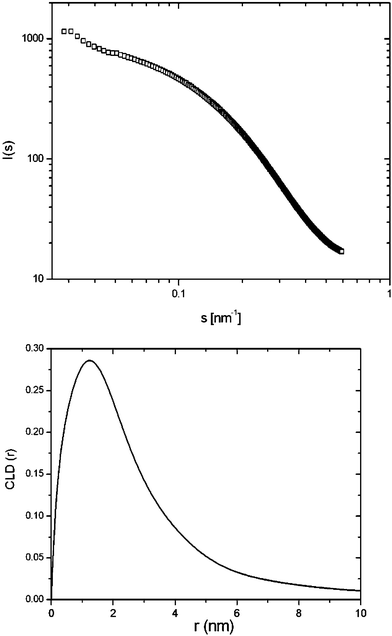 | ||
| Fig. 3 SAXS measurement and cord length distribution of the amine-functionalized MO with TEOS as a comonomer. | ||
As no distinct SAXS peak due to long range order is visible, we attempted to quantify the pore shape and size by employing an algorithm derived by Smarsly et al.24 employing the so-called cord length distribution (CLD). The maximum of the CLD is the Porod length and characterizes the wall and pore size, while the shape of the CLD reflects the architecture of the two-phase system. By taking into account the total pore volume determined by nitrogen adsorption and assuming a density of 1.5 g cm−3 we obtain an averaged pore cross-section of 4.7 nm. The shape of the cord length distribution and the slightly higher value for the average length across the pore than the one determined by nitrogen adsorption provides evidence of an elongated, worm-like pore architecture, in coexistence with a small amount of micropores.
11B and 29Si solid state NMR of the samples were employed to characterize the solid state reactions. For the freshly condensed MO the 11B NMR shows two sharp peaks at −0.15 and 5.62 ppm respectively, which we attribute to two ligating environments with ethanol and water as ligands. No peak is found for the amine-functionalized material, indicating complete aminolysis and showing that all the boron atoms are indeed chemically accessible and most presumably placed at the wall interface. 29Si NMR indicates the presence of about 10% of Si–C bond cleavage as a side product occurring throughout the functionalization process, the products of which turn up as Q sites trapped in the material (Fig. 4). Note that the NMR experiments were performed on the material with the two silsesqusiloxane precursors, that is Q sites can be only generated by side reactions. The T sites are centred around −60 ppm and the Q sites are around −104 ppm.
 | ||
| Fig. 4 29Si NMR of the final amine-functionalized MO in a mixture with bis(triethoxysilyl)ethane as a second monomer. | ||
The presence of amine groups was proven by the Kaiser test, a colour reaction with Ninhydrin25 where the whole sample turned deeply blue. As titration of colloids with high amine content with acids usually does not quantitatively reveal the amount of surface bound amino groups (due to proton sponge effects), we decided to quantify the amine content by the binding of copper ions.26,27 The sample was first suspended in 0.1 M ammonia solution for 1 hour, purged three times with clean water, and a 0.03 M Cu(II) nitrate solution was added (molar amount of copper/amine = 1, as calculated on the base of the known amounts of functional monomer and copper, assuming complete aminolysis of the borane). The Cu-loaded particles were isolated, rinsed with water twice, dried, and the copper content was determined by AAS.
A default experiment with a PMO material made of bis(trimethylsilyl) ethane under similar conditions gives only negligible amounts of copper (<1 mol%), that is unspecific binding of the copper ions to silica is only weak. The amine containing material after adsorption shows a blue coloration indicating the formation of some copper tetramine complexes in the MO. AAS shows 27 mol% of copper in the material with respect to the amine groups. Increasing the relative copper content to 2 : 1 gives a value of 36 mol%, showing that binding even beyond the normal stoichiometry of four amine ligands is possible. It is commonplace in polymer and surface reactions that the ligands are not in the right conformation to realize a geometrically defined four-fold binding motif with copper. This is why we have to expect a distribution of binding sites and binding enthalpies, with the residual coordination sites of Cu(II) open to ligate to water or—if possible—the counterion. This is also why the value of 27 mol% Cu does not prove that all amines are located at the interface; a minority might still reside in the micropores, inaccessible for the hydrated Cu(NO3)2. Nevertheless, when compared to the 5–7% efficiency of Cu(II) binding reported so far, it becomes obvious that the “all-in-one” technique indeed has significantly increased the availability of the amine groups in the structure.
A second, typical “meso effect”28 to be expected is an unusual counterion sensitivity of the binding process. As the pores are just slightly larger than the hydrated copper complexes, and electroneutrality requires the close proximity of at least the majority of counterions to the binding site, we expect the binding equilibrium to be sensitive to the counterion. As a second copper salt, CuCl2 was used, also because of its unique properties to form ladder-type complexes with itself. The values under similar conditions as described for Cu(NO3)2 are 59 mol% and 102 mol%, respectively. As seen from the blue coloration of the MO after adsorption, some copper tetramine complexes still form while the rest of the copper is adsorbed through sub-stoichiometric amine complex formation and chlorine-ladder formation to neighbouring copper complexes.
Conclusion
We have shown that it is possible to increase the accessible functionality of mesoporous organosilica channel walls by a convenient “all-in-one” approach. Here, a commercially available PMO monomer is activated by a hydroboration reaction, and a sterically demanding hydrophobic protection group is fixed to the functional monomer conferring to the molecule porogenic properties. Hydrolysis of the silica precursor results in activated monomers, which can undergo self-organization and hydrolytic polycondensation. This “self process” ensures both the placement of the functional groups along the mesopore wall surface as well as optimal use of the template. The resulting boron-containing MO intermediate enables a rich functionalization chemistry, here exemplified by an aminolysis reaction towards a primary, surface bound amine group. The amount of accessible amine groups was estimated with Cu(II)-binding experiments, thus revealing that, in contrast to most previous experiments, at least the majority of amine groups are accessible. We also found an unusual counterion selectivity of the Cu(II) binding process, which is presumably mainly steric and shows promise for the development of an anion specific exchange process. This is the subject of ongoing work.Acknowledgements
M.A. and R.V. thank the Max Planck Society for continued financial support. GAO wishes to acknowledge the Alexander-von-Humboldt foundation for a Research Fellowship and sustained funding of the Natural Sciences and Engineering Research Council of Canada of the PMO research in his group.References
- C. T. Kresge, M. Leonowicz, W. J. Roth, J. C. Vartuli and J. C. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- G. A. Ozin, E. Chomski, D. Kushalani and M. J. McLachlan, Curr. Opin. Colloid Interface Sci., 1998, 3, 181–193.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950–2963 CrossRef CAS.
- M. H. Lim, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- M. H. Lim, C. F. Blanford and A. Stein, Chem. Mater., 1998, 10, 467–470 CrossRef CAS.
- K. Hossain and L. Mercier, Adv. Mater., 2002, 14, 1053 CrossRef CAS.
- H. Zhu, D. Jones, J. Zajac, R. Dutartre, M. Rhomari and J. Roziere, Chem. Mater., 2002, 14, 4886 CrossRef CAS.
- R. Corriu, A. Mehdi, C. Reye and C. Thieuleux, Chem. Commun., 2002, 1382 RSC.
- R. Corriu, A. Medhi, C. Reye and C. Thieuleux, Chem. Commun., 2003, 1564 RSC.
- P. Corriu, A. Medhi, C. Reye and C. Thieuleux, New J. Chem., 2003, 27, 905 RSC.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- S. Inagaki, S. Guan, T. Oshuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS.
- T. Asefa, M. MacLachlan, H. Grondey, N. Coombs and G. A. Ozin, Angew. Chem., Int. Ed., 2000, 39, 1808 CrossRef CAS.
- C. Beleizao, B. Gigante, D. Das, M. Alvaro, H. Garcia and C. Corma, Chem. Commun., 2003, 1860 RSC.
- M. Kuroki, T. Asefa, W. Whitnall, M. Kruk, C. Yoshina-Ishii, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2002, 124, 13886 CrossRef CAS.
- G. Temtsin, T. Asefa, S. Bittner and G. A. Ozin, J. Mater. Chem., 2001, 11, 3202 RSC.
- C. Y. Ishii, T. Asefa, N. Coombs, M. MacLachlan and G. A. Ozin, Chem. Commun., 1999, 2539 RSC.
- T. Asefa, M. Kruk, M. MacLachlan, N. Coombs, H. Grondey, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2001, 123, 8520 CrossRef CAS.
- T. Asefa, M. Kruk, N. Coombs, H. Grondey, M. MacLachlan, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 11662 CrossRef CAS.
- D. A. Loy, J. P. Carpenter, S. A. Yamanaka, M. D. McClain, J. Greaves, S. Hobson and K. J. Shea, Chem. Mater., 1998, 10, 4129 CrossRef CAS.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef.
- B. Smarsly, M. Antonietti and T. Wolff, J. Chem. Phys., 2002, 116, 2618 CrossRef CAS.
- E. Kaiser, R. Colescot, C. Bossinge and P. I. Cook, Anal. Biochem., 1970, 34, 595 CrossRef CAS.
- S. Dai, M. C. Burleigh, Y. Shin, C. C. Morrow, C. E. Barnes and Z. L. Xue, Angew. Chem., Int. Ed., 1999, 38, 1235 CrossRef CAS.
- H. G. Zhu, D. J. Jones, J. Zajac, R. Dutartre, M. Rhomari and J. Roziere, Chem. Mater., 2002, 14, 4886 CrossRef CAS.
- B. Smarsly, S. Polarz and M. Antonietti, J. Phys. Chem., 2001, 105, 10473–10483 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |