DOI:
10.1039/B504585A
(Application)
J. Mater. Chem., 2005,
15, 4773-4782
Anti-tumor, -viral, and -bacterial activities of polyoxometalates for realizing an inorganic drug
Received 1st April 2005, Accepted 20th June 2005
First published on 21st July 2005
Abstract
Polyoxometalates (PMs) as discrete metal-oxide cluster anions comprise a versatile class of inorganic, anionic clusters that have a wide variety of structures, sizes, and a combination of metals, high solubility in water, and electrochemical activity. Recent investigation of anti-tumor, -viral, and -bacterial activities of PMs shows induced cell-apoptosis, inhibition of binding of virus to receptor, and the enhancement of β-lactam antibiotics as well as the inhibition of bacterial growth, respectively. PMs exemplified for each biological activity are discussed from the chemotherapeutic point of view for the establishment of a novel inorganic drug based on PMs.
 Toshihiro Yamase  A photograph of Chemical Resources Laboratory of Tokyo Institute of Technology where Dr Hideki Shirakawa, a Nobel prize winner in 2000, discovered an electroconductive polyacetylene film. | Toshihiro Yamase was born in Kanazawa, Japan, in 1943. He studied Chemistry at Nagoya Institute of Technology (B.S. in 1965) and at Tokyo Institute of Technology (Ph.D. with Eiichi Inoue in 1970), and had a postdoctoral research position at Fritz-Haber-Institut der Max-Planck-Gesellschaft (with Heinz Gerischer in 1976–1977). He is now Professor of Inorganic Chemistry at Chemical Resources Laboratory of Tokyo Institute of Technology. His research interests include polyoxometalate chemistry, photochemistry, bioinorganic chemistry, inorganic material chemistry, molecular magnetism, and inorganic medicinal chemistry. He is a recipient of a Chemical Society of Japan Award for Creative Work in 1997 and a Rare Earth Society of Japan Academic Award in 2005. |
1 Introduction
Polyoxometalates (PMs) as discrete metal-oxide cluster anions comprise a versatile class of inorganic, anionic clusters that has made a mark on disciplines as diverse as materials science involving electronic, magnetic, electrochemical, and photochemical properties, medicine as anti-tumor, -viral, and -bacterial agents, and homogeneous, heterogeneous, and photo-catalysts.1 Our application of PMs to chemotherapeutic fields originates from our finding of in vivo anti-tumor effect against solid human-cancer xenografts (MX-1, OAT, and CO-4).2 The biological activity of PMs was first noted over 30 years ago in France when [SiW12O40]4−, [Na(SbW7O24)3(Sb3O7)2]18−, and [(AsW9O33)4(WO2)4]28− exhibited their activity against various non-retro RNA and DNA viruses in vitro and in vivo, and the groups of Raynaud and Jasmin demonstrated the inhibition of Friend leukemia virus and Monoley murine sarcoma virus in vitro by [SiW12O40]4− and in vivo by [Na(SbW7O24)3(Sb3O7)2]18−
(a French drug [NH4]17Na[Na(SbW7O24)3(Sb3O7)2]·14H2O historically called HPA-23).3 HPA-23 and other polyoxotungstates inhibited RNA-dependent DNA polymerases of retroviruses, and led to an expectation of antiviral activity against human immunodeficiency virus (HIV). Only HPA-23 was tested in animal models of lentiviral infections and infected humans. Unfortunately, HPA-23 was too toxic to be effective in the French clinical trial and a subsequent one conducted in the US.4,5 The negative result led to a temporary decline in studying polyoxometalates as potential anti-HIV agents. Nevertheless, other researchers familiar with polyoxometalates investigated the possibility of their antiviral activities, and showed that many polyoxotungstates, in particular, K13[Ce(SiW11O39)2]·26H2O (JM1590), K6[BGa(H2O)W11O39]·15H2O (JM2766), [Me3NH]8[(SiNb3W9O37)2O3]
(JM2820), K7[PTi2W10O40]·6H2O (PM-19), K9H5[(GeTi3W9O37)2O3]·16H2O (PM-504), [PriNH3]6H[PTi2W10O38(O2)2]·H2O (PM-523), K10[Fe4(H2O)2(PW9O34)2]·xH2O (HS-058), K7[P2W17NbO62], and K7[P2W17(NbO2)O61] exhibited in vitro activity against HIV and other RNA viruses.6 However, none of these PMs had activity against RNA viruses and cytotoxicity advantageous enough to surpass clinically approved drugs such as 3′-azido-3′-deoxythymidine (AZT) and ribavirin.2d,7 It seems to be required for realizing polyoxometalate-based inorganic drugs that their activity against a biological target is unprecedentedly excellent or can not be replaced by the approved drugs. We recently showed potent antiviral activity of tris(vanadyl)-18-tungsto-2-antimonate(III) anions, [(VIVO)3(SbW9O33)2]12− and [(VVO)(VIVO)2(SbW9O33)2]11−, against a wide variety of enveloped viruses which infect high-risk individuals such as infants born prematurely, cardiovascular failure, pulmonary dysplasia, and HIV: their selective index (SI) values (>104) for HIV are much higher than the values of 3–4 × 103 of AZT and dextran sulfate (in mol. wt. 5000, DS5000).8 In addition, our finding that most of the antiviral polyoxotungstates exhibited a great synergistic effect with β-lactam antibiotics against methicillin-resistant Staphylococcus aureus
(MRSA) strains2f,h has stimulated us to investigate the mechanism of antibacterial activity in relation to the still unclear mechanism of antiviral activities along with exploring the activity against other bacteria such as vancomycin-resistant Staphylococcus aureus
(VRSA) and drug-susceptible and -resistant Streptococcus pneumoniae and Helicobacter pylori.9 VRSA and Streptococcus pneumoniae as well as MRSA are Gram-positive bacteria and pose serious problems of hospital-acquired infections in surgical intensive care units where invasive procedures are performed.10 On the other hand, Helicobacter pylori is a microaerophilic Gram-negative bacterium which colonizes gastric mucosa, and is known to be a principal gastric pathogen of humans.11H. pylori infection in humans is associated with the development of gastritis, gastric ulcers, duodenal ulcers, peptic ulcers, and gastric cancer. It also poses a serious public health problem since approximately half of the world population is infected with H. pylori. The eradication of H. pylori usually requires combination therapy because single antibiotic therapy has not been very effective. The most widely used regimen is triple therapy that is in combination with bismuth compounds (bismuth oxynitrate, bismuth oxycarbonate, bismuth oxysalicylate, bismuth oxygallate, and bismuth oxycitrate) or proton pump inhibitors (omeprazole and lansoprazole) and two drugs from amoxicillin (AMX), clarithromycin (CLR), metronidazole (MTZ) and tetracyclines.12 While this therapy has demonstrated good activity with a high cure rate, the use of CLR and MTZ has resulted in the development of resistance. Thus, drug-resistant H. pylori is prevalent worldwide, and is considered to be the primary reason for failure to eradicate the infection.13 Therefore, it is an important issue to explore new anti-H. pylori drugs that are highly effective against H. pylori. This short review aims to detail some of the recent developments on biological activities of the PMs selected from the viewpoint of the clinical significance. Fig. 1 shows the structures of anions for some of the bio-active PMs, which will be discussed below.![Structures of anions for some of the bio-active PMs which are discussed in this work: [Mo7O24]6−
(a), [PTi2W10O40]9−
(b), [(VO)3(SbW9O33)2]9−
(c), [P2W18O62]6−
(d), [(AsW9O33)4(WO2)4]28−
(e), and [V15O36(CO3)]7−
(f).](/image/article/2005/JM/b504585a/b504585a-f1.gif) |
| | Fig. 1 Structures of anions for some of the bio-active PMs which are discussed in this work: [Mo7O24]6−
(a), [PTi2W10O40]9−
(b), [(VO)3(SbW9O33)2]9−
(c), [P2W18O62]6−
(d), [(AsW9O33)4(WO2)4]28−
(e), and [V15O36(CO3)]7−
(f). | |
2 Anti-tumor activity of polyoxomolybdates by cell apoptosis
Since [NH3Pri]6[Mo7O24]·3H2O (PM-8) was found in 1988 to exhibit potent anti-tumor activity against MX-1 human breast cancer, Meth-A sarcoma, and MM46 adenocarcinoma in vivo,2a the efficacy of PM-8 against solid tumors along with mechanistic investigations has been recognized also for both AsPC-1 human pancreatic cancer and MKN-45 human gastric cancer, which results from the activation of the apoptotic pathway.14 Median inhibitory concentration (IC50) values of PM-8 for 24 h and 48 h proliferations of 2 × 103 AsPC-1 (or MKN-45) cells/well are 1.65 (0.90) and 0.50 (0.50) mg ml−1
[=1.12 (0.62) and 0.34 (0.34) mM], respectively, indicating that the inhibition of the cell growth strongly depends on the cell density: the lower the number of tumor cells, the stronger the growth inhibition by PM-8. The morphology of cells exposed to PM-8 during growth by using Hoechst staining demonstrates the formation of apoptotic small bodies, which arise from DNA fragmentation. Fig. 2(a) shows the apoptotic change of AsPC-1 cells (1 × 106 cells in 25 ml) treated with PM-8 (1.65 mg ml−1) for 24 h. The flow cytometric analysis gives quantitatively an increase in the ratio of AsPC-1 apoptotic cells during the proliferation, as shown in Fig. 2(b). The DNA fragmentation analysis of AsPC-1 cells (after 24 h treatment with PM-8 in 1.65 mg ml−1) reveals the formation of DNA ladders on the electrophoresis of the PM-8-treated cell DNA in 1% agarose gel, as shown in Fig. 2(c). Similar apoptotic change is also observed for the MKN-45 cell line (2 × 106 cells in 25 ml treated by PM-8 of 0.90 mg ml−1). Fig. 3(b) shows the observable DNA ladders for the MKN-45 cells DNA after 24 and 48 h treatments with PM-8, and Fig. 3(a) shows the morphological change (by the Hoechst-staining method) of cells exposed to PM-8 during the 48 h growth due to the formation of apoptotic small bodies. We have proposed that PM-8 would be preferentially taken into tumor cells and converted to approximately ten-fold more toxic species (produced through biological reduction probably in the mitochondrial system) which inhibits ATP generation, and that such a significant difference in toxicity between PM-8 and its reduced species would lead to a tumor-selective inhibition based on the enhanced metabolism in the tumor cells, since the high molecular weight of the reduced species (as a condensed species) would result in a longer stay in the tumor cells.2a,d,e However, the mechanistic details of the anti-tumor activity of PM-8 still remain unclear. The results of an in vivo test for MKN-45 using nude mice (3 mice/group) are shown in Fig. 4. When successive intraperitoneal injections of PM-8 (0.2 g kg−1 day−1) during 14 days were done after 7 days for nu/nu nude mice (on their backs) inoculated with 1 × 106 MKN-45 cells, PM-8 depressed the growth of small MKN-45 neoplasms with inhibition of 50% on day 70, compared to the case of the RPMI-1640 medium (control) for 14 days. Arsenic compounds are considered to be toxic and potently carcinogenic to human skin, lungs, and other organs. Nevertheless, arsenic trioxide, As2O3, can be used clinically without significant side effects at low doses (0.1–5 µM) and is a therapeutic agent for relapsed acute promyelocytic leukemia15 and possibly also human hepatocellular carcinoma HuH7.16 The growth suppression by PM-8 for the MKN-45 bearing mice implies the polyoxomolybdates (especially PM-8) are promising candidates for novel inorganic anti-cancer agents, since both cancer cells of MKN-45 and AsPC-1 are clinically difficult to treat.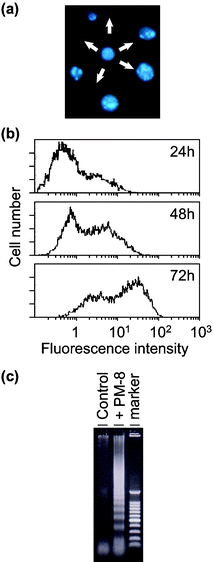 |
| | Fig. 2 Formation of apoptotic small bodies in AsPC-1 (a), variation of fractions of apoptotic cells determined by flow cytometry (b), and DNA ladder formation of AsPC-1 after 24 h treatment with or without PM-8 (c). | |
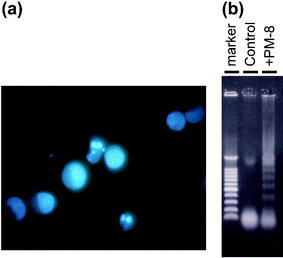 |
| | Fig. 3 Formation of apoptotic small bodies in MKN-45 (a) and DNA ladder formation of MKN-45 after 48 h treatment with or without PM-8 (b). | |
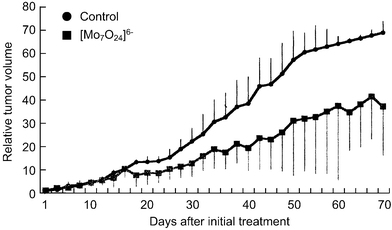 |
| | Fig. 4 Therapeutic effectiveness of PM-8 for MKN-45 implanted nude mice. | |
3. Inhibition of virus adsorption on host cells by polyoxotungstates
K13[Ce(SiW11O39)2]·26H2O (JM1590) and K6[BGa(H2O)W11O39]·15H2O (JM2766), which inhibited HIV-1 and simian immunodeficiency viruses at concentrations as low as 0.008–0.8 µM, seem to be the most potent anti-HIV compounds. They also inhibited the formation of giant cells (syncytium) in co-cultures of HIV-infected HUT-78 cells and uninfected Molt-4 cells. The mechanism of anti-HIV action for the compounds could be attributed to inhibition of virus-to-cell binding, since there was a good correlation between the inhibitory effects on both HIV-1 induced cytopathicity and syncytium formation and also a close correlation between their inhibitory effects on both syncytium formation and the interaction with the viral envelope glycoprotein gp-120.6a Furthermore, it has been proposed that polyoxotungstates act as HIV-1 protease inhibitors by binding to a cationic pocket using molecular modeling computation.6f Recently V/W and Ti/W mixed Keggin derivatives were found to possess a broad spectrum of anti-RNA virus activities.8 Cytotoxicity of some Keggin and lacunary-Keggin sandwiched polyoxotungstates towards Madin–Darby canine kidney (MDCK), Vero, HEp-2, and MT-4 cells was investigated by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetry for cells cultured in the presence of PMs (with a variety of concentrations) at 35 °C during 5 days.17Table 1 lists median cytotoxic concentration (CC50) values of the V/W and Ti/W mixed Keggin derivatives for the above cells. All the compounds did not show any toxicity at concentrations of <200 µM for MDCK, Vero, and HEp-2 cells, which were in a stationary culture condition of cell growth during the examination. On the other hand, K5[SiVW11O40]·xH2O (PM-43), K7[BVW11O40]·xH2O (PM-47), K10Na[(VO)3(SbW9O33)2]·26H2O (PM-1001), and K11H[(VO)3(SbW9O33)2]·27H2O (PM-1002), all of which contain vanadium atoms, showed toxicity at concentrations of 41.9–47.4 µM toward MT-4 cells which were in a growing state of culture during the examination. Table 2 shows inhibitory effects of these polyoxotungstates against dengue virus (DFV), influenza virus (FluV-A), respiratory syncytial virus (RSV), parainfluenza virus (PfluV-2), distemper virus (CDV), and HIV. Median effective concentration (EC50) values (0.03–0.14 µM for the infected MT-4 cells) of PM-1001 and PM-1002 were less than 1 µM against RSV, PfluV-2 and HIV, and lower than those of ribavirin. High anti-HIV activity of PMs-1001 and -1002 was confirmed for HIV-1(IIIb) by multinuclear activation of galactosidase indicator (MAGI) assay.18 EC90 values (0.11 µM) of PMs-1001 and -1002 (with EC50
= 0.0096–0.018 µM) for HeLaCD4/LTR-β-Gal cells were lower than DS5000, and SI (=CC50/EC50) values were more than 5500 and 10000, respectively. Change of the time schedule for administration showed that both compounds worked at very early stages of virus infection (less than 2 h after infection) as well as the cases for DS5000 and bicyclam derivative, octahydrochloride dehydrate of 1,1′-[1,4-phenylenebis(methylene)]-bis-1,4,8,11-tetra-azacyclotetradecane (ADM3100). PMs-1001 (in 10 µM) and -1002 (in 2 µM) also inhibited perfectly the syncytium formation, namely, the multinuclear giant cell formation induced by co-cultivation of Molt-4 cells and HIV-1infected Molt-4/IIIb cells. An analysis of the target molecule which interacts with PMs-1001 and -1002 during the adsorption/penetration process was done: PM-1001 strongly inhibited the binding of anti-gp-120 antibody to HIV-1 infected MT-4 cells, implying the binding of PM-1001 with gp-120. In contrast, AMD3100 strongly inhibited the binding of anti-X4 receptor antibodies (anti-CD4 and anti-CXCR4) to Molt-4 cells.19 Therefore, a combination of PM-1001 or -1002 with AMD3100 let us expect clinically significant synergistic anti-HIV activity. The behavior that PMs bind viral gp-120 and inhibit the HIV-strain dependent syncytium formation due to the interaction with receptors (CD4 and CXCR4) was first observed for K13[Ce(SiW11O39)2]·26H2O (JM1590) and K6[BGa(H2O)W11O39]·15H2O (JM2766)6a and also for K7[PTi2W10O40]·6H2O (PM-19).20 The anti-FluV-A active [PriNH3]6H[PTi2W10O38(O2)2]·H2O (PM-523) inhibited the fusion of the viral envelope into the cellular membrane (at the first step in the entry of FluV into cells), as was revealed by the inhibition of the dequenching of fluorescence from the rhodamine-labeled viral envelope.7b Similarly, K10[Fe4(H2O)2(PW9O34)2]·xH2O (HS-058) inhibited both the adsorption of RSV to cells and the syncytium formation by RSV.21 Not only the above RNA viruses but also DNA viruses such as herpes simple virus (HSV-1 and HSV-2) and human cytomegarovirus (HCMV) were inhibited by the polyoxotungstates.2cIn vivo anti-viral activity was also investigated: PM-19 exhibited the enhancement of the peritoneal macrophage function in mice for HSV-2 infected immune-suppressed mice.22 PM-523 exhibited a therapeutic effect for FluV-A respiratory infection in mice due to a synergistic antiviral activity in combination with ribavirin,7c which inhibits viral RNA synthesis through inhibition of the activities of the multiple enzymes involved in viral RNA synthesis.23 Ti(or V)/W mixed polyoxotungstates (PMs-19, -523, -1001, and -1002) of 125 mg kg−1 day−1 at least was non-toxic for intraperitoneally challenged (with 5 × 106 PFU of HIV-1) Balb/c mice during five days and was potent for the therapeutic use without any HIV-antigen specific immune responses. In vitro antiviral activities of other V/W mixed [XW9O33]9−
(X = SbIII, AsIII, BiIII)- or [PW9O34]9−-sandwiching tris(vanadyl)-18-tungstates were assayed against coronaviruses such as transmissible gastroenteritis Porcine virus (TGEV) and feline infectious peritonitis virus (FIPV) as a family of severe acute respiratory syndrome coronavirus (SARS CoV) using pig kidney cells (CPK) and feline embryonic fibroblast cells (fewf-4), respectively. Table 3 shows potent activities of five tris(vanadyl)-18-tungstate anions against TGEV and FIPV. As shown in Table 3, in contrast, [{Mn(H2O)}3(SbW9O33)2]12−, [Co3(AsW9O33)2]12−, [Co4(H2O)2(PW9O34)2]10−, and [Cu3(AsW9O33)2]12− did not exhibit any efficacy against TGEV and FIPV, implying the significance of the tris(vanadyl) moiety for the antiviral activity.
Table 1 Cytotoxicity of PMs for tissue culture cells
| | CC50a/µM |
|---|
| PMs | MDCK | Vero | HEp-2 | MT-4 |
|---|
| Average of 4 independent experiments. CC50 values were determined by the MTT method. |
|---|
| K5[SiVW11O40]·xH2O (PM-43) | >200 | >200 | >200 | 45.6 ± 0.43 |
| K7[BVW11O40]·7H2O (PM-47) | >200 | >200 | >200 | 47.4 ± 0.48 |
| [Et2NH2]7[PTi2W10O40]·xH2O (PM-518) | >200 | >200 | >200 | >100 |
| [Pri2NH2]5[PTiW11O40]·4H2O (PM-520) | >200 | >200 | >200 | >100 |
| [PriNH3]6H[PTi2W10O40(O2)2]·H2O (PM-523) | >400 | >400 | >400 | >100 |
| K10Na[(VO)3
(SbW9O33)2]·26H2O (PM-1001) | >229.2 ± 36 | 362.7 ± 38.1 | 233.6 ± 21.5 | 41.9 ± 2.9 |
| K10H[(VO)3(SbW9O33)2]·27H2O (PM-1002) | >200 | >200 | >200 | 45.9 ± 0.3 |
Table 2 Antiviral activity of PMs for several RNA viruses in vitro
| | EC50a/µM |
|---|
| PMs | DFVb | FluV-Ab | RSVb | PfluV-2b | CDVc | HIV-1b |
|---|
| Average of 3 to 7 independent experiments. EC50 was determined by the MTT method. EC50 was determined by the plaque reduction method. Average of 2 experiments. Not determined. |
|---|
| K5[SiVW11O40]·xH2O (PM-43) | 10.7 ± 6.7 | 8.4 ± 6.5 | 1.6d | >100 | 7.5 ± 0.95 | 0.3 ± 0.12 |
| K7[BVW11O40]·7H2O (PM-47) | 10.5 ± 6.9 | 11.5 ± 0.6 | 29.0d | 67.1d | 6.0 ± 0.6 | 0.03 ± 0.01 |
| [Et2NH2]7[PTi2W10O40]·xH2O (PM-518) | 36.8 | 62.3 ± 26.5 | 26.5d | 53.2 ± 39.2 | >50 | 2.0 ± 0.8 |
| [Pri2NH2]5[PTiW11O40]·4H2O (PM-520) | 11.7 ± 7.1 | 45.2 ± 25.8 | 0.74 ± 0.58 | 23.2 ± 2.4 | 7.4 ± 0.4 | 2.0 ± 0. 5 |
| [PriNH3]6H[PTi2W10O40(O2)2]·H2O (PM-523) | >61.5 | 5.6 ± 2.0 | 1.3 ± 0.46 | 2.5 ± 1.0 | 7.3 ± 1.1 | 0.3 ± 0.07 |
| K10Na[(VO)3(SbW9O33)2]·26H2O (PM-1001) | 0.45 | 1.75 ± 1.6 | <0.16 | 1.1 ± 0.9 | 5.7 ± 0.5 | 0.14 ± 0.17 |
| K11H[(VO)3(SbW9O33)2]·27H2O (PM-1002) | 1.95 ± 1.4 | 4.6 ± 1.7 | 0.75 ± 0.05 | 0.75 ± 0.05 | 2.8 ± 1.0 | 0.03 ± 0.01 |
| Ribavirin | >100 | 5.0 ± 2.75 | 3.9 ± 3.1 | 14.0 ± 4.8 | 73.6 ± 34.5 | NDe |
Table 3 Antiviral activity of tris(vanadyl)-containing PMs against coronaviruses
| PMs | TGEV | FIPV |
|---|
| EC50/µg ml−1 | CC50/µg ml−1 CPK | SI | EC50/µg ml−1 | CC50/µg ml−1 fewf-4 | SI |
|---|
| K10Na[(VO)3(SbW9O33)2]·26H2O (PM-1001) | 0.8 ± 0.7 | 67 ± 11.8 | 83.8 | 1.1 ± 0.4 | 90 ± 2.1 | 81.8 |
| K11H[(VO)3(SbW9O33)2]·27H2O (PM-1002) | 3.6 ± 2.2 | 67.9 ± 22.0 | 19 | 4.3 ± 4.0 | 26 ± 25 | 6 |
| K12[(VO)3(AsW9O33)2]·17H2O (PM-1207) | 10.9 ± 8.2 | >100 | >9.2 | 0.5 ± 0.2 | 17.6 ± 7.7 | 35.2 |
| K12[(VO)3(BiW9O33)2]·29H2O (PM-1208) | 12.62 ± 11.4 | >100 | >7.9 | >22.3 | 22.3 ± 4.7 | 1 |
| K12[(VO)3(PW9O34)2]·xH2O (PM-1213) | 9.4 ± 5.3 | >100 | 10.6 | >58.0 | 58 ± 15.5 | 1 |
4 Broad spectrum of antibacterial activity against Gram-positive and -negative bacteria
We found that the polyoxotungstates (in fractional inhibitory concentrations FIC = 0.01–0.50, determined by the agar dilution checkerboard method for the combination of PMs with β-lactam antibiotic oxacillin or penicillin) with Keggin, lacunary Keggin, Wells–Dawson, double-Keggin, and Keggin-sandwiched structures enhanced the antibacterial activity of β-lactam antibiotics on methicillin-resistant Staphylococcus aureus
(MRSA), which is a Gram-positive bacterium.2f,h It is known that the β-lactam resistance to MRSA strains is associated with the production of an additional peptidoglycan-synthetic enzyme with low affinity to the β-lactams, called penicillin-binding-protein 2′
(PBP2′) coded by the mecA gene, in addition to usual PBPs1-4.24 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of the membrane proteins separated from MRSA revealed that the polyoxotungstates depressed the formation of PBP2′ which is essential for the cell wall construction during MRSA growth. Qualitative analysis of the inhibitory effect of the polyoxotungstates on the production of all the PBPs was done by [14C]benzylpenicillin's binding with PBPs.2hFig. 5 shows the autoradiography of PBPs for the constitutive MRS394-1 cells cultivated in the presence of K7[PTi2W10O40]·6H2O (PM-19) at various concentrations. Each PBP was identified using prestained molecular weight markers in SDS-PAGE. As shown in Fig. 5 PM-19 has no significant affinity for PBPs but strongly inhibits the emergence of PBPs, especially PBP2′, on the cell membrane. The polyoxotungstates depressed not only the production of PBP2′, but also the production of β-lactamase which hydrolyzes β-lactam antibiotics on the membrane.2h This leads to the synergistic effect of polyoxotungstates against MRSA cells in the presence of β-lactam antibiotics which have high affinities to PBPs 1–4.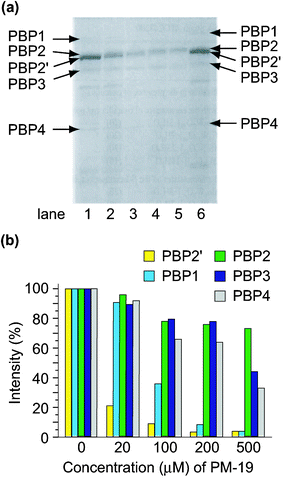 |
| | Fig. 5 Autoradiography of the membrane fractions prepared from the MRAS394-1 cells incubated (at 32 °C for 5 h) in the presence (lane 1, 0; 2, 20; 3, 100; 4, 200; 5, 500 µM) of PM-19 (a) and relative amounts of PBSs against a variety of concentrations of PM-19 (b). Lane 6 in (a) indicates that the membrane fraction prepared from the MRS394-1 cells cultured without any reagents was pre-incubated in the presence of 130 µM PM-19 for 1 h at 32 °C. | |
Interestingly, the enhancement of antibacterial activity of β-lactam antibiotics was observed not only against MRSA but also against vancomycin-resistant S. aureus
(VRSA) for some polyoxometalates such as K6[P2W18O62]·14H2O, K4[SiMo12O40]·3H2O and PM-19. The effectiveness of [SiMo12O40]4− against MRSA and VRSA is surprising, since most of the polyoxomolybdates were not effective against MRSA.2f,h The synergistic effect of PM-19 (in 500 µM) in combination with oxacillin against VRSA-Mu50 strain is exemplified in Fig. 6(a) where an increase of the concentration of PM-19 demonstrates alteration from “homo-resistance of the strain”
(no observation of a growth inhibitory zone) to “susceptibility of the strain to β-lactam”
(observable growth inhibitory zone around the oxacillin disk) through “hetero-resistance of the strain”
(observation of the growth inhibitory zone with colonies inside). The synergistic effect for K6[P2W18O62]·14H2O, K4[SiMo12O40]·3H2O could be observed for lower concentrations of less than 100 µM. Minimum inhibitory concentration (MIC) values of three PMs and oxacillin for a variety of both MRSA and VRSA strains are shown in Fig. 6(b) where the clinically excellent safety of PM-19 (with high values of MIC) is demonstrated. Energy dispersive X-ray analysis of the strain treated with K6[P2W18O62]·14H2O showed the localization of the K6[P2W18O62]·14H2O tungsten atoms at the periphery of the cell. Fig. 7 shows the distribution of W atoms in MRSA MRS394-1 cells cultured for 5 h at 34 °C in the presence of K6[P2W18O62]·14H2O (40 µM). In addition, the biological reduction of K6[P2W18O62]·14H2O (+0.058 V vs. Ag/AgCl for reversible one-electron reduction at the physiological pH level of 7.4) and K4[SiMo12O40]·3H2O (+0.237 V also for reversible one-electron reduction at pH 7.4) was observed, when both MRSA and VRSA cells were incubated in the presence of either polyoxometalate. It is interesting that PMs can penetrate through the cell wall consisting of peptidoglycan layers and can reach cytoplasm membrane with a resultant exertion of the susceptibility to β-lactam antibiotics for MRSA and VRSA. In contrast to K4[SiMo12O40]·3H2O, the same structural Keggin polyoxomolybdate Na3[PMo12O40]·xH2O which is unstable at physiological pH did not exhibit synergistic activity, implying that the structural stability of PMs at the physiological pH level is another factor for the biological activity.
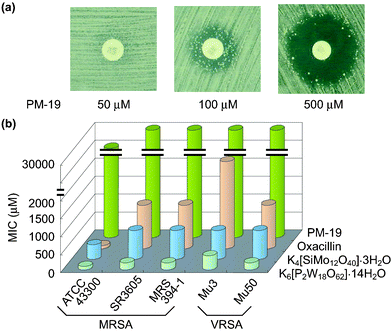 |
| | Fig. 6 Growth inhibitory zone around the oxacillin disk for Mu50 treated with PM-19 at concentrations of 50, 100, and 500 µM (a) and MIC values of three PMs and oxacillin for a variety of both MRSA and VRSA strains (b). | |
![Transmission electron microscope (TEM) and energy-dispersion X-ray (EDX) analysis images for MRS394-1 cells treated with K6[P2W18O62]·14H2O. The cells were harvested by centrifugation (5000g, 15 min, 4 °C), washed twice with PBS, and fixed with 2.5% glutaraldehyde in PBS. TEM image (a), O atoms (b), P atoms (c), K atoms (d), and W atoms (e) due to W-L line.](/image/article/2005/JM/b504585a/b504585a-f7.gif) |
| | Fig. 7 Transmission electron microscope (TEM) and energy-dispersion X-ray (EDX) analysis images for MRS394-1 cells treated with K6[P2W18O62]·14H2O. The cells were harvested by centrifugation (5000g, 15 min, 4 °C), washed twice with PBS, and fixed with 2.5% glutaraldehyde in PBS. TEM image (a), O atoms (b), P atoms (c), K atoms (d), and W atoms (e) due to W-L line. | |
The depression of the PBP2′ expression by PMs would originate from one (or a combination) of four possibilities: (i) missing the mecA gene, (ii) transcription inhibition from mecA gene to mRNA, (iii) translation inhibition from mecA-induced mRNA to PBP2′, and (iv) post-translation inhibition of the folding of the translated polypeptide chain.25 The inhibitory effect of PMs on (i) and (ii) for the expression of mecA gene was at the outset investigated: the tested strains were cultured for 24 h in a Mueller Hinton (MH) broth containing K6[P2W18O62]·14H2O, K4[SiMo12O40]·3H2O, and PM-19 (in MIC = 0.10–0.40, 0.40–0.80, and 12.8–25.6 mM, respectively), and thereafter aliquots of the broth were added to MH agar broth in the absence of PMs according to the standard oxacillin-disk method, which resulted in no growth inhibitory zone around the disk. This demonstrates that there is no missing of the mecA gene ((i)) in the strains treated with PMs, since the PM-treated strain would exhibit oxacillin susceptibility if the mecA gene was missed by PMs. The reverse transcription polymerase chain reaction (RT-PCR) analysis of the strains (except for ATCC43300 because of inducible strain) cultured for 2 h in the presence of PMs at various concentrations was carried out in order to examine the effect of PMs on the expression of mecA-induced mRNA. Fig. 8 shows the electrophoresis results for the mecA-induced mRNA (258 bp) extracted from MRSA SR3605 and VRSA Mu50 strains, together with 16S rRNA (528 bp) as a control. While the intensities of the 16S rRNA band did not change at any concentrations of PMs, the effect on the expression of the mecA-induced mRNA was different among the three kinds of PMs. As shown in Fig. 8(a), K6[P2W18O62]·14H2O gave a decrease in the band intensities of mecA-induced mRNA at concentrations of more than 100 µM for SR3605 and Mu50, indicating the inhibition of the transcription process ((ii)) from the mecA gene by K6[P2W18O62]·14H2O. Similar results were obtained for other strains. As shown in Fig. 8(b), however, K4[SiMo12O40]·3H2O did not depress the mecA-induced mRNA expression for both strains even at a concentration of 1000 µM, despite the observation of the synergistic activity at a concentration of less than 500 µM. Thus, the mechanism of the synergistic activity of K4[SiMo12O40]·3H2O is different from that of K6[P2W18O62]·14H2O, suggesting that the synergistic effect of K4[SiMo12O40]·3H2O is connected to the translation ((iii)) and/or post-translation ((iv)) processes. As shown in Fig. 8(c), PM-19 suppressed the mecA-induced mRNA expression for VRSA Mu50 at the concentration of more than 500 µM, but did not for MRSA SR3605 even at the high concentration of 1000 µM. A similar suppression of the mecA-induced mRNA expression by PM-19 was observed for VRSA Mu3, but not for MRSA MRS394-1. Thus, PMs on the cytoplasm membrane interact with the proteins related to the signal-transducer system leading to modifications of the metabolisms inside the cytoplasm such as transcription inhibition from the mecA gene to mRNA (in the case of K6[P2W18O62]·14H2O), translation inhibition from mecA-induced mRNA to PBP2′, and post-translation inhibition of the folding of the translated polypeptide chain (in the case of K4[SiMo12O40]·3H2O). PM-19 shows dual mechanisms dependent on the strain (the former inhibition for VRSA and the latter for MRSA).
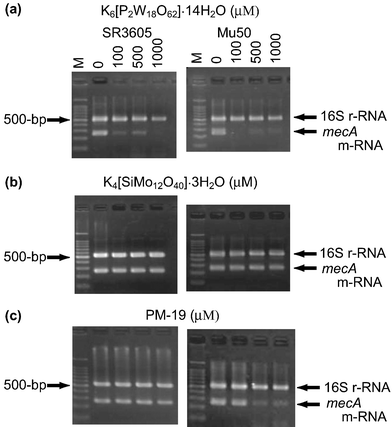 |
| | Fig. 8 RT-PCR analysis of the mRNA expression for both MRSA and VRSA strains incubated in MH broth for 2 h in the presence of three PMs (a–c). | |
Highly negatively-charged polyoxotungstates such as K28[(AsW9O33)4(WO2)4]·xH2O and K18[K(SbW7O24)3(Sb3O7)2]·xH2O, and Keggin-structured K5[SiVW11O40]·xH2O exhibited potent antibacterial activity with MIC values of less than 256 µg ml−1 against ATCC43504 (as a standard H. pylori strain), ID3023 (as a MTZ-resistant strain), and 15 clinically isolated H. pylori strains.26 In particular, K28[(AsW9O33)4(WO2)4]·xH2O was the most active, and superior to MTZ against MTZ-susceptible and -resistant strains and also to CLR against CLR-resistant strains, as shown in Fig. 9(a). Most of the polyoxomolybdates showed little anti-H. pylori activity with MIC values of more than 256 µg ml−1. Fig. 9(b) shows a morphological change from bacillary form to U-shaped or coccoid form for the K28[(AsW9O33)4(WO2)4]·xH2O (or K18[K(SbW7O24)3(Sb3O7)2]·xH2O)-treated H. pylori cells. It is pointed out that the Keggin structure and/or highly negatively-charged polyoxotungstates are important factors for the antibacterial activity against H. pylori, probably with a mechanism different from those of AMX (as an inhibitor of the synthesis of peptideglycan in cell membrane),27 CLR (as an inhibitor of the synthesis of protein at 23S rRNA within cytoplasm),28 tetracyclines (as inhibitors of protein synthesis),28 and MTZ (as an inhibitor of DNA replication).29
![MIC values of MTZ, CLR, and PMs for drug-susceptible, MTZ-resistant, and CLR-resistant H. pylori strains (a) and scanning electron micrographs of H. pylori ATCC43504 strain exposed to K28[(AsW9O33)4(WO2)4]·xH2O (more than MIC) for 36 h (b).](/image/article/2005/JM/b504585a/b504585a-f9.gif) |
| | Fig. 9 MIC values of MTZ, CLR, and PMs for drug-susceptible, MTZ-resistant, and CLR-resistant H. pylori strains (a) and scanning electron micrographs of H. pylori ATCC43504 strain exposed to K28[(AsW9O33)4(WO2)4]·xH2O (more than MIC) for 36 h (b). | |
Most of the vanadate and vanadyl compounds showed potent antibacterial activity against Streptococcus pneumoniae: the MIC values of the vanadium compounds were 4–32 µg ml−1, while the MIC values of tungstates and molybdates were in the range 128 to more than 8000 µg ml−1.2g Scanning electron microscopy showed the elongation of S. pneumoniae strains under low concentrations (<MIC) of vanadium compounds such as [tert-BuNH3]4[V4O12], K5H2[V15O36(CO3)]·15.5H2O, Na3VO4, VOSO4, as shown in Fig. 10 where IID554 strain was incubated for 15 h in the presence of [tert-BuNH3]4[V4O12]
(with a variety of concentrations). The vanadium compounds non-selectively inhibited the incorporation of thymidine, uridine, leucine, and glucose into the cells of S. pneumoniae and led to an efflux of potassium ions from the cells, implying the interference of the transport of substrates and ions through the cell membrane in or on the cell membrane.
![Morphological change of S. pneumoniae IID554 strain by incubation in the presence of [tert-BuNH3]4[V4O12]: 0 mg ml−1
(a), 4 mg ml−1
(b), and 100 mg ml−1
(c).](/image/article/2005/JM/b504585a/b504585a-f10.gif) |
| | Fig. 10 Morphological change of S. pneumoniae IID554 strain by incubation in the presence of [tert-BuNH3]4[V4O12]: 0 mg ml−1
(a), 4 mg ml−1
(b), and 100 mg ml−1
(c). | |
5 Conclusions
We can state that by now considerable insight has been gained into the bio-activity of PM molecules. The anti-tumor activity of polyoxomolybdates, in particular heptamolybdate [Mo7O24]6−, against solid tumors has been extended to both AsPC-1 human pancreatic cancer and MKN-45 human gastric cancer along with the mechanistic investigation which provides the activation of the apoptotic pathway. V/W and Ti/W mixed Keggin polyoxotungstates exhibit a broad spectrum of anti-RNA virus activities. In particular, the tris(vanadyl)-18-tungsto-2-antimonate(III) anions, [(VIVO)3(SbW9O33)2]12− and [(VVO)(VIVO)2(SbW9O33)2]11−, are significantly potent against a variety of RNA and DNA viruses due to the inhibition of the virus-to-cell binding. Most of the antiviral polyoxtungstates are taken up into the periphery of the Gram-positive MRSA and VRSA cells, and strongly enhance the activity of β-lactam antibiotics due to the depression of the production of PBP2′, which results from transcription inhibition from the mecA gene to mRNA or translation inhibition from mRNA to PBP2′. Low (namely more positive) electrochemical-redox potential of PMs (as exemplified by [P2W18O62]6− and [SiMo12O40]4−) together with large chemical stability under physiological conditions are other factors for the synergistic activity (due to depression of the PBP2′ expression) with the coexistence of β-lactam antibiotics. Antibacterial activities of both highly negatively-charged polyoxotungstates and V/W mixed Keggin polyoxotungstates against drug-resistant Gram-negative H. pylori strains and strong activity of polyoxovanadates against Gram-positive S. pneumoniae strains also provide a chemotherapeutic method based on these PMs for serious public health problems.Both potency and broad spectrum of anti-tumor and -viral activities of PMs are important for the clinical diagnosis of cancer diseases and virus infections at the initial stage, as treated by 5-fluorouracil (5-FU), interferon, and ribavirin. It is noteworthy that we quite recently found some of the polyoxotungstates to be sulfotransferase-specific inhibitors. Since the sulfated glycans are involved not only in various cell-to-cell interactions but also in cancer progression and other diseases to be overcome, PMs would serve as a novel inorganic drug, as well as against worldwide major nosocomial agents (MRSA, VRSA, S. pneumoniae, and H. pylori). In vivo anti-tumor, -viral and -bacterial experiments are in progress.
Acknowledgements
I would like to thank Professors Masazumi Eriguchi and Hironobu Yanagië
(Univ. Tokyo), Professor Kenji Okuda and Dr Kiyohiko Matsui (Yokohama City Univ.), Drs Katsuko Yamashita and Akira Seko (Sasaki Inst.), Professor Shiroh Shigeta (Fukushima Med. Univ.), and Dr Yutaka Tajima (Juntendo Univ.) for their valuable discussions and help in my research to date. Also, I acknowledge Grants-in-Aid for Scientific Research, No. 14204067 from the Ministry of Education, Science, Sports, and Culture and No. 99P01201 from RFTF/JSPS, and “Creation of bio-devices and bio-systems with chemical and biological molecules for medical use”, CREST, Japan Science and Technology Agency (JST).References
-
(a) Polyoxometallates: From Platonic Solids to Anti-Retroviral Activity, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, Dordrecht, 1994 Search PubMed;
(b) T. Yamase, in Polymeric Materials Encyclopedia: Synthesis, Properties, and Applications, ed. J. C. Salamone, CRC Press, Inc., Boca Raton, FL, 1996, p. 365 Search PubMed;
(c) C. L. Hill (editor), Chem. Rev., 1998, 98, 1 Search PubMed;
(d) Polyoxometallates: From Topology to Industrial Applications, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed;
(e) Polyoxometalate Chemistry for Nanocomposite Design, ed. T. Yamase and M. T. Pope, Kluwer Academic/Plenum Publishers, Dordrecht, 2002 Search PubMed;
(f) Polyoxometalate Molecular Science, NATO Science series, ed. J. J. Borrás-Alamenar, E. Coronado, A. Müller, and M. T. Pope, Kluwer Academic Publishers, Dordrecht, 2003 Search PubMed;
(g) T. Yamase, Catal. Surv. Asia, 2003, 7, 203 CrossRef CAS.
-
(a) T. Yamase, H. Fujita and K. Fukushima, Inorg. Chim. Acta, 1988, 151, L15;
(b) Y. Inouye, Y. Take, Y. Tokutake, T. Yoshida, A. Yamamoto and T. Yamase, Chem. Pharm. Bull., 1990, 38, 285 CAS;
(c) M. Fukuma, Y. Seto and T. Yamase, Antiviral Res., 1991, 16, 327 CrossRef CAS;
(d) T. Yamase, K. Tomita, Y. Seto and H. Fujita, in Polymers in Medicine: Biomedical and Pharmaceutical Applications, Technomic Pub. Company Inc., Lancaster, PA, 1992, ch. 13, p. 187 Search PubMed;
(e) T. Yamase, Molecular Engineering, Kluwer Academic Publishers, Dordrecht, 1994, vol. 3, p. 241 Search PubMed;
(f) T. Yamase, N. Fukuda and Y. Tajima, Biol. Pharm. Bull., 1996, 19, 459 CAS;
(g) N. Fukuda and T. Yamase, Biol. Pharm. Bull., 1997, 20, 927 CAS;
(h) N. Fukuda, T. Yamase and Y. Tajima, Biol. Pharm. Bull., 1999, 22, 463 CAS;
(i) K. Dan, K. Miyashita, Y. Seto, H. Fujita and T. Yamase, Pharmacol. Res., 2002, 46, 357 CrossRef CAS.
-
(a) N. Raybaud, C. Jasmin, J. Huppert, J. C. Chermann, G. Mathě and M. Raynaud, Rev. Eur. Études Clin. Biol., 1972, XVII, 295 Search PubMed;
(b) C. Jasmin, M. Raybaud, J. C. Chermann, D. Haapala, F. Sinoussi, C. B. Loustau, C. Bonissol, P. Kona and M. Raynaud, Biomedicine, 1973, 18, 319 Search PubMed;
(c) D. K. Haapala, C. Jasmin, F. Sinoussi, J. C. Chermann and M. Raynaud, Biomedicine, 1973, 19, 7 Search PubMed;
(d) C. Jasmin, J. C. Chermann, G. Herve, A. Teze, P. Souchay, C. Boy-Loustau, N. Raybaud, F. Sinoussi and M. Raynaud, J. Nat. Cancer Inst., 1974, 53, 469 Search PubMed;
(e) J. C. Chermann and F. C. Sinoussi, Biochem. Biophys. Res. Commun., 1975, 65, 1229 CAS;
(f) N. Larnicol, Y. Augery, C. L. Bousse-Kerdiles, V. Degiorgis, J. C. Chermann, A. Teze and C. Jasmin, J. Gen. Virol., 1981, 55, 17 Search PubMed.
- W. Rozenbaum, D. Dormont, B. Spire, E. Vilmer, M. Gentilini, L. Montagnier, F. Barre-Sinoussi and J. C. Chermann, Lancet, 1985, 450 CrossRef CAS.
- HPA-23 Cooperative Study Group and B. L. Moskovitz, Antimicrob. Agents Chemother., 1988, 32, 1300 Search PubMed.
-
(a) N. Yamamoto, D. Schols, E. De Clercq, Z. Debyser, R. Pauwels, J. Barlzarini, H. Nakashima, M. Babaa, M. Hosoya and R. Snoeck, Mol. Pharmacol., 1992, 42, 1109 CAS;
(b) Y. Inouye, Y. Tokutake, J. Kunihara, T. Yoshida, T. Yamase, A. Nakata and S. Nakamura, Chem. Pharm. Bull., 1992, 40, 805 CAS;
(c) M. S. Weeks, C. L. Hill and R. F. Schinazi, J. Med. Chem., 1992, 35, 1216 CrossRef CAS;
(d) J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327 CrossRef CAS;
(e) M. Witvrouw, H. Weigold, C. Pannecouque, D. Schols, E. De Clercq and G. Holan, J. Med. Chem., 2000, 43, 778 CrossRef CAS;
(f) D. A. Judd, J. H. Nettles, N. Nevins, J. P. Snyder, D. C. Liotta, J. Tang, J. Ermolieff, R. F. Schinazi and C. L. Hill, J. Am. Chem. Soc., 2001, 123, 886 CrossRef CAS.
-
(a) Y. Take, Y. Tokutake, Y. Inouye, T. Yoshida, A. Yamamoto, T. Yamase and S. Nakamura, Antiviral Res., 1991, 15, 113 CrossRef CAS;
(b) S. Shigeta, S. Mori, J. Watanabe, T. Yamase and R. F. Schinazi, Antiviral. Chem. Chemother., 1996, 7, 346 Search PubMed;
(c) S. Shigeta, S. Mori, J. Watanabe, S. Soeda, K. Takahashi and T. Yamase, Antimicrob. Agents Chemother., 1997, 41, 1423 CAS.
- S. Shigeta, S. Mori, E. Kodama, J. Kodama, K. Takahashi and T. Yamase, Antiviral Res., 2003, 58, 265 CrossRef CAS.
- M. V. da Silva Coimbra, M. C. Silva-Carvalho, H. Wisplinghoff, G. O. Hall, S. Tallent, S. Wallace, M. B. Edmond, A. M. Figueiredo and R. P. Wenzel, J. Hosp. Infect., 2003, 53, 103–110 CrossRef.
-
(a) H. Hanaki, K. Kuwahara-Arai, S. Boyle-Vavra, R. S. Daum, H. Labischinaki and K. Hiramatsu, J. Antimicrob. Chemother., 1998, 42, 199 CrossRef CAS;
(b) H. Hanaki, H. Labischinski, Y. Inaba, N. Kondo, H. Murakami and K. Hiramatsu, J. Antimicrob. Chemother., 1998, 42, 315 CrossRef CAS.
-
(a) B. E. Dunn, H. Cohen and M. J. Blaser, Clin. Microbiol. Rev., 1997, 10, 720 CAS;
(b) A. Leodolter, M. Kulig, H. Brasch, W. Meyer-Sabellek, S. N. Willich and P. Malfertheiner, Aliment. Pharmacol. Ther., 2001, 15, 1949 Search PubMed;
(c) R. T. Wang, T. Wang, K. Chen, J. Y. Wang, J. P. Zhang, S. R. Lin, Y. M. Zhu, W. M. Zhang, Y. X. Cao, C. W. Zhu, H. Yu, Y. J. Cong, S. Zheng and B. Q. Wu, World J. Gastroenterol., 2002, 8, 1103 Search PubMed;
(d) L. Marzio, L. Cellini and D. Angelucci, Dig. Liver Dis., 2003, 35, 20 Search PubMed.
-
(a) R. P. Logan, P. A. Gummet, J. J. Misiewicz, Q. N. Karim, M. M. Walker and J. H. Baron, Lancet, 1991, 338, 1249 CrossRef CAS;
(b) D. Y. Graham, G. M. Lew, H. M. Malaty, D. Evans, P. Klein and L. Alpert, Gastroenterology, 1992, 102, 493 CAS;
(c) E. Hentchel, G. Brandsätter, B. Dragosics, A. M. Hirschl, H. Nemec, K. Schutze and N. Engl, J. Med., 1993, 328, 308 Search PubMed;
(d) F. Bazzoli, R. M. Zagari, S. Fossi, P. Pozzato, G. Alampi, P. Simoni, S. Sottili, A. Roda and E. Roda, Eur. J. Gastroenterol. Hepatol., 1994, 6, 773.
-
(a) T. K. Ling, A. F. Cheng, J. J. Sung, P. Y. Yiu and S. S. Chung, Helicobacter, 1996, 1, 57 CrossRef CAS;
(b) V. Ellenrieder, W. Boeck, C. Richter, R. Marre, G. Adler and B. Glasbrenner, Scand. J. Gastroenterol., 1999, 34, 750 CrossRef CAS.
- A. Ogata, S. Mitsui, H. Yanagie, H. Kasano, T. Hisa, T. Yamase and M. Eriguchi, Biomed. Pharmacother., 2005, 59, 240 Search PubMed.
- Y. Shen, Z. X. Shen, H. Yan, J. Chen, X. Y. Zeng, J. M. Li, W. Wu, S. M. Xiong, W. I. Zhao, W. Tang, F. Wu, Y. F. Liu, C. Miu, Z. Y. Wang, S. J. Chen and Z. Chen, Leukemia, 2001, 15, 735 CrossRef CAS.
- M. Kito, K. Matsumoto, N. Wada, K. Sera, S. Futatsugawa, T. Naoe, Y. Nozawa and Y. Akao, Cancer Sci., 2003, 94, 1010 Search PubMed.
-
(a) R. Pauwels, J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijin, J. Desmyter and E. De Clercq, J. Virol. Methods, 1988, 20, 309 CrossRef CAS;
(b) W. Watanabe, K. Konno, K. Ijichi, H. Inoue, T. Yokota and S. Shigeta, J. Virol. Methods, 1994, 48, 257 CrossRef CAS;
(c) S. Mori and S. Shigeta, Tohoku J. Exp. Med., 1995, 177, 315 Search PubMed.
- J. Kimpton and M. Emerman, J. Virol., 1992, 66, 2232 CAS.
- G. A. Donzella, D. Schols, S. W. Lin, J. A. Esté, K. A. Nagashima, P. J. Maddon, G. P. Allaway, T. P. Sakmar, G. Henson, E. De Clercq and J. P. Moore, Nature Med., 1998, 4, 72 CAS.
- Y. Inouye, Y. Fujimoto, M. Sugiyama, T. Yoshida and T. Yamase, Biol. Pharm. Bull., 1995, 18, 996 CAS.
- S. Shigeta, S. Mori, J. Watanabe, M. Baba, A. M. Khenkin, C. L. Hill and R. F. Schinazi, Antiviral Chem. Chemother., 1995, 6, 114 Search PubMed.
-
(a) D. O. Andersen, B. K. Murray and J. A. North, Antiviral Chem. Chemother., 1993, 4, 249 Search PubMed;
(b) B. F. Gilbert and V. Knight, Antimicrob. Agents Chemother., 1986, 30, 201 CAS.
- S. Ikeda, S. Nishiya, A. Yamamoto, T. Yamase, C. Nishimura and E. De Clercq, J. Med. Virol., 1993, 41, 191 CrossRef CAS.
-
(a) K. Ubukata, N. Yamashita and M. Konno, Antimicrob. Agents Chemother., 1985, 27, 851 CAS;
(b) M. Matsuhashi, M. D. Song, F. Ishino, M. Wachi, M. Doi, M. Inoue, K. Ubukata, N. Yamashita and M. Konno, J. Bacteriol., 1986, 167, 975 CAS;
(c) K. Hiramatsu, Microbiol. Immunol., 1995, 39, 531 Search PubMed;
(d) H. F. Chambers, Clin. Microbiol. Rev., 1997, 10, 781 CAS.
- M. Matsuhashi, M. D. Song, F. Ishino, M. Wachi, M. Doi, M. Inoue, K. Ubukata, N. Yamashita and M. Konno, J. Bacteriol., 1986, 167, 975 CAS.
- M. Inoue, K. Segawa, S. Matsunaga, N. Matsumoto, M. Oda and T. Yamase, J. Inorg. Biochem., 2005, 99, 1023 CrossRef CAS.
- D. J. Waxman and J. L. Strominger, Ann. Rev. Biochem., 1983, 52, 825 CrossRef CAS.
- S. Pestka, Annu. Rev. Microbiol., 1971, 25, 487 CrossRef CAS.
-
(a) R. C. Knight, I. M. Skolimowski and D. I. Edwards, Biochem. Pharmacol., 1978, 27, 2089 CrossRef CAS;
(b) M. Müller, Surgery, 1983, 93, 165 CAS.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.