Elucidating the porous and chemical structures of ZnCl2-activated polyacrylonitrile on a fiberglass substrate
Zhongren
Yue
a,
Kelly R.
Benak
a,
Jinwen
Wang
a,
Christian L.
Mangun
b and
James
Economy
*a
aUniversity of Illinois Dept. of Materials Science, 1304 W. Green St., Urbana, IL 61801, USA
bEKOS Materials Corporation, 101 Tomaras Avenue, Savoy, IL 61874, USA
First published on 23rd June 2005
Abstract
ZnCl2-activated polyacrylonitrile (PAN) coated onto a fiberglass substrate was prepared in N2 at 450 °C. The porous and chemical structures were characterized by using N2 adsorption at 77 K, XPS, FTIR, competitive adsorption of CO2/CH4 and the adsorption of Cs+, Sr2+ and Ag+. The activated PAN displays a high BET surface area up to 1012 m2 g−1, a broad mesopore size distribution as well as a major micropore size distribution. Up to 19.3 wt% of nitrogen, which is probably in the form of pyridinic and pyrrolic structures, is incorporated in the chemically activated PAN. HCl uptake results show that such a material has a higher amount of weakly basic functional groups compared to a commercially available ACF15. The activated PAN also exhibits a higher selectivity coefficient for CO2/CH4 at STP and higher adsorption amounts for Cs+, Sr2+ and Ag+ than ACF15. It is proposed that nitrogen-containing, weakly basic functional groups positioned on the suitable-sized pore walls improve the adsorption ability of the activated PAN toward CO2, Cs+, Sr2+ and Ag+.
Introduction
Water contamination and greenhouse gases are of increasing concern in the United States and throughout the world.1–3 They have prompted industry to develop advanced new materials and improve the performance of existing processes to address these problems. In many cases, activated carbons (AC) have been determined to provide the best available technologies for the removal of trace contaminants from water and air.4Over the past 30 years, a vast literature has appeared focused on the preparation, characterization, and application of activated carbons.5–8 Recently, nitrogen-containing activated carbons have been given considerable attention for potential use, because of their potential basic surface character.9–12 Nitrogen-containing activated carbons were prepared by grafting nitrogen-containing chemicals onto the surface of AC,13 activating AC with ammonia gas9,10 and using a nitrogen-rich precursor, such as polyacrylonitrile (PAN).10,11 It was reported that the ammonia activated carbon fibers displayed sharply increased capacities for HCl gas.9 The presence of surface nitrogen functional groups in active carbons incorporated by ammonia treatment at high temperature markedly increased the adsorption capacities for transition metal ion species such as Cd2+, Ni 2+ and Cu2+. The amount of transition metal ions adsorbed correlated directly with the nitrogen content.10
In recent years, a number of new, low cost, high surface area fibers were prepared at relatively low temperatures (250–500 °C), by coating fiberglass with a polymer infused with a chemical activation reagent.14–16 Using this method, a very high nitrogen content material coupled with high surface areas was synthesized for the first time by the activation of PAN with ZnCl2 at low temperature.16 Its unique surface chemistry and porous structure are expected to facilitate the adsorption of trace contaminants from air and water. The fibrous nature of this material will also provide excellent contact efficiency with contaminated media.
In the present study, ZnCl2-activated PAN on fiberglass was prepared in N2 at 450 °C. The pore structure and surface chemistry of activated PAN were investigated by using N2 adsorption analysis, X-ray photoelectron spectroscopy (XPS), FTIR, elemental and chemical analyses. The adsorption capability was demonstrated by comparing the activated PAN with a commercially available ACF15 in the competitive adsorption of CO2/CH4 and the removal of Cs+, Sr2+ and Ag+ in water.
Experimental
Materials
Polyacrylonitrile (PAN) was employed as the starting material in this study. Zinc chloride (98+%) and N,N-dimethylformamide (DMF) (99.8%) were used as the chemical reagent and solvent, respectively. They were obtained from Aldrich Chemical Company. Hydrochloric acid (37.2%) and deionised water were used to wash the samples. The substrate fiber was a non-woven fiberglass mat, Craneglas® 230, (nominal mat width and average filament diameter 381 and 6.5 µm, respectively), obtained from Crane & Co. (Dalton, MA). The activated carbon fiber used for this study, designated ACF15, is commercially available from Nippon Kynol and is prepared by carbonizing and activating a phenolic fiber precursor (Kynol™) under steam/CO2.Preparation of activated PAN on fiberglass
PAN was first dissolved in DMF at 70 °C with electromagnetic stirring, then ZnCl2 was dissolved in the solution at ambient temperature to give a viscous and homogeneous (PAN–ZnCl2–DMF) solution having a concentration of 3.2 wt% PAN and 9.4 wt% ZnCl2. A glass mat was dip-coated with the solution. The coated fiber was passed through a bath containing a 5 wt% ZnCl2 aqueous solution at room temperature, in order to remove the DMF and to better coagulate the PAN coating. The coated mat was then dried and stabilized for 6 h by placing it in an air convection oven preheated to 200 °C. The stabilized material was cooled in air and transferred to a separate furnace, where it was activated in flowing N2 by heating it at ∼30 °C min−1 to 450 °C and holding that temperature constant for 30 min. After cooling in flowing N2, the sample was thoroughly washed with D.I. water, followed by thorough washing with 0.5 M HCl and rinsing with D.I. water. The activated PAN on fiberglass was obtained after the washed sample was dried in an air convection oven at 150 °C for 1 h, and then transferred to a vacuum oven and dried further at 120 °C under vacuum for at least 12 h.As a comparison, an activated PAN without using a fiberglass substrate was prepared by putting the above PAN–ZnCl2–DMF solution into a beaker, removing the DMF by placing the mixed solution in an air convection oven preheated to 160 °C for 3 h. The dried PAN with ZnCl2 was then stabilized and activated under the same conditions as indicated above.
Characterization
HCl uptake. HCl solutions (1 mM) were prepared with boiled distilled water to remove dissolved carbon dioxide. Approximately 0.01 g of sample was immersed for 4 h in 50 ml of HCl solution in a plastic vial. The HCl concentration changes were measured with a pH meter (MP220, Mettler Toledo).
Ag+ adsorption. A weighed amount of sample (∼0.04 g) was immersed in 50 ml of AgNO3 solution (∼5 mM) and shaken at 25 °C for 12 h in the dark. The initial pH value of the AgNO3 solution was 5.1. Another initial solution was adjusted with NH3·H2O to 8.2. The change in Ag+ concentration after adsorption was determined by KSCN titration using Fe(NH4)(SO4)2 as the indicator.17 Before titration, the pH of all of the adsorbates was adjusted to an acidic state (pH = 2–4).
Cs+ and Sr2+ adsorption. A weighed amount of sample (∼0.04 g) was immersed in 25 ml of CsCl solution (∼8 mM) or SrCl2 (∼4 mM) and shaken at 25 °C for 12 h. The concentration of Cs+ and Sr2+ was measured with an Atomic Absorption Spectrophotometer (3030B, Perkin-Elmer).
The equilibrium isotherms of CO2 and CH4 were measured at room temperature. The CO2/CH4 selectivity coefficient was then calculated by dividing the amount adsorbed by the pure gas isotherms at standard temperature and pressure (STP).
XPS spectra were obtained using an achromatic Mg Kα (1253.6 eV) X-ray source operated at 300 W. Survey scans were collected from 0–1100 eV with a pass energy equal to 178.95 eV. High-resolution scans were performed with the pass energy adjusted to 35.75 eV. The pressure inside the vacuum system was maintained at approximately 10−9 Torr during all XPS experiments.
A non-linear least squares curve fitting program (XPSPEAK4.1 software) with an asymmetric Gaussian–Lorentzian sum function and Shirley background subtraction was used to deconvolute the XPS peaks. The carbon 1s electron binding energy corresponding to graphitic carbon was referenced at 284.5 eV for calibration.18
Results and discussion
Porous structures
In previous research we showed that ZnCl2 acts as a dehydration agent to promote the thermal cross-linking of stabilized (oxidized) PAN at a much lower temperature, leading to activated PAN with much higher char yields and very high surface areas.16 The porosity was also created in part by dissolution of the ZnCl2 leftover in the charred coating. The effect of activation temperature on the micropore and mesopore volumes of ZnCl2-activated PAN on fiberglass is shown in Fig. 1. In these early experiments, the coating solution had a concentration of 3.2 wt% PAN and 6.4 wt% ZnCl2 which is lower than the value of 9.4 wt% ZnCl2 used in the following experiments. The micropore volumes (Fig. 1) display a sharp increase from 300 to 350 °C and a dramatic decrease after 650 °C. The mesopore volumes show a peak value at 450 °C over the temperature range between 300 and 650 °C, and then increase with temperature above 650 °C. Early experiments showed that the optimum temperature range for surface area development of ZnCl2-activated PAN on fiberglass is from 350 to 650 °C.16 It was found that the ZnCl2-activated PANs on fiberglass would become somewhat brittle as the activation temperature increased above 450 °C, though they are more flexible as compared to most other precursor-based porous materials obtained from ZnCl2 activation. By combining the above results with the previous surface area tests, 450 °C was determined to be the optimum temperature for ZnCl2-activated PAN on fiberglass. | ||
| Fig. 1 Normalized pore volumes of activated PAN as a function of temperature. | ||
The typical pore structure of ZnCl2-activated PAN on fiberglass at 450 °C is listed in Table 1. This material has 30 wt% of coating content on fiberglass. The normalized (calculation was based on the coating only) surface area and total pore volume are 1012 m2 g−1 and 0.574 cm3 g−1, respectively. They are much higher than 306 m2 g−1 and 0.170 cm3 g−1 obtained from the activated PAN without using the fiberglass substrate. The latter sample without the fiberglass is a small particulate in shape but its size (several mm) is much larger than that of the coating (film) on the fiberglass mat. This demonstrates that the activated PAN coating (film) on the fiberglass substrate is necessary to obtain high surface area and pore volume. Similar results were also found for the preparation of H3PO4-activated PVA on fiberglass.19 Presumably a thin layer coating (film) allows for more intimate contact of the activation reagent and for ease of outgassing during activation. Also, the fiberglass mat restricts the shrinkage of PAN or PVA during heat-treatment, leading to tenting and micro-cracking which create a huge amount of internal porosity.
| Sample | BET SSA/m2 g−1 | Total pore volume (P/P0 = 0.95)/cm3 g−1 | Micropore volume/cm3 g−1 | Mesopore volume/cm3 g−1 |
|---|---|---|---|---|
| Activated PAN without fiberglass substrate | 306 | 0.170 | 0.122 (71.8%) | 0.048 (28.2%) |
| Activated PAN coating (30 wt%) on fiberglass | 1012 | 0.574 | 0.384 (66.9%) | 0.190 (33.1%) |
| ACF15 | 1394 | 0.593 | 0.543 (91.6%) | 0.050 (8.4%) |
The typical morphologies of the activated PAN on fiberglass are shown in Fig. 2. The SEM image (Fig. 2A) shows the non-uniformity of the coating, such as some bridging between fibers which formed as a thin film of activated PAN. A thin coating (∼2 µm) on the surface of one side of the glass fiber can be seen in Fig. 2B. The non-uniformity of the coating might occur from the action of gravity on the PAN solution during the preparation.
 | ||
| Fig. 2 SEM photographs of activated PAN (450 °C) on fiberglass. | ||
ZnCl2-activated PAN has a higher percentage (33.1%) of mesopore volume (shown in Table 1) in contrast to a commercially available activated carbon fiber ACF15 which has a low percentage (8.4%) of mesopore volume. The distribution of pore volume with respect to pore size was deduced for both ACF15 and activated PAN from their N2 adsorption isotherms at 77 K using a method based on density functional theory (DFT). Fig. 3 shows that ACF15 is mainly microporous in nature and has a typical size distribution with a major peak at 11.7 Å. The activated PAN has a broad yet discernible mesopore size distribution from 50 to 500 Å as well as a narrow and dominant micropore size distribution with a peak at 11.6 Å.
 | ||
| Fig. 3 Pore size distributions of activated PAN (450 °C) and ACF15. | ||
Chemical structures
The chemical compositions of activated PAN and ACF15 were determined using an Elemental Analyzer and the results are listed in Table 2. In contrast with ACF15, activated PAN has higher N, H and O contents, especially note the N content which is typically in the range of 19.3 wt%. It is clear that heat treatment of PAN would result in the formation of cyclized structures containing conjugated –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– sequences which look like condensed polypyridine rings.20,21 It was felt that the nitrogen in the polypyridine rings would display a basic character. Table 2 shows a much higher HCl uptake on activated PAN versus ACF15, consistent with the presence of weakly basic functional groups in activated PAN.
N– sequences which look like condensed polypyridine rings.20,21 It was felt that the nitrogen in the polypyridine rings would display a basic character. Table 2 shows a much higher HCl uptake on activated PAN versus ACF15, consistent with the presence of weakly basic functional groups in activated PAN.
| Sample | Elemental analysis (wt%) | HCl uptake/mmol g−1 | |||
|---|---|---|---|---|---|
| C | N | H | Oa | ||
| a Data calculated from the results of elemental analysis (EA) and TGA. The elemental analyses of C, H and N have associated errors of ± 0.40%, thus the %O results determined by difference are subject to a cumulative error. | |||||
| Activated PAN | 63.1 | 19.3 | 3.0 | 14.6 | 3.6 |
| ACF15 | 93.2 | 0.29 | 0.17 | 6.34 | 0.5 |
The changes in functionality from PAN to activated PAN were characterized by FTIR as shown in Fig. 4. A sharp absorption band at 2240 cm−1 which is assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N structure disappears after the PAN (along with ZnCl2) was heated in air at 200 °C. An intense absorption peak, assigned to conjugated –CN– and –CC– structures, appears in the 1600 cm−1 range for the stabilized PAN and activated PAN, indicating the presence of structures similar to polypyridine rings. The carbonyl absorption at 1744 cm−1 is due to the acid group in the PAN (here PAN should be a copolymer). After stabilization and activation, the carbonyl absorption has shifted to lower frequencies and is observed as a shoulder at 1672 cm−1 on the main conjugation absorption.20 It is also observed that the intensity of this band decreased from stabilized PAN to activated PAN, due to the carbonization of the stabilized PAN at an elevated heat-treatment temperature in N2. This suggests that the carbonyl group has become part of the conjugated structure.
N structure disappears after the PAN (along with ZnCl2) was heated in air at 200 °C. An intense absorption peak, assigned to conjugated –CN– and –CC– structures, appears in the 1600 cm−1 range for the stabilized PAN and activated PAN, indicating the presence of structures similar to polypyridine rings. The carbonyl absorption at 1744 cm−1 is due to the acid group in the PAN (here PAN should be a copolymer). After stabilization and activation, the carbonyl absorption has shifted to lower frequencies and is observed as a shoulder at 1672 cm−1 on the main conjugation absorption.20 It is also observed that the intensity of this band decreased from stabilized PAN to activated PAN, due to the carbonization of the stabilized PAN at an elevated heat-treatment temperature in N2. This suggests that the carbonyl group has become part of the conjugated structure.
 | ||
| Fig. 4 FTIR spectra of PAN powder, stabilized PAN and activated PAN on fiberglass. | ||
XPS experiments showed that all three samples have carbon, oxygen, and nitrogen atoms on the surface. High-resolution XPS spectra of the N 1s region (Fig. 5) show that only one peak at 398.8 eV is observed in PAN, which represent the nitrile groups (CN*). After stabilization in air at 200 °C, all the nitrile groups have disappeared. The deconvolution of the N 1s spectrum shows that the nitrile groups were converted to pyridinic (398.3–399 eV), pyrrolic (∼400 eV) and quaternary (∼401 eV) nitrogen.22–28 The quaternary nitrogen is probably due to the formation of protonated amine. After activation with ZnCl2 in N2 at 450 °C, two peaks are observed and can be ascribed to pyridinic and pyrrolic structures. An additional peak at ∼403 eV is probably due to N-oxide22 or shake-up effects. As compared to the N 1s spectrum of stabilized PAN, the relative intensity of the pyridinic structure in activated PAN increases, probably due to the conversion of pyrrolic to pyridinic groups. The relative intensity of quaternary nitrogen also increases, suggesting that activated PAN has more protonated basic groups than stabilized PAN.
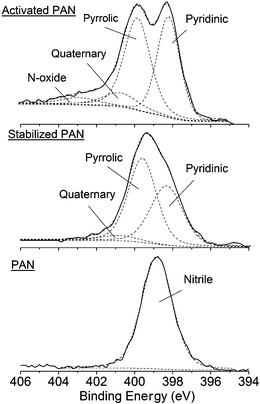 | ||
| Fig. 5 High-resolution XPS N 1s spectra of PAN powder, stabilized PAN and activated PAN on fiberglass. | ||
The high-resolution XPS spectrum of the C 1s region (Fig. 6) shows that PAN has a relatively symmetrical peak. The deconvolution of this C 1s spectrum gives two peaks that represent carbon in C*N and carbon in C*–C or C*–H. After stabilization and activation, both C 1s regions show obvious shoulder peaks which shifted to higher binding energies. Based upon the literature,29,30 the four peaks in the spectra represent carbon in C*–H and C*–C (284.5 eV), carbon present in C*–O (phenolic, alcohol and ether) and C–N groups (around 286 eV), C*O (carbonyl or quinone) and O–C*–O groups (287.3–287.7 eV), and C*OO (carboxyl or ester) groups (around 288.5–289.7 eV).
 | ||
| Fig. 6 High-resolution XPS C 1s spectra of PAN powder, stabilized PAN and activated PAN on fiberglass. | ||
In general, XPS, IR and elemental analysis show that the ZnCl2-activated PAN at 450 °C has not only N-containing but also O-containing functional groups on the surface.
Adsorption of CO2 and CH4
It is well known that the adsorption of CO2 and its separation from CH4 is a continuing area of interest within the industrial and scientific community. Herein lies an opportunity for designing new advanced materials as an economical means of separating these gases for industrial use.It was felt that the weakly basic functional groups in the porous activated PAN might facilitate the adsorption of CO2 over methane. This is in contrast to the typical method of using molecular sieving to achieve separation. The comparative adsorption isotherms for CO2 and CH4 are shown in Fig. 7 for activated PAN and ACF15. As expected, the adsorbed amounts of carbon dioxide are always higher than the amounts of methane since at the same pressure and temperature the adsorption potential (considering only London dispersion forces) is higher for CO2 than for CH4. There is typically little evidence of hysteresis for methane since it is such a low boiling point gas that quickly desorbs as the relative pressure is lowered. In contrast, hysteresis effects are observed for these two samples with CO2, especially for activated PAN. It was found that such hysteresis effects typically occur when good selectivity coefficients are present. Table 3 lists the selectivity coefficients at STP for these two samples and other commercially available materials for this application. Note that activated PAN has better selectivity than ACF15, pillared clays31 and zeolite materials32 while being second to the carbon molecular sieve.33 Another interesting aspect of activated PAN can be gleaned from plotting the selectivity factor versus pressure (Fig. 8). This graph shows that as the pressure decreases, the selectivity factor of CO2/CH4 on activated PAN begins to increase quickly, whereas the factor on ACF15 remains nearly constant. This is not entirely unexpected since the adsorbate–adsorbent interaction for CO2 is higher for activated PAN than for ACF15. The possible interpretation for the improved comparative adsorption of CO2 is most likely due to the presence of weakly basic functional groups on the pore surface of activated PAN.
 | ||
| Fig. 7 Adsorption–desorption isotherms of CO2 and CH4 at room temperature. | ||
 | ||
| Fig. 8 CO2/CH4 selectivity factor versus pressure. | ||
Adsorption of Cs+, Sr2+ and Ag+
The adsorption of Cs+, Sr2+ and Ag+ from aqueous solutions was done to further elucidate the chemical structure of activated PAN. Table 4 compares the adsorption amounts of Cs+, Sr2+ and Ag+ onto activated PAN and ACF15 under the same experimental conditions. 1.5 mmol g−1 of Cs+ and 2.2 mmol g−1 Sr2+ were adsorbed on activated PAN, respectively, whereas the amount of Cs+ or Sr2+ adsorbed on ACF15 is negligible. Enhanced metal ion adsorption could be attributed to the nitrogen atoms with lone electron pairs in activated PAN that can coordinate to metal ions. In addition, the suitably sized pores might have another benefit, namely, the small pores resemble cages with nitrogen atoms on the internal surface. This kind of configuration could act as a crown ether to sequester Cs+ or Sr2+. It was reported that some crown ethers have a higher selective adsorption for Cs+ and Sr2+ from nuclear and chemical waste, due to the excellent size match between the ions and the ether loop.34,35 For the activated PAN, it is proposed that nitrogen atoms on the surface of small pores (cages) might exhibit a high affinity for metal ions, through the donation of electrons to the positively charged metal ions.
Table 4 shows a markedly higher adsorption amount of Ag+ onto activated PAN than ACF15. It can also be seen that the adsorption amount is higher at pH 8.3.1 than at pH 5.1 of the original solution. These results suggest that in addition to the nitrogen surface functional groups, some other functional groups on the surface of activated PAN and ACF15 could react with Ag+, such as ion exchange (COOH + Ag+
→ COO–Ag + H+) and/or redox reactions (C–OH + Ag+
→ CO + Ag0 + H+). Evidence from X-ray diffraction showed that silver metal exists in both samples after adsorption. In addition, nano-sized silver particles were observed on the surface of the samples by means of SEM.
Conclusions
A new, low-cost adsorbent fiber was prepared by activating PAN on fiberglass with ZnCl2 in N2 at 450 °C. This material display a surface area of 1012 m2 g−1. It has a broader pore size distribution with a higher percentage (33.1%) of mesopore volumes, in contrast with a commercially available activated carbon fiber ACF15 (8.4%). It was found that the fiberglass substrate facilitates the development of pore structure of the PAN coating. Elemental analysis shows that activated PAN contains up to 19.3 wt% of nitrogen. HCl uptake results indicate that some weakly basic functional groups are present in activated PAN. IR and XPS show N-containing (probably pyridinic, pyrrolic and quaternary nitrogen), and O-containing functional groups incorporated in activated PAN. The weakly basic functional groups in porous activated PAN facilitate the adsorption of CO2, which displays a better selectivity for CO2/CH4 than ACF15 and most other adsorbents. The adsorption amounts of Cs+, Sr2+ and Ag+ from aqueous solutions are much higher on activated PAN than on ACF15, suggesting that the nitrogen atoms with a lone electron pair in activated PAN can coordinate to metal ions.Acknowledgements
We thank the STC program of NSF under agreement number CTS-0120978 and the Defense Advanced Research Project Agency/DSO (Grant # DABT-63-98-C-0053), NSF (Grant # DMR 97-12489). SEM and XPS analyses were carried out in the Center for Microanalysis of Materials, University of Illinois, which is partially supported by the U.S. Department of Energy under grant DEFG02-91-ER45439. We thank Crane & Co. for the free samples and Raymund W. M. Kwok, Department of Chemistry, The Chinese University of Hong Kong, for free use of the XPSPEAK software.References
- C. J. Vörösmarty, P. Green, J. Salisbury and R. B. Lammers, Science, 2000, 289, 284–288 CrossRef CAS.
- P. Aldhous, Nature, 2003, 422, 251 CrossRef CAS.
- R. A. Kerr, Science, 2004, 16, 303–306.
- I. N. Najm, V. L. Snoeyink, B. W. Lykins and J. Q. Adams, J. Am. Water Works Assoc., 1991, 83, 65–76 CAS.
- J. F. Byrne and H. Marsh, in Porosity in carbons: characterization and applications, ed. J. W. Patrick, Halsted Press, New York, 1995, p. 15 Search PubMed.
- B. McEnaney and T. J. Mays, in Introduction to Carbon Science, ed. H. Marsh, Butterworth, London, 1989 Search PubMed.
- M. Jaroniec and J. Choma, in Chemistry and Physics of Carbon, ed. P. A. Thrower, Marcel Dekker, New York, 1989, vol. 22, ch. 3 Search PubMed.
- P. N. Cheremisinoff and F. Ellerbusch, Carbon Adsorption Handbook, Ann Arbor Science, Michigan, 1978 Search PubMed.
- C. L. Mangun, K. R. Benak, J. Economy and K. L. Foster, Carbon, 2001, 39, 1809–1820 CrossRef CAS.
- Y. F. Jia, B. Xiao and K. M. Thomas, Langmuir, 2002, 18, 470–478 CrossRef CAS.
- K. Laszlo, E. Tombacz and K. Josepovits, Carbon, 2001, 39, 1217–1228 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- W. Yantasee, Y. Lin, G. E. Fryxell, K. L. Alford, B. J. Busche and C. D. Johnson, Ind. Eng. Chem. Res., 2004, 43, 2759–2764 CrossRef CAS.
- J. Economy and M. A. Daley, US patent 5834114, 1998.
- J. Economy, C. L. Mangun and Z. R. Yue, US Patent 6517906, 2001 Search PubMed.
- Z. R. Yue, C. L. Mangun and J. Economy, Carbon, 2002, 40, 1181–1191 CrossRef CAS.
- W. F. Hillebrand, G. E. F. Lundell, H. A. Bright and J. I. Hoffman, Applied Inorganic Analysis, 2nd edn., John Wiley & Sons, Inc., New York, 1953, p. 207 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, in Handbook of X-ray Photoelectron Spectroscopy, ed, J. Chastain, Perkin-Elmer Corporation, Eden Prairie, MN, 1992, p. 216 Search PubMed.
- Z. R. Yue, C. L. Mangun and J. Economy, Carbon, 2003, 41, 1809–1817 CrossRef CAS.
- N. Grassie and R. McGuchan, Eur. Polym. J., 1972, 8, 257–269 CrossRef CAS.
- E. Fitzer and D. J. Mueller, Carbon, 1975, 13, 63–69 CrossRef CAS.
- K. Stanczyk, R. Dziembaj, Z. Piwowarska and S. Witkowski, Carbon, 1995, 33, 1383–1392 CrossRef CAS.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021–1027 CrossRef CAS.
- A. Ishitan, Carbon, 1981, 19, 269–275 CrossRef CAS.
- R. H. Bradley, X. Ling, I. Sutherland and G. Beamson, Carbon, 1994, 32, 185–186 CrossRef CAS.
- J. Mittal, H. Konno, M. Inagaki and O. P. Bahl, Carbon, 1998, 36, 1327–1330 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- K. Laszlo, E. Tombacz and K. Josepovits, Carbon, 2001, 39, 1217–1228 CrossRef CAS.
- Y. Xie and P. M. A. Sherwood, Chem. Mater., 1990, 2, 239–299.
- Z. R. Yue, W. Jiang, L. Wang, S. D. Gardner and C. U. Pittman, Jr., Carbon., 1999, 37, 1785–1796 CrossRef CAS.
- P. R. Pereira, J. Pires and M. Brotas de Carvalho, Langmuir, 1998, 14, 4584–4588 CrossRef.
- D. W. Breck, Zeolite Molecular Sieves, John Wiley, New York, 1974 Search PubMed.
- Y. Kawabuchi, H. Oka, S. Kawano, I. Mochida and N. Yoshizawa, Carbon, 1998, 36, 337–382.
- W. W. Schulz and L. A. Bray, Sep. Sci. Technol., 1987, 22, 191–214 CrossRef CAS.
- L. X. Dang, Chem. Phys. Lett., 1994, 227, 211–214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |