X-Ray photoelectron spectroscopy studies of graphite powder and multiwalled carbon nanotubes covalently modified with Fast Black K: evidence for a chemical release mechanism via electrochemical reduction
Gregory G.
Wildgoose
a,
Nathan S.
Lawrence
b,
Henry C.
Leventis
a,
Li
Jiang
b,
Timothy G. J.
Jones
b and
Richard G.
Compton
*a
aPhysical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK, OX1 3QZ. E-mail: richard.compton@chemistry.ox.ac.uk; Fax: +44 (0)1865 275410; Tel: +44 (0)1865 275413
bSchlumberger Cambridge Research, High Cross, Madingley Road, Cambridge, UK, CB3 0EL
First published on 4th February 2005
Abstract
The development of new materials from which to construct controlled chemical-release systems has been an active area of research for the past four decades. Using X-ray photoelectron spectroscopy (XPS) we demonstrate that graphite powder and multiwalled carbon nanotubes (MWCNTs) covalently derivatised with 2,5-dimethoxy-4-[4-(nitrophenyl)azo]benzenediazonium chloride (FBK) or a derivative of FBK are important new micro and nano-scale materials for use as voltammetrically controlled chemical-release reagents in applications where the small size of the material is advantageous. By examining the N1s and O1s regions of the XPS spectra we can identify functionalities within the FBK moiety as well as hydroxyl, quinonyl and carboxylic acid functional groups present on the carbon surface. Comparison of the XPS spectra of the FBK derivatised carbon (FBKcarbon) and FBK derivatised MWCNTs (FBK-MWCNTs) before and after electrochemical reduction reveals that cleavage of the azo-linkage within the FBK moiety occurs upon reduction in aqueous solution. The voltammetric cleavage of the azo-linkage induces chemical release of 1,4-phenylenediamine from the carbon surface, demonstrating the proof of concept for these novel materials. It is envisaged that derivatives of these materials could be used in vivo in a wide range of areas including medical diagnosis and targeted drug-delivery systems as well as in in vitro applications such as analytical chemistry, sensor technology and industrial process monitoring and control.
1. Introduction
Systems involving the controlled release of chemicals are common in many everyday products including foods, cosmetics and pesticides.1 Research into controlled-release systems for drug delivery—arguably one of the most important fields that use controlled chemical release—and the materials from which to construct them first began in the 1960s.1 Since then there has been a dramatic increase in the types of materials used in chemical-release systems. A common approach is to use polymers with specific physical or chemical properties such as thermoresponsive swelling, pH responsiveness and biodegradability.1,2 The chemical to be released is then incorporated into the polymer as either a colloidal suspension or within microspheres, with release being initiated by leaching of the chemical from the polymer or by degradation of the polymer matrix.2,3 Polymeric prodrugs include a large variety of materials where the basic principle is to chemically attach the drug to a polymer “carrier” in a specific way so that controlled cleavage of the drug from the polymer carrier can be achieved as desired.4 Other examples of materials used in controlled-release drug-delivery systems include hydrogels,2 hydroxyapatite and carbonated apatite,5 bioactive glass6 and even wood.7Conventional drug delivery such as tablets or injections often result in a sharp increase in the concentration of the drug within the body, usually above the therapeutic concentration threshold, followed by a rapid decrease in the drug concentration until the drug falls below the therapeutic range. One common solution to this problem is to use a “pulsative” delivery system where the chemical or drug is released in a pulse-train over time. This has been achieved using some of the materials presented above, in particular polymers that respond to changes in electric or magnetic fields, exposure to ultrasound, light, enzymes, changes in pH or temperature (see reference 1 and references contained therein for a review of pulsative drug delivery). Recently the advent of microchip controlled drug-delivery devices has provided a powerful tool for clinical therapeutics and biological researches as this has allowed sustained, controlled release of a drug so that the concentration profile of the drug remains constant with time, or allowing programmed pulsative drug release to be carried out in a specific manner through integration into a microelectronic circuit.1,8
Whilst the small size of such microchip and microelectronic devices is advantageous, particularly if the chip were to be implanted in the body, there is increasing interest in the field of nanotechnological devices, and in particular in the use of carbon nanotubes (CNTs) and modified CNT electrodes for bioelectrochemical applications.9 The use of graphitic materials such as graphite powder or CNTs for chemical-release reagents in drug-delivery systems suffers from the same problem that early approaches using non-biodegradable polymers encountered, namely that the graphite powder or CNTs may build up in the tissue requiring surgical removal and thus limiting their applicability.3 Furthermore the toxicological effects of CNTs within living tissue are not yet fully understood.
A solution to this problem has recently been proposed by Strano et al. who developed a glucose sensor by incorporating CNTs into a dialysis capillary.10 The capillary provides a stable matrix for the CNTs and can be inserted under the skin whilst still allowing the diffusion of the relevant biomolecules or drugs to and from the CNTs.
In this report we covalently derivatise graphite powder and MWCNTs with the azo-dye Fast Black K (2,5-dimethoxy-4-[4-(nitrophenyl)azo]benzenediazonium chloride, FBK) using hypophosphorous acid.11,12 We use X-ray photoelectron spectroscopy (XPS) and voltammetric techniques, to examine samples of FBK derivatised graphite powder (FBKcarbon) and FBK derivatised MWCNTs (FBK-MWCNTs) before and after electrochemical reduction. XPS characterisation of these novel materials was undertaken via analysis of the elemental composition of the surface region, and a detailed study of the N1s and O1s regions confirmed that FBKcarbon and FBK-MWCNTs undergo chemical release upon electrochemical reduction in aqueous media.
Having demonstrated proof of concept, we propose that this novel approach could be used to develop novel materials for use in chemical-release and drug-delivery systems such as nano-scale intra-cellular drug-delivery agents13 or molecular tag agents in nano-sensors for use in molecular or chemical analysis.14,15 Biological applications could make use of the existing technologies such as microchip devices1,8 and/or the methods of Strano et al.10 for the targeted, controlled release of drugs and bioactive molecules in therapeutic medicine and biological research applications.
2. Experimental
2.1 Reagents and equipment
With the exception of potassium chloride (purchased from Riedel de Haën, Seelze, Germany), all reagents were obtained from Aldrich (Gillingham, UK), were of the highest grade available and used without further purification. All solutions were prepared using deionised water from an Elgastat (Elga, UK) UHQ grade system with a resistivity of not less than 18.2 MΩ cm.Electrochemical measurements were recorded using a μAutolab computer controlled potentiostat (Ecochemie, Utrecht, Netherlands) with a standard three-electrode configuration. Electrochemical experiments were carried out in a glass cell of volume 25 cm3. A basal plane pyrolytic graphite (bppg, 0.79 cm2, Le Carbone Ltd, Sussex, UK) electrode acted as the working electrode (see below). A platinum coil (99.99% Goodfellow, Cambridge, UK) acted as the counter electrode. The cell assembly was completed using a saturated calomel electrode (SCE, Radiometer, Copenhagen, Denmark) as the reference electrode. All electrochemical experiments were carried out after degassing the solution using pure N2 gas (BOC gases, Guildford, Surrey, UK) for 30 minutes and were conducted at 20 ± 2 °C.
Solutions of known pH in the range pH 1.0 to pH 12.0 were prepared in deionised water as follows: pH 1.0, 0.10 M HCl; pH 1.7, 0.1 M potassium tetraoxalate; pH 4.6, 0.10 M acetic acid + 0.10 M sodium acetate; pH 6.8, 0.025 M Na2HPO4 + 0.025 M KH2PO4; pH 9.2, 0.05 M disodium tetraborate; pH 10.5, 0.1 M disodium tetraborate; pH 12.0, 0.01 M sodium hydroxide. These solutions contained in addition 0.10 M KCl as supporting electrolyte. pH Measurements were performed using a Hanna pH213 pH meter.
The synthetic graphite powder used consisted of irregularly shaped particles of between 2 and 20 µm diameter and was purchased from Aldrich. Bamboo multiwalled carbon nanotubes (purity >95%) were of diameter 10–40 nm, length 5–20 µm and were purchased from NanoLab Inc. (Brighton, MA) and used without further purification.
2.2 Covalent derivatisation of graphite powder and MWCNTs with FBK
Derivatisation of graphite powder (2 g) or MWCNTs (50 mg) was achieved by mixing with a 10 cm3 solution containing 5 mM FBK, to which 50 cm3 hypophosphorous acid (H3PO2, 50%) was added (Scheme 1). A radical species of FBK (species A, Scheme 1) is generated from the parent diazonium ion upon reduction with the hypophosphorous acid, with subsequent loss of nitrogen gas providing a strong entropic driving force for the reaction. Note that voltammetric data clearly show that the diazonium salt is reduced at less cathodic potentials than the nitro group which remains intact during this procedure.12 The reaction mixture was then stirred continuously at 5 °C for 30 minutes during which time the radical species A in Scheme 1 is free to form a covalent bond, predominantly at edge-plane or edge-plane-like defect sites on the graphite powder or MWCNT surface.11,12 Finally the solution was filtered by water suction in order to remove any unreacted species from the graphite powder or the MWCNTs. Further washing with deionised water was carried out to remove any remaining acid and finally with acetonitrile to remove any unreacted diazonium salt from the mixture. The resulting FBKcarbon and FBK-MWCNTs were then air-dried by placing inside a fume hood for a period of 12 hours after which they were stored in an airtight container in the dark.11,12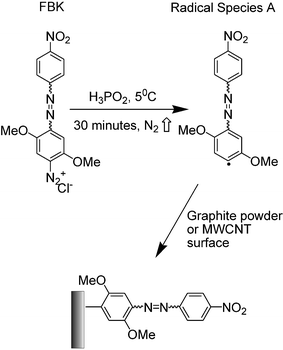 | ||
| Scheme 1 The structure of Fast Black K and its derivatisation of graphite powder or MWCNTs using hypophosphorous acid. | ||
2.3 Immobilisation onto basal plane pyrolytic graphite
The newly derivatised FBKcarbon and FBK-MWCNTs were abrasively immobilised onto the surface of a bppg electrode prior to experiments being performed. This was done by initially polishing the electrode on glass polishing paper (H00/240) after which they it was polished on silicon carbide paper (P1000C) for smoothness. The FBKcarbon or FBK-MWCNTs were then abrasively immobilised onto the bppg electrode by gently rubbing the electrode surface on a fine filter paper (Whatman) containing either material.2.4 X-Ray photoelectron spectra
X-Ray photoelectron spectroscopy (XPS) experiments were carried out on a Scienta ESCA30016,17 instrument at the NCESS laboratory, Daresbury, UK using X-ray radiation from the aluminium Kα band (hν = 1486.7 eV), source setting 14 hν, 200 mA. All spectra were recorded using a pass energy of 150 eV and a take off angle of 90°. A slit width of 0.8 mm was used, unless otherwise stated. The base pressure in the analysis chamber was maintained at not more than 2.0 × 10−9 mbar.XPS experiments on FBKcarbon abrasively immobilised on a bppg electrode were carried out by cleaving the electrode and mounting a slice (of thickness 0.5 mm) onto a stub using non-conducting double-sided adhesive tape. The FBKcarbon or FBK-MWCNT powders used without electrochemical pre-treatment were simply adhered to the non-conducting double-sided adhesive tape and thus onto the stub. The stub was then mounted in the ultra-high vacuum sample analysis chamber of the spectrometer. To prevent the samples from becoming positively charged when irradiated due to emission of photoelectrons, the sample surface was bombarded with an electron beam from a “flood gun” within the spectrometer's analysis chamber (35% maximum filament current, 35% deflection, 1 eV kinetic energy, 10% emission current). Curve fitting was performed using a Gaussian sum function within the Scienta ESCA300 data system.18
3. Results and discussion
3.1 Voltammetric behaviour of FBKcarbon and FBK-MWCNTs
In this section we briefly summarise the voltammetric behaviour of FBKcarbon and FBK-MWCNTs which is discussed in more detail elsewhere.11,12 Both FBKcarbon and FBK-MWCNTs have been voltammetrically characterised as being covalently bound to the surface of graphite powder and MWCNTs over the pH range 1.0 to 12.0.11,12 At all pH's studied the voltammetric response was found to consist of three cathodic waves which are shown at ca. 0 V, −0.1 V and −0.3 V vs. SCE in Fig. 1, the exact peak potential varied with pH in a Nernstian fashion.11,12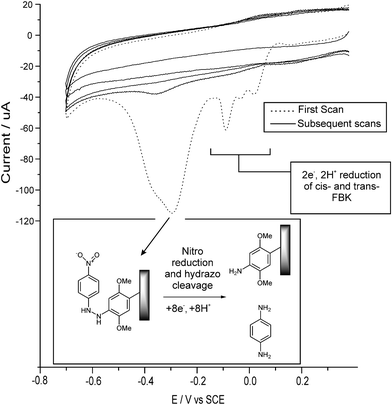 | ||
| Fig. 1 Five consecutive cyclic voltammograms showing the voltammetric behaviour of FBK-MWCNTs in aqueous solution (pH 1.0), see text. | ||
The cathodic waves at ca. 0 V and −0.1 V vs. SCE in Fig. 1 have been shown to correspond to an electrochemically reversible system comprising the two-electron two-proton reduction of the azo-linkage to the hydrazo form for cis-FBK and trans-FBK moieties respectively.12 The small peak at ca. −0.05 V in Fig. 1 is due to the protonation of the azo-linkage at pH 1.0.12 It was found that the trans form could be irreversibly converted to the cis form voltammetrically, see Fig. 2.12 Our previous studies focused primarily on this system to develop a nano-scale voltammetric switching device for use in nano-circuitry or in high density memory storage devices.12 As such this aspect will not be discussed further in this report.
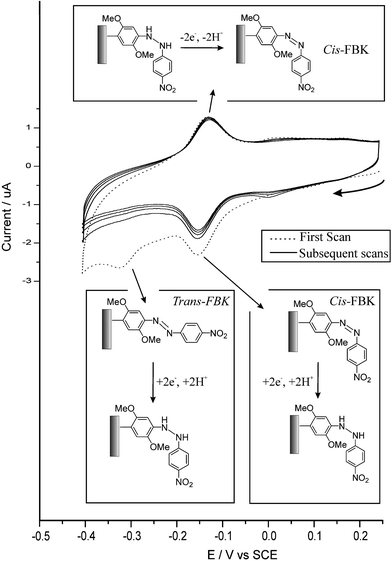 | ||
| Fig. 2 Five consecutive cyclic voltammograms showing the irreversible electrochemical conversion of trans-FBKcarbon to cis-FBKcarbon at pH 4.6, see text. | ||
If the potential is swept to more negative values then a second, large, electrochemically irreversible reduction wave is observed at ca. −0.3 V in Fig. 1, after which no further voltammetric features are observed. A slight shoulder at more negative potentials is always observed on this reduction wave. We proposed that this wave corresponded to the six-electron, six-proton reduction of the nitro group within the FBK molecule, followed by simultaneous cleavage of the hydrazo-linkage in a further two-electron, two-proton step at a similar potential, producing the shoulder on the reduction wave. Cleavage of the hydrazo-linkage would effect chemical-release of a 1,4-phenylenediamine fragment (fragment B, Scheme 2) whilst a 2,5-dimethoxyaniline moiety (surface moiety C, Scheme 2) would remain covalently bound to the carbon surface. This would account for no further voltammetric features being observed which corresponded to either the nitro group or the azo-linkage.12
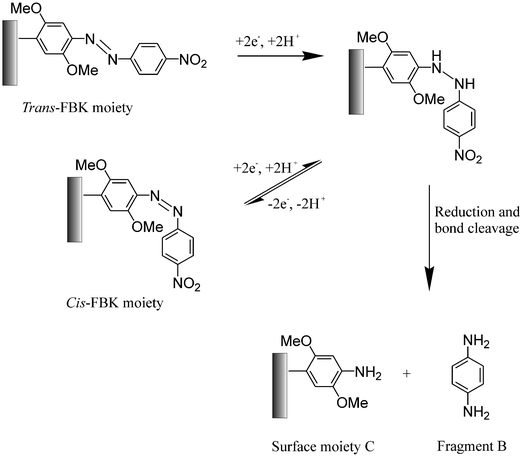 | ||
| Scheme 2 The proposed electro-reduction mechanism for cis- and trans-FBK moieties on derivatised graphite powder or MWCNTs and the proposed chemical release mechanism. | ||
In the next section we use XPS to first confirm that the FBK has derivatised the surface of the graphite powder and the MWCNTs and then to confirm whether our proposed mechanism is correct and that we have indeed voltammetrically induced chemical release from the FBKcarbon and FBK-MWCNTs.
3.2 XPS studies of FBKcarbon, FBK-MWCNT and unmodified carbon samples
XPS was used to analyse the compositions of the FBKcarbon, FBK-MWCNT and unmodified carbon samples. In each case, an initial broad scan was performed (1300 eV–0 eV) to establish the gross elemental composition of the samples' surface. As shown in Fig. 3, the broad scans all showed carbon to predominate in terms of elemental composition as expected, with a C1s emission at ca. 285 eV and a CKLL Auger emission at ca. 1200 eV. The presence of oxygen was confirmed in all three samples with a small O1s peak at ca. 540 eV and an OKLL Auger emission observed at ca. 1000 eV. The presence of nitrogen was confirmed in the FBKcarbon and FBK-MWCNT samples whilst no nitrogen was found to be present on the surface of the underivatised carbon samples. The FBK-MWCNT sample also exhibited a minor feature at 688.2 eV, assigned to the F1s signal resulting from the PTFE19 used in electrode manufacture which may have been abraded into the sample during previous electrochemical studies.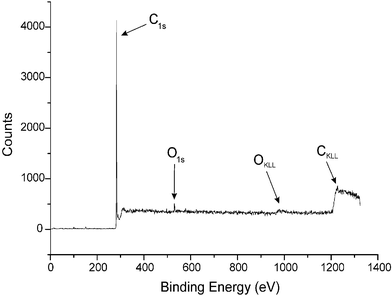 | ||
| Fig. 3 The XPS spectrum of FBKcarbon recorded from 0 to 1300 eV (slit width 0.8 mm). | ||
Elemental analysis of the unmodified carbon samples revealed that the total oxygen signal accounted for 1.8% of the surface elemental composition. This oxygen corresponds to surface groups such as hydroxyl, carbonyl and carboxylic moieties. The figure of ca. 2% elemental surface coverage for such groups is in good agreement with previous studies.20 After derivatisation with FBK the total oxygen signal found in FBKcarbon had increased to 2.7%, indicating that FBK contributes ca. 1% to the total elemental surface coverage. The total nitrogen signal which is entirely due to the presence of FBK molecules on the surface was also found to be ca. 1% indicating that the O1s results and the N1s results are in good agreement with each other. Whilst it is not possible to calculate a quantitative value for the surface coverage of FBK molecules, this result does give us a qualitative indication as to the extent of surface coverage of the FBK molecules. Note that in the light of voltammetric evidence the act of abrasively immobilising the FBKcarbon onto the bppg electrode has no chemical consequences.
In the following sections deconvolution of the O1s and N1s spectra into individual components was achieved by fitting the data curves using the Gaussian sum function described in section 2.4. This enabled the determination not only of the relative atomic compositions of the surface region, but also the binding energies of electrons in those atoms, and thus we could assign spectral peaks to functional groups on the carbon surface by direct comparison with the literature values.20–22 The results presented hereafter in parentheses give details of the calculated electron binding energies and the percentage of the overall N1s or O1s signal arising from the respective functional groups. Values for binding energies shown include a consideration of the effect of the flood gun on the measured binding energies, and have all been corrected to the position of the C1s signal, measured by performing one scan over the C1s region (300 eV–275 eV), relative to the value in pure graphite (284.60 eV).19
The FBKcarbon sample yielded two broad superposed Gaussian peaks in the O1s region (532.4 eV, 66.2%; 533.7 eV, 31.0%) which are mainly attributed to the nitro and methoxy moieties respectively, with some contribution from the hydroxyl and carboxylic signals which unfortunately could not be fully resolved due to their similar binding energies.19,23,24 The FBK-MWCNT sample yielded three superposed Gaussian waves (530.5 eV, 8.3%; 533.0 eV, 37.5%; 534.8 eV, 52.0%) shown in Fig. 4. Comparison with literature values allows us to assign these signals as surface quinone moieties (530.5 eV), the nitro moiety (533.0 eV) and methoxy (534.8 eV) functionalities.19,24 Again full resolution of the signals arising from surface hydroxyl and carboxylate groups was not possible.
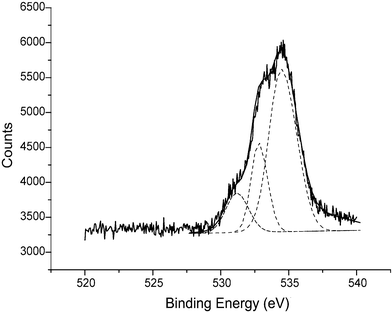 | ||
| Fig. 4 The cumulative XPS spectrum (ten scans) corresponding to the O1s region of FBK-MWCNTs with the data-fitted spectrum (overlaid). Note that this spectrum has not been corrected to the C1s peak position (see text). | ||
The FBKcarbon N1s signals were clearly resolved using a slit width of 1.9 mm, whereupon two peaks were observed (400.2 eV, 68.2% and 405.9 eV, 31.0%) shown in Fig. 5, which were assigned to the azo and nitro moieties of Fast Black K respectively.22,24 The ratio of nitrogen atoms corresponding to the azo and nitro moieties was found to be 2 : 1 ± 0.1, a figure which is consistent with the stoichiometry of the molecule. The FBK-MWCNT sample also yielded two distinct N1s signals (400.4 eV, 63.6%; 405.3 eV, 28.9%) and again, these are assigned as azo and nitro signals respectively in the ratio of 2 : 1 ± 0.2. As mentioned above no nitrogen signals were observed in the unmodified carbon sample. Thus using XPS we have provided conclusive evidence that FBK derivatises the surface of graphite powder and MWCNTs using XPS, in good agreement with previous voltammetric studies.12 In the next section we show that we can effect voltammetrically controlled chemical release from the FBKcarbon and FBK-MWCNTs.
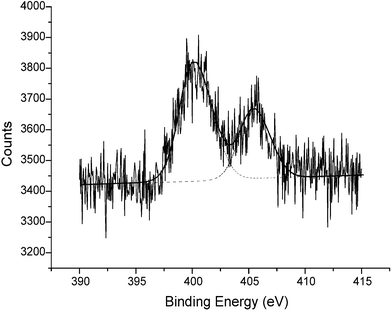 | ||
| Fig. 5 The cumulative XPS spectrum (ten scans) corresponding to the N1s region of FBKcarbon (slit width 1.9 mm) with the data-fitted spectrum (overlaid). Note that this spectrum has not been corrected to the C1s peak position (see text). | ||
Fig. 6 shows that a single N1s signal was observed for the reduced FBK sample (400.2 eV), consistent with the aromatic amine group19 on the surface moiety C, Scheme 2, which results from the fragmentation of the parent FBK molecule.
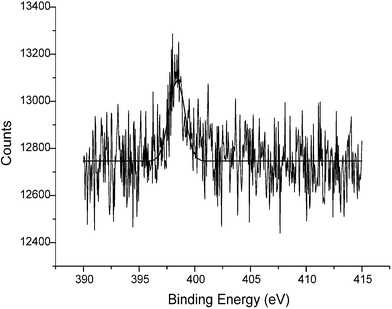 | ||
| Fig. 6 The cumulative XPS spectrum (ten scans) corresponding to the N1s region of FBKcarbon after electrochemical reduction (slit width 1.9 mm) with the data-fitted spectrum (overlaid). Note that this spectrum has not been corrected to the C1s peak position (see text). | ||
These results confirm our proposed mechanism that at sufficiently reducing potentials, the hydrazo-linkage in the FBK molecules, which are themselves covalently bound to the surface of graphite and MWCNTs, is cleaved. This cleavage results in the chemical release of 1,4-phenylenediamine into the solution phase with the 2,5-dimethoxyaniline fragment remaining on the carbon surface.
4. Conclusion
In this report we have characterised graphite powder and MWCNT as being covalently derivatised with Fast Black K using XPS. Furthermore we have demonstrated the proof of concept that these novel materials can be used as voltammetrically controlled chemical release reagents. We envisage that this approach could be used to develop chemical release systems, e.g. for controlled drug delivery, by coupling the target molecules to suitable derivatives of FBK. This is similar to certain prodrug approaches where the drug molecules are attached to polymer carriers using azoxy linkages which then selectively degraded to initiate chemical release.4 However, our approach has the advantage that the derivatised graphite powder and MWCNTs are excellent electrical conductors. As such these novel materials can easily be incorporated onto a microchip device, for example as an array of microelectrodes. Such devices allow the programmable controlled release of the desired chemical via controlling the applied potential, with the advantages discussed above. Furthermore such devices could be manufactured rapidly and relatively inexpensively in bulk quantities through techniques such as screen-printing or pad-printing. Finally the small size of MWCNTs could be utilised to miniaturise chemical-release devices down to the nano-scale, and as such we envisage that materials such as FBK derivatised MWCNTs will find important nanotechnological applications.Acknowledgements
GGW would like to thank the BBSRC for financial support. The authors gratefully acknowledge Schlumberger Cambridge Research for additional financial support. The authors thank Dr. Graham Beamson and Dr. Daniel S-L Law at the NCESS Laboratory, Daresbury and Professor R. G. Egdell at the University of Oxford for their supervision and guidance in carrying out the XPS experiments and for their invaluable help with interpreting the spectra.References
- J. T. Santini, A. C. Richards, R. Scheidt, M. J. Cima and R. Langer, Angew. Chem., Int. Ed., 2000, 39, 2396 CrossRef.
- D. C. Coughlan, F. P. Quilty and O. I. Corrigan, J. Controlled Release, 2004, 98, 97 CrossRef CAS.
- S. Freiberg and X. X. Zhu, Int. J. Pharm., 2004, 282, 1 CrossRef CAS.
- A. Joseph, M. D'Souza and E. M. Topp, J. Pharm. Sci., 2004, 93, 1962 CrossRef.
- A. J. Melville, L. M. Rodriguez-Lorenzo, J. S. Forsythe and K. A. Gross, Key Eng. Mater., 2004, 254–256(Bioceramics), 529 CrossRef CAS.
- Z. R. Domingues, M. E. Cortes, T. A. Gomes, H. F. Diniz, C. S. Freitas, J. B. Gomes, A. M. C. Faria and R. D. Sinisterra, Biomaterials, 2003, 25, 327 CrossRef.
- L. Mortain, I. Dez and P.-J. Madec, Compt. Rend. Chim., 2004, 7, 635 Search PubMed.
- M. C. Peterman, J. Noolandi, M. S. Blumenkranz and H. A. Fishman, Proc. Natl. Acad. Sci. USA, 2004, 101, 9951 CrossRef CAS.
- E. Katz and I. Willner, ChemPhysChem, 2004, 5, 1084 CrossRef CAS.
- P. W. Barone, S. Baik, D. A. Heller and M. S. Strano, Nat. Mater., 2005, 4, 86 CrossRef CAS.
- H. C. Leventis, I. Streeter, G. G. Wildgoose, N. S. Lawrence, L. Jiang, T. G. J. Jones and R. G. Compton, Talanta, 2004, 63, 1039 CAS.
- H. C. Leventis, G. G. Wildgoose, I. G. Davies, L. Jiang, T. G. J. Jones and R. G. Compton, ChemPhysChem, 2005 DOI:10.1002/cphc.200400536 , in press.
- C. D. Potter, J. Coated Fabrics, 1989, 18, 259 Search PubMed.
- S. M. Bernard and D. R. Walt, Science, 1991, 251, 927 CAS.
- R. W. Giese, Trends Anal. Chem., 1983, 2, 166 CrossRef CAS.
- Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs and J. T. Grant, IM Publications and Surface Spectra Ltd, London, UK, 2003 Search PubMed.
- G. Beamson, D. Briggs, S. F. Davies, I. W. Fletcher, D. T. Clark, J. Howard, U. Gelius, B. Wannberg and P. Balzer, Surf. Interface Anal., 1990, 15, 541 CrossRef CAS.
- Scienta ESCA300 Users' Manual, Scienta, Uppsala Search PubMed.
- G. Beamson and D. Briggs, in High Resolution X-Ray Photoelectron Spectroscopy of organic polymers, John Wiley and Sons Ltd, London, 1992 Search PubMed.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard and M. Winter, J. Power Sources, 2001, 97–98, 171 CrossRef CAS.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard, P. Golob and M. Winter, Appl. Surf. Sci., 2000, 167, 99 CrossRef CAS.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713 CrossRef CAS.
- L. Qu, R. B. Martin, W. Huang, K. Fu, D. Zweifel, Y. Lin, Y.-P. Sun, C. E. Bunker, B. A. Haruff, J. R. Gord and L. F. Allard, J. Chem. Phys., 2002, 117, 8089 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sorol and K. D. Bomben, Handbook of X-ray Photoelectron Spectra, Perkin-Elmer, Minnesota, 1979 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |