Layered solids as a “molecular container” for pharmaceutical agents: L-tyrosine-intercalated layered double hydroxides
Min
Wei
a,
Qi
Yuan
a,
David G.
Evans
a,
Zhiqiang
Wang
b and
Xue
Duan
*a
aKey Laboratory of Science and Technology of Controllable Chemical Reactions, Ministry of Education, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: duanx@mail.buct.edu.cn; Tel: +86 10 64425395; Fax: +86 10 64425385
bDepartment of Chemistry, Tsinghua University, Beijing 100084, P. R. China
First published on 25th January 2005
Abstract
L-Tyrosine has been intercalated into NiAl, MgAl and ZnAl layered double hydroxides by coprecipitation. The structure and composition of the intercalated materials have been characterized by X-ray diffraction (XRD) and elemental analysis. It is found that intercalation can inhibit racemization of L-tyrosine under the influence of sunlight, high temperature or ultraviolet light. Therefore, this layered material may have potential application as a “molecular container” for storing or transporting unstable chiral biomolecules or pharmaceutical agents. In addition, the thermal decomposition of the L-tyrosine intercalated NiAl-LDH has been investigated in detail by means of in situ HT-XRD, in situ FT-IR and TG-DTA. Loss of interlayer water occurs between room temperature and 150 °C while decomposition of intercalated L-tyrosine and dehydroxylation of the host layers begin at about 250 °C and 300 °C respectively.
Introduction
Layered double hydroxides (LDHs), also known as hydrotalcite-like materials and anionic clays, are available as both naturally occurring minerals and synthetic materials.1 LDHs may be described by the general formula [M1−xIIMxIII(OH)2]x+(An−)x/n·mH2O, where MII and MIII are di- and trivalent metals respectively and An− is an anion.1 Their structure is based on brucite-like layers, where a divalent metal cation is located in the center of oxygen octahedra and two dimensional infinite layers are formed by edge-sharing of octahedra. Partial substitution of isomorphous trivalent cations for divalent ones results in a positive charge on the layers. Organic or inorganic anions are intercalated between the layers in order to maintain charge balance, and water of crystallization is also found in the interlayer galleries. The structure as outlined above was first described by Allmann2 and Taylor.3 Various anionic species have been intercalated into the gallery region of LDHs mainly by coprecipitation or ion-exchange, e.g., inorganic acids,4 organic acids,5 anionic polymers,6 cyclodextrin,7 and reduced C60.8These layered solids based upon the alternation of inorganic and organic layers have received considerable attention because of their many practical applications, including as catalysts,9 functional materials,10 and nanocomposite materials.11 The attractive feature of such materials is that they serve as a template for the formation of supramolecular structures.5 The host layers can impose restricted geometry on the interlayer guests leading to enhanced control of stereochemistry, rates of reaction, and product distributions. Many species can be assembled by reaction of guest species in the LDH matrices.1,10 Therefore the study of advanced materials based on LDHs is a rapidly growing field and has application in areas such as separation science,12,13in situ polymerization,6,14 photochemistry,15 and electrochemistry.5
Recently, much attention has been paid to the intercalation of biomolecules or pharmaceutical agents into LDHs.16–19 In general, most of these are chiral,20 and their optical activity is readily lost by racemization under relatively mild conditions.20–23 Importantly, the chirality usually influences the properties and efficacy of drugs. As a rule, one enantiomer has the desired therapeutic effect whilst the other has not or is even deleterious.20 How to conserve such substances effectively, therefore, has attracted much attention.21–23 To the best of our knowledge, there has been no report of the effect of intercalation in LDHs on the rate of racemization of chiral species. L-Tyrosine (4-hydroxyphenylalanine, represented as L-Tyr) is a non-essential amino acid that is normally synthesized in the body from phenylalanine. Deficiencies in L-Tyr have been associated with depression and L-Tyr supplements in the diet have shown a beneficial effect.24L-Tyr is also used in the treatment of dementia,25 vitiligo26 and in easing the adverse effects of stress.27 In addition to racemization, L-Tyr also undergoes oxidation to a quinone28 and intercalation of the amino acid in an LDH host may reduce the rate of both of these processes. The structure of L-Tyr is very similar to that of pharmaceuticals used in the treatment of Parkinson's disease29,30 such as L-dopa (L-3,4-dihydroxyphenylalanine) and methyldopa (L-3-(3,4-dihydroxyphenyl)-2-methylalanine) and the amino acid can also serve as model for the more expensive drugs in intercalation studies. In addition to reducing the rate of racemization or decomposition of pharmaceutical molecules, intercalation in the layered host has another potential benefit since several recent papers have demonstrated that LDHs are effective as a matrix for controlled release drug delivery systems.31 We were therefore interested to study the intercalation of L-Tyr in NiAl-, MgAl- and ZnAl-LDHs and in this paper we report the results of our study of the effect of this process on the thermal and photo-stability of the amino acid with respect to racemization. There has been only one previous report of the intercalation of L-Tyr in an LDH17 and the stability of the intercalated L-Tyr was not compared with that of the pristine amino acid.
Experimental
Synthesis
Measurement
Characterization
The in situ powder X-ray diffraction (in situ XRD) measurements were performed on a Rigaku D/MAX2500VB2+/PC diffractometer in the temperature range 25–700 °C under vacuum conditions, using Cu Kα radiation (λ = 0.154 nm) at 40 kV, 200 mA, a scanning rate of 5° min−1, and a 2θ angle ranging from 2° to 70°. The rate of temperature increase was 10 °C min−1 with a holding time of 5 min before each measurement.The in situ Fourier transform infrared (in situ FT-IR) spectra were recorded using a Nicolet 605XB FT-IR spectrometer in the range 4000 to 400 cm−1 with 4 cm−1 resolution under flowing N2 (65 mL min−1) with a heating rate of 5 °C min−1 in the range 25–450 °C. The standard KBr disk method (1 mg of sample in 100 mg of KBr) was used.
Thermogravimetric analysis and differential thermal analysis (TG-DTA) were measured on a PCT-1A thermal analysis system with a heating rate of 5 °C min−1.
Microanalysis of metals was performed by inductively coupled plasma (ICP) emission spectroscopy on a Shimadzu ICPS-7500 instrument using solutions prepared by dissolving the samples in dilute HNO3. Carbon, hydrogen and nitrogen analyses were carried out using an Elementarvario elemental analysis instrument.
The UV–visible spectra were recorded on a Shimadzu UV-2501PC spectrometer after dissolution of the L-Tyr in HCl solution.
13C Nuclear magnetic resonance (NMR) spectra were run on a Bruker AV600 spectrometer operating at a frequency of 150.9194 MHz for 13C with a 3 s pulse delay. The samples of L-Tyr were dissolved in 20% DCl–D2O.
Optical rotation measurements were carried out on a WZZ-1S automatic polarimeter at 589.3 nm (Na D-line).
Results and discussion
Characterization of L-Tyr–LDHs
 | ||
| Fig. 2 In situ variable temperature FT-IR spectra for L-Tyr–NiAl-LDH. The four absorption bands of L-Tyr between 1600 and 1250 cm−1 show a shift in position at low temperatures associated with changes in host–guest interactions with the layers. Decomposition begins at about 250 °C and there is a major structural change at 400 °C. | ||
The O–H stretching vibration becomes weaker with a slight shift to high frequency (3428 cm−1) on increasing the temperature, and disappears at 450 °C, indicative of the loss of water molecules as well as the dehydroxylation of the LDH layers. At 25 °C, the asymmetric and symmetric stretching vibrations of the carboxylate groups are observed at 1582 and 1395 cm−1 respectively and on raising the temperature to 150 °C the difference, Δν, between their position increases from 187 to 201 cm−1. According to Nakamoto,34 the value of Δν gives information about the symmetry of the interaction between the carboxylate groups and the hydroxylated layers. Therefore, it may indicate there is some change in the interaction between the guest and host associated with the loss of hydrogen bonding space as a result of liberation of interlayer water. No significant changes in the positions of the four absorption bands of L-Tyr between 1600 cm−1 and 1250 cm−1 are observed between 150 °C and 200 °C, indicating that the arrangement of the intercalated guest is stable in this temperature range. However, obvious changes are observed at about 250 °C. The four characteristic bands of L-Tyr become weaker and the value of Δν shows a further increase. Eventually the band at 1395 cm−1 disappears completely at 400 °C. This implies that the decomposition of intercalated L-Tyr begins at about 250 °C, and there is a major structural change at 400 °C. These bands become even weaker at 450 °C, corresponding to further decomposition of L-Tyr.
 | ||
| Fig. 3 In situ variable temperature XRD patterns for L-Tyr–NiAl-LDH. The decrease in (00l) spacings below 150 °C is associated with the deintercalation of interlayer water molecules. Collapse of the layer structure at 400 °C is associated with the appearance of reflections from a cubic NiO phase (*). | ||
 | ||
| Fig. 4 The relationship between d003 basal spacing of L-Tyr–NiAl-LDH and temperature. The initial decrease in basal (003) spacing below 150 °C is associated with loss of interlayer water molecules and the second decrease is due to dehydroxylation of the host layers. | ||
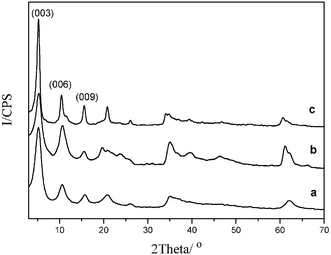
It can be observed in Fig. 3 that the (003), (006) and (009) diffraction peaks of L-Tyr–NiAl-LDH move to higher angle 2θ with increasing temperature. The value of d003 decreases in two steps as shown in Fig. 4. The first is from 1.71 nm at 20 °C to 1.52 nm at 150 °C, which is related to the destruction of the hydrogen bonding space as a result of deintercalation of interlayer water molecules. This is in accordance with the in situ FT-IR data, and also provides supporting evidence for the structural model of L-Tyr–LDH discussed above. The observed contraction on heating from 20 °C to 150 °C (0.19 nm) is similar to the calculated dimension of the hydrogen bonding space (0.22 nm). A further decrease in the value of d003 is observed between 200 °C (1.52 nm) and 350 °C (1.38 nm), which can be attributed not only to the decomposition of the intercalated L-Tyr anions, but also to the dehydroxylation of the host layers beginning at 300 °C because the intensities of the reflections decline significantly from 300 to 350 °C.
The intensities of all reflections associated with the LDH phase decrease gradually with further increase in temperature, and the layered structure collapses completely at a temperature of 400 °C with the first appearance of cubic NiO reflections at about 43.8° and 51.0° 2θ. The diffraction peaks of NiO become stronger as the temperature increases from 400 to 700 °C. It has been demonstrated by X-ray photoelectron spectra (XPS)35 and 27Al MAS NMR36,37 for related LDH systems that in this temperature range the chemical environment of the aluminium changes from the exclusively octahedral coordination that is present in the parent LDH to partly tetrahedral.
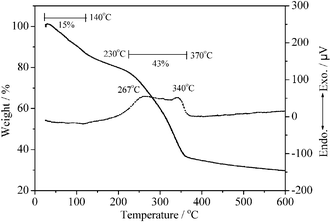 | ||
| Fig. 5 TG and DTA curves for L-Tyr–NiAl-LDH. The first weight-loss step involves loss of surface and interlayer water molecules, whilst the second involves dehydroxylation of the layers and decomposition of the organic guest. | ||
Investigation of extent of racemization of L-Tyr
In order to study the stabilizing effect of the LDHs when used as a “molecular container” for the amino acid, the influence of three factors, i.e., sunlight, heat, and UV light were investigated. The relationship between the exposure time to sunlight and the optical activity of L-Tyr (Fig. 6a) and L-Tyr intercalated NiAl-, MgAl- and ZnAl-LDHs (Fig. 6b, 6c and 6d, respectively) are dramatically different. The specific optical rotation of pristine L-Tyr is 10.38° under the determination conditions. During the first eight hours exposure to sunlight, the specific optical rotation of pristine L-Tyr decreases sharply by 38%, and then remains constant up to 48 h exposure. However, there is no significant decrease in the specific optical rotation of the L-Tyr intercalated NiAl, MgAl- and ZnAl-LDHs over the same time. Furthermore, when the optical rotations of the three L-Tyr–LDHs were measured after exposure for one month they were only slightly less than the initial value.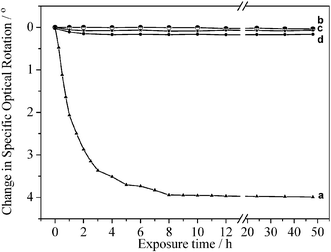 | ||
| Fig. 6 The relationship between the optical activity and the exposure time to sunlight for (a) L-Tyr, (b) L-Tyr–NiAl-LDH, (c) L-Tyr–MgAl-LDH and (d) L-Tyr–ZnAl-LDH. Intercalation leads to a marked increase in photostability. | ||
The influence of temperature on the optical activity of L-Tyr and L-Tyr–LDHs is illustrated in Fig. 7. The value of specific optical rotation of L-Tyr decreases rapidly with increasing temperature, and is reduced by about half on heating at 180 °C (Fig. 7a). In contrast, the specific optical rotations of the L-Tyr intercalated NiAl-, MgAl- and ZnAl-LDHs only decrease by 4.2%, 2.2% and 5.5% under the same conditions (Fig. 7b, 7c and 7d, respectively).
 | ||
| Fig. 7 The relationship between the optical activity and the heating temperature for (a) L-Tyr, (b) L-Tyr–NiAl-LDH, (c) L-Tyr–MgAl-LDH and (d) L-Tyr–ZnAl-LDH. Intercalation leads to a marked increase in thermal stability. | ||
The effects of UV radiation on the optical activity of L-Tyr and intercalated L-Tyr have also been studied (see Fig. 8). A similar result is obtained. The specific optical rotation of pristine L-Tyr decreases rapidly by 44% over 24 h exposure to UV irradiation (Fig. 8a). However, the three intercalated L-Tyr materials only show very slight decrease (less than 3%) in its optical activity (Fig. 8b, 8c and 8d).
 | ||
| Fig. 8 The relationship between the optical activity and the exposure time to UV radiation for (a) L-Tyr, (b) L-Tyr–NiAl-LDH, (c) L-Tyr–MgAl-LDH and (d) L-Tyr–ZnAl-LDH. Intercalation leads to a marked increase in photostability. | ||
In order to confirm the reason for the decrease of optical activity of pristine L-Tyr, FT-IR, UV-Vis and 13C NMR spectra were recorded for samples exposed to sunlight for 72 h, heated at 180 °C for 1 h, and irradiated by UV light for 24 h (Fig. 9, 10 and 11, respectively). In each case, the spectra of the samples after different treatment are identical to that of fresh L-Tyr, confirming that loss of optical activity is due to racemization rather than oxidation or other decomposition processes.
 | ||
| Fig. 9 FT-IR spectra of (a) L-Tyr, (b) L-Tyr exposed in light for 72 h (c) L-Tyr heated at 180 °C for 1 h and (d) L-Tyr irradiated by UV light for 24 h. | ||
 | ||
| Fig. 10 UV–Visible spectra of (a) L-Tyr, (b) L-Tyr exposed in light for 72 h (c) L-Tyr heated at 180 °C for 1 h and (d) L-Tyr irradiated by UV light for 24 h. | ||
 | ||
| Fig. 11 13C MAS spectra of (a) L-Tyr, (b) L-Tyr exposed in light for 72 h (c) L-Tyr heated at 180 °C for 1 h and (d) L-Tyr irradiated by UV light for 24 h. | ||
The results above indicate that the specific optical rotation of pristine L-Tyr decreases significantly on exposure to sunlight, heat or UV light because of partial racemization. However, intercalation of L-Tyr into NiAl-, MgAl- and ZnAl-LDHs can prevent its racemization under these conditions, i.e., the photochemical stability against racemization of L-Tyr is extremely improved by hybridization with LDHs. The observed increase in photochemical stability may be due to racemization of the chiral guest being restricted significantly in the galleries of the host layers as a result of the host–guest and guest–guest interactions. Moreover, the obstruction of light penetration by the anion clay in the UV region also accounts for this improvement under exposure to sunlight or UV radiation. Therefore, such layered materials may have potential applications as the basis of a novel storage or delivery system for chiral biomolecules or pharmaceutical molecules.
Conclusions
L-Tyr intercalated NiAl, MgAl and ZnAl layered double hydroxides have been obtained by coprecipitation and were characterized by powder X-ray diffraction, Fourier transform infrared spectroscopy, inductively coupled plasma emission spectroscopy and elemental analysis.In situ FT-IR, in situ HT-XRD and TG-DTA measurements allow a detailed understanding of the thermal decomposition process for L-Tyr–NiAl-LDH. From room temperature to 150 °C, loss of the interlayer water molecular results in the destruction of the hydrogen bonding space as well as a decrease in the value of the basal spacing d003 from 1.71 nm to 1.52 nm. In situ FT-IR shows an increase in the difference between asymmetric and symmetric stretching vibrational frequencies for the carboxylate group, indicating some change in the interaction between the guest and host occurs as a result of loss of interlayer water. Decomposition of intercalated L-Tyr occurs at about 250 °C, as has been confirmed by in situ FT-IR, in situ HT-XRD and TG-DTA analysis. A further decrease in the d003 value of L-Tyr–NiAl-LDH from 1.52 nm at 200 °C to 1.38 nm at 350 °C can be attributed not only to the decomposition of the intercalated L-Tyr anions, but also to the dehydroxylation of the host layers beginning at 300 °C because the intensity of reflections declined significantly from 300 to 350 °C. Finally, the layer structure collapses completely at 400 °C.
The specific optical rotation of L-Tyr decreases significantly on exposure to sunlight, heat, or UV light because of partial racemization. However, intercalation of L-Tyr in LDHs can prevent racemization under these conditions. The observed increase in photochemical stability may be due to racemization of the chiral guest being restricted significantly in the galleries of the host layers as a result of the host–guest and guest–guest interactions. Moreover, the obstruction of light penetration by the anion clay in the UV region also accounts for this improvement. Therefore, such layered materials may have potential applications as the basis of a novel storage or delivery system for chiral biomolecules or pharmaceutical molecules.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant No. 90206004) and the Beijing Nova Program (No. 2004A13).References
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS
.
- R. Allmann, Acta Crystallogr., Sect. B, 1968, 24, 972 CrossRef CAS
- H. F. W. Taylor, Mineral. Mag., 1973, 39, 377 Search PubMed
- F. Malherbe and J. P. Besse, J. Solid State Chem., 2000, 155, 332 CrossRef CAS
- S. P. Newman and W. Jones, New J. Chem., 1998, 22, 105 RSC
- F. Leroux and J. P. Besse, Chem. Mater., 2001, 13, 3507 CrossRef CAS
- H. T. Zhao and G. F. Vance, Clays Clay Miner., 1998, 46, 712 CAS
- W. P. Ding, G. Gu, W. Zhong, W. C. Zang and Y. W. Du, Chem. Phys. Lett., 1996, 262, 259 CrossRef CAS
- B. Sels, D. De Vos, M. Buntinx, F. Pierard, A. Kirsch-De Mesmaeker and P. Jacobs, Nature, 1999, 400, 855 CrossRef CAS
- M. Ogawa and K. Kuroda, Chem. Rev., 1995, 95, 399 CrossRef CAS
-
S. P. Newman and W. Jones, Supramolecular Organization and Materials Design, Cambridge University Press, Cambridge, UK, 2001, p. 295 Search PubMed
- A. M. Fogg, V. M. Green, H. G. Harvey and D. O'Hare, Adv. Mater., 1999, 11, 1466 CrossRef CAS
- F. Millange, R. I. Walton, L. X. Lei and D. O'Hare, Chem. Mater., 2000, 12, 1990 CrossRef CAS
- M. Moujahid El, M. Dubois, J. P. Besse and F. Leroux, Chem. Mater., 2002, 14, 3799 CrossRef
- H. Tagaya, S. Sato, T. Kuwahara, J. Kadokawa, K. Masa and K. Chiba, J. Mater. Chem., 2002, 4, 1907 Search PubMed
- N. T. Whilton, P. J. Vickers and S. Mann, J. Mater. Chem., 1997, 7, 1623 RSC
- Á. Fudala, I. Pálinkó and I. Kiricsi, Inorg. Chem., 1999, 38, 4653 CrossRef CAS
- S. Aisawa, S. Takahashi, W. Ogasawara, Y. Umetsu and E. Narita, J. Solid State Chem., 2001, 162, 52 CrossRef CAS
- J. H. Choy, S. Y. Kwak, J. S. Park and Y. J. Jeong, J. Mater. Chem., 2001, 11, 1671 RSC
- R. N. Patel, R. L. Hanson, A. Banerjee and L. J. Szarka, J. Am. Oil Chem. Soc., 1997, 74, 1345 CrossRef CAS
- R. Griffith, J. Soria and J. G. Wood, Exp. Neurol., 2000, 161, 297 CrossRef CAS
- B. A. Cohen and C. F. Chyba, Icarus, 2000, 145, 272 CrossRef CAS
- M. Shinitzky, F. Nudelman, Y. Barda, R. Haimovitz, E. Chen and D. W. Deamer, Origins Life Evol. Biosphere, 2002, 32, 285 CrossRef CAS
- A. J. Gelenberg, J. D. Wojcik, W. E. Falk, R. J. Baldessarini, S. H. Zeisel, D. Schoenfeld and G. S. Mok, J. Affective Disord., 1990, 19, 125 Search PubMed
- J. H. Growdon, E. Melamed, M. Logue, F. Hefti and R. J. Wurtman, Life Sci., 1982, 30, 827 CrossRef CAS
- D. P. Chakraborty, S. Roy and A. K. Chakraborty, Pigm. Cell Res., 1996, 9, 107 Search PubMed
- G. S. Kelly, Altern. Med. Rev., 1999, 4940, 249 Search PubMed
- M. Luthra, D. Ranganathan, S. Ranganathan and D. Balasubramanian, J. Biol. Chem., 1994, 269, 22678 CAS
- J. E. Ahlskog, R. J. Uitti, P. A. Low, G. M. Tyce, J. F. O'Brien and K. K. Nickander, Neurology, 1996, 46, 796 CAS
- P. Lautala, B. T. Ethell, J. Taskinen and B. Burchell, Drug Metab. Dispos., 2000, 28, 1385 Search PubMed
- A. I. Khan, L. X. Lei, A. J. Norquist and D. O'Hare, Chem. Commun., 2001, 2342 RSC
- S. P. Newman, T. D. Cristina, V. Coveney and W. Jones, Langmuir, 2002, 18, 2933 CrossRef CAS
-
Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw–Hill, New York, 15th edn., 1998 Search PubMed
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 5th edn., 1997 Search PubMed
- T. L. Barr, S. Seal, K. Wozniak and J. Klinowski, J. Chem. Soc., Faraday Trans., 1997, 93, 181 RSC
- M. J. Hudson, S. Carlino and D. C. Apperly, J. Mater. Chem., 1995, 5, 323 RSC
- S. Velu, V. Ramkumar, A. Narayanan and C. S. Swamy, J. Mater. Sci., 1997, 32, 957 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2005 |