Altered cellular response to adsorbed matrix protein by chemoselective ligation of small molecules
Paul A. De Banka, Barrie Kellama, David A. Kendallb and Kevin M. Shakesheff*a
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: kevin.shakesheff@nottingham.ac.uk
bSchool of Biomedical Sciences, University of Nottingham, Queen's Medical Centre, Nottingham, UK NG7 2UH
First published on 20th April 2005
Abstract
In the field of tissue engineering, there is a constant drive to develop materials that enable control over cell adhesion, growth and differentiation for optimal tissue development. In many cases, these materials incorporate or are coated with extracellular matrix proteins to increase cell adhesion and spreading. Here we show that chemoselective modification of an adsorbed matrix protein can be used to selectively alter the response of cultured cells. Using the glycoprotein laminin-1, it was shown that oxidation of terminal sialic acid moieties resulted in the generation of aldehyde groups which could be employed to chemoselectively ligate hydrazide-bearing biomolecules. The hydrazide derivative of the tripeptide Leu–Arg–Glu, a “stop” signal for motoneuron outgrowth, was ligated to periodate-treated laminin and shown to inhibit the mean neurite length of primary spinal cord motoneurons by 39% in comparison to controls. These results demonstrate that adsorbed glycoproteins can be readily modified in order to alter the cell response and could enable matrices to be tailored to different cell types.
Introduction
The aim of tissue engineering is to exchange a patient's damaged or diseased tissue or organ with an indistinguishable, laboratory-fabricated replacement, ideally using cells from the same individual. Great strides have been made in the pursuit of this goal, although there are a number of challenges to overcome.1–3 To convert a population of single cells into a functional tissue, it is important that we can control all aspects of the tissue's development. Since current tissue engineering strategies usually involve seeding cells on to a polymer scaffold, it is therefore imperative that we can control cell adhesion, growth and differentiation by manipulating the properties of the cells and the biomaterial. One aspect of tissue engineering that has been largely overlooked, however, is the problem of innervating engineered tissue and organs, thus enabling correct nervous control. To do so, a neuron must be directed to form a synapse at a specific site within the tissue. We hypothesize that this can be achieved by chemically engineering the desired synaptic region with a “stop” signal that arrests neuron outgrowth and enables synapse development. Our model system of interest for investigating this possibility is the neuromuscular junction (NMJ); the synapse between skeletal muscle and a motoneuron.In vivo, if the NMJ is destroyed by injury, there is a remarkable propensity for the muscle to be reinnervated at the original synaptic site by a regenerating motor axon, even though the original synapses represent less than 0.1% of the muscle surface.4 Detailed studies in which the muscle was destroyed, leaving the surrounding sheath of basal lamina (BL), intact demonstrated that this phenomenon still occurred, with motor axons contacting the BL almost exclusively at the original synaptic site.5 Hence, BL components direct reinnervation independent of the presence of an underlying myofiber. The BL is largely composed of glycoproteins such as laminin, collagen and fibronectin and is involved in the formation, maintenance and repair of muscle in addition to providing structural support and guiding axonal extension and synaptogenesis.6 The distribution of BL components varies between synaptic and extrasynaptic sites,7 and this is particularly true of laminins.8 Fifteen years ago, a new member of the laminin family was discovered. This molecule, recognized by motoneurons, was found to be concentrated in the synaptic cleft9,10 and named s-laminin (later renamed laminin-11).11,12 In adult muscle, extrasynaptic BL is rich in laminin-2 on which motor axons grow freely. Of the laminins concentrated in the synaptic BL (4, 9 and 11), laminin-11 was shown to arrest motor axon growth upon contact.13
To investigate the molecular determinant responsible for motoneuron adhesion to laminin-11, Hunter et al. cultured motoneurons on recombinant fragments of the glycoprotein and demonstrated that the adhesive site is present in the C-terminal 10% of the molecule. Using chick ciliary ganglion (CG) neurons, it was shown that this sequence was motoneuron-specific with poor adhesion of other neuronal cell types, while all cell types adhered well to laminin. What is more, using successively smaller synthetic peptides spanning this fragment, it was revealed that the amino acid sequence of the adhesive site is leucine–arginine–glutamate (LRE).14 Further studies revealed that recombinant LRE-containing peptides promoted adhesion while inhibiting neurite outgrowth from chick CG neurons and NSC-34 cells, a murine motoneuron-like cell line. When mixtures of an LRE fusion peptide and laminin were used as a growth substrate, neurite outgrowth from motoneurons was reduced in comparison to laminin alone.15 Furthermore, when CG neurons on laminin encountered patterned areas of s-laminin, neurite outgrowth from a majority of cells stopped at the border between the two proteins.16 These data suggest that LRE is an adhesive “stop” signal for motoneuron outgrowth and may play an important role in innervation of NMJs.
We have previously demonstrated that living cell surfaces can be chemically modified with bioactive molecules of our choosing via the selective oxidation of sialic acid residues by sodium periodate.17 This reaction generates an aldehyde moiety on the cell surface and this functional group, not normally present, can be used to specifically ligate certain chemical species, such as hydrazides, to oxidized cells via a covalent hydrazone bond (Scheme 1).18 As mentioned above, the extrasynaptic surface of a muscle fiber is rich in laminins which act as an effective substrate for motoneuron outgrowth and, as glycoproteins, possess sialic acid residues that can potentially be used for chemoselective modification via periodate oxidation.
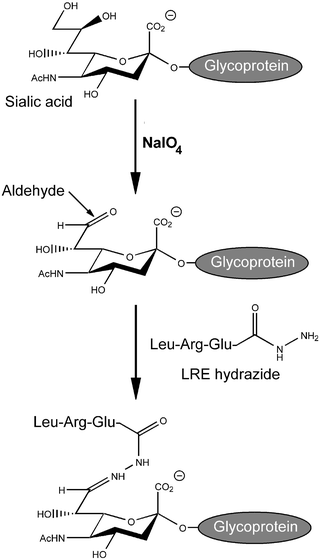 | ||
| Scheme 1 Reaction Scheme for the modification of a glycoprotein with the LRE motoneuron “stop” signal. Oxidation of glycoprotein sialic acid residues with sodium periodate generates aldehyde groups which can be used to selectively ligate LRE hydrazide to the protein by the formation of a hydrazone bond. | ||
In this paper we describe our investigation into the feasibility of using this technique. Using culture plates coated with laminin from the murine Engelbreth–Holm–Swarm (EHS) tumour as a model basal lamina,19 we demonstrate that we can successfully engineer this ECM protein directly using a chemoselective ligation strategy. What is more, modification of this laminin with a simple LRE “stop” signal elicits the desired cellular response in cultured primary motoneurons. This selective modification of ECM proteins in this manner has the potential to be used for a number of other tissue engineering applications, such as increasing cell adhesion to ECM-coated scaffolds by ligation of sequences such as RGD.
Experimental
Materials
Amino acids and peptide coupling reagents were obtained from Novabiochem. All other reagents for chemical synthesis were from Aldrich, while solvents were from Fisher Scientific Ltd. All reagents for cell culture and analysis were from Sigma-Aldrich with the exception of PBS (Oxoid Ltd.), antibiotic/antimycotic, DMEM, trypsin/EDTA and HBSS (Invitrogen) and goat anti-choline acetyltransferase (Chemicon, AB144).Synthesis of H–Glu(OtBu)–NH–NH–Boc
Fmoc–Glu(OtBu)–OH (1.00 g, 2.35 mmol), TBTU (0.834 g, 2.60 mmol), HOBt (0.200 g, 1.31 mmol) and DIPEA (0.45 ml, 2.58 mmol) were dissolved in dichloromethane (60 ml). The solution was stirred at room temperature for 15 min and tert-butylcarbazate (2.841 g, 21.50 mmol) was then added. The reaction was stirred at room temperature for 22 h and concentrated in vacuo to yield a pale yellow oil, which was dissolved in EtOAc (30 ml). The solution was filtered to remove the resulting white crystalline precipitate and washed with saturated KHSO4 (3 × 30 ml), saturated NaHCO3 (3 × 30 ml), distilled water (40 ml) and saturated NaCl (30 ml). The solution was then dried over MgSO4, filtered and concentrated in vacuo to yield a white foam which was partially purified by dry flash column chromatography using DCM : MeOH (95 : 5) as eluent to yield 1.156 g of a white foam. A portion of the Fmoc-protected product (900 mg) was dissolved in dichloromethane (16 ml) and piperidine (4 ml) was added. After 45 min stirring at room temperature, the solvent and piperidine were removed under high vacuum to yield a white foam which was purified by dry flash column chromatography using DCM : MeOH (95 : 5 to 80 : 20) as eluent to yield a white solid (0.477 g, 76.9%); δH (250 MHz, CDCl3) 1.36 (s, 9H), 1.39 (s, 9H), 1.70–2.06 (br m, 2H,), 2.32 (t, 2H, J = 7.47 Hz), 3.44 (t, 1H, J = 6.49 Hz); δC (62.9 MHz, CDCl3): 28.4, 28.5, 30.5, 32.0, 53.81, 80.9, 81.7, 155.9, 173.18, 174.4. NMR spectra were obtained using a Brüker ARX250 nuclear magnetic resonance spectrometer and chemical shifts are quoted as parts per million on the δ scale.Synthesis of H–Leu–Arg–Glu–NH–NH2 (LRE hydrazide)
H–Leu–Arg–Glu–NH–NH2 was synthesized in the solution phase using standard peptide coupling techniques. H–Glu(OtBu)–NH–NH–Boc (361 mg, 1.14 mmol) was used as the protected hydrazide-containing starting material with Fmoc–Arg(Pbf)–OH and Boc–Leu–OH residues sequentially coupled using DCC/HOBt. The Fmoc protecting group was cleaved from the Fmoc–Arg(Pbf)–Glu(OtBu)–NH–NH–Boc intermediate by treatment with piperidine, and the final protected tripeptide hydrazide was deprotected with TFA to yield a white solid (0.311 g, 28.8%); δH (400 MHz, CDCl3) 0.919 (t, 6H, J = 6.98 Hz), 1.56 (m, 6H), 1.68 (m, 1H), 1.86 (m, 2H), 2.27 (m, 2H), 3.14 (m, 2H), 3.84 (m, 1H), 4.31 (m, 1H), 4.41 (m, 1H), 7.83 (t, 1H, J = 5.52 Hz), 8.19 (s, 3H), 8.42 (d, 1H, J = 7.32 Hz), 8.71 (d, 1H, J = 7.80 Hz), 10.64 (s, 1H); δC (100 MHz, CDCl3): 22.43, 23.04, 23.94, 25.46, 27.67, 29.45, 30.33, 40.71, 40.92, 50.93, 51.15, 52.65, 157.29, 169.33, 170.82, 174.08; high resolution mass spectrum calculated for C17H35N8O5 (MH+) 431.2730, found 431.2721. NMR spectra were obtained using a Bruker 400 Avance nuclear magnetic resonance spectrometer and chemical shifts are quoted as parts per million on the δ scale. High resolution mass spectrometry was performed on a Waters Micromass LCT Mass Spectrometer.Coating and chemical modification of culture plates
Falcon Primaria 24 well plates were incubated with 2 µg cm−2 poly-L-ornithine for 30 min, the solution aspirated and wells allowed to air-dry. Plates were then incubated with phosphate-buffered saline (PBS) or 1–3 µg cm−2 laminin in PBS at 37 °C for 2.5 h. After washing with 1 ml PBS, wells were incubated for 30 min at 37 °C with 200 µl of PBS adjusted to pH 5.5 or 1.0 U ml−1 sialidase (Clostridium perfringens) in pH 5.5 PBS. Each well was then washed twice with 1 ml PBS and incubated for 15 min in the dark at 4 °C with 0.5 ml cold PBS or 1 mM sodium periodate solution. Wells were washed again with 1 ml PBS and incubated for 90 min at 37 °C with 0.5 ml of 5 mM biotin hydrazide in PBS, adjusted to pH 5.5. Wells were washed twice with 1 ml PBS and, to visualize biotinylated laminin, double-stained by incubating twice for 10 min each in the dark with 0.5 ml of 5 µg ml−1 FITC-avidin in PBS. After washing twice more with 1 ml PBS, plates were imaged using fluorescence microscopy to detect bound FITC–avidin. Images were analyzed by generating a histogram to determine the mean level of green fluorescence of an 800 × 800 pixel region at the centre of each image using Paint Shop Pro software (Jasc Software Inc.). Data were collected from three different wells per assay plate and are expressed as the mean ± SEM of three separate experiments.Cell culture
Results
Chemical modification of laminin-coated tissue culture plates
To demonstrate the ability to chemically modify a laminin-containing substrate, we coated tissue culture plates successively with poly-L-ornithine and laminin and then subjected them to oxidation with sodium periodate. As a model biomolecule that could be readily detected, biotin, as its hydrazide derivative, was then ligated to the resulting aldehyde groups on the surface-bound laminin molecules. To fluorescently label the ligated biotin, wells were treated with fluorescein isothiocyanate–avidin (FITC–avidin) and subsequently examined by fluorescence microscopy. Control wells of poly-L-ornithine with no laminin coating, that were treated with PBS and subsequently incubated with biotin hydrazide, displayed low levels of non-specific FITC–avidin binding, while laminin-coated wells exhibited a significant shift in fluorescence (Fig. 1). This difference was statistically significant for all laminin densities tested with P values of 0.0481, 0.0012 and 0.0138 for 1, 2 and 3 µg cm−2 laminin, respectively. Differences in fluorescence intensity between the three laminin densities were not significant. The ligation of biotin to the wells was also shown to be dependent on both sodium periodate treatment and the presence of laminin, with background green intensity recorded when either treatment was absent, although the fluorescence of PBS-treated laminin was significantly lower than that observed for poly-L-ornithine-only surfaces (P = 0.001). What is more, sialidase treatment of laminin prior to periodate oxidation resulted in background levels of fluorescence, suggesting the ligation of biotin was chemoselective and occurred via oxidized sialic acid residues. Additionally, no decrease in fluorescence was observed when the treated plates were incubated for 24 h in PBS at 37 °C (not shown), suggesting that the resulting hydrazone bond is stable over this time period.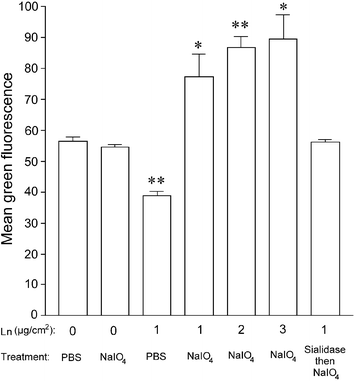 | ||
| Fig. 1 The extent of biotinylation of different surfaces after treatment with PBS or sodium periodate and subsequent incubation with biotin hydrazide. Tissue culture plastic was coated with poly-L-ornithine followed by different concentrations of laminin (Ln). Wells were then incubated with buffer or sialidase to cleave sialic acid moieties, then oxidized where appropriate. After ligation of biotin hydrazide, wells were incubated with FITC–avidin to stain ligated biotin molecules and subsequently analyzed by fluorescence microscopy. A histogram of green fluorescence intensity was generated for each well to determine the mean value. Data represent the mean ± SEM of three individual experiments, each performed in triplicate. * P < 0.05, ** P < 0.01. | ||
Adhesion of primary motoneurons to modified laminin surfaces
Laminin-coated tissue culture plates were oxidized with sodium periodate, then incubated with PBS, biotin hydrazide or LRE hydrazide to generate wells that displayed laminin modified with aldehydes, biotin molecules or the LRE stop signal. To determine whether these treatments affected the adhesion of motoneurons to the substrate, primary spinal cord motoneuron cultures were isolated and partially purified from embryonic rats and plated in the wells at a density of 1 × 104 cm−2. After 24 h, wells were washed and the cells fluorescently stained for choline acetyltransferase (ChAT), a motoneuron marker. ChAT-positive cells were quantified by fluorescence microscopy and the cell densities in treated wells compared to PBS-treated controls. Wells coated with unaltered laminin contained a mean of 10082 ± 1014 ChAT-positive cells cm−2. In comparison, aldehyde- and biotin-modified wells contained a mean of 10432 ± 533 and 9600 ± 358 ChAT-positive cells cm−2, respectively, both within 5% of the control value. However, wells modified with the LRE stop signal contained a mean of 12929 ± 937 ChAT-positive cells cm−2, an increase of 28% compared to unaltered laminin (Fig. 2). These data suggest that modification of laminin-coated surfaces with LRE increases motoneuron adherence with a moderate effect, while the presence of aldehydes or biotin molecules on laminin has little or no effect.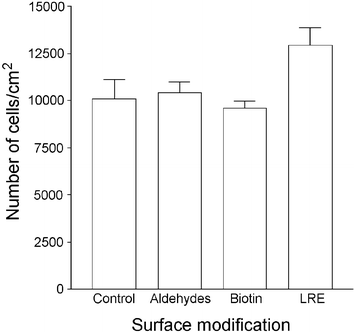 | ||
| Fig. 2 The effect of chemically modifying laminin-coated tissue culture plates on motoneuron adhesion. Tissue culture plates were coated with poly-L-ornithine and laminin, then treated with PBS or sodium periodate solution. Wells were then incubated with PBS, biotin hydrazide or synthetic LRE hydrazide to generate laminin surfaces that were unaltered, aldehyde-modified, biotinylated or LRE-modified. Primary rat spinal cord cells were plated at a density of 1 × 104 cm−2 and, after 24 h, fixed and stained for the motoneuron marker choline acetyltransferase (ChAT). The density of ChAT-positive cells on each surface was quantified by fluorescence microscopy. Each data point represents the mean ± SEM of triplicate wells. | ||
Effects of surface modification on neurite outgrowth
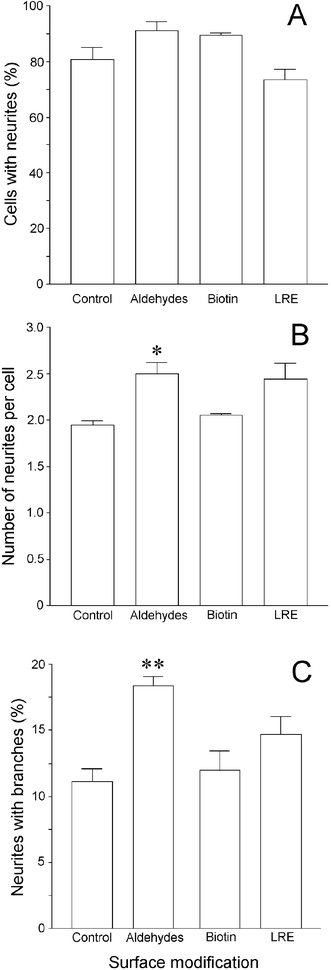 | ||
| Fig. 3 The effect of different chemically-modified laminin surfaces on the number of neurites extended by primary motoneurons and their degree of branching. Primary rat spinal cord cells were plated on the different surfaces and, after 24 h, were fixed and stained for the motoneuron marker choline acetyltransferase (ChAT). The number of ChAT-positive cells with neurites (A), the number of neurites per cell (B) and number of branched neurites per cell (C) were determined using phase contrast microscopy. Each data point represents the mean ± SEM of triplicate wells. * P < 0.05; ** P < 0.01. | ||
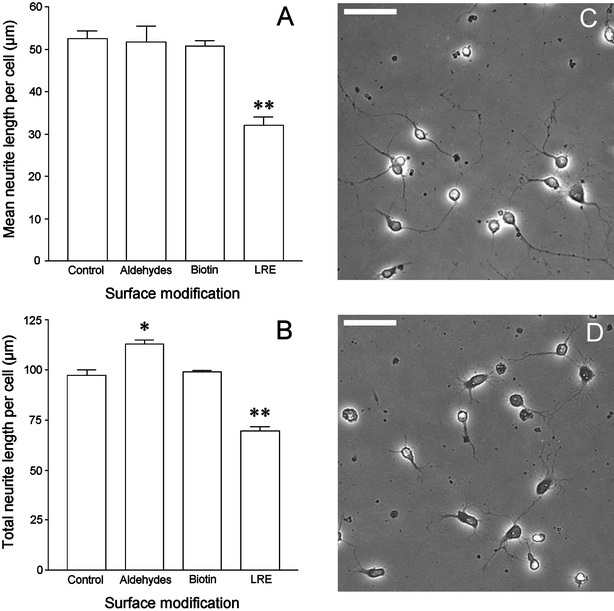 | ||
| Fig. 4 The effect of chemically modifying laminin surfaces on the length of neurites extended by primary motoneurons. Primary rat spinal cord cells were plated on the different surfaces and, after 24 h, were fixed and stained for the motoneuron marker choline acetyltransferase (ChAT). The length of each neurite extending from ChAT-positive cells was measured as described in the Experimental section and used to calculate the mean neurite length per cell (A) and the total length of neurites per cell (B). Each data point represents the mean ± SEM of triplicate wells. * P < 0.05; ** P < 0.01. Micrographs C and D demonstrate the difference between primary motoneurons on the control and LRE-modified laminins, respectively. Bars = 50 µm. | ||
When the number of neurites per motoneuron was excluded, instead examining the total length of processes per cell, ChAT-positive cells on control and biotinylated laminin had mean total neurite lengths of 97.41 ± 2.70 µm and 98.88 ± 0.73 µm, respectively. As expected, motoneurons on LRE-modified laminin surfaces displayed a significant reduction in total neurite length, with a mean of 69.67 ± 1.94 µm (P = 0.0011), a reduction of 28.5% in comparison to untreated laminin. Aldehyde-modified surfaces caused a significant increase in the total cellular neurite outgrowth, with a mean of 112.87 ±2.06 µm (P = 0.0104), a 15.9% increase compared to the control (Fig. 4B). Again, total neurite length was unaffected by non-ligated LRE hydrazide in comparison to control laminin (not shown).
When the distribution of mean neurite lengths was analyzed it was apparent that there was little difference between ChAT-positive cells on the control, aldehyde-modified and biotinylated laminin surfaces (Fig. 5). However, modification of laminin with LRE resulted in a higher proportion of motoneurons extending shorter neurites, while fewer cells extended long processes. These differences were particularly apparent in the 1–20 and 21–40 µm ranges, with 89.1% and 21.7% more cells, respectively, than on unmodified laminin (P = 0.0088 and P = 0.0090) and the 61–80 and >100 µm ranges, with 63.9% and 82.5% fewer cells than on unmodified laminin, respectively (P = 0.0092 and 0.0027). It is noteworthy that the number of cells in the >100 µm range was also reduced on both aldehyde-modified and biotinylated surfaces, with the biotinylated surfaces displaying a significant reduction of 41.0% (P = 0.0293).
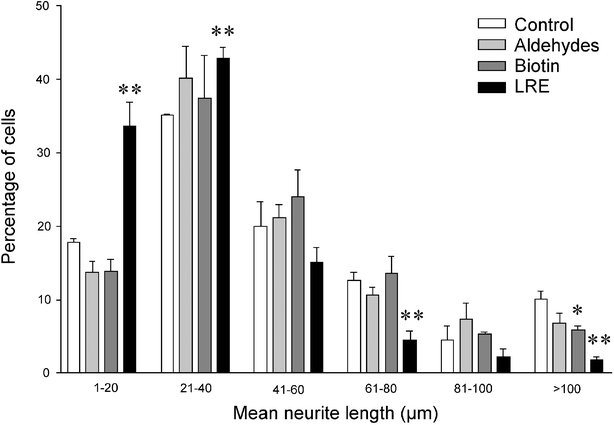 | ||
| Fig. 5 The distribution of mean neurite lengths extended by primary motoneurons on different chemically modified laminin surfaces. Primary rat spinal cord cells were plated on the modified surfaces and, after 24 h, were fixed and stained for the motoneuron marker choline acetyltransferase (ChAT). The length of each neurite extending from ChAT-positive cells was measured as described in the Experimental section and used to calculate the mean neurite length per cell. The proportion of cells with mean neurite lengths within arbitrary length ranges was then determined. Each data point represents the mean ± SEM of triplicate wells. * P < 0.05; ** P < 0.01. | ||
Discussion
In the present study we have investigated the feasibility of chemically modifying surface-bound laminin via oxidation with sodium periodate in order to examine the effect of the LRE “stop” signal on neurite outgrowth from primary spinal motoneurons.Using tissue culture plates successively coated with poly-L-ornithine and various concentrations of laminin, we utilized sodium periodate to specifically oxidize terminal sialic acid residues on the laminin molecules and subsequently used the resulting aldehyde groups to chemoselectively ligate biotin hydrazide, as a model biomolecule, to the protein. On staining with FITC-avidin, it was apparent that biotinylation only occurred in wells that were both treated with sodium periodate and coated with laminin. Wells that were coated with poly-L-ornithine, but not laminin exhibited the same biotinylation as control wells. However, laminin-coated surfaces that were incubated with PBS instead of periodate exhibited a significantly lower level of fluorescence than poly-L-ornithine-only controls. The reason for this difference is unclear, and cannot be explained by decreased electrostatic interaction between FITC-avidin and laminin since they carry net positive and negative charges, respectively. Significantly, when laminin surfaces were incubated with sialidase prior to oxidation and biotinylation, the resulting fluorescence levels were reduced to that of the controls. Overall, these data demonstrate that biotin can be chemoselectively ligated to laminin via sialic residues on its carbohydrate component following the generation of aldehydes by mild oxidation with sodium periodate. These results indicate that this technique is an effective way of labelling or modifying surface-bound glycoproteins. Although similar methods have been used to label proteins in solution,22 we are not aware of previous studies that have chemically modified adsorbed proteins.
Using this technique to chemically modify laminin surfaces with different groups, we generated culture wells containing unaltered laminin, laminin oxidized to display aldehyde-modified sialic acids and laminins that were modified by the ligation of biotin or LRE via a covalent hydrazone bond. To investigate whether the LRE-modified laminin could affect neurite outgrowth from motoneurons, the effects of the different surface modifications on primary rat spinal cord cells was examined. After a period of 24 h on these surfaces, cells were fixed, stained for choline acetyltransferase and analyzed microscopically. When the effect of laminin modifications on cell adhesion was investigated, it was observed that there were 28% more ChAT-positive cells on LRE-modified laminin than on unaltered laminin. This increase in cell number is consistent with previous observations that show the postulated LRE motoneuron stop signal is specifically adhesive for motoneurons.14,15 The presence of an aldehyde group on the sialic acid residues of laminin did not affect cell adhesion, and this was also the case when biotin was ligated to sialic acids. This suggests that sialic acid oxidation does not affect the adhesion of motoneurons, nor is an adhesive site on laminin blocked when a small molecule is ligated. Thus, the increase in adhesiveness of the LRE-modified laminin is specifically due to the presence of the LRE tripeptide.
When the effect of modified laminins on neurite growth was examined, it was clear that none of the treatments affected the proportion of motoneurons that extended processes. However, the number of neurites per cell was affected by modifying laminin, with the presence of aldehydes or the LRE group increasing the number of processes per cell. Aldehydes also significantly increased the branching of neurites in comparison to the other treatments, suggesting that oxidized sialic acids may promote neurite extension. It is known that a number of neuronal receptors bind to laminin-1, including members of the integrin family, non-integrin laminin binding proteins, α-dystroglycan and β-1,4-galactosyltransferase.23 The latter receptor initiates neurite outgrowth by recognition of specific carbohydrate epitopes on laminin,24,25 and it is possible that modifying sialic acids within these sequences with aldehydes increases the receptor's affinity for the substrate, while other modifications assayed have no effect.
The length of neurites extended on the modified laminins was greatly affected by changes in surface chemistry. The mean neurite length per cell was approximately 50 µm on all laminins except the LRE-modified substrate, where the mean was 32 µm. This highly significant reduction in outgrowth demonstrates that the presence of the LRE sequence was inhibitory to neurite extension and, as far as we are aware, is the first time that this tripeptide has been shown to inhibit the outgrowth of neurites from primary mammalian motoneurons. The effect of LRE hydrazide was also shown to be a result of its chemoselective ligation to oxidized laminin, with no inhibitory effect observed without prior periodate treatment. When the total neurite length per cell was analyzed, a similar result was observed. However, the total neurite length per cell for motoneurons grown on aldehyde-modified surfaces was significantly higher than that of the control. This can be explained by surface aldehydes promoting an increase in the number of neurites per cell as described above, thereby increasing the total cellular neurite length while leaving the mean length unaffected. The effects of the modifications of laminin are clearly shown by the plot of mean neurite length distributions on Fig. 5. It is clear that, although LRE did not totally prevent neurite extension, motoneurons on LRE-modified laminin extended a greater number of shorter neurites than the other surfaces, while extending fewer long neurites. Thus LRE inhibited neurite outgrowth. The reason for the reduction in neurites >100 µm on biotinylated laminin, however, is unclear.
When compared to previous studies that investigated motoneuron behaviour on LRE-containing peptides the results of this study are not as pronounced. For example, when MSC-34 cells were plated on a mixture of laminin and a carrier protein expressing an LRE-containing hexapeptide, the number of cells with processes was reduced by more than 80% in comparison to laminin alone.15 However, high concentrations of hexapeptide conjugate were employed and the presence of processes assayed after only 3 h. In the present study, the amount of laminin adsorbed on to the culture wells is likely to be low, with previous studies indicating approximately 200 ng cm−2 using the conditions we employed.26 It has been previously demonstrated that the sialic acid content of purified laminin from the murine EHS sarcoma is 2.4% by mass.27 Based on 200 ng cm−2 laminin and ligation of LRE to every sialic acid residue, the maximum density of the tripeptide would be 15.5 pmol cm−2. A significant number of sialic acids will be inaccessible due to their unfavourable orientation on the adsorbed laminin, so the LRE density is likely to be much less than this. What is more, we observed the motoneurons after 24 h, during which time they would have had the opportunity to extend neurites, even on LRE surfaces. In addition, this study employed primary rat motoneurons and there are likely to be differences in their growth characteristics when compared to a motoneuron-like cell line. However, a later study compared neurite extension from primary rat motoneuronson laminin-1, laminin-2/4/8 and laminin-11.28 It was observed that motoneurons adhered well to all substrates, with a greater number of cells on laminin-11 than laminin-1. Motoneurons extended processes on all three substrates at early time-points but, as the time in culture progressed, cells on laminin-11 were shown to have shorter neurites. The data presented in the present study are in agreement with these observations, suggesting that chemoselective modification of laminin-1 with the LRE stop signal found in laminin-11 effectively inhibits the outgrowth of neurites from motoneurons cultured on this substrate.
In conclusion, we have demonstrated that glycoproteins adsorbed on to tissue culture plastic can be effectively chemoselectively modified via oxidation with sodium periodate. We have used this technique to study the effects of modifying laminin, a glycoprotein known to promote neurite outgrowth from motoneurons, with a tripeptide motoneuron “stop” signal, LRE. We demonstrate that LRE-modified laminin promotes cell adhesion and is inhibitory to neurite extension in comparison to native laminin. In addition we demonstrate that generation of aldehydes on native laminin by periodate oxidation increases the number of neurites extended per cell. This is the first demonstration that a simple tripeptide can, by itself, inhibit primary mammalian motoneuron outgrowth. In effect, a commercially available matrix molecule has been made to behave like one that is only obtainable by isolation from a cellular source. In addition to applications in neural and neuromuscular tissue engineering, this chemoselective technique is applicable to the modification of any adsorbed glycoprotein for altering cellular adhesion and behaviour.
Acknowledgements
We thank the BBSRC for funding.References
- L. G. Griffith and G. Naughton, Science, 2002, 295, 1009–1014 CrossRef CAS.
- A. Persidis, Nat. Biotechnol., 1999, 17, 508–510 CrossRef CAS.
- M. J. Lysaght and A. L. Hazlehurst, Tissue Eng., 2004, 10, 309–320 CrossRef.
- S. Ramon y Cajal, in Degeneration and Regeneration of the Nervous System, ed. R. M. May, Oxford University Press, London, 1928, pp. 265–280 Search PubMed.
- J. R. Sanes, L. M. Marshall and U. J. McMahan, J. Cell Biol., 1978, 78, 176–198 CrossRef CAS.
- J. R. Sanes, J.Biol. Chem., 2003, 278, 12601–12604 CrossRef CAS.
- J. R. Sanes, J. Cell Biol., 1982, 93, 442–451 CrossRef CAS.
- B. L. Patton, Microsc. Res. Tech., 2000, 51, 247–261 CrossRef CAS.
- D. D. Hunter, V. Shah, J. P. Merlie and J. R. Sanes, Nature, 1989, 338, 229–234 CrossRef CAS.
- P. T. Martin, A. J. Ettinger and J. R. Sanes, Science, 1995, 269, 413–416 CAS.
- J. H. Miner and B. L. Patton, Int. J. Biochem. Cell Biol., 1999, 31, 811–816 CrossRef CAS.
- A. Y. Chiu, M. Ugozolli, K. Meiri and J. Ko, J. Neurochem., 1992, 59, 10–17 CAS.
- B. L. Patton, J. H. Miner, A. Y. Chiu and J. R. Sanes, J. Cell Biol., 1997, 139, 1507–1521 CrossRef CAS.
- D. D. Hunter, B. E. Porter, J. W. Bulock, S. P. Adams, J. P. Merlie and J. R. Sanes, Cell, 1989, 59, 905–913 CrossRef CAS.
- D. D. Hunter, N. Cashman, R. Morrisvalero, J. W. Bulock, S. P. Adams and J. R. Sanes, J. Neurosci., 1991, 11, 3960–3971 CAS.
- B. E. Porter, J. Weis and J. R. Sanes, Neuron, 1995, 14, 549–559 CrossRef CAS.
- P. A. De Bank, B. Kellam, D. A. Kendall and K. M. Shakesheff, Biotechnol. Bioeng., 2003, 81, 800–808 CrossRef CAS.
- G. A. Lemieux and C. R. Bertozzi, Trends Biotechnol., 1998, 16, 506–513 CrossRef CAS.
- M. Manthorpe, E. Engvall, E. Ruoslahti, F. M. Longo, G. E. Davis and S. Varon, J. Cell Biol., 1983, 97, 1882–1890 CrossRef CAS.
- R. I. Schnaar and A. E. Schaffner, J. Neurosci., 1981, 1, 204–217 CAS.
- W. Camu and C. E. Henderson, J. Neurosci. Methods, 1992, 44, 59–70 CrossRef CAS.
- K. C. Ingham and S. A. Brew, Biochim. Biophys. Acta, 1981, 670, 181–189 CAS.
- S. K. Powell and H. K. Kleinman, Int. J. Biochem. Cell Biol., 1997, 29, 401–414 CrossRef CAS.
- W. A. Thomas, A. W. Schaefer and R. M. J. Treadway, Development, 1990, 110, 1101–1114 Search PubMed.
- Q. Huang, B. D. Shur and P. C. Begovac, J. Cell Sci., 1995, 108, 839–847 Search PubMed.
- M. Balcells and E. R. Edelman, J. Cell. Physiol., 2002, 191, 155–161 CrossRef CAS.
- R. N. Knibbs, F. Perini and I. J. Goldstein, Biochemistry, 1989, 28, 6379–6392 CrossRef CAS.
- S. I. Cho, J. Ko, B. L. Patton, J. R. Sanes and A. Y. Chiu, J. Neurobiol., 1998, 37, 339–358 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |