Combining one-, two- and three-dimensional polyphenylene nanostructures
Jishan
Wu
,
Andrew C.
Grimsdale
and
Klaus
Müllen
*
Max-Planck-Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
First published on 29th November 2004
Abstract
The achievement of a true nanotechnology is regarded as one of the Holy Grails of modern science. To achieve this will require the construction by synthesis or self-assembly of complex functional nanoarchitectures. Such structures might contain combinations of one-, two-, and three-dimensional phenylene nanostructures. In this paper we provide an overview of recent progress in the synthesis and characterisation of new materials containing various combinations of one-, two-, and three-dimensional phenylene structural units. Though synthetically driven the ultimate aim of this work is to design and prepare new functional materials for use in electronic or other devices. Control of their optical and electronic properties by synthetic design and the evaluation of their potential for electronic applications are thus key features of our work. A vital aspect of this is the control of the supramolecular order of the materials in the solid state, by regulation of their intermolecular interactions. The attachment of bulky three-dimensional phenylene dendrimers to one-dimensional conjugated polyphenylenes or two-dimensional discotic graphite molecules enables the control of their intermolecular interactions and thus of their optical and electronic properties, with potential importance for their application in electronic devices. Linking two-dimensional graphite units together into linear and non-linear chains provides a new way to control the self-assembly of discotic materials and thus opens a way towards more new complex superstructures. Another way is demonstrated to combine elements of one- and two-dimensional polyphenylenes by the synthesis of graphite ribbons. Finally partial cyclodehydrogenation of polyphenylene dendrimers has been used to produce three-dimensional arrays of two-dimensional graphenes. Such materials have potential applications in lithium and hydrogen storage. Taken together these results present a useful step in the development of the complex architectures necessary for a true nanotechnology.
 Jishan Wu | Jishan Wu was born in Hubei province, China in 1975. He received his BSc degree from Wuhan University in 1997. He then worked on his masters thesis on conjugated molecular wires under the supervision of Professor Xianhong Wang and Professor Fosong Wang at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences where he obtained a masters degree in polymer science in 2000. He performed his doctoral work on the synthesis of new discotic graphitic materials under the supervision of Professor Müllen, and was awarded his PhD in 2004 from Johannes-Gutenberg University in Mainz. He is currently project leader in charge of research into the synthesis, characterisation and applications of discotic materials at the Max-Planck Institute for Polymer Research. |
 Andrew C. Grimsdale | Andrew Grimsdale was born in Waiouru, New Zealand and graduated from the University of Auckland. He received his PhD there under the supervision of Professor R. C. Cambie in 1990 for work on the synthesis of analogues of biologically active drimane sesquiterpenes. He then undertook postdoctoral work on photochromic and electroactive materials with Professor A. Pelter at the University of Wales, Swansea, and with Professor A. B. Holmes at the University of Cambridge on electroluminescent polymers. Since 1999 he has been working with Professor Müllen at the Max-Planck Institute for Polymer Research at Mainz, where he is currently project leader in charge of research into conjugated polymers. His current research interests are in the synthesis of polyphenylene-based materials for optoelectronic applications. |
 Klaus Müllen | Klaus Müllen received his Diplom Chemiker degree (1969) at the University of Cologne after work with Professor E. Vogel and his PhD degree from the University of Basel, Switzerland (1972), where he undertook research with Professor F. Gerson on EPR spectroscopy. He pursued postdoctoral research at the Swiss Federal Institute of Technology in Zurich in the group of Professor J. F. M. Oth, where he worked in the field of dynamic NMR spectroscopy and electrochemistry. He received his habilitation there (1977) and was appointed Privatdozent. In 1979 he became a Professor in the department of Organic Chemistry, University of Cologne, and in 1983 he accepted a chair in Organic Chemistry at the University of Mainz. He joined the Max-Planck Society in 1989 as one of the directors of the Max-Planck Institute for Polymer Research. His research interests are centred around synthetic macromolecular chemistry and materials science. |
1. Introduction
A major challenge in modern materials science is to create complex functional supramolecular nanoscale architectures, thus mimicking the similarly sized active structures found inside cells. Such architectures, like their biological counterparts, might be used to control and manipulate the transfer of e.g. energy and/or charge, and are essential for the realisation of the dream of so-called nanotechnology, which involves the construction of functional optoelectronic devices with dimensions in the nanometer range, in other words devices based upon single molecules or, more realistically, small ensembles of molecules.Polyphenylenes1–3 are an important class of organic materials, as active materials in optoelectronic devices including light-emitting diodes (LEDs),4 field-effect transistors (FETs),5 and solar cells.6 They exist as linear one-dimensional conjugated polymers, with conjugation along the backbone giving them the character of a molecular wire, as two-dimensional discotic polycyclic aromatic hydrocarbons (PAHs), which can form long columnar stacks with very high charge carrier mobilities along the columnar axis, or as three-dimensional quasi-spherical shape-persistent polyphenylene dendrimers, which can act as functional nanoobjects. As part of our ongoing investigations into one-, two-, and three-dimensional polyphenylenes we are examining ways in which to obtain more complex supramolecular architectures combining polyphenylene nanostructures of different dimensionalities for use in devices. We now present an overview of our progress in preparing and characterising new materials containing combinations of such polyphenylene nanostructures, and of the effects that combining different dimensionalities has upon the materials properties. A particular aim is to obtain materials that can be readily processed for use in electronic and optoelectronic devices.
The combination of one- and three-dimensional architectures is exemplified by dendronised polymers in which the central conjugated chain is surrounded by a dendrimer shell, which shields the chains from aggregation. The combination of one- and two-dimensional architectures is illustrated by the linking together of two or more hexa-peri-hexabenzocoronene (HBC) units so as to enable the binding of their columns together, and by the synthesis of graphite ribbons. Finally methods for the combination of two- and three-dimensional structures are presented: the substitution of an HBC with dendrons to control the packing of the discs and the preparation of molecular propellers which contain a three-dimensional array of two-dimensional graphene units. The preparation of new carbon materials by the pyrolysis of two-dimensional polyphenylenes is also discussed.
2. Combining one- and three-dimensional structures
One-dimensional polyphenylenes, particularly polyfluorenes7 and polyindenofluorenes,8 are of considerable interest as blue-emitting materials for use in LEDs. A major problem with these materials however is that their emission colour is not stable, with the rapid appearance of an emission band in the green part of the spectrum. Originally this was attributed to the effects of aggregation of the polymer chains,9 but it has now been demonstrated that the green emission from polyfluorenes arises from ketone defects produced either during polymer preparation and processing or during device operation.10 Attachment of dendrimer groups to polyfluorenes or polyindenofluorenes was anticipated to suppress the green emission as the aryl sidechains were expected to be more resistant to oxidation to form ketones, and the bulky substituents should hinder exciton migration to any defect sites that might be formed.Two routes to prepare dendronised polyfluorenes (1) have been developed. In the first a convergent approach was used (Scheme 1).11 A bromo-dendron 2 was lithiated, then added to the ester 3. Ring-closure of the diadduct 4 then gave the dendronised dibromomonomer 5 which was polymerised with nickel(0) (Yamamoto conditions). The resulting polymer was of modest molecular weight (Mn = 1.0 × 104, Mw/Mn = 3).
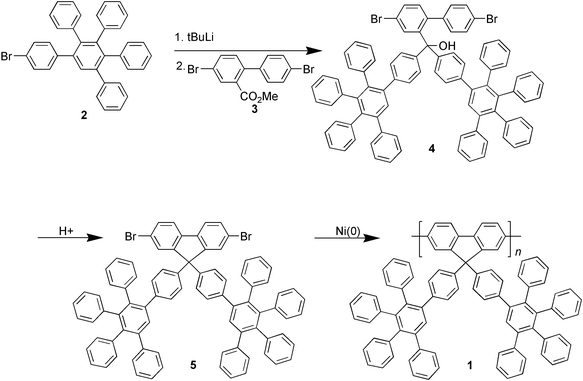 | ||
| Scheme 1 Convergent route to dendronised polyfluorenes. | ||
This route has the disadvantages that second-generation dendrons will be difficult to attach due to steric hindrance at the ester, while the range of functionalised dendrons that are attachable is restricted to those compatible with the reaction conditions. Accordingly a divergent approach (Scheme 2) has been developed as a more general route to dendronised polyfluorenes.12 In this route the ester 3 is converted to the bisethyne 6, which then undergoes Diels–Alder cycloaddition with suitably functionalised cyclopentadienones followed by ring-closure to the monomer. Both first- and second-generation dendrons can be built up from 6. Both chloro (5b) and bromomonomers (5a) have been made by this route. The molecular mass of polymer 1 made from the chloromonomer was found to be significantly higher (Mn = 3.2 × 104, Mw/Mn = 2.7). than that prepared from the bromomonomer.
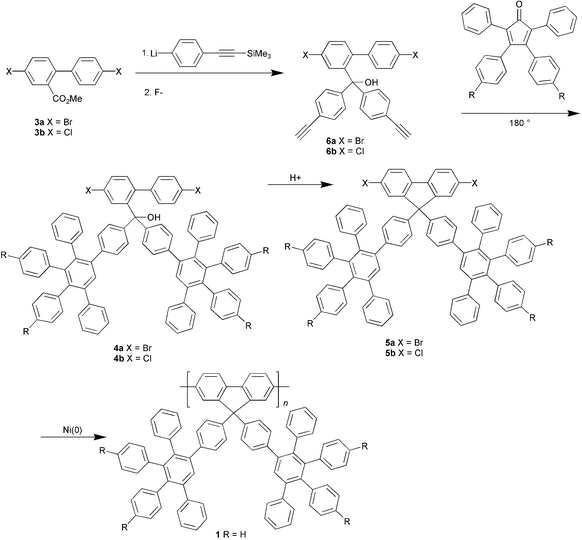 | ||
| Scheme 2 Divergent route to dendronised polyfluorenes. | ||
The dendronised polymer 1 displayed more stable blue emission than alkylated polyfluorenes in an LED.13 Studies14–16 on the comparative photophysics of the dendronised polymer 1 and of alkylated polyfluorenes have shown that the dendronisation has profound effects on the optical properties, in particular the behaviour of the excitons. Interestingly the effects seem to be stronger for triplet excitons than for singlets. Thus the triplet lifetime in 1 (24 ms) is increased by an order of magnitude over that of an alkylfluorene polymer (2.4 ms).15 This is associated with a decrease in triplet exciton migration.15,16 So far it is unclear if singlet exciton migration is hindered by the dendrons,14,16 but a buildup of triplet excitons in 1 might have an adverse effect on LED efficiency. The reduced interactions between the chains may also reduce the efficiency of charge transport, but this effect might be counteracted by the use of dendrons functionalised on the periphery with charge transporting units. The synthesis and characterisation of polyfluorenes bearing such groups is now underway, as is the extension of this approach to other polymer backbones.
The attachment of three-dimensional dendrimers to one-dimensional polymers clearly leads to enhanced colour purity and stability, at the cost of potentially reduced charge transport and EL efficiency, and further fundamental research into the photophysics of dendronised polymers will be required before the optimum balance between these effects can be attained.
3. Combining one- and two-dimensional nanostructures
Discotic two-dimensional polyphenylenes are of great interest as potential active materials in electronic devices because of their ability to form ordered columnar mesophases, with very high charge carrier mobilities along the columnar axis.17,18 This has been utilised to make efficient field-effect transistors19,20 and solar cells.21 The anisotropy of the charge conduction is well demonstrated by the much lower source-drain currents obtained from FETs with source and drain perpendicular to the columnar axis compared with those in devices with the electrodes parallel to it. In order to maximise the charge carrier mobilities, considerable research effort has been devoted to optimising the columnar order in the mesophases and into ordering the columns themselves into larger structures.22 One way to accomplish the latter objective is to form combined one- and two-dimensional structures consisting of linear arrays of HBCs, made by linking the HBC units together either directly or via short conjugated spacers. This would permit charge transfer along the chains as well as up and down the columnar stacks of the discs, thus creating “bypasses” for the movement of charge in large networks. For such ideas to work, the phase-forming properties of these HBC oligomers are thus of paramount importance. Dimers and trimers of hexa-peri-hexabenzocoronene (HBC) 7–10 (Fig. 1) of different geometries have been prepared, and their self-assembly has been studied by wide-angle X-ray diffraction (WAXD) in the bulk. Remarkably enough, an ordered columnar stacking has been detected in the dimer 7, the para-trimer 8, and the superfluorene 9.23 Due to the strong out-of-plane twisting of the ortho-HBC units such stacking is not seen in the ortho-trimer 10.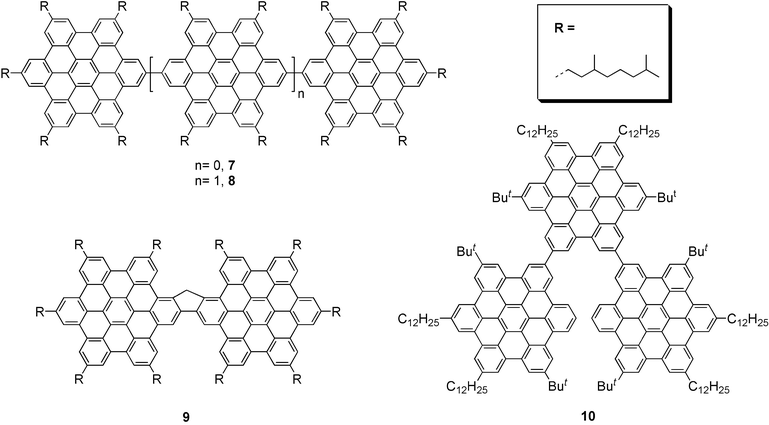 | ||
| Fig. 1 Dimers and trimers of HBC. | ||
Two different synthetic approaches have been used. The first is to couple suitably functionalised HBCs. Thus the dimer 6 was prepared by Yamamoto coupling of the corresponding mono-bromo-substituted HBC 11 (Scheme 3).
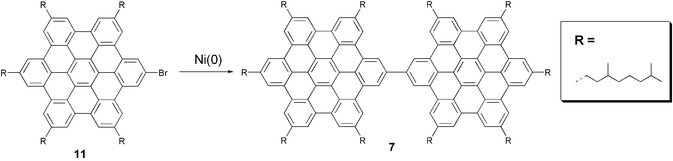 | ||
| Scheme 3 Synthesis of HBC-dimer 6. | ||
The alternative approach is to synthesise oligo(hexaphenylbenezene)s and then convert the hexaphenylbenzene units into HBCs by oxidative cyclodehydrogenation. Thus the para- and ortho-trimers 8 and 10 were prepared by cyclodehydrogenation of the corresponding tris(hexaphenylbenzene) precursors, e.g. 12 (Scheme 4). These in turn were obtained by Diels–Alder cycloaddition of functionalised cyclopentadienones to hexaphenylbenzenes 13 and 14 with diphenylethyne substituents respectively “para” and “ortho” to each other. Similarly the “superfluorene” 9 was made by cycloaddition of the diethynyl fluorene 15 with a cyclopentadienone and cyclodehydrogenation of the resulting compound 16.
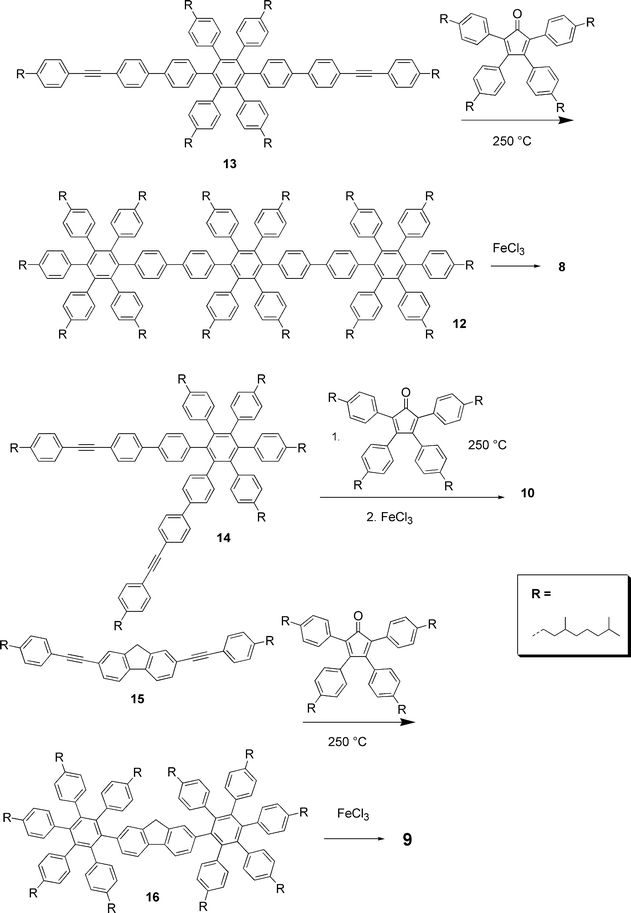 | ||
| Scheme 4 Synthesis of HBC-trimers and of “superfluorene”. | ||
The dimer and trimers represent a new kind of electronically decoupled oligoarylene and serve as model compounds for the corresponding poly-para-(ortho)-“super-phenylenes”. Sterically induced torsion about the bonds between the HBC units means that they are not coplanar with poor conjugation between neighbouring units. As a result their UV and PL spectra look essentially identical to those of the individual HBC units. By contrast the methylene bridge in 9 induces coplanarity of the two HBC units which improves the π-conjugation and suppresses the geometrical relaxation of the backbone upon electronic excitation, leading to a more prominent 0–0 transition band in the fluorescence spectrum than is seen for HBCs.
An extension of the graphene structure of HBC in one dimension results in graphite ribbons, which are expected to show high in-plane conductivity along the axis of the ribbon. A homologous series of discrete graphite nanosheets (Fig. 2) has been synthesized, by different multi-step procedures.2 More recently, one-dimensional polymeric graphite ribbons (17) were synthesized by intramolecular oxidative cyclodehydrogenation of soluble branched polyphenylenes (18), obtained by Diels–Alder polymerization of 1,4-bis(2,4,5-triphenylcyclopentadienone-3-yl) benzene (19) and diethynylterphenyl (20) in good yield (Scheme 5).24
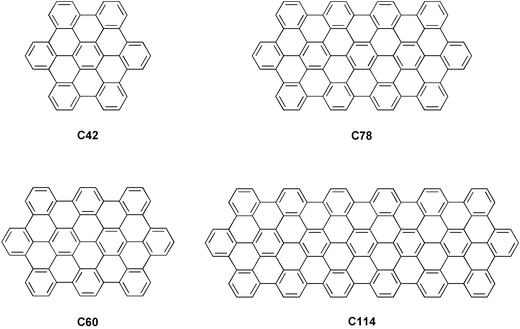 | ||
| Fig. 2 Well-defined ribbon-like graphitic structures. | ||
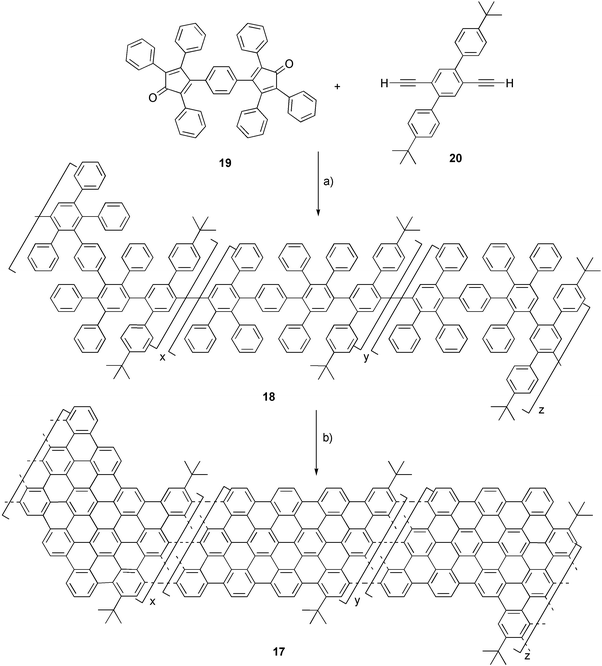 | ||
| Scheme 5 Synthesis of one-dimensional graphite ribbons: a) diphenyl ether, 250 °C; b) FeCl3, CH2Cl2, CH3NO2. | ||
While the insolubility of the resulting graphite ribbons 17 precluded standard spectroscopic structure elucidation, their electronic and vibrational properties were probed by solid-state UV-Vis, Raman and infrared spectroscopy. A wide and unstructured absorption band covering the visible range of the electronic spectrum (λmax ∼ 800 nm) was observed, confirming the highly extended conjugated framework. The profile of the visible Raman spectrum of the material is characterized by two strong bands (at 1603 cm−1 and 1322 cm−1), corresponding to the G and D bands of graphite. The obtained graphite ribbons are not linear, but rather contain “kinks” due to the structural design of the polyphenylene precursor. High resolved transmission electron microscopy (HRTEM) images of the black precipitate of 17 disclosed two different domains: one contains an ordered graphite layer structure with a layer distance of ca. 3.45 Å, while the other appears as a disordered area as would be expected due to the nonlinear structure of the obtained graphite ribbons.
The above results show that one-dimensional arrays of two-dimensional graphite units can be made which still display well-ordered columnar stacking, but the development of processable graphite ribbons, which might act as nano-wires due to high in-plane conductivity along the axis of the ribbon, still remains an elusive goal.
4. Combining two- and three-dimensional nanostructures
Two combinations of two- and three-dimensional polyphenylene nanostructures have been investigated. First, dendrons have been attached to the periphery of an HBC. It was anticipated that, as was shown for one-dimensional polymers above, the attachment of bulky sidegroups would hinder the π-stacking of the discs.The synthetic route to the dendronised HBCs is shown in Scheme 6. The insoluble hexaiodo-HBCs 20 and 21 undergo six-fold Sonogashira–Hagihara couplings to produce soluble hexa-ethynyl-HBCs 22 and 23.25–27 The dendronised HBCs 24 and 25 were then obtained by Diels–Alder cycloadditions.
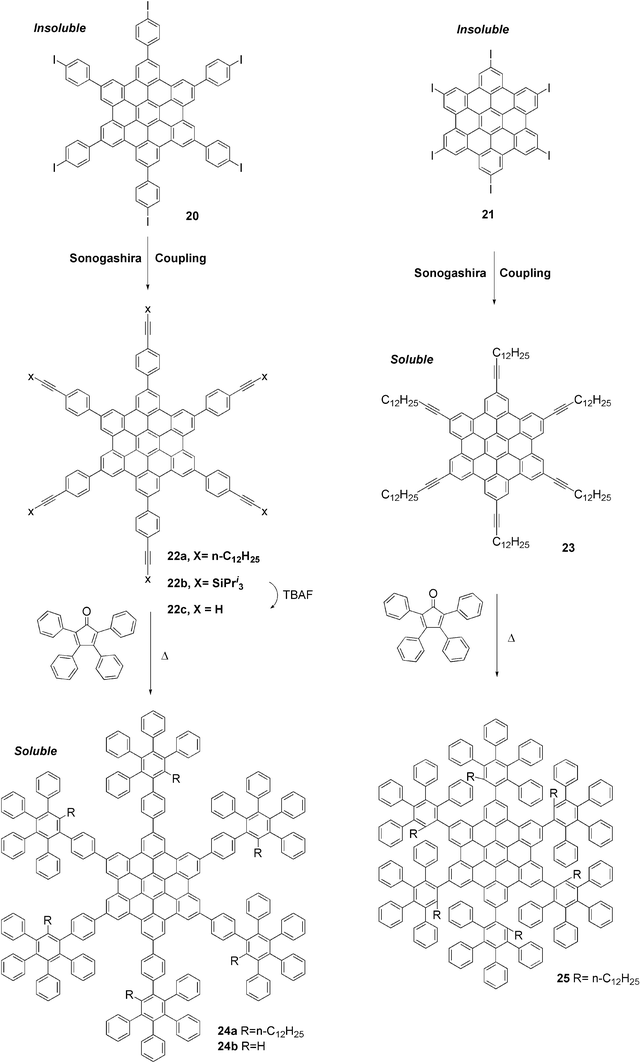 | ||
| Scheme 6 Synthesis of dendronised HBCs. | ||
The degree of steric hindrance between the dendrons and the HBC cores was controlled by varying the distance between the dendrons and the cores (24vs. 25) and by the substituents in the bay positions of the dendrons (24avs.24b). The self-assembly behaviour of 24 and 25 was studied by NMR techniques in combination with quantum chemical calculations. An interesting monomer–dimer dynamic equilibrium which is slow on the NMR time-scale was observed for 24a. This equilibrium was controllable by temperature, concentration and choice of solvent. By contrast, compound 24b shows a stable dimer due to the diminished steric hindrance arising from there being only a hydrogen in the bay positions, and compound 25 with the even greater steric hindrance only displays a discrete monomer structure.28 These results show that the interactions between discotic molecules can be controlled and even suppressed by means of the use of substituents of varying steric bulk. Though these monomeric and dimeric structures have no immediately obvious utility in optoelectronic devices, more subtle control of the interactions between graphite discs offers a way to fine-tune their self-assembly and electronic properties.22
In a second approach to combining two- and three-dimensional nanostructures, propeller-like structures with graphene sheets linked through a non-planar core have been prepared.29 It has been shown that disordered carbon materials containing graphene units, obtained by heat treatment of one-dimensional polyphenylenes, can store lithium for use in secondary batteries,30 while materials containing graphene sheets have been shown to be able to adsorb hydrogen, which is important for the development of new energy producing technology (e.g. fuel cells).31,32 Structures containing well-defined three-dimensional arrays of two-dimensional graphite subunits are thus obvious candidates for investigation as new materials for lithium or hydrogen storage applications.
The synthetic procedure is exemplified in Scheme 7. Dehydrogenation of the C474 dendrimer 26 with iron(III) chloride produces a mixture of the fully dehydrogenated planar hydrocarbon 27 and the propeller molecules 28 and 29 with triphenylbenezene and benzene cores respectively.
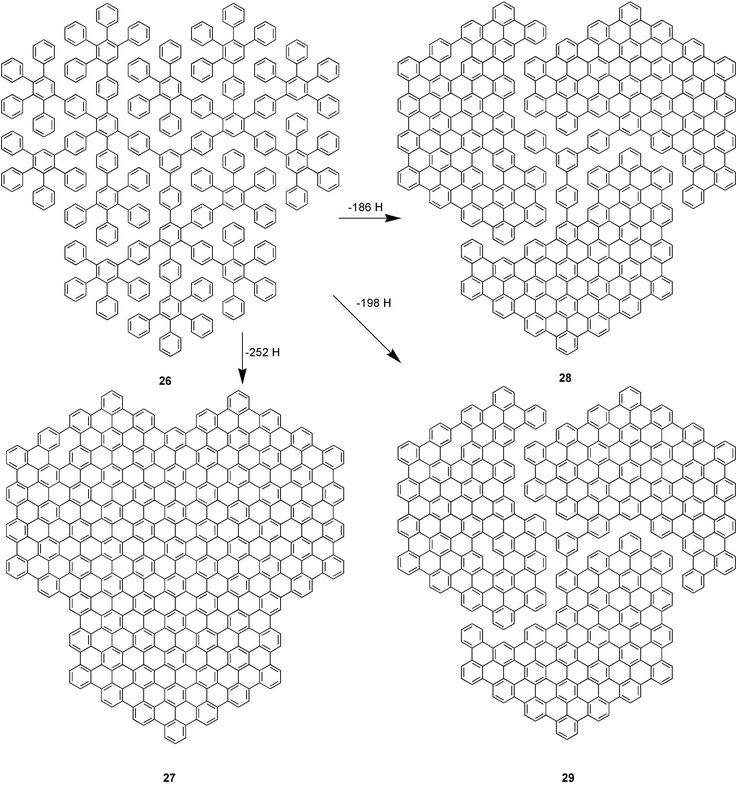 | ||
| Scheme 7 Cyclodehydrogenation of a C474 dendrimer to “propeller” molecules and a graphite disc. | ||
Due to their insolubility these materials could not be characterised by solution methods, but the structures could be determined by means of a combination of solid-state UV-Vis, fluorescence, and Raman spectroscopy and mass spectrometry. A soluble model compound 30 was made by dehydrogenation of dendrimer 31 to assist in this characterisation process (Scheme 8).
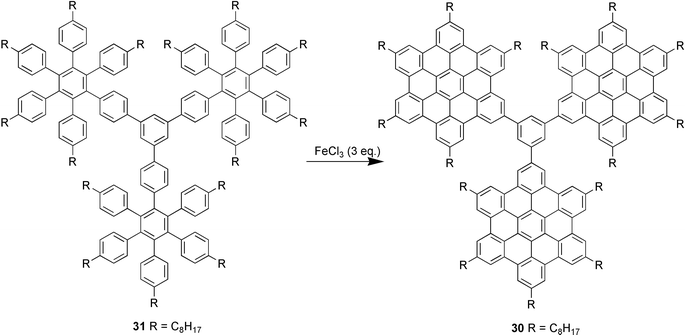 | ||
| Scheme 8 Partial cyclodehydrogenation of a C132 dendrimer to a model “propeller” molecule. | ||
The positions of the main bands in the absorption spectra of the tris-HBC 30 were almost unchanged with respect to hexaalkyl-HBC, thus showing that the HBC units were not conjugated to each other, confirming it was non-planar.29 The next objective of this research is to make larger processable systems to more deeply investigate the behaviour of such well-defined arrays of graphene sheets and to evaluate them for lithium and hydrogen storage applications.
An alternative approach towards making materials containing three-dimensional arrays of two-dimensional graphite units is to pyrolyse discotic graphite molecules or their precursors. Here the aim is to utilise the ordered mesophases of these materials to produce materials with controllable and well-defined structures by pyrolysis under controlled temperatures and conditions, i.e. to transform the ordered starting materials into ordered products. We have recently shown that pyrolysis of HBCs produces carbon nano- and microstructures of novel structures via large mesomorphic structures.33 These products are much less well-defined than those discussed previously, and the product nanomorphologies are characterised by scanning and transmission electron microscopy techniques. Structures, many of them novel, ranging from discrete microspheres of diameters between a few hundred nm and 2 µm, through branched nanosticks, to nanowires with diameters of 20 nm and lengths of up a few µm have been detected. The effects of systematic variation of the temperature profile and the substrate structure have been investigated, and although a variety of structures are always seen after each experiment, the overall range of structures obtained is determined by the pyrolysis temperature.
We have investigated ways to use the ordering of graphite molecules such as HBC to induce order in the pyrolysis products. Thus a well-controlled pyrolysis of HBC molecules has been performed in a porous alumina membrane which acts as a template.34 The HBC molecules were introduced to the nanochannnels within the alumina template by a simple wetting process, and then subjected to pyrolysis by various temperature procedures. After dissolution of the template uniform carbon nanotubes with graphene layers perpendicular to the nanotube axis were obtained. It was found that the ordered pre-organization of the liquid crystalline HBC molecules on the surface of the membrane played a key role in obtaining carbon nanotubes with controlled graphene layer orientation.
Very recently, we found that pyrolysis of a HBC-cobalt complex 32 in the solid state yielded nearly quantitative yields of uniform either “bamboo-shaped” or straight nanotubes (Scheme 9).35 The shape and yield of the carbon nanostructures can be finely controlled by regulation of the heating process. This provides an alternative high-yielding synthesis of carbon nanotubes to supplement the existing methods such as carbon vapour deposition, electron discharge and laser ablation. The precursor 32 contains both an as-ready graphite carbon source (HBC) and a catalyst precursor (Co2(CO)6), and at high temperatures the graphite islands form tube-like graphene structures on the cobalt catalyst units.
 | ||
| Scheme 9 High-yielding synthesis of carbon nanotubes by pyrolysis of an HBC-cobalt complex in the solid state. | ||
The above results show that it is possible to construct well-defined arrays of two-dimensional phenylene units, which are of great interest due to their potential applications in hydrogen or lithium storage, but further work is still needed to obtain processable materials for testing. Clearly the successful conversion of graphite islands such as HBC to well-defined carbon nanotubes also has promising applications in nanoscale electronics.
Conclusions
Methods have been developed for preparation of polyphenylene compounds incorporating combinations of one-, two- and three-dimensional polyphenylene units in order to obtain materials with novel architectures and properties. Attachment of bulky three-dimensional polyphenylene dendrimer units to one-dimensional conjugated phenylene polymers or to two-dimensional discotic graphite molecules hinders interactions between the chains or discs, thus enabling control of their optical and electronic properties. For example, the dendronised polymers display reduced exciton migration and longer triplet lifetimes, which affect their luminescence efficiency. Further, deeper understanding of the effects of the bulky substituents may lead to new ways to improve the performance of such polymers in electronic devices. Similarly dendronisation of two-dimensional discotic graphite molecules hinders stacking of these systems so that they can only form at most dimers and not extended columns. By controlling the degree of steric bulk of the substituents it is possible to control the degree of dimer formation.HBC units can be linked together by σ-bonds to form dimers and trimers. By varying the geometry of these molecules it is possible to obtain either electronically coupled or decoupled systems which show markedly different optical properties and self-assembly behaviour, thus providing control of their electronic properties. In a related approach more linear-like extended graphite molecules have been made. This has been extended to the oxidation of branched polyphenylenes to produce graphite ribbons. These materials are kinked due to isomers introduced during synthesis of the precursors, and so further improvement of this approach to obtain completely linear ribbons is still required. Such materials would be expected to act like one-dimensional graphite.
Partial cyclodehydrogenation of a polyphenylene dendrimer produces propeller-like molecules containing a three-dimensional array of planar graphitic discs around a benzene or triarylbenezene core. Materials containing such nanoarchitectures have potential for storage of lithium or hydrogen and so the development of large but soluble propeller molecules for testing in such applications is an important target for future research. The controlled pyrolysis of graphite molecules has also been used to make new three-dimensional carbon materials containing two-dimensional phenylene nanostructures.
The above results open the way towards the development of means for the controllable preparation of complex nanoscale architectures with tunable optoelectronic properties for use in putative microelectronic or nanoelectronic devices. There is still considerable work to be done before such devices are feasible, but the first steps towards the sort of complex nano-architectures required have been taken. As a result a true nanotechnology while still in the future is no longer the preserve of science fiction, but a plausible and increasingly realistic goal for serious scientific research.
Acknowledgements
We wish to thank all our many colleagues and collaborators who have assisted in the progress of this research. Financial assistance from the European Commission (TMR Project SISITOMAS, Projects DISCEL and MACMES), the Deutsche Forschungsgemeinschaft (Schwerpunktprogramm organische Feldeffekttransistoren, SFB 625), the Fonds der Chemischen Industrie, the European Science Foundation (Project fund Bionics), and the Bundesministerium für Bildung and Forschung (Multifunctional Materials and Miniaturised Devices Centre 03N 6500, Project 13N8165 OLAS) is also gratefully acknowledged.References
- A. J. Berresheim, M. Müller and K. Müllen, Chem. Rev., 1999, 99, 1747 CrossRef CAS.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS.
- A. C. Grimsdale and K. Müllen, Chem. Record, 2001, 1, 243 CrossRef CAS.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew Chem., Int. Ed., 1998, 37, 402 CrossRef.
- C. D. Dimitrakopoulos and P. R. L. Malefant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15 CrossRef CAS.
- D. Neher, Macromol. Rapid Commun., 2001, 22, 1345 CrossRef.
- S. Setayesh, D. Marsitzky and K. Müllen, Macromolecules, 2000, 33, 2016 CrossRef CAS.
- A. C. Grimsdale, P. Leclère, R. Lazzaroni, J. D. MacKenzie, C. Murphy, S. Setayesh, C. Silva, R. H. Friend and K. Müllen, Adv. Funct. Mater., 2002, 12, 729 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946 CrossRef CAS.
- J. Jacob, L. Oldridge, J. Zhnag, M. Gaal, E. J. W. List, A. C. Grimsdale and K. Müllen, Curr. Appl. Phys., 2004, 4, 339 Search PubMed.
- A. Pogantsch, F. P. Wenzl, E. J. W. List, G. Leising, A. C. Grimsdale and K. Müllen, Adv. Mater., 2002, 14, 1061 CrossRef CAS.
- J. M. Lupton, P. Schouwink, P. E. Keivanidis, A. C. Grimsdale and K. Müllen, Adv. Funct. Mater., 2003, 13, 154 CrossRef CAS.
- A. Pogantsch, F. P. Wenzl, U. Scherf, A. C. Grimsdale, K. Müllen and E. J. W. List, J. Chem. Phys., 2002, 119, 6904 CrossRef.
- A. Pogantsch, C. Gadermaier, G. Cerullo, G. Lanzani, U. Scherf, A. C. Grimsdale, K. Müllen and E. J. W. List, Synth. Met., 2003, 139, 847 CrossRef CAS.
- A. M. van de Craats, J. M. Warman, J. D. Brand, Y. Geerts and K. Müllen, Adv. Mater., 1998, 10, 36 CrossRef CAS.
- A. M. van de Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand, M. A. Harbison and K. Müllen, Adv. Mater., 1999, 11, 1469 CrossRef CAS.
- A. M. van de Craats, N. Stutzmann, O. Bunk, M. M. Nielsen, M. Watson, K. Müllen, H. D. Chanzy, H. Sirringhaus and R. H. Friend, Adv. Mater., 2003, 15, 495 CrossRef CAS.
- J. Piris, M. G. Debye, N. Stutzmann, A. M. van de Craats, M. D. Watson, K. Müllen and J. M. Warman, Adv. Mater., 2003, 15, 1736 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS.
- C. D. Simpson, J. Wu, M. D. Watson and K. Müllen, J. Mater. Chem., 2004, 14, 494 RSC.
- J. Wu, M. D. Watson, N. Tchebotareva, Z. Wang and K. Müllen, J. Org. Chem., 2004, 69, 8194 CrossRef CAS.
- J. Wu, L. Gherghel, M. D. Watson, J. Li, Z. Wang, C. D. Simpson, U. Kolb and K. Müllen, Macromolecules, 2003, 36, 7082–7089 CrossRef CAS.
- J. Wu and M. D. Watson, Angew. Chem., Int. Ed., 2003, 42, 5329 CrossRef CAS.
- J. Wu, M. D. Watson, L. Zhang, Z. Wang and K. Müllen, J. Am. Chem. Soc., 2004, 126, 177 CrossRef CAS.
- J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kasteler, G. Fechtenkötter, M. Wagner and K. Müllen, J. Am. Chem. Soc., 2004, 126, 11311 CrossRef CAS.
- J. Wu, M. Baumgarten, M. G. Debije, J. M. Warman and K. Müllen, Angew. Chem., Int. Ed., 2004, 43, 5331 CrossRef CAS.
- C. D. Simpson, G. Mattersteig, K. Martin, L. Gherghel, R. E. Bauer, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2004, 126, 3139 CrossRef CAS.
- K. Sato, M. Noguchi, A. Demachi, N. Ok and M. Endo, Science, 1994, 264, 556 CrossRef CAS.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787 CrossRef CAS.
- L. Gherghel, C. Kübel, G. Lieser, H.-J. Räder and K. Müllen, J. Am. Chem. Soc., 2002, 124, 13130 CrossRef CAS.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Müllen, Angew. Chem., Int. Ed., 2004 Search PubMed , in press.
- J. Wu, B. El Hamaoui, J. Li, L. Zhi, U. Kolb and K. Müllen, Small, 2004 Search PubMed , in press.
| This journal is © The Royal Society of Chemistry 2005 |