Boron-modified polysilazane as a novel single-source precursor for SiBCN ceramic fibers: synthesis, melt-spinning, curing and ceramic conversion
S. Bernard*a, M. Weinmannb, P. Gerstelb, P. Miele†a and F. Aldingerb
aLaboratoire des Multimateriaux et Interfaces (UMR CNRS 5615), Université Claude Bernard, 43 bd du 11 Novembre 1918, F-69622 Villeurbanne Cedex, France. E-mail: Samuel.Bernard@univ-lyon1.fr
bMax-Planck-Institut für Metallforschung and Institut für Nichtmetallische Anorganische Materialien, Universität Stuttgart, Pulvermetallurgisches Laboratorium, Heisenbergstrasse 5, D-70569 Stuttgart, Germany
First published on 5th November 2004
Abstract
A novel boron-modified polysilazane functionalised with Si- and N-bonded methyl groups has been synthesised and characterised by means of FT-IR and NMR spectroscopies as well as elemental analysis. Both Si- and N-bonded methyl groups inhibited extensive cross-linking to yield a tractable polymer which was successfully processed into polymer green fibers by a melt-spinning process. After the shaping processing, the N-bonded methyl groups offered the capability of facile polymer cross-linking in an ammonia atmosphere at 200 °C to increase the ceramic yield of the polymer and avoid inter-fiber fusion. The as-obtained cured fibers were subsequently pyrolysed at 1400 °C in a nitrogen atmosphere to provide amorphous and stable Si3.0B1.0C5.0N2.4 ceramic fibers with ca. 1.3 GPa in tensile strength, ca. 170 GPa in Young's modulus and ca. 23 μm in diameter.
Introduction
The polymer-derived ceramic (PDC) approach is a chemical process in which pre-ceramic polymers are thermally decomposed in a controlled atmosphere into non-oxide covalently-bonded ceramics.1–4 The main interest of this route lies in the feasibility of tailoring polymers with adjustable viscoelastic properties for the preparation of fine-diameter fibers.5–7 For example, Si-based polymers including polysilanes8 and polysilazanes9 have proven to be promising tractable compounds for the elaboration of fibers by a spinning process. After a curing step using suitable agents, which is required for rendering green fibers infusible, and following an appropriate thermally-induced conversion of the cured fibers, Si-based ceramic fibers were obtained in the desired composition and microstructure. In particular, commercially available SiC and Si3N4 fibers have found broad applications as reinforcing materials in Ceramic Matrix Composites (CMCs) for high-temperature exposure up to 1200 °C.10,11 Above 1200 °C, the grain coarsening and crystal growth as well as the release of oxygen-containing gaseous species CO and/or SiO caused the breakdown of fibers and the loss of shape integrity.12,13 Since properties of CMCs depend highly upon the nature and type of the fibrous reinforcement, Si-based ceramic fibers should exhibit heat resistance and high mechanical properties if they are exposed to temperatures higher than 1200 °C. Such properties require that the amorphous state is retained at high temperature and that incorporation of undesirable oxygen, for example during the curing step, is suppressed. Improvement of the thermal stability of non-oxide ceramic fibers in comparison to conventional binary SiC and Si3N4 fibers can be achieved using polycarbosilazane-derived SiCxNy fibers.14,15 For example, Ziegler and co-workers14 reported that the amorphous state of SiCN fibers is retained at higher temperatures (T ≈ 1400 °C) than that of SiC fibers. However, recent publications showed that multi-component advanced ceramics exhibit improved properties compared to SiCN ceramics. In particular, ceramic fibers as well as bulk materials based on the elements silicon, boron, carbon and nitrogen remain amorphous even at very high temperatures (T ≈ 1700 °C).16–21 Initial work for the preparation of SiBCN fibers was done by Takamizawa et al. in 1986.16 They used a mixture of organopolysilane and organoborazine and achieved the spinning in the molten state. Ceramic fibers of ca. 11 μm in diameter, ca. 3.0 GPa in tensile strength and ca. 250 GPa in Young's modulus were produced after a curing step by oxidation or electron beam irradiation and a subsequent pyrolysis in a nitrogen atmosphere. These fibers remained amorphous up to 1500 °C in a nitrogen environment and their tensile strength was maintained at 2.0 GPa at this temperature. Lu et al.17 used polycarbosilazane (SiCN precursor) as the starting polymer and BCl3 as the curing agent to elaborate SiBCN fibers which were thermally stable up to 1600 °C. More recently, they investigated the production of SiBCN(O) fibers of 15.6 μm in diameter and 1.6 GPa in tensile strength from a mixture of polyorganosilane and polyborosilazane. However, this procedure, i.e. physical mixing of different polymers or heterogeneous introduction of elements during the curing and pyrolysis, does not lead to ideal compositional and structural homogeneities in the final ceramic. In order to (1) control the composition on an atomic level, (2) avoid phase separation, and (3) increase the stability of these materials, single-source precursors are required. Such molecules contain all elements in a desired stoichiometry and chemical environment. Following this approach, Sneddon and co-workers synthesised polyborosilazanes with appropriate rheological properties and thermal stability for a melt-spinning process.18 These polymers were prepared by chemical modification of hydridopolysilazanes (HPZ) with various monofunctional borane or borazine derivatives [pinacolborane (PIN) or B-diethylborazine (DEB)]. For example, 30–40 μm PIN–HPZ green fibers were cured with HSiCl3 acid following by exposure to humid air and subsequently pyrolysed in an argon atmosphere into SiBCN fibers. The final material was amorphous at 1600 °C. Jansen and co-workers prepared N-methylpolyborosilazanes by aminolysis of trichlorosilylaminodichloroborane (TADB) via a monomer route (Fig. 1).19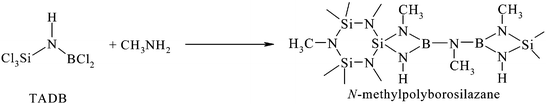 | ||
| Fig. 1 Synthesis of single-source N-methylpolyborosilazanes via Jansen's route. | ||
These polyborosilazanes can be tailored to be meltable and spinnable by melt- or solution-spinning processes. The resulting green fibers were cured in HSiCl3 and pyrolysed up to 1500 °C in a nitrogen atmosphere. Amorphous SiBCN fibers of 3.0 GPa in tensile strength, 8–15 μm in diameter and a length exceeding 200 m were obtained. Such fibers remained thermally stable up to ca. 1750 °C in a non-oxidizing atmosphere (0.1 MPa, He) and retained their tensile strength around 3.0 GPa at 1400 °C in oxidizing conditions.
An alternative access to SiBCN precursors was developed by Riedel et al. (Fig. 2).20
![Synthesis of single-source precursors of the type [B(C2H4SiCH3NH)3]n (3, C2H4 = CHCH3, CH2CH2).](/image/article/2005/JM/b408295h/b408295h-f2.gif) | ||
| Fig. 2 Synthesis of single-source precursors of the type [B(C2H4SiCH3NH)3]n (3, C2H4 = CHCH3, CH2CH2). | ||
From dichloromethylvinylsilane 1, the monomeric tris(dichloromethylsilylethyl)borane B(C2H4SiCH3Cl2)32 (C2H4 = CHCH3, CH2CH2) was synthesised by quantitative hydroboration using borane dimethylsulfide. Subsequent ammonolysis yielded the boron-modified polysilazane [B(C2H4SiCH3NH)3]n3. It represents a polysilazane which is cross-linked via C–B–C bridges. We described recently that B(C2H4SiHCl2)3 is an alternative synthon for the preparation of boron-modified polysilazanes. After ammonolysis, it gave a precursor with a significantly higher ceramic yield.21 In this synthesis route, the polymerisation step proceeds quickly and provides polymers with a high degree of cross-linking due to the polycondensation ability of the N–H functions. Furthermore, the Si–H sites supply latent reactivity to thermoset the resulting polymer during further pyrolysis by dehydrogenative Si–N coupling in an argon atmosphere. Both the highly cross-linked structure and the capability of these polymers to thermally cross-link allow us to obtain high-performance bulk materials in high ceramic yields. For example, bulk amorphous ceramics derived from such polymers can be thermally stable up to 2000 °C in an argon environment.20,21 In return, an important disadvantage of such polymers is their insufficient softening on heating and their insolubility, each of which prevents the preparation of fibers requiring either melt- or solution-processing.21 These observations point out that one of the demanding problems for processing fibers is to develop polymers with the appropriate rheology and melt-stability to allow for continuous melt-spinning. The required properties may be provided by the introduction of specific groups to the polymer backbone. With this aim in mind, our strategy was to avoid excessive reactive Si–H and N–H units in the pre-ceramic polymer. Therefore, we used B(C2H4SiCH3Cl2)32 as the monomer which was polymerised with methylamine to introduce N-methyl groups into the polymer backbone. The present paper is devoted to a detailed study on the synthesis of a C–B–C-bridge-containing polyborosilazane without Si–H and N–H sites. It will be shown that rheology and melt-stability can be ideally tailored for the melt-spinning process and that green fibers derived from [B(C2H4SiCH3NCH3)3]n may be rendered infusible by a curing step in an ammonia atmosphere at 200 °C. As-cured fibers are then pyrolysed in a nitrogen atmosphere at 1400 °C yielding high-temperature stable amorphous SiBCN fibers.
Experimental
1. General comments
All reactions were carried out in a purified argon atmosphere using standard Schlenk techniques.22 Dichloromethylvinylsilane CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiCH3Cl21 was obtained from ABCR chemicals and freshly distilled from magnesium before use. Borane dimethylsulfide BH3·S(CH3)2
(2 M solution in toluene) was obtained from Sigma Aldrich. Methylamine (methylamine anhydrous, 98+% from Sigma Aldrich) was dried with KOH prior to its use. Tetrahydrofuran (THF) and toluene were purified by distillation from potassium. Argon, nitrogen and ammonia with 99.999% purity were used during the specimen preparation.
CHSiCH3Cl21 was obtained from ABCR chemicals and freshly distilled from magnesium before use. Borane dimethylsulfide BH3·S(CH3)2
(2 M solution in toluene) was obtained from Sigma Aldrich. Methylamine (methylamine anhydrous, 98+% from Sigma Aldrich) was dried with KOH prior to its use. Tetrahydrofuran (THF) and toluene were purified by distillation from potassium. Argon, nitrogen and ammonia with 99.999% purity were used during the specimen preparation.2. Polymer synthesis
The synthesis of the tris(dichloromethylsilylethyl)borane B(C2H4SiCH3Cl2)3 (2, C2H4 = CHCH3, CH2CH2) was performed from 1 according to procedures described in the literature.23 In a 2000 ml Schlenk flask equipped with a water-cooled reflux condenser and a gas inlet tube, 30.80 g (70.5 mmol) of 2 were dissolved in 500 ml of THF and cooled to 0 °C. 20.43 g (634.5 mmol) of methylamine were slowly introduced into the solution under vigorous stirring whereby methylamine hydrochloride precipitation was immediately observed. During the reaction, the temperature was maintained below 2 °C. After the addition of methylamine was completed, the reaction mixture was allowed to warm up to room temperature. The polymer solution was then separated from the precipitated methylamine hydrochloride by filtration through a pad of Celite. The precipitate was thoroughly extracted three times with 50 ml of solvent and then disposed of. The filtrate and the extract were combined and concentrated in a high vacuum (room temperature/10−2 mbar) to produce 20.90 g (67.10 mmol, 96%) of [B(C2H4SiCH3NCH3)3]n (4) as a white powder that is sensitive to moisture and air. Tg ≈ 38 °C. Anal. Found (wt%): C, 47.5; H, 9.9; N, 16.7; B, 3.0; Si, 22.9 [C12H30N3BSi3]n ([311.46]n). Calculated (wt%): C, 46.28; H, 9.71; N, 13.49; B, 3.47; Si, 27.05. Chemical shift values in NMR spectra are reported in ppm relative to (CH3)4Si (1H, 13C; δ = 0). All resonance signals appear broadened. 1H NMR (C6D6) (ppm): δ 0.38 (br,![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiCH3), 0.59 (vbr, SiCH2CH2B), 0.98 (br, CH3CHBSi), 1.03 (br, SiCH2CH2B), 1.33 (br, CH3CHBSi), 2.78 (vbr, N–CH3); 13C NMR (C6D6)
(ppm): δ
−3.03 (SiCH3), 12.94 (SiCH2CH2B), 25.58 (CH3CHBSi), 27.91 (–NHCH3), 30.70 (CH3CHBSi), 32.56 (NCH3); IR (KBr): ν(CN–H)
(terminating N–H)
= 3425, 3313, 3235 (w); ν(SiC–H)
= 2951, 2889 (s); ν(NC–H)
= 2800 (m); δ(CN–H)
(terminating N–H)
= 1596 (m); δ(NCH3)
= 1460 (m); δas(CHCH3)
= 1407 (m); δs(CHCH3)
= 1359 (m); δs(SiCH3)
= 1254 (s); δ(C–B–C)
= 1181 (m); δ(NH)
(SiNHSi)
= not observed; δ(SiCH2C)
= 1145; ν(N–CH3)
= 1076 (s); δ(N–Si–N)
= 913 (sh)–870 (vs).
SiCH3), 0.59 (vbr, SiCH2CH2B), 0.98 (br, CH3CHBSi), 1.03 (br, SiCH2CH2B), 1.33 (br, CH3CHBSi), 2.78 (vbr, N–CH3); 13C NMR (C6D6)
(ppm): δ
−3.03 (SiCH3), 12.94 (SiCH2CH2B), 25.58 (CH3CHBSi), 27.91 (–NHCH3), 30.70 (CH3CHBSi), 32.56 (NCH3); IR (KBr): ν(CN–H)
(terminating N–H)
= 3425, 3313, 3235 (w); ν(SiC–H)
= 2951, 2889 (s); ν(NC–H)
= 2800 (m); δ(CN–H)
(terminating N–H)
= 1596 (m); δ(NCH3)
= 1460 (m); δas(CHCH3)
= 1407 (m); δs(CHCH3)
= 1359 (m); δs(SiCH3)
= 1254 (s); δ(C–B–C)
= 1181 (m); δ(NH)
(SiNHSi)
= not observed; δ(SiCH2C)
= 1145; ν(N–CH3)
= 1076 (s); δ(N–Si–N)
= 913 (sh)–870 (vs).3. Polymer green fiber preparation
Polymer green fibers were prepared in a nitrogen atmosphere using a lab-scale melt-spinning apparatus set up in a glove-box. Compound 4 was molten by heating within a heater block until an appropriate viscosity was obtained and progressively compacted by a piston. The molten polymer flowing was then driven through heated elements containing a filter and spinneret having a single 0.2 mm capillary. The resulting polymer emerging from the capillary as an endless filament was stretched and continuously collected on a rotating spool.4. Polymer-to-ceramic conversion
Green fibers were converted by appropriate curing and pyrolysis processes leading to SiBCN ceramic fibers. Green fibers were first cured in an ammonia–nitrogen atmosphere (70 : 30) (25 °C–200 °C) for ca. 15 h: from T = 25 to 70 °C at a heating rate of 25 °C h−1, then held for 2h; T = 70 to 200 °C at 15 °C h−1, then held for 2 h in a silicate tube furnace. The following heat-treatment was carried out in the same furnace in a pure nitrogen atmosphere to 1000 °C: T = 200 to 1000 °C at 50 °C h−1, then held for 30 min. An additional heat-treatment was performed in a high-temperature graphite furnace in a nitrogen atmosphere from T = 25 to 1400 °C at 100 °C h−1, then held at this temperature for 2 h to ensure the complete ceramic conversion of the fibers. As-obtained SiBCN fibers were collected together.5. Characterisation
As the polyborosilazane as well as cured fibers are reactive towards moisture and oxygen, the following sample preparations were performed within a glove-box in an argon environment. In contrast, no precaution was necessary with SiBCN fibers after pyrolysis at 1400 °C.The chemical structure of 4 was determined by FT-IR spectroscopy using a Bruker IF66 spectrometer in KBr pellets. 1H and 13C NMR spectra were obtained using a Bruker AM 300 spectrometer in C6D6 operating at 300 and 62.5 MHz, respectively. Tetramethylsilane (TMS) was used as a reference for the NMR data. Elemental analyses were performed using various apparatus (ELEMENTAR, Vario EL CHN-Determinator; ELTRA, CS 800, C/S Determinator; LECO, TC-436, N/O Determinator) and by atom emission spectrometry (ISA JOBIN YVON JY70 Plus). Thermal properties (polymer softening and decomposition) were studied by differential scanning calorimetry (DSC, Mettler Toledo DSC TA 8000) in an argon atmosphere between −50 and 150 °C at a heating rate of 10 °C min−1 in alumina crucibles. Melt-behaviour was investigated by thermomechanical analysis (TMA, Mettler Toledo TMA/SDTA 840) on polymer pellets in a nitrogen atmosphere from 30 to 160 °C at a heating rate of 5 °C min−1. Thermogravimetric analyses (TGA, Setaram TGA 92 16–18) of the polymer-to-ceramic conversion was carried out in a nitrogen atmosphere (50 cm3 min−1) from 25 to 1000 °C at a heating rate of 1 °C min−1 in silicate crucibles. High-temperature thermogravimetric analysis (HT-TGA, Mettler Toledo TGA/SDTA 851 equipment) of amorphous SiBCN fibers was performed in air from 25 to 1500 °C at a heating rate of 10 °C min−1 using platinum crucibles. The crystallisation of amorphous SiBCN fibers was investigated in graphite furnaces in a nitrogen atmosphere from 1400 to 2000 °C at a heating rate of 10 °C min−1 (100 °C steps up to 1600 °C; 50 °C steps from 1600 to 1750 °C for 1 h each). XRD was performed on ground-up fibers using a Siemens D5000/Kristalloflex diffractometer (CuKα1 radiation), equipped with an OED and a quartz primary monochromator. Fiber morphology was observed by scanning electron microscopy (SEM) with field emission equipment, Hitachi S800. The cross-section as well as the surface of the fibers were investigated. Fibers were glued onto a aluminium support and polymer green fibers were metallized with an Au/Pd film prior to their observation. Tensile tests and diameter values were obtained at room temperature from single filaments with a gauge length of 10 mm. 42 single filaments were taken from bundles throughout to incorporate any possible property variation within the lot in fiber fracture statistics. Each filament was centreline mounted and both tips were glued onto special slotted tabs for handling and testing. The diameter ϕ of each filament was measured by laser interferometry in two positions along the filament axis and an average diameter of SiBCN fibers was calculated from the 42 as-obtained values. Single filament tensile properties were determined using a standard tensile tester (Adamel Lhomargy DY 22) including two grips, a displacement transducer and a load cell (5 N). The cross-head speed was fixed at 0.1 mm min−1. Filament cross-sectional areas (A) were determined from the values of both diameters and initial lengths of the filament (lo = 10 mm). Failure strain ε, Young's modulus E, and tensile strength σ were measured from data of breaking load–elongation curve records and cross-sectional area calculations. Each value of failure strain and Young's modulus was modified with system compliance. The average failure strain and Young's modulus were calculated from the 42 tests. The statistical variability of the strength was reported in terms of Weibull statistics.24 An average tensile strength σ of the SiBCN fibers was then estimated for a failure probability P = 0.632.
Results and discussion
1. Polymer synthesis
Hydroboration of vinyldichlorosilanes and subsequent ammonolysis of resulting hydroborated compounds is a well-established procedure for the preparation of SiBCN polymers.20,21,23 According to this two-step approach, Si-methyl- and N-methyl-containing polyborosilazane 4 was obtained by the quantitative hydroboration of the vinyl group of the starting dichloromethylvinylsilane 1 with borane dimethylsulfide and the subsequent aminolysis of the resulting monomeric tris(dichloromethylsilylethyl)borane 2 (Fig. 3).![Synthesis of [B(C2H4SiCH3NCH3)3]n (4, C2H4 = CHCH3, CH2CH2).](/image/article/2005/JM/b408295h/b408295h-f3.gif) | ||
| Fig. 3 Synthesis of [B(C2H4SiCH3NCH3)3]n (4, C2H4 = CHCH3, CH2CH2). | ||
H2CCHSiCH3Cl21 was dissolved in toluene and a 2 M solution of BH3·S(CH3)2 in toluene was carefully added at 0 °C. After evaporation of the solvent and (CH3)2S at 60 °C, a moisture- and air-sensitive tris(dichloromethylsilylethyl)borane B(C2H4SiCH3Cl2)32 was obtained as a colourless liquid which was used without further purification. Spectroscopic data are consistent with those well-described in the literature23,25 and therefore are not discussed here. A 70.5 mmol portion of B(C2H4SiCH3Cl2)32 was dissolved in THF and an excess of methylamine was added slowly at 0 °C. The formation of large quantities of solid by-product methylamine hydrochloride necessitated vigorous stirring of the reaction mixture. The reaction between B(C2H4SiCH3Cl2)3 and CH3NH2 proceeded quantitatively; the separation of 4 from the by-product methylamine hydrochloride by filtration resulted in a minor low product loss. Compound 4 was purified by vacuum evaporation of all volatile compounds at room temperature. It was generated as a very highly soluble white powder in 96% yield. Remarkably, once it has been completely dried in vacuum, 4 can be redissolved easily in contrast to the less soluble ammonolysed analogs of the type [B(C2H4SiRNH)3]n
(R = CH320 and H;21 C2H4
= CHCH3, CH2CH2).
2. Chemical analysis and spectroscopy characterisation
The chemical composition and structure of compound 4 were investigated by NMR in solution, FT-IR spectroscopy and microanalysis.It is difficult to structurally characterise C–B–C-containing polyborosilazanes because of the limitation of applicable spectroscopic methods. For example, it has already been published that the hydroboration of CH2CHSiRCl2
(R = CH3, H) with borane dimethylsulfide is not regioselective and a mixture of the various regioisomers due to α
(CH3CHBSi)- as well as β
(SiCH2CH2B)-hydroboration is produced.26 Consequently, in NMR solution of the monomeric B(C2H4SiRCl2)3, the appearance of a series of spectroscopically differentiable isomers causes sets of resonance signals that overlap. Moreover, the ammonolysis reaction of B(C2H4SiRCl2)3 generally yields polymers with a poor solubility. Therefore, high-resolution NMR spectroscopy solution of the C–B–C-containing polyborosilazanes cannot generally be applied.
Here, polymer 4 is soluble in C6D6 but both 1H and 13C NMR spectra are very complex and display broad signals. The broad resonances indicated that the magnetic environments around H and C are not unique, suggesting previously quoted features as well as branching and cyclic/polycyclic silazane structures.
In the 1H NMR spectrum, the polymer 4 shows a broad signal centred at 0.38 ppm which is attributed to the Si-bonded methyl groups. The very broad Si–CH2 resonance signal due to the β-hydroboration is observed at 0.59 ppm. Both signals for the formation of an alkyl bridge through the incorporation of boron by α- and β-hydroboration of vinyl groups are crowded together in the 0.80–1.72 ppm region. In particular, the 1H spectrum shows a resonance assigned to the CH3CHBSi protons centred at 0.98 ppm whereas the signals attributed to SiCH2CH2B and CH3CHBSi protons are centred at 1.03 and 1.33 ppm, respectively. The incorporation of N–CH3 units by aminolysis of B(C2H4SiCH3Cl2)3 is characterised by a broad signal emerging at 2.78 ppm. In contrast, no signals assigning unreacted vinyl side groups which would point out an incomplete addition of the borane to the CC double bond during hydroboration reactions are observed.
The integration of the 1H signals is in the appropriate ratio calculated from the reaction depicted in Fig. 3. In particular, the Si–CH3 : N–CH3 integration ratio is 0.95, indicating that the ammonolysis of 3 occurs via the expected pathway. The structure of 4 is confirmed by 13C NMR. The spectrum shows a broad signal at −3.03 ppm assigned to SiCH3 sites. The formation of a C–B–C bridge by α- and β-hydroboration is identified in the 9.0–35.0 ppm region: the hydroboration reaction of borane to the α-vinyl carbon atoms is characterised by signals at 25.58 ppm (CH3CHBSi units) and 30.70 ppm (CH3CHBSi units) whereas the SiCH2CH2B units resulting from β-addition are characterised by a broad signal centred at 12.94 ppm. Owing to the broadening of the signal, no distinction between SiCH2CH2B and SiCH2CH2B carbon atoms can be made.
The introduction of methylamino groups by aminolysis is shown by signals at 27.91 (–NHCH3) and 32.56 ppm (N–CH3). The signal at 27.91 ppm indicates the presence of –NHCH3 ending groups in the polymer which reflects that N-bonded methyl groups inhibit extensive condensation reactions. Therefore, the polymer in part is a linear silazane terminated by –NHCH3 groups as illustrated in Fig. 4. The small amounts of –NHCH3 units are probably responsible for the high solubility of 4.
![Terminal –N(H)CH3 groups in [B(C2H4SiCH3NCH3)3]n (4, C2H4 = CHCH3, CH2CH2).](/image/article/2005/JM/b408295h/b408295h-f4.gif) | ||
| Fig. 4 Terminal –N(H)CH3 groups in [B(C2H4SiCH3NCH3)3]n (4, C2H4 = CHCH3, CH2CH2). | ||
In addition to the NMR data, some absorptions in the FT-IR spectrum are useful indications for determining the structure of the investigated compound. The IR spectrum of 4 [seen later in Fig. 8(a)] shows the characteristic stretching and deformation bands of all expected motifs and supports previous interpretations.
The series of weak absorption signals for the N–H stretching modes in the 3235–3425 cm−1 region confirm the presence of –NHCH3 ending groups in the polymer structure. Strong and very broad aliphatic C–H bands are also observed in the 2800–2951 cm−1 range. In particular, N-bonded C–H [ν(NC–H)
= 2800 cm−1] and Si-bonded C–H [ν(SiC–H)
= 2951 and 2889 cm−1] vibrations can be differentiated. The band assigned to N–H bond deformation in the –NHCH3 ending groups is located at 1596 cm−1 whereas those which are assigned to the deformation and stretching of the NCH3 units are located at 1460 [δ(NCH3)] and 1076 cm−1 [ν(N–CH3)], respectively. Other bands are characteristic of common C–B–C-bridge-containing polyborosilazanes. In particular, a Si–CH3 deformation band at 1254 cm−1 and a broad N–Si–N asymmetric stretch at 870–913 cm−1 as well as δ(C–B–C) at 1181 cm−1 are observed. The latter indicates, in accordance with NMR investigations, a trigonal planar coordination of boron by three carbon atoms due to α- and/or β- hydroboration reactions. The assigned major absorptions in the infrared spectrum are compiled in the Experimental section.
According to the elemental analysis, polyborosilazane 4 has an empirical formula Si3.0B1.0C14.5N4.4H36.3 per monomer unit (oxygen values were determined to be <2 wt% and are omitted). The elemental analysis data for [B(C2H4SiCH3NCH3)3]n definitively prove that the synthesis of the precursor via the monomer route occurs in the expected pathway since they agree reasonably well with the theoretical values. In particular, the Si : B ratio of 3 : 1 in the polymer is retained. There is, in contrast, a deviation of the determined and calculated nitrogen, carbon and hydrogen values. The found values are higher than expected. The –NHCH3 ending groups are most likely the reason for excessive carbon, nitrogen and hydrogen contents as previously shown by NMR and IR spectroscopies.
3. Melt-spinning studies – preparation of polymer green fibers
Once the polyborosilazane is prepared, it is necessary to assess its capability to form polymer fibers by melt-spinning. Differential scanning calorimetry [DSC, Fig. 5(a)] provides valuable data on the softening properties of the polymer as well as its thermal degradation. The DSC curve is completed by the thermomechanical analysis curve [TMA, Fig. 5(b)] using a compressive load of 3 × 10−1 N. The latter technique was used to observe the dimensional change of the polymer under load upon heating.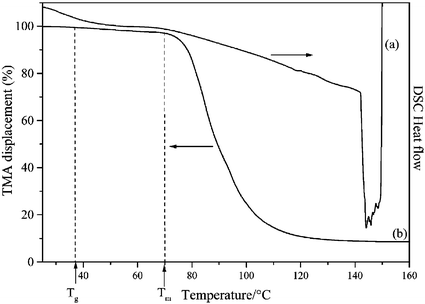 | ||
| Fig. 5 Differential scanning calorimetry (heating rate 10 °C min−1, argon atmosphere) (a) and thermomechanical analysis (heating rate 5 °C min−1, nitrogen atmosphere) (b) curves. | ||
Upon heating, 4 transforms into a softened compound through the glass transition which extends over 25 °C going from 30 to 55 °C. The wide range of the glass transition may be interpreted as a high molar mass dispersion in the polymer. The glass transition temperature of 4 is centred at 38 °C. The observed low softening point of 4 results from the low cross-linking density of the polymer network. Above the glass transition, the softened polymer remains stable up to 140 °C after which point a strong exothermic peak due to the polymer decomposition. Therefore, the polymer can be heated in the temperature range 25–140 °C without change in its physical properties and chemical composition. From these results, the polymer can be considered stable as a melt from 38 to 140 °C.
Fig. 5(b) shows the TMA curve of 4 measured between 25 and 160 °C. From the TMA, it is clear that the sample strongly shrinks in thickness from 70 °C (Tm) and the large dimensional change, reflecting the melting of the polymer, extends over 80 °C from 70 to 150 °C.
The obtained DSC and TMA data suggest that [B(C2H4SiCH3NCH3)3]n appears to be a potential candidate for the preparation of polymer green fibers. It exhibits a low glass transition temperature and, owing to the absence of potential cross-linking motifs, neither thermal cross-linking nor decomposition occur in the 38–140 °C temperature range in which the polymer is in the molten state. Obviously, both Si- and N-bonded methyl groups act as plasticizers and increase the chain flexibility compared to its hydrogen-substituted derivative [B(C2H4SiHNH)3]n. Owing to its controlled rheology (low Tg) and the high dimensional change upon heating, [B(C2H4SiCH3NCH3)3]n exhibits good melt-processability (i.e. melt-viscosity) qualifying it as a potential polymer for melt-spinning in the viscous region shown on the TMA curve (70–150 °C). As an illustration, using a lab-scale melt-spinning apparatus, 4 could be easily spun at 82 °C, where an optimal viscosity is attained, through a capillary of a single-hole spinneret of 200 μm in diameter. The resulting colourless and flexible endless filament fell under gravity and was immediately taken up on a rotating spool. Without optimising the conditions, typical endless polymer green fibers were extruded from the melt at 1.3 mm min−1. They could be stretched by the spool at a low rotation rate of ca. 50 m min−1 to produce flexible endless fibers ca. 55 μm in diameter which were continuously collected on the spool (Fig. 6).
 | ||
| Fig. 6 SEM micrographs of the as-spun polyborosilazane fibers. | ||
In accordance with the melt-stability of 4 up to 140 °C, its spinning remained stable leading to a good reproducibility of the extrusion operation. Consequently, uniform and defect-free green fibers were obtained. However, the following features are certainly responsible for the medium stretchability of the polymer filament: (1) the low softening temperature of 4 which is close to room temperature, (2) the large glass transition, and (3) the high dimensional change upon heating.
4. Polymer-to-ceramic conversion
As-spun fibers were cured and subsequently pyrolysed to produce black ceramic SiBCN fibers. During heat-treatment, fibers were maintained on the spool to prevent crimping due to shrinking effects caused by the weight loss of the polymer and the density increase of the fibers.First, the weight change of 4 was recorded using thermogravimetric analysis (TGA) upon heating up to 1000 °C in a nitrogen atmosphere [Fig. 7(a)].
![TGA curves for [B(C2H4SiCH3NCH3)3]n (a), [B(C2H4SiCH3NCH3)3]n treated in NH3 at 70 °C (b), and in NH3 at 200 °C (c).](/image/article/2005/JM/b408295h/b408295h-f7.gif) | ||
| Fig. 7 TGA curves for [B(C2H4SiCH3NCH3)3]n (a), [B(C2H4SiCH3NCH3)3]n treated in NH3 at 70 °C (b), and in NH3 at 200 °C (c). | ||
The as-obtained polyborosilazane rapidly decomposes and provides SiBCN ceramics in only 29.6 wt% ceramic yield. In comparison with previous works,21 the TGA data suggest that the substitution of hydrogen atoms in untractable [B(C2H4SiHNH)3]n with methyl units in [B(C2H4SiCH3NCH3)3]n significantly decreases the ceramic yield. For example, [B(C2H4SiHNH)3]n transformed into an amorphous ceramic in 88% yield (Ar, 1050 °C). This is a consequence of its higher degree of cross-linking as well as the presence of Si–H motifs with latent reactivity which inhibit depolymerisation and thus volatilisation of low molecular weight species. The low ceramic yield of [B(C2H4SiCH3NCH3)3]n is thus a consequence of both its low dimensional (linear) structure and the lack of potential cross-linking sites which are pre-requisites for high ceramic yields. As a consequence, one of the limitations of the spinnable polymer as a potential precursor is certainly the volatilisation of lower molecular weight species in addition to the evolution of typical gaseous by-products (dihydrogen, hydrocarbons, amines, etc.). Owing to the very low ceramic yield, the integrity of green fibers derived from [B(C2H4SiCH3NCH3)3]n cannot be preserved during heat-treatment. It is therefore necessary to investigate an appropriate curing process to render the green fibers infusible by improving the cross-linking density of the polymer and, therefore, its ceramic yield during the polymer-to-ceramic conversion.
Si–CH3 and/or N–CH3 units with thermally reactive Si–H and/or N–H units in [B(C2H4SiCH3NCH3)3]n should improve ceramic yields and thus render green fibers infusible. A chemical approach was to introduce potential cross-linking sites such as Si–H and/or N–H into the polymer structure after spinning by replacing Si–CH3 and/or N–CH3 units.It is known that carbon-containing organic groups in various precursors including AlN27 and BN28 precursors or polycarbosilanes29 as well as polysilazanes30 can be removed through pyrolysis in a reactive atmosphere of ammonia. To interlock the polymer backbones and increase the cross-linking density of [B(C2H4SiCH3NCH3)3]n, a curing process was developed in which the green fibers are slowly heated to 200 °C in an ammonia atmosphere. The effect of this treatment on the polymer decomposition was monitored by TGA [Fig. 7(b) and (c)], FT-IR measurements [Fig. 8(b) and (c)], and elemental analysis (Table 1). To observe the progress in the curing step in an ammonia atmosphere, an intermediate fiber sample was taken after heating to 70 °C.
![FT-IR spectra for [B(C2H4SiCH3NCH3)3]n (a), green fibers treated in NH3 at 70 °C (b), green fibers treated in NH3 at 200 °C, (c) and as-obtained ceramic fibers (d).](/image/article/2005/JM/b408295h/b408295h-f8.gif) | ||
| Fig. 8 FT-IR spectra for [B(C2H4SiCH3NCH3)3]n (a), green fibers treated in NH3 at 70 °C (b), green fibers treated in NH3 at 200 °C, (c) and as-obtained ceramic fibers (d). | ||
| Content (wt%)a | Found formulab | |||||
|---|---|---|---|---|---|---|
| Si | B | C | N | H | ||
| a Referenced to 100%; oxygen values were determined to be <2 wt%.b Empirical formula normalized to three Si atoms. | ||||||
| [B(C2H4SiCH3NCH3)3]n | 22.9 | 3.0 | 47.5 | 16.7 | 9.9 | Si3.0B1.0C14.5N4.4H36.3 |
| Green fibers heated at 70 °C | 28.1 | 3.8 | 42.6 | 16.0 | 9.5 | Si3.0B1.0C10.6N3.4H28.5 |
| Green fibers heated at 200 °C | 30.6 | 4.3 | 41.8 | 14.6 | 8.7 | Si3.0B1.1C9.6N2.9H24.0 |
| SiBCN fibers heated at 1400 °C | 44.6 | 6.1 | 31.6 | 17.7 | — | Si3.0B1.0C5.0N2.4 |
Examination of the IR spectrum of the fibers after annealing in ammonia at 70 °C [Fig. 8(b)] shows absorption bands similar to those of the starting polymer with only small changes in intensity [Fig. 8(a)].
In particular, the decreasing intensity in the series of N–H bands in the 3235–3425 cm−1 region, C–H bond stretching (2951, 2889 and 2800 cm−1), H3CN–H deformation (1596 cm−1) as well as H3C–N deformation (1460 cm−1) and stretching (1076 cm−1) suggests the loss of methylamino groups during cross-linking. The concurrent increasing of bands centred at 3425 and 1160 cm−1 are respectively assigned to stretching and deformation bands of N–H units bridging two silicon atoms (Si–NH–Si) in the pre-ceramic network. These findings directly reflect the substitution of the N–CH3 units with N–H functions producing Si–NH–Si chains. Moreover, a small shoulder to the left of the N–H bond deformation in the –NHCH3 ending groups appears at 1631 cm−1 in Fig. 8(b) resulting from the scissors deformation of –NH2 groups bonded to Si atoms. The symmetric deformation of –NH2 groups usually occurs in the region 1590–1650 cm−1.31 No bands which could be assigned to Si–H units (2144 cm−1) are detected after heating at 70 °C. Moreover, the intensity of the band corresponding to Si–CH3
(1254 cm−1) remains unchanged. Chemical analyses of the fibers heated at 70 °C support the interpretations performed on the basis of infrared spectroscopy (Table 1).
In particular, the Si : B atomic ratio of 3 : 1 in the polymer is sustained in the green fibers heated at 70 °C, indicating that no reactions involving boron or silicon species occur. Volatilisation of lower molecular weight Si- and/or B-based species is therefore not observed in an ammonia atmosphere. Against this, carbon, nitrogen and hydrogen contents decrease.
These findings may support the conclusion that cross-linking of [B(C2H4SiCH3NCH3)3]n in an ammonia atmosphere at 70 °C predominantly occurs via amine-exchange reactions. The general pathway (Fig. 9) most probably involves several steps according to previous investigations. Initially, NCH3 groups are displaced by amido groups (–NH2) whereby silylamine units (Si–NH2) form. These moieties subsequently condense through transamination reactions with the elimination of ammonia and/or methylamine as indicated in Fig. 9 leading to secondary silazanes (Si–NH–Si).
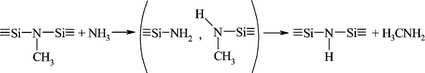 | ||
| Fig. 9 Transamination mechanism in the polymer during curing in an ammonia atmosphere. | ||
By further increasing the temperature, N–H units allow for further cross-linking through the development or tertiary silazanes via additional elimination of ammonia or methylamine.
Transaminations depicted in Fig. 9 are responsible for increasing the cross-linking density of [B(C2H4SiCH3NCH3)3]n. From the TGA investigations in Fig. 7, it is remarkable to find that the ceramic yield increases by ca. 28.3% from 29.6 wt% [Fig. 7(a)] to 57.9 wt% [Fig. 7(b)] simply through the introduction of ammonia as the curing gas. This points to the fact that ammonia is highly effective for cross-linking and making significant improvements to the ceramic yield of the polymer. This is unambiguously attributed to the presence of methylamino groups which are chemically sufficiently reactive towards ammonia even at comparably low temperature. As an illustration, FT-IR spectroscopy and elemental analysis investigations show that the transamination reactions involving NCH3 groups are preferred at low temperature, whereas, for example, the substitution of Si-bonded methyl groups with –NH2 groups did not take place. Replacement of the latter should involve a gain in nitrogen content in the resulting material which is not observed here from a chemical composition point of view.29,30
Increasing the curing temperature from 70 to 200 °C results in a continuous decrease in the IR bands assigned to –NHCH3 deformation and C–H stretching along with a noticeable increase in the intensity of a broad stretching band of Si2N–H units centred at 3429 cm−1 [Fig. 8(c)]. Accordingly, the intensity of deformation and stretching bands of H3C–N bonds (1460 and 1076 cm−1, respectively) decreases to the detriment of Si2NH deformation, typically for secondary silazanes (Si–NH–Si, 1160 cm−1). Against this, the intensity of the band assigned to Si–CH3 stretching remains unchanged by the curing process.
The chemical composition of fibers cured at 200 °C gives a calculated formula Si3.0B1.1C9.6N2.9H24.0, which confirms that the curing step at 200 °C is mainly associated with transamination reactions as suggested with the decrease in the carbon, nitrogen and hydrogen contents.
TGA profiles indicate that the major difference in weight loss between as-obtained [B(C2H4SiCH3NCH3)3]n and [B(C2H4SiCH3NCH3)3]n heated at 200 °C occurs in the low temperature range in which volatile Si-containing compounds and condensation products are usually volatilised. The TGA curve in Fig. 7(c) shows that fibers cured at 200 °C are thermally stable up to ca. 300 °C. The loss of gaseous by-products occurs between 300 and 650 °C. No weight loss is observed above 650 °C. At 1400 °C, the ceramic yield is well-enhanced and the evolution of volatile by-products during pyrolysis is considerably inhibited. Compared to the as-prepared polyborosilazane, the mass loss of the cross-linked polymer is reduced by ca. 44% to 26.5 wt%. In conclusion, exposure of green fibers to ammonia allows an efficient adsorption of the gaseous ammonia in the fiber, and chemical reactions, i.e. transaminations, which increase the degree of cross-linking of the polymer by the introduction of thermal reactive N–H sites, thus enhance ceramic yields.
A comparison of the empirical formulae of cured fibers and ceramic fibers (Table 1) suggests that the thermolytic conversion occurs with the liberation of hydrocarbons, ammonia, and amine as well as dihydrogen. Table 1 shows that the Si : B ratio ≈ 3 : 1 in the starting polymer is retained in the final ceramic fibers while the Si : N ratio slightly decreases. It is obvious that the higher carbon content in the starting polymer (47.5 wt%) compared with [B(C2H4SiCH3NH)3]n (40.5 wt%)20 and [B(C2H4SiHNH)3]n (34.9 wt%)21 is directly reflected in a higher amount of carbon in the final ceramic fibers (31.6 wt%) despite the use of ammonia during the curing process. As expected, adsorption signals for the N–H and C–H bond vibrations gradually disappear in the FT-IR spectra from 200 °C [Fig. 8(c)] to 1400 °C [Fig. 8(d)]. Only relatively broad absorption bands indicating the predominantly amorphous state of the materials are found. Absorptions at 1385 and 815 cm−1 point out the presence of amorphous BN segregation in the SiBCN fibers. Indeed, referring to the literature, the band centred at 1385 cm−1 is assigned to B–N bond vibrations.31 A second band assigned to the deformation of the B–N bond is observed at 815 cm−1. The band assigned to Si–C vibrations which should emerge at around 815 cm−1 is certainly superimposed with that of B–N deformation.
In order to determine the identity and total amount of the major gaseous by-products evolved during the polymer-to-ceramic conversion, further spectroscopic analysis (TGA coupled with mass spectra and/or with gas chromatography) will be investigated and the results published separately. These informations, along with microstructural investigations (solid-state NMR) of the fiber residue at various stages of the curing and pyrolysis processes, will enable identification of the chemical mechanisms and microstructural evolution.
5. Characterisation of [B(C2H4SiCH3NCH3)3]n-derived Si3.0B1.0C5.0N2.4 fibers
Fig. 10 shows the typical morphology of the as-obtained Si3.0B1.0C5.0N2.4 ceramic fibers after curing at 200 °C then pyrolysis at 1400 °C. The fibers exhibit a dense texture with a glassy-like section, indicating an amorphous state of the ceramic. The uniform surface is free of apparent defects. Fibers are almost circular which means that the curing and pyrolysis processes retain the fiber shape and occur without inter-fiber fusion. | ||
| Fig. 10 SEM micrographs of the cross-section and the surface morphology of the as-obtained Si3.0B1.0C5.0N2.4 fibers. | ||
The fiber typical diameter is in the range 20–25 μm. Using the laser interferometry technique, an average diameter from 42 measurements is precisely found to be 22.8 μm. Despite a large average diameter, mechanical properties at room temperature and especially, tensile strengths, are found to be suitable for use as fibrous reinforcement in CMCs. As shown in Table 2, Si3.0B1.0C5.0N2.4 fibers display average tensile strengths and Young's modulus of 1.3 and 172 GPa, respectively. Remarkably, values as high as 2.2 GPa can be found in the distribution of the tensile strength which reflects the high potential of these fibers for reinforcing CMCs.
| σ/GPa | E/GPa | ε (%) | ϕ/μm | |
|---|---|---|---|---|
| Si3.0B1.0C5.0N2.4 fibers | 1.3 ± 0.4 | 172 ± 32 | 0.69 ± 0.26 | 22.8 ± 2.2 |
Considering the dependence of fiber strength with the population of flaws and the processing conditions,6,11 significant progress in fiber strengths are particularly expected by improving the stretchability, i.e decreasing the green fiber diameter, during the spinning process.
6. High-temperature studies of [B(C2H4SiCH3NCH3)3]n-derived Si3.0B1.0C5.0N2.4 fibers
The high-temperature stability of the as-obtained Si3.0B1.0C5.0N2.4 fibers was investigated in a nitrogen atmosphere by XRD analysis from 1400 to 2000 °C (Fig. 11).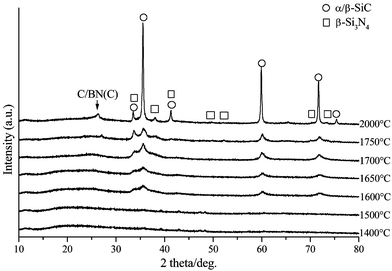 | ||
| Fig. 11 XRD patterns of ground-up Si3.0B1.0C5.0N2.4 fibers after annealing at 1400–2000 °C in a nitrogen atmosphere. | ||
The as-pyrolysed Si3.0B1.0C5.0N2.4 fibers are X-ray amorphous and appear amorphous even after annealing at 1700 °C for 1 h. Nevertheless, XRD patterns of samples annealed between 1400 and 1700 °C exhibit very broad reflections indicating segregation of nanometer-sized silicon carbide crystals. In contrast, there is no evidence for the formation of other crystalline phases such as silicon nitride, boron nitride or graphite. Heat-treatment above 1700 °C leads to a reduction of the half-width of all SiC reflections and the appearance of reflections with very low intensity which can be assigned to silicon nitride.
The presence of a mixture of silicon nitride/silicon carbide at this temperature is correlated with the thermal degradation of Si–N units of the amorphous phase according to eqn. (1)
| Si3N4 + 3C → 3SiC + 2N2 | (1) |
The XRD results are nicely reflected in the SEM micrographs of SiBCN fibers annealed from 1400 to 1900 °C (Fig. 12).
 | ||
| Fig. 12 SEM micrographs of the cross-sections of the Si3.0B1.0C5.0N2.4 fibers annealed at 1550 °C (a), 1650 °C (b), 1750 °C (c) and 1900 °C (d). | ||
The glassy-like texture of the Si3.0B1.0C5.0N2.4 fibers is retained at 1550 [Fig. 12(a)] and 1650 °C [Fig. 12(b)] whereas amorphous-to-crystalline transition occurs in sample heated between 1650 and 1750 °C as indicated with the appearance of a granular texture in the fibers heated at 1750 °C [Fig. 12(c)]. The appearance of a coarse-grained texture in the fibers annealed at 1900 °C [Fig. 12(d)] well-reflects the progress of the crystallisation process in the material at very high temperature.
The oxidation behaviour at high-temperature of the ceramic fibers derived from 4 was studied under air by means of HT-TGA from 25 to 1500 °C (10 °C min−1) (Fig. 13).
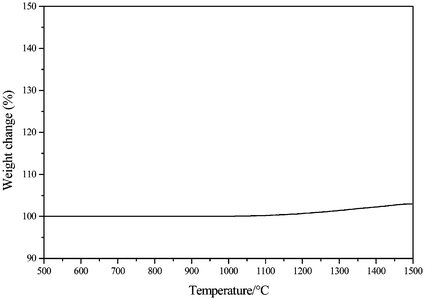 | ||
| Fig. 13 Oxidation behaviour of the Si3.0B1.0C5.0N2.4 fibers (heating rate: 10 °C min−1; flowing air). | ||
Non-oxide ceramic fibers are generally creep-resistant but lack chemical stability upon heating. In particular, one of their more noticeable disadvantages is generally their low resistance toward oxidative attack. Consequently, stress oxidation limits the durability of non-oxide composites under cyclic loading conditions. For example, the oxidative attack of conventional binary SiC and Si3N4 ceramic fibers involves the breakdown of the fibers above 1000–1100 °C.11
Oxidation in air proceeds by a passive oxidation mechanism in which diffusion of oxygen through a growing silica layer is often the rate-determining step. The mechanism occurring during oxidation of SiC and Si3N4 materials is depicted in eqns. (2) and (3):
| 2SiC + 3O2 → 2SiO2 + 2CO | (2) |
| Si3N4 + 3O2 → 3SiO2 + 2N2 | (3) |
As reported in our previous work, the weight gain of pure SiC and Si3N4 materials during oxidation starting from 1000 and 1130 °C represented ca. 28 and ca. 26 wt%, respectivley, at 1600 °C.21 The oxidation resistance of the Si3.0B1.0C5.0N2.4 fibers was remarkably enhanced in comparison to those of conventionally produced SiC- and Si3N4-based ceramics since there is only a slight weight increase (+2.9 wt%) detectable during the oxidation treatment from 1100 to 1500 °C. As previously observed with SiBCN bulk ceramics,21 oxidative attack occurs at the surface of the fibers by formation of a passivating amorphous oxide layer which offers some resistance to further oxidation by hindering diffusion-controlled oxidation. These observations are entirely in accordance with works of Jansen et al. in which an increase of only 2–4 wt% was observed during the TGA experiment of SiBCN ceramics in a pure oxygen atmosphere up to 1400 °C.33
Conclusion
The boron-modified polysilazane [B(C2H4SiCH3NCH3)3]n obtained by hydroboration of the dichloromethylvinylsilane and subsequent aminolysis of the as-obtained monomeric boron-modified chlorosilane offers processing advantages compared with ammonolysed analogs. Si- and N-bonded methyl groups have a major impact on the polymerisation kinetics by limiting the progress of the condensation reactions and therefore by improving the required polymer processability, i.e. solubility and fusibility. [B(C2H4SiCH3NCH3)3]n exhibits controlled viscoelastic properties to be readily melt-spinnable into flexible and uniform fine-diameter green fibers. After the spinning operation, the N-bonded methyl groups offer the capability of facile polymer cross-linking (curing) through a transamination pathway using an ammonia atmosphere up to 200 °C to yield an infusible Si3.0B1.1C9.6N2.9H24.0 fiber. Subsequent pyrolysis of the cured fibers in a nitrogen atmosphere up to 1400 °C releases amorphous Si3.0B1.0C5.0N2.4 ceramic fibers of 1.3 GPa in tensile strength, 172 GPa in Young's modulus and 23 μm in diameter, which remain thermally and chemically stable at 1500 °C.Acknowledgements
The authors gratefully acknowledge supporting co-workers at the PML in MPI of Stuttgart: Gerhard Kaiser (Elemental analysis), Horst Kummer (HT-TGA), Harmut Labitzke (SEM), Martina Thomas (XRD). In addition, they wish to thank the members of the LMI in Lyon: Dr David Cornu, Marie-Paule Berthet (Mechanical testing), Dr Jean-Marie Létoffé (DSC) and Dr Catherine Sigala (TMA) for their contributions. The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for financial support.References
- P. G. Chantrell and E. P. Popper, in Special Ceramics-4, E. P. Popper, ed., Academic Press, New York, 1964, p. 87 Search PubMed.
- M. Peuckert, T. Vaahs and M. Brück, Adv. Mater., 1990, 2, 398 CrossRef CAS.
- M. Weinmann and F. Aldinger, Precursor-Derived Ceramics, in The Handbook of Advanced Materials: Materials, Applications, Processing and Properties, S. Somiya, F. Aldinger, M. Kaneno, K. Uchino and R. M. Spriggs, eds., Academic Press, London, 2003, p. 265 Search PubMed.
- J. Bill and F. Aldinger, Adv. Mater., 1995, 7, 775 CAS.
- K. J. Wynne and R. W. Rice, Annu. Rev. Mater. Sci., 1984, 14, 297 Search PubMed.
- J. Lipowitz, Fiber Synthesis Processes, in Carbide, Nitride and boride Materials, Synthesis and Processing, A. W. Weimer, ed., Chapman and Hall, London, 1997, p. 433 Search PubMed.
- B. G. Penn, F. E. Ledbetter, III, J. M. Clemons and J. G. Daniels, J. Appl. Polym. Sci., 1982, 27, 3751 CrossRef CAS.
- S. Yajima, T. Shishido and H. Kayano, Nature, 1978, 273, 525 CAS.
- G. E. LeGrow, T. F. Lim, J. Lipowitz and R. S. Reaoch, Am. Ceram. Soc. Bull., 1987, 66, 363 CAS; G. Winter, W. Verbeek and M. Mansmann, US Pat., 3 892 583, 1975 Search PubMed.
- Fine Ceramic Fibres, A. R. Bunsell and M.-H. Berger, eds., Marcel Dekker Inc., New York, 1999 Search PubMed.
- K. Okamura, Composites, 1987, 18, 107 Search PubMed.
- S. M. Johnson, R. D. Brittain, R. H. Lamoreaux and D. J. Rowcliffe, J. Am. Ceram. Soc., 1988, 71, C-132 CAS.
- T. Mah, N. L. Hecht, D. E. McCullum, J. R. Hoenigman, H. M. Kin, A. P. Katz and H. A. Lipsitt, J. Mater. Sci., 1984, 19, 1191 CAS.
- G. Motz, J. Hacker, G. Ziegler, B. Clauss and D. Schawaller, Low-Cost-Ceramic SiCN Fibers by an Optimized Polycarbosilazane and Continuous Processing, in Inorganic Structural Fiber Composites, P. Vincenzini and C. Badini, eds., Techna Srl, Faenza, Italy, 2003, p. 47 Search PubMed.
- D. Schawaller and B. Clauss, Preparation of Non-Oxide Ceramic Fibers in the System Si–C–N and Si–B–C–N, in High Temperature Ceramic Matrix Composites, W. Krenkel, R. Naslain and H. Schneider, eds., Wiley-VCH, Weinheim, 2001, p. 56 Search PubMed.
- M. Takamizawa, T. Kobayashi, A. Hayashida and Y. Takeda, US Pat., 4 604 367, 1986 Search PubMed.
- L. Lu, C. X. Feng and Y. C. Song, J. Mater. Sci. Lett., 1998, 17, 481 CrossRef CAS; L. Lu, C. X. Feng and Y. C. Song, J. Mater. Sci. Lett., 1998, 17, 599 CrossRef CAS; Z.-Y. Chu, C.-X. Feng and Y.-C. Song, J. Mater. Sci. Lett., 2003, 22, 725 CrossRef CAS.
- T. Wideman, E. Cortez, E. E. Remsen, G. A. Zank, P. J. Carroll and L. G. Sneddon, Chem. Mater., 1997, 9, 2218 CrossRef CAS; T. Wideman, P. J. Fazen, K. Su, E. E. Remsen, G. A. Zank and L. G. Sneddon, Appl. Organomet. Chem., 1998, 12, 681 CrossRef CAS.
- H. P. Baldus, O. Wagner and M. Jansen, Mater. Res. Soc. Symp. Proc., 1992, 271, 821 CAS; H. P. Baldus, M. Jansen and O. Wagner, Key Eng. Mater., 1994, 89–91, 75 CAS; H. P. Baldus and M. Jansen, Angew. Chem., 1997, 109, 338; H. P. Baldus, M. Jansen and D. Sporn, Science, 1999, 285, 699 CrossRef CAS; H. P. Baldus, A. Thierauf, R. Henborn and D. Sporn, US Pat., 5 885 519, 1999 Search PubMed.
- R. Riedel, A. Kienzle, W. Dressler, L. Ruwisch, J. Bill and F. Aldinger, Nature, 1996, 382, 796 CAS.
- F. Aldinger, M. Weinmann and J. Bill, Pure Appl. Chem., 1998, 70, 439 CrossRef CAS; M. Weinmann, High Temperature Stable Ceramics from Inorganic Polymers, in Precursor-Derived Ceramics: Synthesis, Structures and High Temperature Mechanical Properties, J. Bill, F. Wakai and F. Aldinger, eds., Wiley-VCH, Weinheim, 1999, p. 83 Search PubMed; M. Weinmann, J. Schuhmacher, H. Kummer, S. Prinz, J. Peng, H. J. Seifert, M. Christ, K. Müller, J. Bill and F. Aldinger, Chem. Mater., 2000, 12, 623 Search PubMed.
- This preparative method is based on experiments developed by the German chemist Wilhelm Schlenk. All apparatus are equipped with sidearms for pumping out the air and moisture and introducing inert gas. See also: D. F. Shriver and M. A. Drezdzon, The Manipulation of Air-Sensitive Compounds, 2nd edn., Wiley, New York, 1986 Search PubMed.
- A. Kienzle, Thesis, Universität Stuttgart, 1994(in German) Search PubMed.
- S. N. Patankar, J. Mater. Sci. Lett., 1991, 10, 1176 CAS.
- L. M. Ruwisch, P. Dürichen and R. Riedel, Polyhedron, 2000, 19, 323 CrossRef CAS.
- M. Weinmann, T. W. Kamphowe, P. Fischer and F. Aldinger, J. Organomet. Chem., 1999, 592, 115 CrossRef CAS.
- Z. Jiang and L. V. Interrante, Chem. Mater., 1990, 2, 439 CrossRef CAS.
- S. Bernard, D. Cornu, P. Miele, H. Vincent and J. Bouix, J. Organomet. Chem., 2002, 357, 91 CrossRef CAS.
- G. Burns and G. Chandra, J. Am. Ceram. Soc., 1989, 72, 333 CAS.
- R. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Chem. Mater., 1992, 4, 711 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fately and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press Limited, London, 1991 Search PubMed.
- H. J. Seifert, H.-L. Lukas and F. Aldinger, Ber. Bunsen-Ges. Phys. Chem., 1998, 102, 1309 CAS.
- M. Jansen, B. Jäschke and T. Jäschke, Amorphous Multinary Ceramics in the Si–B–N–C System, in High Performance Non-Oxide Ceramics I, Structure and Bonding, Vol. 101, Springer-Verlag, Berlin, Heidelberg, 2002, p. 137 Search PubMed.
Footnote |
| † Member of the IUF (Institut Universitaire de France). |
| This journal is © The Royal Society of Chemistry 2005 |