Comparison of Ga+ and SF5+ primary ions for the molecular speciation of oxysalts in static secondary ion mass spectrometry (S-SIMS)
Rita
Van Ham
a,
Luc
Van Vaeck
a,
Freddy
Adams
a and
Annemie
Adriaens
*b
aUniversity of Antwerp, Department of Chemistry, Universiteitsplein 1, B 2610 Antwerp, Belgium
bGhent University, Department of Analytical Chemistry, Krijgslaan 218–S12, B 9000 Ghent, Belgium
First published on 4th August 2005
Abstract
Speciation of inorganic compounds by means of static secondary ion mass spectrometry (S-SIMS) aims at deriving the molecular composition of the analyte in the solid. The recent use of polyatomic primary ions, instead of the traditionally applied monatomic or diatomic projectiles, potentially offers an increase in secondary ion intensity and an increase in molecular specificity. This paper focuses on the comparison of SF5+ and Ga+ primary ion bombardment for the molecular speciation of inorganic oxysalts. The polyatomic primary ions are found to increase the total ion yield by a factor of 4–10. Furthermore, the relative intensities of the most structure-specific signals are increased in comparison with the low m/z peaks under SF5+ bombardment relative to Ga+. Although the formation of oxide fragments is promoted with polyatomic projectiles, the mass spectra significantly gain in information concerning the molecular specificity.
Introduction
The identification of the organic and inorganic constituents within one monolayer of some specific molecules at the surface of solids is vital to the understanding of many material properties. The direct determination of the molecular composition of inorganic compounds by means of signals referring to the intact analyte is called molecular speciation. At this moment, mass spectrometry (MS) in combination with microbeam ionization of solids, by means of photons in laser microprobe MS (LMMS) or by means of a limited dose of primary ions in S-SIMS, plays an important role in the continuing development of methods for molecular speciation with high lateral and/or depth resolution.The specific advantage of S-SIMS is the capability of generating molecular information from the outer (few) monolayers at the surface of the sample.1 The limitation of the ion dose inherently implies that a relatively small number of secondary ions become available for analysis. Hence, nowadays, research in S-SIMS focuses on the improvement of this ion yield. In this respect, the relatively recent development of polyatomic primary ion bombardment as an alternative to the traditionally used monoatomic or small diatomic ions receives increasing attention. Polyatomic projectiles involve the simultaneous impact of several atoms within a distance of a few tenths of a nanometre. The simultaneous generation of overlapping collision cascades is expected to improve the recoil of adequate momentum to the surface component. On the one hand, the damage cross section determining the decay rate of molecular signals in S-SIMS is likely to increase for polyatomic projectiles compared with monatomic primary ions.2,3 On the other hand, polyatomic ion bombardment may ultimately lead to a FAB-like phase explosion in the subsurface,4 reminiscent of fast atom bombardment, and thereby improve the release of molecular species. Recent simulation supports the latter concept.5
The literature data available on inorganic speciation using polyatomic bombardment is rather limited; nevertheless, a few studies are worth mentioning here. The study by Groenewold et al. focused on the analysis of CuCl and CuCl2 by using Ga+ and ReO4− primary ions.6 The polyatomic projectiles proved to be more adequate to yield information on the surface oxidation state. This was attributed to the lower penetration depth of the ReO4− projectile in comparison with Ga+. The study was further elaborated by Van Ham et al. including various binary salts and the use of SF5+ as the primary ion beam.7
Van Stipdonk et al. showed increased chemical damage in the surface layers of NaIO3 under polyatomic, as opposed to monatomic, primary ion bombardment.3 A low dose (104–106 ions cm−2) of 20 keV CsI·Cs+ primary ions yielded only IO3− and NaIO3·IO3− as iodate specific anions. Most of the total ion current was carried by ions to be expected from NaI. Since XPS analysis of the iodate sample revealed only a minor amount of NaI, the observed (NaI)n·I− peaks were assigned to beam-induced chemical damage. Comparison of different projectiles (CsI)n·Cs+ (n = 0–2) at the same impact energy showed that the relative yield of the iodate type ions decreased with n. The reverse was true for the iodide-type ions. This confirmed increased chemical damage by larger polyatomic primary ions seen for NaNO3.
The same research group also studied the changes in the negative ion mass spectra of NaBF4 by the use of Cs+ and CsI·Cs+, (CsI)2·Cs+ and (CsI)3·Cs+ (all with 20 keV energy).8,9 The results with Cs+ reflected the competition between two ion formation pathways observed in plasma desorption MS.10 One route involved emission of intact BF4− and its incorporation into larger ions, whereas the other generated NaF·F− type ions. Polyatomic primary ions proved to be more efficient for producing both high and low mass secondary ions.8 Interestingly, the relative yield enhancement of ions BF4− and NaBF4·BF4− reached a plateau value at high primary ion mass, whereas that of (NaF)n·F− continued to increase. This trend pointed to overlapping collision cascades. The intensity ratios of NaF·F− and NaBF4·BF4− over BF4− were more sensitive to the number of atoms in the primary projectile than to the total energy deposited by the incident projectile.9 The secondary ion yield normalised on the number of atoms in the primary projectile showed a similar non-linear dependence on the primary projectile energy per mass, as described earlier for iodates and nitrates.3,11,12 Interestingly, the yield enhancement by larger polyatomic primary ions was larger for NaF·F− ions, associated with beam-induced decomposition, than for the BF4− and NaBF4·BF4− ions.
The purpose of this paper is to study the characteristic features of the mass spectra of oxysalts generated by Ga+ and SF5+ primary ions. The following questions are to be addressed: (1) does the overall yield improve? (2) is there an increase in the relative importance of high m/z adducts? and (3) are low m/z fragments and/or unspecific cluster ions promoted by the use of polyatomic ions? Attention is focused on a set of well-selected molecules such as alkali nitrates, nitrites, phosphates and hydrogen phosphates. Apart from the beam-induced damage, the nitrate and nitrite analogues have been chosen to compare the two guns in the case of so-called fine-speciation, i.e., the distinction between salts with the same elements in a different oxidation state. The series of alkali hydrogen phosphates allows the speciation capabilities of both guns to be delineated for salts with the same elements in the same oxidation states but in different ratios. The possible effect of surface water on the S-SIMS data may cause confusion between the phosphates and the hydrogen phosphates. The works follows a previous study on the use of polyatomic primary ions for the analysis of binary salts.7
Experimental
Analyses were performed with a TOF-SIMS IV (ION-TOF, Germany) instrument, equipped with an ion reflector. Spectra were taken using a Ga+ liquid metal ion source and an SF5+ electron ionization gun. The liquid metal ion source for the production of Ga+ was operated at 25 kV beam voltage in bunched mode with a pulse width of 20 ns. This provided a sufficient mass resolution for better than nominal separation for ions with a mass-to-charge (m/z) ratio of up to 1000. The 9 keV SF5+ primary ions were produced by an electron ionization source filled with SF6 at 3 × 10−6 mbar. The diameter of the Ga+ and SF5+ beam on the sample was 2–5 and 25 μm, respectively. The analysis time was 300 s, allowing 3 × 106 spectra to be accumulated. The area of analysis was 300 × 300 μm2. The total ion dose was typically kept under 4 × 1011 ions cm−2. Electron flooding was applied to compensate for the charging.The samples were analyzed as pellets (surface 1 cm2, thickness 1–2 mm), which were prepared by pressing pure salts in a dial under a pressure of 5–8 tons cm−2. The surface roughness was within 0.1 mm. In order to avoid the adsorption of water from the air, the pellets were stored in a desiccator under vacuum or made just before analysis. A small piece with a flat surface was mounted in the TOF S-SIMS sample holder so that a vacuum of typically 10−8 mbar could be reached in a reasonable time.
The oxysalts NaNO2a, NaNO3b, K3HPO4c, K2HPO4d, and KH2PO4b were purchased from Janssen Chimicaa, Merckb, Aldrich Chemical Companyc and UCBd.
Results and discussion
To facilitate the detailed comparison of SF5+ and Ga+ data for the various inorganic salts, we have made use of the total ion current (TIC) and the relative intensity of specific ions per given analysis time, defined as the B ratio: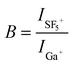 | (1) |
 | (2) |
To assist in the interpretation of the figures, it has been attempted to separate the “winners” (ions for which the polyatomic ions are beneficial) from the “losers”. Therefore, whenever R < 1, the inverted value is given a negative sign before plotting.
NaNO3 and NaNO2
The mass spectra taken with Ga+ primary ions are given in Figs. 1 and 2 for NaNO3 and NaNO2, respectively. The mass spectral peak pattern for the cations reflects the features to be expected. Specifically, the generation of NaOH and Na2O related ions refers to the characteristic structural decomposition observed in all oxysalts:| 2NaNO3 → Na2O + N2O5 | (3) |
 | ||
| Fig. 1 Positive (a) and negative (b) ion mass spectra of NaNO3 under Ga+ bombardment. | ||
 | ||
| Fig. 2 Positive (a) and negative (b) ion mass spectra of NaNO2 under Ga+ bombardment. | ||
As to the distinction between sodium nitrate and nitrite, Fig. 2 shows that the nitrite has a substantially lower yield of the high m/z ions than the nitrate. This holds true for the positive and negative ions. In particular, the signal from the cationised analyte does not even reach 3 cpsat for nitrite as opposed to the cationised ion of nitrate. In the negative mode, molecular adducts in both cases reach only the threshold for detection in pure products. The comparison of the relative intensities of the analyte adducts over those of the other form shows that both ratios, NaNO2·Na+/NaNO3·Na+ as well as NaNO2·NO3−/NaNO3·NO3−, are substantially in favour of the form analysed. Otherwise stated, the availability in the solid state improves the yield of the corresponding adduct. Hence, these ratios can be used to achieve fine speciation but reference spectra must be recorded under identical conditions. Spectra from a database are inadequate because the balance between availability in the sample and formation of the other stable form is rather delicate. Specifically, charge compensation is critical. It has been shown that the intensity ratio NaNO3·NO3−/NaNO3·NO2− in nitrates goes from 0.41 to 0.72 without and with electron flooding. Also, the ratio NO2−/NO3− for nitrate rises from 0.71 to 1.14 without and with use of the flood gun. A reduction of the nitrate to the nitrite form is logically explained by the electrons striking the surface.
The mass spectra taken with SF5+ are given in Figs. 3 and 4 for NaNO3 and NaNO2, respectively. With SF5+ the total ion current (TIC) substantially increases, which allows the series of detected adducts to be extended. In the positive mode, dimers exceed the 3 cpsat criterion for nitrates with SF5+ (only monomers with Ga+) and also trimers are practically useful for nitrates (only dimers with Ga+).
 | ||
| Fig. 3 Positive (a) and negative (b) ion mass spectra of NaNO3 under SF5+ bombardment. | ||
 | ||
| Fig. 4 Positive (a) and negative (b) ion mass spectra of NaNO2 under SF5+ bombardment. | ||
Quantitatively expressed, the B factor for NaNO2·Na+ from nitrite is about 1400 and about 70 for NaNO3·Na+ from nitrate. Also for the negative ions, one additional adduct is seen (dimers against monomers for Ga+) for nitrate and nitrite. The B factor for NaNO2·NO2− and NaNO3·NO3− is about 2000 and 200 for nitrite and nitrate, respectively. The potential gain in molecular specificity of the mass spectra taken with SF5+ compared with Ga+ is illustrated by the R factor (relative peak intensities SF5+/Ga+) in Figs. 5 and 6 for NaNO2 and NaNO3, respectively. To avoid confusion between isobars such as NaNO2·NO3− and NaNO3·NO2−, the NaNO2·NO2− and NaNO3·NO3− ions have been used for normalisation of the signal intensities in nitrite and nitrate, respectively.
 | ||
| Fig. 5 Comparison of relative intensities (R-factor) of diagnostic ions from NaNO3 under SF5+ bombardment referenced to the ones under Ga+ impact. | ||
 | ||
| Fig. 6 Comparison of relative intensities (R-factor) of diagnostic ions from NaNO2 under SF5+ bombardment referenced to the ones under Ga+ impact. | ||
As to the positive ions, the relative importance of the ions based on the oxide fragments significantly increases with SF5+ projectiles. This observation confirms the data of Van Stipdonk et al.12 On the other hand, the strong decrease of the Na+ and Na2+ intensities reflects the increase of the monomeric adduction contribution to the TIC. The relative importance of the dimeric adducts also decreases in comparison with the monomeric ones. A roughly similar picture is seen for the negative ions. The low m/z ions NO− and, to a less extent, also NO2− and NO3− are serious “losers” for both analytes. The molecular ion production is disfavoured in the case of NaNO2, not for NaNO3. However, in both cases the monomeric adduct ion (NaNO2·NO2− and NaNO3·NO3− for nitrite and nitrate, respectively) of the original analyte is the main “winner”.
Summarizing, bombardment with SF5+ projectiles improves the capability to distinguish between both analyets in comparison the Ga+. Table 1 lists the ratios of the characteristic ions to be used for molecular speciation. In particular, negative ion mass spectra taken with SF5+ produce diagnostic peak ratios more different for nitrate and nitrite analytes than Ga+ does. This holds true also for the signal intensity ratio NO2−/NO3−. The improvement in molecular specificity by SF5+ projectiles is less dramatic for the positive than for the negative ion mass spectra.
| Ga+ | SF5+ | |||
|---|---|---|---|---|
| NaNO2 | NaNO3 | NaNO2 | NaNO3 | |
| NO2−/NO3− | 2.9 ± 0.3 | 0.9 ± 0.2 | 3.5 ± 1.2 | 0.53 ± 0.06 |
| NaNO2·NO2−/NaNO3·NO3− | 3.8 ± 0.5 | 0.12 ± 0.02 | 19 ± 10 | 0.23 ± 0.01 |
| NaNO2·Na+/NaNO3·Na+ | 3.9 ± 0.9 | 0.56 ± 0.03 | 10 ± 5 | 0.71 ± 0.01 |
Potassium (hydrogen) phosphates
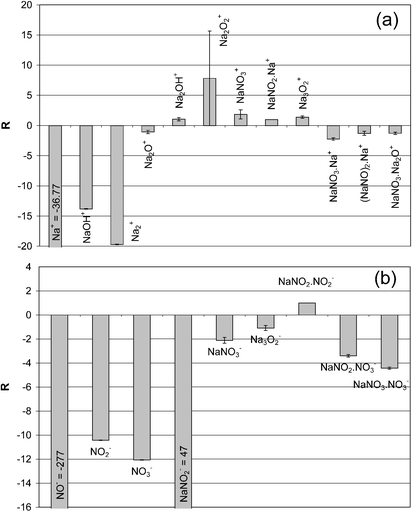
Pellets of K3PO4, K2HPO4 and KH2PO4 were measured under Ga+ and SF5+ ion bombardment on the same day. Fig. 7 lists the positive ion spectra of K3PO4 using SF5+ as found with Ga+ and shows a similar pattern for both primary ions. The main diagnostic information comes from K3PO4·K+ and (K3PO4)2·K+. Once again, the SF5+ primary ion bombardment increases the intensity of adduct ions from both the analyte (K3PO4·K+) and the contaminant K2CO3·K+ with an average factor of 10. Negative ion mass spectra contain little diagnostic information apart from POx− signals. The SF5+ bombardment does not yield additional information for speciation analysis in comparison with Ga+. The absolute intensity of the POx− ions increase substantially (B = 15) and the ratio of PO2−/PO3− inverts. | ||
| Fig. 7 Positive ion mass spectrum of K3PO4 under SF5+ (a) and Ga+ (b) bombardment. | ||
Fig. 8 describes the differences in ion yield by using SF5+ and Ga+ bombardment in a more quantitative manner. The B-factors (ISF5+/IGa+) show that the intensity gain of the oxide related species equals that of the molecular adducts K3PO4·K+. The huge RSD on the former B factors for oxides is due to the variability in the Ga+ spectra. The enhanced fragmentation into oxides of the analyte under SF5+ bombardment is noted again. The R factors, using the adduct K3PO4·K+ as reference evidence the increased molecular specificity of SF5+ mass spectra. In fact, only the monomeric adduct resides at the “winning” side of the plot while all the other signals manage to be “losers”. The depreciation of the oxide species is small in comparison with KnH(3−n)PO4 adducts. Hence, it looks as though the intensity increase of the K3PO4·K+ happens at the expense of the less specific KnH(3−n)PO4 adducts, the presence of which is to be associated with surface water. Also, the dimeric adducts are “losers” as before for NaNO2, NaNO3 and CuCl2.
 | ||
| Fig. 8 Comparison of absolute (B-factor, (a)) and relative intensity ratios (R-factor, (b)) of diagnostic ions from K3PO4 under SF5+ bombardment referenced to the ones under Ga+ impact. | ||
Figs. 9 and 10 compare the positive ion mass spectra taken with SF5+ and Ga+ from K2HPO4 and KH2PO4, respectively. Polyatomic projectiles produce essentially the same positive ions as Ga+ but with higher intensity. Specifically, the gain for the monomeric adducts is a factor 4 and 40 for K2HPO4 and KH2PO4, respectively. However, as opposed to nitrates and nitrites, no additional high m/z signals are observed with polyatomic projectiles in comparison to the Ga+ ions. The same holds true for the anion spectra, where the yield is also increased but no additional ions are found. Specifically, in the case of KH2PO4, the KPO3·PO3− signal increases by a factor of 10 under SF5+ bombardment and can now be considered for practical applications. The P− and PO− signals almost disappear under SF5+ bombardment. If these ions are to be considered as indicative for beam-induced damage or destructive ionisation, polyatomic ions create a milder regime.
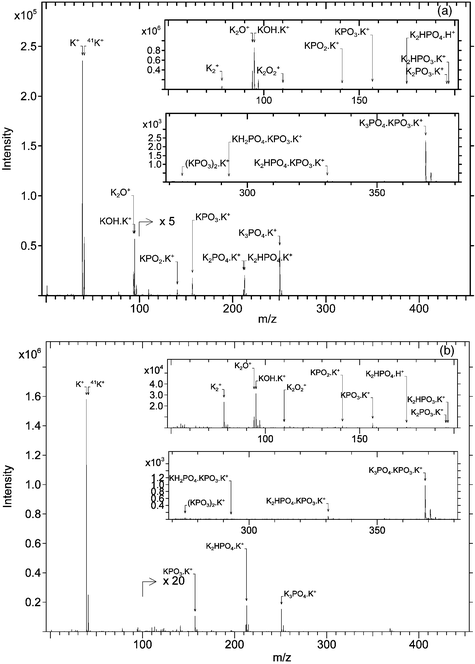 | ||
| Fig. 9 Positive ion mass spectrum of K2HPO4 under SF5+ (a) and Ga+ (b) bombardment. | ||
 | ||
| Fig. 10 Positive ion mass spectrum of KH2PO4 under SF5+ (a) and Ga+ (b) bombardment. | ||
 | ||
| Fig. 11 Comparison of relative intensity ratios by R-factor of diagnostic ions from K2HPO4 (a) and KH2PO4 (b) for the positive ions under SF5+ bombardment referenced to the ones under Ga+ impact. | ||
The evolution of relative intensities when changing from Ga+ to SF5+ is plotted in Fig. 11 for both analytes. Peak intensities are referenced on the cationised analyte molecules (K2HPO4·K+ and KH2PO4·K+, respectively). Once again, the elemental ions and K2+ are serious “losers”. A remarkable feature is the increased contribution of all ions derived from K2O(2) or KOH species. Amongst these ions, the KOH·K+ is a significant winner. The good reproducibility of the corresponding R-factor contrasts with its variability seen before in the case of K3PO4. We explain this by the role of adsorbed surface water in the formation of these ions from K3PO4. It seems like the cleavage of the oxide moieties is favoured by the presence of the hydrogen in the analyte and this feature is intensified with the use of SF5+. As has been argued before, this does not reflect beam-induced damage since the fragments are structural in nature. Together with the increased contribution of the oxide species comes, of course, enhanced importance of the cationised KPO3, which is the complementing part to the oxide in the analyte. Note that the normalised peak intensity of KPO3·K+ gains a factor 4 against only 1.5 in the case of KH2PO4 and K2HPO4, respectively.
The distinction between KH2PO4 and K2HPO4 must be based on the peaks from the cationised molecules. From Fig. 11 it can be seen that the molecular specificity of the mass spectrum increases with SF5+ for K2HPO4 because the intensity ratio of the “wrong analyte” form (KH2PO4·K+/K2HPO4·K+) decreases substantially. However, for KH2PO4, the situation becomes problematic because the K2HPO4·K+/KH2PO4·K+ increases, suggesting the “wrong” analyte. Although the normalised K3PO4·K+ is a significant winner, confusion with the pure phosphate sample is unlikely since KnH3−nPO4 adducts are absent. However, problems are to be expected with mixtures. Once again, the dimeric adducts are all losers with SF5+ (up to a factor of 8). In conclusion, SF5+ bombardment seems to stimulate the formation of oxide species and disfavour the generation of dimeric adducts. The molecular specificity is increased in the case of K2HPO4 by the increased generation of the relevant adducts but KH2PO4 shows the opposite trend.
Conclusions
The potential benefits arising from the use of polyatomic SF5+ primary ions have been evaluated in comparison the Ga+ bombardment for selected oxysalts. It has been demonstrated that polyatomic ions give a substantial gain in the total ion current by a factor 4–10, depending on the analogue under study. Equally important is the significant improvement in molecular specificity of the mass spectra taken under polyatomic bombardment. Specifically, the adduct ions are of particular diagnostic interest for speciation. Their relative contribution to the total ion current systematically increases when SF5+ instead of Ga+ ions are used. As a result, bombarding the surface with SF5+ primary ions is beneficial in distinguishing between the different hydrogen phosphates and between nitrites and nitrates. At the same time, a polyatomic primary ion beam tend to decrease the relative importance of the elemental ions.The reported experiments have focused on a preliminary assessment of the practical advantages of SF5+ primary ions in comparison with Ga+ for the speciation of inorganic oxysalts. Further and fundamental work will be needed to pinpoint the most interesting type of primary ion but our experimental assessment already confirms that the use of polyatomic primary ions becomes one of the most promising developments in the continuing search for yield improvement in S-SIMS.
Acknowledgements
This work was supported in part by the Belgian Office for Scientific, Technical and Cultural Affairs (IUAP 5) and by FWO, Brussels, Belgium (research projects G.0090.98 and G.0172.00).References
- R. Van Ham, L. Van Vaeck, F. Adams and A. Adriaens, Anal. Chem., 2004, 76(9), 2609 CrossRef CAS.
- G. S. Groenewold, A. K. Gianotto, J. E. Olson, A. D. Appelhans, J. C. Ingram, J. E. Delmore and A. D. Shaw, Int. J. Mass Spectrom. Ion Processes, 1998, 174, 129 CrossRef CAS.
- M. J. Van Stipdonk, V. Santiago, E. A. Schweikert, C. C. Chusuei and D. W. Goodman, Int. J. Mass Spectrom., 2000, 197, 149 CrossRef CAS.
- J. Sunner, A. Morales and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1988, 86, 169 CrossRef CAS.
- K. D. Krantzmann, Z. Postawa, B. J. Garrison, N. Winograd, J. S. Stuart and J. A. Harrison, Nucl. Instrum. Meth. Phys. Res. B, 2001, 180, 159 CrossRef.
- G. S. Groenewold, A. D. Appelhans, G. L. Gresham, J. C. Ingram and A. D. Shaw, Int. J. Mass Spectrom. Ion Processes, 1998, 178, 19 CrossRef CAS.
- R. Van Ham, L. Van Vaeck, A. Adriaens, F. Adams, B. Hodges and G. Groenewold, J. Anal. Atom. Spectrom., 2002, 17(8), 753 RSC.
- M. J. Van Stipdonk, R. D. Harris and E. A. Schweikert, Rapid Commun. Mass Spectrom., 1997, 11, 1794 CrossRef CAS.
- M. J. Van Stipdonk, V. Santiago and E. A. Schweikert, Int. J. Mass Spectrom., 1999, 34, 554 CrossRef CAS.
- M. J. Van Stipdonk, J. B. Shapiro and E. A. Schweikert, Vacuum, 1995, 46, 1227 CrossRef CAS.
- M. J. Van Stipdonk, D. R. Justus, V. Santiago and E. A. Schweikert, Rapid Commun. Mass Spectrom., 1998, 12, 1639 CrossRef CAS.
- M. J. Van Stipdonk, in TOF-SIMS: Surface Analysis by Mass Spectrometry, eds. J. C. Vickerman and D. Briggs, IM Publications, Chichester, UK, 2001, p. 309 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |