Detection of phosphorus tagged carboxylic acids using HPLC-SF-ICP-MS
Received
14th October 2004
, Accepted 17th December 2004
First published on 10th January 2005
Abstract
High performance liquid chromatography (HPLC) has been coupled with sector field inductively coupled plasma mass spectrometry (SF-ICP-MS) for the determination of maleic, sorbic and fumaric acids after derivatisation with the phosphorus containing reagent tris(2,4,6-trimethoxyphenyl)phosphonium propylamine (TMPP). This allowed 31P+ selective detection to be performed for these compounds, which are normally invisible to detection by ICP-MS at low concentrations. Optimal reagent conditions for the derivatisation of 0.1 mM maleic acid were: 1 mM TMPP; 10 mM 2-chloro-1-methylpyridinium iodide (CMPI); 11 mM triethylamine. The efficiency of the derivatisation reaction was estimated to be between 10–20% and detection limits, estimated as 3 times baseline noise, were 0.046 nmol for TMPP and 0.25 nmol for derivatised maleic acid, for a 5 μl injection.
Introduction
The vast majority of organic compounds are essentially invisible to detection by inductively coupled plasma mass spectrometry (ICP-MS) because they only contain H, C, O and N. These elements have extremely high background signals because they are either present in the solvents used during sample introduction or present in atmospheric air entrained into the plasma, hence trace analysis is impossible. The exceptions to this are the determination of C at minor and trace levels1–3 and the element selective detection of organic compounds which contain a hetero-atom such as P, S, Si, Cl, Br, I, a metal or metalloid. For example, detection of phosphorus containing compounds has previously been performed for the determination of phosphorylated peptides4–6 and sulfur-containing pharmaceuticals.7 An alternative has recently been proposed by Smith et al.8 which uses aqueous eluents only (at temperatures of 60 °C and 160 °C) or isotopically enriched solvents to detect carbon in organic compounds by LC-ICP-MS. If a means of making all organic compounds detectable by ICP-MS could be achieved, many new applications would be realised. For example, high accuracy quantitative analysis of organic compounds could be achieved using external calibration; and many biological molecules could be determined in metabolic studies.9
Leavens et al.10 have previously reported on the synthesis of a large molecular weight phosphorus reagent, tris(2,4,6-trimethoxyphenyl)phosphonium propylamine bromide (TMPP), which was used to derivatise organic compounds to make them amenable to detection by positive ion electrospray ionization mass spectrometry (ESI-MS). We have used the TMPP reagent for the determination of low molecular weight carboxylic acids in pharmaceutical samples, and have determined the limits of detection using ESI-MS-MS.11 This reaction incorporates a coupling reaction with 1-chloro-4-methylpyridinium iodide and triethylamine to activate the carbonyl group on the carboxylic acid prior to nucleophilic attack by the amine group on the coupling reagent. The resulting derivatised carboxylic acid contains a stable amide bond linking the derivatising reagent with the carboxylic acid. The majority of carboxylic acids do not contain a heteroatom so cannot be detected using ICP-MS. However, by derivatising these acids with TMPP they have effectively been tagged with phosphorus, so 31P+ selective detection using ICP-MS can be performed. This has useful applications in pre-screening for quality control and investigations involving intellectual property rights.7 In this paper, detection of some pharmaceutically important carboxylic acids (maleic, fumaric, salicylic and sorbic) by LC-ICP-MS, after derivatisation, is described. The derivatisation reaction was also examined to further enhance the derivatisation procedure to lead to more sensitive detection, and hence lower detection limits.
Experimental
Chemicals and reagents
Carboxylic acids, 2-chloro-1-methylpyridinium iodide (CMPI), triethylamine (TEA) and formic acid were obtained from Sigma–Aldrich (Poole, Dorset, UK). HPLC grade acetonitrile was obtained from Fisher Scientific UK Ltd. (Loughborough, Leicestershire, UK). Distilled deionised water (18.2 MΩ) was obtained using an Elga Maxima water purifying system.
Synthesis of tris(2,4,6-trimethoxyphenyl)phosphonium propylamine bromide (TMPP)
(Scheme 1)
TMPP was synthesised in-house using an adaptation of the procedure detailed by Leavens et al.10 Initial attempts to synthesize this derivatising reagent using the literature method were unsuccessful, possibly due to the propylamine side chain not attaching to the tertiary phosphonium. This may be because the 3-bromopropylamine free base was unstable and tended to condense with itself by nucleophilic displacement of bromide by the primary amine. Hence, it was decided to perform the liberation of the free amine from the salt, and the subsequent clarification, as quickly as possible. Thus, the following method was developed successfully: To a solution of 3-bromopropylamine hydrobromide (4.98 g, 22.74 mmol, now in large excess) in water (20 ml) was added potassium carbonate (2.24 g) and toluene (20 ml) with stirring. The resultant toluene phase was isolated, clarified with saturated brine (20 ml), and dried over magnesium sulfate. This dry toluene phase was filtered directly into a pre-refluxing solution of tris(2,4,6-trimethoxyphenyl)phosphine (2.12 g, 3.98 mmol) in toluene (40 ml) to minimise the time for the 3-bromopropylamine free base to condense with itself. The mixture was refluxed for a further 30 min and the resulting white precipitate was isolated by filtration, washed with toluene (2 × 5 ml) and diethyl ether (25 ml) and dried overnight at 30 °C to give the title compound. A schematic of the reaction is shown in Scheme 1.
 |
| | Scheme 1 Synthesis of tris(2,4,6-trimethoxyphenyl)phosphonium propylamine bromide (TMPP). | |
Preparation of coupling reagent
Solutions of CMPI were prepared by dissolving the amount of CMPI in approximately 20 ml of acetonitrile in a 25 ml volumetric flask. The corresponding amount of triethylamine was added, and the solution made up to volume with acetonitrile.
To 500 μl of carboxylic acid in 90 ∶ 10%
(v/v) water ∶ acetonitrile were added 500 μl of CMPI/TEA coupling reagent (prepared as above). After thorough mixing for 5 min at room temperature, 500 μl of a TMPP propylamine solution in acetonitrile was added. The solution was left to react for 30 min in an ultrasonic bath at room temperature. A schematic of the reaction is shown in Scheme 2.
HPLC-SF-ICP-MS analyses
For HPLC-SF-ICP-MS analyses, a HP1050 modular chromatography system (Agilent Technologies, Stockport, UK) equipped with a Phenomenex Luna C18(2) reversed phase column (100 × 4.6 mm, 3 μm particle size) was used. The mobile phase comprised a binary system of: eluent A, water ∶ acetonitrile (90 ∶ 10% v/v) containing 0.05% formic acid (v/v); and eluent B, water–acetonitrile (10 ∶ 90% v/v) containing 0.05%
(v/v) formic acid. The linear gradient employed started at 100% A, changing to 40% A and 60% B over 20 min. The flow rate was 1 ml min−1 with an injection volume of 5 μl. All experiments were performed using a sector-field inductively coupled plasma mass spectrometer (SF-ICP-MS, Thermo Elemental Axiom, Winsford, UK), using a mass resolution setting of 3000. Operating conditions are shown in Table 1.
Table 1 Operating conditions for SF-ICP-MS
| ICP |
|
| Nebuliser gas flow/l min−1 |
1.10 |
| Auxiliary gas flow/l min−1 |
0.85 |
| Coolant gas flow/l min−1 |
14.0 |
| Nebuliser |
Micromist (Glass Expansion, Switzerland) |
| Spray chamber |
Jacketed quartz cyclonic, cooled to 5 °C |
| Torch |
Quartz Fassel-type with quartz bonnet, without shield |
| |
| Interface |
|
| Sampling cone |
Nickel 1 mm id |
| Skimmer cone |
Nickel, 0.7 mm id |
| |
| Single ion monitoring |
|
| Resolution |
3000 |
| Slit settings |
Source, 330; collector, 220 |
| Masses monitored |
Single ion monitoring for 31P+ at 30.974 m/z |
| Dwell time/ms |
500 |
| |
| Scanning |
|
| Resolution |
3000 |
| Slit setting |
Source, 330; collector, 220 |
| Masses monitored |
Scanning between 30.838 and 31.109 m/z |
| Dwell time/ms |
50 |
| Points |
20 |
It is common, in large pharmaceutical companies, to adopt a standard HPLC elution method which can be used in the majority of cases. The method is often standardised to a simple linear elution of acetonitrile–water which can run up to 100%
(v/v). Such a high concentration of acetonitrile is incompatible with the normal operation of an ICP-MS unless an extremely low flow nebuliser is used. One solution to this, which was adopted in this work, is to use a membrane desolvation system, which removes the vast majority of solvent and allows routine operation of gradient elution up to 100% acetonitrile at a standard 1 ml min−1 flow rate. However, it should be noted that low molecular weight polar analytes may also be removed by the desolvation system, so caution should be used when adopting this approach. In this work the HPLC column was coupled with the SF-ICP-MS simply by inserting the end of the tubing from the column into the sample uptake inlet of the nebuliser, and the output from the spray chamber was introduced into a Universal Interface Model B desolvation device (Vestec Corporation, Houston, USA). The sweep gas was optimised daily at approximately 2.0 l min−1 to ensure that the maximum signal for 31P+ was achieved. The dry aerosol exiting the desolvator was then transferred to the ICP torch via 1 m of 0.25 in. id Tygon tubing.
HPLC was performed using a P580A binary pump (Dionex–Softron GmbH, Germering, Germany) coupled with a Phenomenex Luna C18(2) reversed phase column (100 × 4.6 mm, 3 μm particle size) with the same gradient elution as is used for HPLC-SF-ICP-MS. The 1 ml min−1 flow rate was split post-column (high pressure micro-splitter valve; Upchurch Scientific Ltd, Oak Harbor, WA, USA), and ∼200 μl min−1 was diverted to the mass spectrometer and the residue to waste. Sample injections (5 μl) were made manually with a metal-free Rheodyne injector (model number 9125, CA, USA). Mass spectrometry analysis was performed using an ion trap mass spectrometer fitted with an electrospray interface (ThermoQuest Finnigan Mat LCQ, San Jose, CA). Data were acquired and processed with Xcalibur 1.0 software. Instrument optimization was performed by infusing a 100 ng ml−1 solution of TMPP at 3 μl min−1, monitoring for the characteristic positive ion at 590 m/z. The following instrument parameters were used: source voltage, +4.50 kV; capillary voltage, +20 V; tube lens offset, −10.00 V; capillary temperature, 220 °C; nitrogen sheath gas flow rate, 60 (arbitrary units) and nitrogen auxiliary gas flow rate 20 (arbitrary units). Mass spectra were recorded in the positive ion mode within m/z 190–1000.
Results and discussion
Phosphorus selective detection
Chromatograms of maleic acid (MA), after derivatisation with TMPP, are shown in Fig. 1. Detection was performed using SF-ICP-MS with 31P+ selective detection at 30.974 m/z
(Fig. 1A and 1B), and ESI-MS (Fig. 1C and 1D). As can be seen, the TMPP derivatising reagent contained many phosphorus containing impurities (Fig. 1A), so it was necessary to optimise the chromatography such that the derivatised MA was separated from these peaks (Fig. 1B), and particularly the large peak for unreacted TMPP. For comparison, and confirmatory purposes, detection was also performed using ESI-MS and the major peak at 8.43 min was found to have a mass of 590 m/z, which corresponds to the mass of TMPP. Likewise, the peak at 12.65 min was found to have a mass of 688 m/z, indicating it to be MA derivatised with TMPP (Fig. 1D). A comparison of the chromatogram obtained using ICP-MS (Fig. 1B) with the total ion chromatogram obtained using ESI-MS detection (Fig. 1C) indicates that the former method yielded a slightly improved signal-to-noise ratio, which should result in better detection limits. However, in this case, ESI-MS showed better signal-to-noise when single ion monitoring at the base peak of 688 m/z was performed. This highlights both the advantages and disadvantages of the two techniques, namely that ICP-MS is useful for high sensitivity screening purposes, when confirmation that a compound contains phosphorus (or any other element for that matter) is required. Hence, once these peaks in the chromatogram have been identified, then ESI-MS can be used in single ion mode to perform further qualitative analysis. In this case the TMPP was synthesised in-house so was not particularly pure: however, it is envisaged that a purer form of the reagent would alleviate some of the problems caused by interfering peaks in the ICP-MS chromatogram.
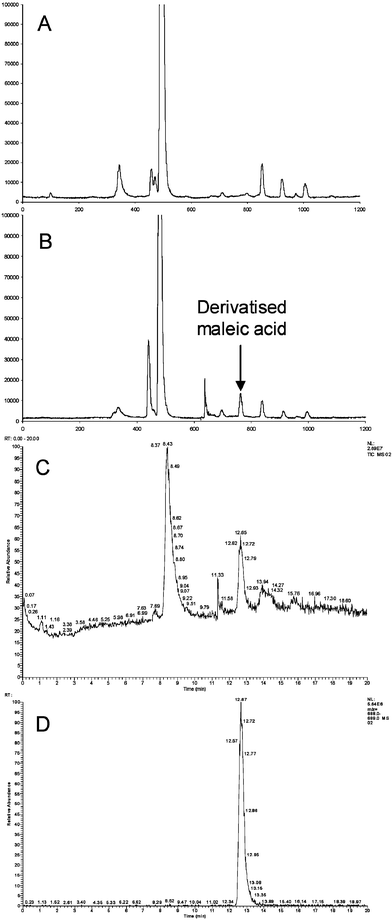 |
| | Fig. 1 Chromatograms of derivatised maleic acid: A, blank injection of TMPP and 31P+ selective detection using SF-ICP-MS; B, derivatised maleic acid and 31P+ selective detection using SF-ICP-MS; C, derivatised maleic acid and total ion current using positive ion ESI-MS; D, derivatised maleic acid and selective ion monitoring at 688 m/z using positive ion ESI-MS. | |
Optimisation of derivatisation reaction
In order to achieve optimal sensitivity the concentrations of the derivatisation reagents were optimised using a 2-factor, 3-level factorial design (Table 2). Concentrations of TMPP and CMPI were set at 0.1 mM, 1 mM and 10 mM for the derivatisation of 0.1 mM of MA. Triethylamine (TEA) was also present as the base for the reaction so its concentration was adjusted to equal the sum of the concentrations of TMPP and MA, in order to maintain the stoichiometry of the reagents. At higher concentrations of TMPP, many impurities were observed, thus making it difficult to identify the peak due to derivatised MA (see Fig. 2B). It is desirable to achieve a situation where the derivatisation of MA is at a maximum for the lowest concentration of reagents, but the reagents are always in sufficient excess to ensure constant reaction efficiency for varying concentrations of acid. In this case, maximum signal for derivatised MA (at 0.1 mM), and hence maximum reaction efficiency, was achieved at concentrations 1 mM (TMPP) and 10 mM (CMPI), with the concentration of TEA at 11 mM. The effect of TMPP on the derivatisation of MA is shown in Fig. 2. At a TMPP ∶ MA ratio of 10 ∶ 1 (1.0 mM TMPP, 0.1 mM MA) a net peak height signal of approximately 1500 for derivatised MA was observed (Fig. 2A). At a TMPP ∶ MA ratio of 100 ∶ 1 (10 mM TMPP, 0.1 mM MA) the peak height signal was similar at approximately 1700; however, impurities in the TMPP eluted close to the analyte peak (Fig. 2B), which made identification of the derivatised MA more difficult. A rough estimate of the efficiency of the reaction was obtained by ratioing the peak height of the derivatised MA to the sum of the derivatised acid plus the residual TMPP reagent peak, then expressing this as a percentage of the expected theoretical ratio calculated from the known concentrations. This assumes equal instrumental response for the two compounds, which should hold roughly true in this case given that a desolvator was used and the nebuliser efficiency was similar over the duration of the gradient in which the peaks eluted. This is illustrated in Fig. 2A, where it is evident that the baseline signal, resulting from phosphorus impurities in the eluent, increased in a linear manner by only a factor of 2 over the course of the chromatographic run, and only by a factor of approximately 1.5 between the elution of TMPP and MA. Hence, for equimolar concentrations of MA and TMPP (1 mM) the reaction efficiency was 22%, and when TMPP was in 10-fold excess the reaction efficiency was 16%. Hence, it is likely that only between 10–20% of MA was derivatised under the conditions used and detection limits could be improved further by improving the efficiency of the reaction.
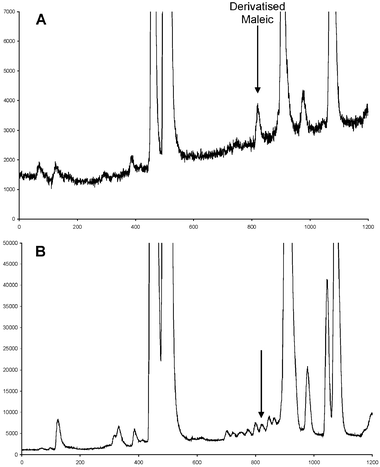 |
| | Fig. 2 HPLC-ICP-MS chromatograms measuring 31P+ at 30.974 m/z for the derivatisation of 0.1 mM maleic acid with: (A) 10 mM CMPI and 1.0 mM TMPP; and (B) 10 mM CMPI and 10 mM TMPP. | |
Table 2 Two factor, three level full factorial design experiment to determine the optimal conditions for derivatisation of 0.1 mM maleic acid
| Solution number |
CMPI concentration/mM |
TMPP concentration/mM |
TEA concentration/mM |
| 1 |
1.0 |
1.0 |
1.1 |
| 2 |
0.1 |
1.0 |
1.1 |
| 3 |
1.0 |
0.1 |
0.2 |
| 4 |
1.0 |
10.0 |
10.1 |
| 5 |
10.0 |
0.1 |
0.2 |
| 6 |
0.1 |
10.0 |
10.1 |
| 7 |
10.0 |
10.0 |
10.1 |
| 8 |
0.1 |
0.1 |
0.2 |
| 9 |
10.0 |
1.0 |
1.1 |
An approximate limit of detection for derivatised MA, using 31P+ selective detection by ICP-MS, was calculated as the concentration of MA which resulted in a peak height of 3× baseline noise. This resulted in a detection limit of 0.05 mM for a 5 μl injection. This is confirmed by Fig. 2A, where a 0.1 mM injection can clearly be seen to be close to the detection limit. For comparison, detection limits for TMPP obtained in this work, and for malathion from previous work on the same instrument, are shown in Table 3. As can be seen, the compound specific detection limit for TMPP was approximately 5–6 times lower than that for derivatised MA (in molar terms), reflecting the 10–20% reaction efficiency of the latter. The 31P+ specific detection limit for malathion achieved previously12 was approximately 10-times lower than the detection limit for TMPP determined in this work. The probable explanation for this is that a plasma shield was not used on this occasion, which resulted in a 10-fold reduction in sensitivity, with a consequent increase in the detection limit.
Table 3 Approximate absolute limits of detection for a 5 μl injection
| |
Compound specific |
31P specific |
| |
/nmol |
/ng |
/ng |
Ref. |
|
100 μl injection.
|
| TMPP (bromide salt) |
0.046 |
30 |
1.4 |
This work |
| MA (derivatised) |
0.25 |
29 |
7.8 |
This work |
| Malathiona |
|
|
0.16 |
12 |
The usefulness of the derivatisation reaction was tested for the detection of several other carboxylic acids, namely, fumaric, sorbic and salicylic acids. Detection using both HPLC-SF-ICP-MS (31P+ selective) and LC-ESI-MS (total ion and extracted ion ranges) was performed to verify which of the phosphorus containing peaks was due to the derivatised acids (Fig. 3). Derivatised MA was observed at 12.6 min (Fig. 2), sorbic acid at 14.4 min and fumaric acid at 12.0 min (Fig. 3). No peak was observed with either 31P+ selective ICP-MS or ESI-MS detection for derivatised salicylic acid. This is probably due to strong internal hydrogen bonding making salicylic acid a poor nucleophile, leading to an inefficient reaction with the CMPI activating reagent. It should be possible to derivatise a range of carboxylic acids using this method, as described by Leavens et al.10 They did not give any indication as to the efficiency of the reaction or detection limits: however, this has been addressed by us in another paper.11 It should be possible to improve reaction efficiency by optimising the derivatisation chemistry, and utilising a solid phase analytical derivatisation approach as described by Pilus et al.13
 |
| | Fig. 3 Chromatograms of derivatised sorbic (left) and fumaric (right) acids: A, 31P+ selective detection using SF-ICP-MS; B total ion current using positive ion ESI-MS; C, selective ion monitoring using positive ESI-MS. | |
Conclusions
HPLC-SF-ICP-MS has been used for the determination of maleic, sorbic and fumaric acids after derivatisation with the phosphorus containing reagent tris(2,4,6-trimethoxyphenyl)phosphonium propylamine. This allowed 31P+ selective detection to be performed on organic compounds which are normally not amenable to detection by ICP-MS at low concentrations. The derivatisation reaction was partially optimised for MA: however, there is scope for further improving the reaction efficiency to achieve lower detection limits and a more quantitative analysis. Work is currently underway, in our laboratory, on the preparation of a multiply brominated TMPP reagent, which will allow simultaneous bromine and phosphorus selective detection. Multiple bromination should improve detection limits and make possible the use of this reagent to detremine the degree of phosphorylation of peptides. The scope of application for reagents such as these is great, particulalrly in proteomics and genomics where ever more selective and sensitive methods of analysis are required. The application of isotopically tagged reagents is a particular focus of our work and is now close to publication.
Acknowledgements
The authors would like to thank Dr. Peter Marshall, Dr. William J. Leavens and Dr. Richard Carr (all GlaxoSmithKline) for developing the TMPP reagent and their useful discussions and suggestions. The authors are also grateful to the EPSRC and GlaxoSmithKline for providing an Industrial CASE award to A.J.C.
References
- E. T. Luong and R. S. Houk, J. Am. Soc. Mass Spectrom., 2003, 14(4), 295 CrossRef CAS.
- J. Vogl and K. G. Heumann, Fresenius’ J. Anal. Chem., 1997, 359(4–5), 438 CrossRef CAS.
- J. Vogl and K. G. Heumann, Anal. Chem., 1998, 70(10), 2038 CrossRef CAS.
- M. Wind, M. Edler, N. Jakubowski, M. Linscheid, H. Wesch and W. D. Lehmann, Anal. Chem., 2001, 73(1), 29 CrossRef CAS.
- M. Wind, H. Wesch and W. D. Lehmann, Anal. Chem., 2001, 73(13), 3006 CrossRef CAS.
- M. Wind, O. Kelm, E. A. Nigg and W. D. Lehmann, Proteomics, 2002, 2(11), 1516 CrossRef CAS.
- E. H. Evans, J. C. Wolff and C. Eckers, Anal. Chem., 2001, 73(19), 4722 CrossRef CAS.
- C. Smith, B. P. Jensen, I. D. Wilson, F. Abou-Shakra and D. Crowther, Rapid Commun. Mass Spectrom., 2004, 18(13), 1487 CrossRef CAS.
- P. Marshall, O. Heudi, S. McKeown, A. Amour and F. Abou-Shakra, Rapid Commun. Mass Spectrom., 2002, 16(3), 220 CrossRef CAS.
- W. J. Leavens, S. J. Lane, R. M. Carr, A. M. Lockie and I. Waterhouse, Rapid Commun. Mass Spectrom., 2002, 16(5), 433 CrossRef CAS.
- A. J. Cartwright, E. H. Evans, P. Jones, J. C. Wolff, P. Marshall and W. J. Leavans, Rapid Commun. Mass Spectrom., 2004 Search PubMed , submitted for publication.
- J. Carter, L. Ebdon and E. H. Evans, J. Anal. At. Spectrom., 2003, 18(2), 142 RSC.
-
R. Pilus, J. M. Rosenfeld, J. Terlouw and W. Leavens, Eighth International Symposium on Hyphenated Techniques in Chromatography, February 4–6, 2004, Bruges, Belgium Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.