Strecker intermediates as non-pollutant scavengers for cyanides
Fernando
Godinez-Salomon
ab,
Jose M.
Hallen-Lopez
b,
Herbert
Höpfl
c,
Adela
Morales-Pacheco
a,
Hiram I.
Beltrán
*a and
Luis S.
Zamudio-Rivera
*a
aPrograma de Ingeniería Molecular, Instituto Mexicano del Petróleo, Eje Central Lázaro Cárdenas 152, Col. San Bartolo Atepehuacan, México D.F. E-mail: lzamudio@imp.mx. hbeltran@imp.mx; Fax: +52 5591756239; Tel: +52 5591757399
bDepartamento de Ingeniería Metalúrgica, Escuela Superior de Ingeniería Química e ndustrias Extractivas, I.P.N. Unidad Profesional Adolfo López Mateos, México D.F
cCentro de Investigaciones Químicas, U.A.E.M., Av. Universidad 1001, C. P. 62210 Cuernavaca, Morelos, México
First published on 30th August 2005
Abstract
Using a series of five bis(aminomethyl)ethers, a fast and efficient transformation of sodium cyanide to sodium N-2-hydroxyethylglycinates is reported, where the starting materials are prototypes for application in the mining and oil industries to diminish the pollution of the tailings derived from their processes.
Introduction
For the petroleum industry, hydrogen sulfide and metal cyanides are among the most common corrosive pollutants in crude oil.1,2 Of these two reagents cyanide ions are especially problematic, since their damage is twofold: firstly, the H2S induced corrosion is enhanced by −CN ions through the interaction they have with iron sulfide; and secondly, cyanide pollution affects the petroleum feedstock for further processes.2,3 On the other hand, the cyanide process of gold recovery enables a higher percentage of not only gold but silver to be extracted from hard rock, making many operations viable that would otherwise have had to close.4,5 During the ore extraction, environmental harm is caused through the tailings, which have unavoidably high amounts of cyanide ions.4,5Tailings, also called gangue, are the rejected material from mining and screening operations. These tailings are the uneconomic remainders from mining; as mining techniques and the price of minerals improve it is not unusual for tailings to be reprocessed, mainly to recover minerals other than those originally mined.
One of the strategies to solve these problems is to diminish the concentration of the pollutants, which in industrial application is commonly achieved by the addition of scavenger molecules.1–3,6 Characteristics required for a good scavenger are (i) that the scavenging reaction takes place under the industrial process conditions, (ii) that the product derived from the scavenging reaction is no longer contaminating and/or does not interfere in such a process, and (iii) that the resulting compounds can be reused within the industrial facilities or used for other important side applications.
So far, only a few scavengers that are selective towards cyanide ions are known; among them are hydroxocobalamin,7 dicyano-cobalt(III)-porphyrins8 and other vitamin B12 analogues,8 metallophthalocyanines8 and the hexahydrated dichloride compounds of cobalt(II) and nickel(II), which have shown good performance, but are rather toxic.8
Results and discussion
One pot reactions between the β-aminoalcohols 1a–1e and paraformaldehyde (Scheme 1) led to five cyanide scavengers 2a–2e in yields ranging from 80 to 99%. The title compounds are isolated intermediates of the well known Strecker synthesis.9 Because of ecological reasons the reactions were carried out in the absence of any solvent. When compounds 2a–2e were treated with two equivalents of sodium cyanide and heated in water or different water/solvent mixtures to a temperature of 40 °C, the expected cyanide induced hydrolysis reaction took place, giving the N-substituted N-2-hydroxyethylglycine sodium salts 3a–3e (Scheme 1) in yields between 66 and 94%.10 It was noticeable that the n-butyl, 2b (66%), and tert-butyl, 2c (80%), Strecker scavengers were the less efficient for the scavenging action. It is also worth mentioning that in the mining industry, most of the washing operations are carriers of harmful ions, where the water molecules physically scavenge the cyanide counterparts. For that reason, the present scavengers imply an important chemical strategy since they take their action in the presence of water. In order to evaluate the potential of compounds 2a–2e for an application directed to the petroleum industry, the reactions outlined in Scheme 1 have also been successfully carried out in 50 ∶ 1 heptane–water and in a second trial in 50 ∶ 1 chloroform–water solvent mixtures. For application purposes, the temperature required for the transportation and general processing of crude oil is sufficient to reach the activation energy required for the scavenger reaction that is fast and easily accomplished at 40 °C. The application of compounds 2a–2e in real petroleum currents is under study and a patent development with a large number of derivatives for these means is being carried out.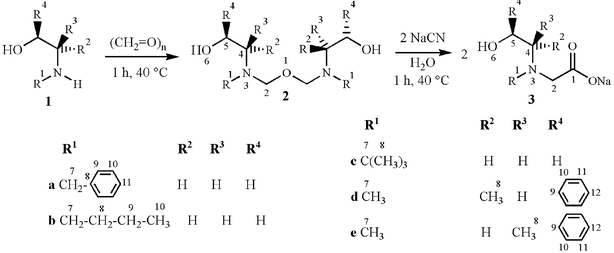 | ||
| Scheme 1 Reaction for the preparation and scavenger activity of bis(semiaminals) 2a–2e for sodium cyanide. | ||
Compounds 3a–3e were isolated in yields ranging from 56 to 94% by filtration of the precipitates that were formed after slow addition of acetone to the reaction mixture. All products were completely characterized by IR, 1H, 13C and 2D NMR spectroscopic methods and elemental analyses, certifying the formation of the scavenger molecules as well as the corresponding sodium salts.
As an insight into the supramolecular architectures formed by these chemical products, further structural evidence was provided by the X-ray diffraction analysis of compound 3d.†11–14 Two solutions were obtained for the collected data in different space groups, trigonal, P32, and hexagonal, P62; both of them were refined resulting in better statistical values for the latter† which hence was used for the forthcoming discussion. Moreover, an incoming problem was the possibility of a crystal as a racemic twin, since the Flack parameter was 0.4(10) as well as the thermal ellipsoids for the water molecules and the final R indices were high in both pseudosymmetric solutions when the collection was carried out at room temperature due to the disorder present in the uncoordinated water moiety. Attempts to refine as a racemic twin were unsuccessful.15 Finally, the collection of X-ray data at 100 K led to a congruent Flack parameter near to zero and an uncoordinated water moiety was easily situated. Fig. 1a shows one of the independent molecular fragments present in the crystal lattice of 3d, which is the repeating unit of a linear coordination polymer (similar to transition metal coordination polymers) as shown in Fig. 1b. Each sodium is coordinated to the three donor atoms of the chelating tridentate ligand in a meridional fashion, and furthermore to two water molecules and the carbonyl group of a neighbouring complex molecule, giving rise to a coordination polymer along c. The sodium ions possess distorted octahedral coordination geometries; the Na–N bond length is 2.572 Å, the Na–O bond lengths are: for Na–OH, 2.414, for Na–OOCintramol 2.320 Å, for Na–OH2, 2.391, 2.604 Å, and for Na–OOCintermol, 2.335 Å. Due to the coordination of the tridentate ligand towards the sodium ion, a five-five-membered rings fused structure is constructed. The O(2)–C(4)–C(3)–N(1)–Na(1) five-membered ring has an envelope conformation, where the C(1) atom is placed away from the plane due to the presence of the phenyl moiety. In the crystal lattice, the two independent linear polymers form double chains through three different hydrogen-bonding interactions.
 | ||
| Fig. 1 (a) The crystal lattice of 3d contains six molecular units, one of which is shown here. (b) Fragment of one of the linear coordination polymers, showing the octahedral coordination geometry of the sodium ions. | ||
Six molecular slabs of the infinite chain were selected to mimic the supramolecular structure organized through further hydrogen-bonding interactions (two per individual complex molecule) around a crystallographic 62 symmetry axis to give a chiral tubular structure as shown in Fig. 2. The hydrogen bonds are formed between the sodium-coordinated water molecules, the hydroxyl groups of the β-aminoalcohol fragments in the ligands and the carboxylate groups (O⋯O = 2.72–2.83 Å) as well as with the uncoordinated water molecules. The nano-sized tubes have overall diameters of 22.9 Å and are organized in a pseudosymmetric hexagonal honeycomb-like arrangement (Fig. 2b). They possess a hydrophobic outer-sphere formed by the phenyl and methyl groups of the β-aminoalcohol fragments and contain hydrophilic channels. The distance between opposite sodium ions is 10.3 Å. The pores, whose hydrophilic interior is formed by the sodium-coordinated water molecules, extend throughout the whole crystal lattice and are filled by uncoordinated water molecules; the water molecules mainly govern this type of architectures.16 A search of the CCDC database17 revealed that there are only very few structurally characterized sodium complexes containing tridentate ligands of the NO2 type.
 | ||
| Fig. 2 (a) Perspective view of the crystal lattice of compound 3d along c, showing one of the chiral tubes. (b) Hexagonal arrangement of the tubes shown before; special attention should be paid to the hydrophobic outer-sphere and the hydrophilic core (for clarity, the sodium-coordinated and uncoordinated water molecules have been omitted. (c) Lateral view of the chiral assembly of the two independent (dark and light grey) coordination polymers present in the supramolecular structure. | ||
Since the Strecker intermediates, 2a–2e, and the resulting ligands, 3a–3e, are going to be tested as anthropogenic chemicals, they might be evaluated as environment-friendly compounds. Following a well established protocol,3,18–19 fast screening toxicity tests were performed for compounds 2a–2e and 3a–3e.18–19 The EC50 values measured at incubation times of 5 and 15 min range from 416–1127 ppm (Table 1), so that these chemicals can be classified as only slightly toxic, in comparison to the high toxicity in the case of sodium cyanide and other cyanide derivatives.
| Compound | Yield (%) | EC50 (ppm) | |
|---|---|---|---|
| 5 min | 15 min | ||
| a Concentration range in ppm, classification, category: 0.01–0.10, 5, extremely toxic; 0.1–1.0, 4, highly toxic; 01–10, 3, moderately toxic; 010–100, 2, slightly toxic; 0100–1000, particularly non toxic, more than 1000, 0, non toxic. See ref. 3 and 17 for further details.2 | |||
| 2a | 99 | 681 | 420 |
| 2c | 97 | 660 | 499 |
| 2b | 86 | 551 | 416 |
| 2e | 80 | 646 | 659 |
| 2d | 85 | 794 | 455 |
| 3a | 91 | 959 | 744 |
| 3c | 66 | 1127 | 841 |
| 3b | 80 | 596 | 463 |
| 3e | 92 | 659 | 583 |
| 3d | 94 | 863 | 636 |
| NaCN | — | 0.017 | 0.011 |
Conclusions
The Strecker intermediates described herein can be prepared in fast and efficient one-step reactions without the requirement of a solvent. They can function as scavengers for sodium cyanide in water and solvent mixtures that have a composition similar to crude oil and the tailings derived from the mining processes. The Strecker intermediates themselves and the resulting tridentate ligands coordinated with sodium are environment-friendly and water-soluble compounds. These ligands coordinate to sodium ions through their carboxylate, hydroxyl and amino functional groups, thus giving insight into the coordination chemistry of aminoalcohol and aminoacid derivatives, which indeed construct interesting nano-rod motifs.Experimental
Paraformaldehyde and β-aminoalcohols were purchased from Aldrich Co. The acetone, chloroform and heptanes solvents were purchased as reagent grade from Fermont Co. All reactions and operations were carried out under atmospheric conditions.Instrumentation
NMR experiments were performed on a VARIAN Mercury 200-BB spectrometer. 1H and 13C chemical shifts [ppm] are relative to internal SiMe4 (TMS). Coupling constants are quoted in Hz. The IR spectra were recorded in the range of 4000–400 cm−1 on a Bruker Tensor-27 FT-IR spectrometer by using KBr pellets or the ATR technique. The X-ray structure determination was performed in a BRUKER-AXS APEX diffractometer with a CCD area detector (λMo Kα = 0.71073 Å, monochromator: graphite).General method for the preparation of compounds 2a–2e and 3a–3e
The 2a–2e and 3a–3e series of compounds have each been prepared by analogous methods, therefore, only the preparation of compounds 2c and 3c is described in detail: 2.00 g (8.50 mmol) of 1c and 0.51 g (8.50 mmol) of paraformaldehyde were heated to 40 °C for 1 h without using a solvent. The water formed during the condensation reaction was removed at reduced pressure giving 2c as a light greenish liquid in a yield of 86%. Then, 1.80 g of 2c (6.47 mmol) and 0.64 g of sodium cyanide (13.03 mmol) were heated to 40 °C for 1h in 30 mL of bidistilled water. When the reaction time was accomplished, the solvent was partially removed at reduced pressure and 3c was precipitated by addition of 30 mL of acetone. The resulting beige powder was filtered and identified as 3c (yield: 59%).Characterization and spectroscopic data
2a: Yield 99%. IR (ν/cm−1) 3465, 3062, 3028, 2878, 1494, 1453, 1154, 733, 697. 1H NMR (δ/ppm, J/Hz): 7.33–7.31 (m, 10H, H-9, 10, 11), 4.10 (s, 4H, H-2), 3.85 (t, J = 6.8, 4H, H-5), 3.71 (s, 4H, H-7), 2.93 (t, 4H, J = 6.8, H-4). 13C NMR (δ/ppm): 138.2 (C-8), 128.3–126.6 (C-9, 10, 11), 86.3 (C-2), 63.2 (C-5), 58.0 (C-7), 52.1 (C-4).3a: Yield 91%, mp 85–87 °C. IR (ν/cm−1) 3062, 2878, 1612, 1406, 1000, 733, 697. 1H NMR (δ/ppm, J/Hz): 7.24 (m, 5H, H-9, 10, 11), 3.61 (s, 2H, H-7), 3.48 (t, 2H, J = 6.0, H-5), 2.82 (s, 2H, H-2), 2.58 (t, 2H, J = 6.0, H-4). 13C NMR (δ/ppm): 179.5 (C-1), 137.7 (C-8) 137.2–127.7 (C-9, 10, 11), 59.0 (C-5), 58.4 (C-2), 57.5 (C-7), 55.3 (C-4).
2b: Yield 97%. IR (ν/cm−1) 3446, 2932, 2872, 1460, 1021, 738. 1H NMR (δ/ppm, J/Hz): 4.16 (s, 4H, H-2), 3.67 (t, 4H, J = 7.6, H-5), 2.83 (t, 4H, J = 7.6, H-4), 2.41 (t, 4H, J = 7.4, H-7), 1.33 (m, 8H, H-8,9), 0.83 (t, 6H, J = 7.6, H-10). 13C NMR (δ/ppm): 86.4 (C-2), 63.4 (C-5), 53.8 (C-4), 52.3 (C-7), 31.7 (C-8), 30.7 (C-9), 14.3 (C-10).
3b: Yield 66%, mp > 400 °C. IR (ν/cm−1) 3446, 2932, 2872, 1596, 1460, 1021, 738. 1H NMR (δ/ppm, J/Hz): 3.33 (t, 2H, J = 5.2, H-5), 2.80 (s, 2H, H-2), 2.44 (t, 2H, J = 5.2, H-4), 2.42 (t, 2H, J = 4.6, H-7), 1.27 (m, 4H, H-8,9), 0.83 (t, 3H, H-10). 13C NMR (δ/ppm): 176.2 (C-1), 60.8 (C-5), 60.4 (C-2), 59.5 (C-4), 55.2 (C-7), 29.3 (C-8), 20.8 (C-9), 14.7 (C-10).
2c: Yield 86%. IR (ν/cm−1) 2970, 1657, 1470, 1393, 1091. 1H NMR (δ/ppm, J/Hz): 4.34 (s, 4H, H-2), 3.78 (t, 4H, J = 6.6, H-5), 2.89 (t, 4H, J = 6.6, H-4), 1.06 (s, 18H, H-8). 13C NMR (δ/ppm): 80.6 (C-2), 66.1 (C-5), 52.4 (C-4), 45.1 (C-7), 26.9 (C-8).
3c: Yield 59%, mp 171–172 °C. IR (ν/cm−1) 3255, 2971, 2813, 1602, 1408, 1359, 1084. 1H NMR (δ/ppm, J/Hz): 4.65 (s, 1H, H-6), 3.42 (t, 2H, J = 6.0, H-5), 3.02 (s, 2H, H-2), 2.89 (t, 2H, J = 6.0, H-4), 1.06 (s, 9H, H-8). 13C NMR (δ/ppm): 182.0 (C-1), 60.8 (C-5), 55.6 (C-2), 52.3 (C-7), 51.8 (C-4), 25.7 (C-8).
2d: Yield 80%. IR (ν/cm−1) 3032, 2968, 2875, 1674, 1494, 1053, 810, 750. 1H NMR (δ/ppm, J/Hz, 200 MHz): 7.34 (bs, 10H, H-10,11,12), 4.75 (d, 2H, J = 3.3, H-2a), 4.48 (d, 2H, J = 8.2, H-5), 4.29 (d, 2H, J = 3.3, H-2b), 2.43 (dq, 2H, J = 6.2, 2.0, H-4), 2.36 (s, 6H, H-7), 1.17 (d, 6H, J = 6.2, H-8). 13C NMR (δ/ppm): 140.4 (C-9), 128.2-126.0 (C-10, 11, 12), 88.6 (C-5), 85.8 (C-2), 68.0 (C-4), 36.8 (C-7), 14.2 (C-8).
3d: Yield 92%, mp = 172–173 °C. IR (ν/cm−1) 3356, 3031, 1731, 1572, 1494, 1043, 763, 700. 1H NMR (δ/ppm, J/Hz): 7.25 (bs, 5H, H-10,11,12), 4.19 (d, 2H, J = 3.2, H-5), 2.98 (d, 1H, J = 16.2, H-2a), 2.97 (d, 1H, J = 16.2, H-2b), 2.69 (dq, 1H, J = 6.7, 3.2, H-4), 2.12 (s, 3H, H-7), 0.49 (d, 3H, J = 6.7, H-8). 13C NMR (δ/ppm): 180.1 (C-1), 140.1 (C-9), 128.7-127.7 (C-10,11,12), 72.3 (C-5), 64.2 (C-2), 57.5 (C-7), 39.9 (C-4), 7.8 (C-8).
2e: Yield 85%. IR (ν/cm−1): 3064, 3028, 2786, 1604, 1493, 1454, 1224, 751, 698. 1H NMR (δ/ppm, J/Hz): 7.29 (bs, 10H, H-10,11,12), 5.11 (d, 2H, J = 7, H-5), 4.88 (bs, 2H, H-2a), 4.06 (bs, 2H, H-2b), 2.87 (t, 2H, J = 6.6, H-4), 2.37 (s, 6H, H-7), 0.67 (d, 6H, J = 6.4, H-8). 13C NMR (δ/ppm): 139.8 (C-9), 127.8-126.8 (C-10,11,12), 88.1 (C-2), 81.8 (C-5), 63.3 (C-4), 37.6 (C-7), 14.2 (C-8).
3e: Yield 94%, mp > 172 °C (dec.). IR (ν/cm−1) 3250, 2952, 2834, 1888, 1581, 1017, 760, 694. 1H NMR (δ/ppm, J/Hz): 7.25 (bs, 5H, H-10, 11, 12), 3.46–3.35 (m, 2H, H-2a, H-5), 2.99 (bs, 1H, H-2b), 2.65 (bs, 1H, H-4), 2.23 (s, 3H, H-7), 0.71 (bs, 3H, H-8). 13C NMR (δ/ppm): 177.5 (C-1), 145.2 (C-9), 128.6–126.7 (C-10, 11, 12), 72.4 (C-5), 64.7 (C-2), 59.6 (C-7), 40.8 (C-4), 10.5 (C-8).
The 8 eV EI mass spectra of compounds 2a–2e do not show the molecular ion peak; all show a C–O bond rupture leading to iminium fragments.
X-ray crystallography
An X-ray diffraction study of 3d was carried out on a BRUKER-AXS APEX diffractometer with a CCD area detector (λMo Kα = 0.71073 Å, monochromator: graphite). Frames were collected at T = 100 K or 298 K viaω- and φ-rotation at 10 s per frame (SMART11). The measured intensities were reduced to F2 and corrected for absorption with SADABS (SAINT-NT12). Structure solution, refinement, and data output were carried out with the SHELXTL-NT,13 SHELXS-97,14 and SHELXL-9714 programs. Non-hydrogen atoms were refined anisotropically. All data were corrected for Lorentz and polarization effects. All additional treatments were done through the WIN-GX20 program set with the PARST21 utility; the corresponding molecular graphs were prepared with the ORTEP 322 and Mercury 1.223 programs.Acknowledgements
The present study was supported by Instituto Mexicano del Petróleo, Programa de Ingeniería Molecular with project number D.00178.References
- D. Brodel, R. Edwards, A. Hayman, D. Hill, S. Mehta and T. Semerad, Oilfield Rev., 1994, 35, 4 Search PubMed.
- L. A. C. J. Garcia, C. J. B. M. Joia, E. M. Cardoso and O. R. Mattos, Electrochim. Acta, 2001, 46, 3879 CrossRef CAS; D. Urban, S. Frisbie and S. Croce, Environ. Prog., 1997, 16, 171 CrossRef CAS.
- G. F. Whale and T. S. Whitham, Soc. Pet. Eng., 1991, 23357, 355 Search PubMed.
- M. R. Ally, J. B. Berry, L. R. Dole, J. J. Ferrada and J. W. Van Dyke, Economical Recovery of by-Products in the Mining Industry,ORNL/TM-2001/225, Oak Ridge National Laboratory, Oak-Ridge Tennessee, 37831-6285, UT-Batelle LLC & U.S. Department of Energy, 2001, p. 33. Web page: www.dole.nu/lesdole/TM225.pdf Search PubMed.
- A. Smith and T. Mudder, The Chemistry and Treatment of Cyanidation Wastes, Mining J. Books Ltd. Pub., London, 1991 Search PubMed.
- L. S. Zamudio-Rivera, A. Estrada-Martinez, A. Morales-Pacheco, J. L. R. Benitez-Aguilar, R. Roldan-Perez, J. L. Yañez-Ayuso, J. Marin-Cruz and M. G. Guzman-Pruneda, Mex. Pat. Req., 2002 Search PubMed Pa/a/2002/006434.
- S. W. Sauer and M. E. Keim, Ann. Emerg. Med., 2001, 37, 635 CrossRef CAS.
- S. Mosseri, P. Neta, A. Harriman and P. Hambright, J. Inorg. Biochem., 1990, 39(2), 93 CrossRef CAS; R. Rahimi and P. Hambright, J. Porphyrins Phthalocyanines, 1998, 2, 493 CrossRef CAS.
- A. Strecker, Liebigs. Ann. Chem., 1850, 75, 27; D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC.
- N. Farfan, L. Cuellar, J. M. Aceves and R. Contreras, Synthesis, 1987, 10, 927 CrossRef.
- Bruker Analytical X-ray Systems, SMART: Bruker Molecular Analysis Research Tool, version 5.618, 2000 Search PubMed.
- Bruker Analytical X-ray Systems, SAINT+NT, version 6.04, 2001.
- Bruker Analytical X-ray Systems, SHELXTLNT, version 6.10, 2000.
- G. M. Sheldrick, SHELX-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1993 Search PubMed.
- R. I. Cooper, R. O. Gould, S. Parsons and D. J. Watkin, J. Appl. Crystallogr., 2002, 35, 168 CrossRef CAS ; ROTAX program.
- P. Román-Bravo, M. López-Cardoso, P. García y García, H. Höpfl and R. Cea-Olivares, Chem. Commun., 2004, 1940 RSC.
- CSD version 5.25, November 2003, http://www.ccdc.cam.ac.uk.; F. M. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380 Search PubMed; I. J. Bruno, J. C. Cole, P. R. Edington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B, 2002, 58, 389 CrossRef.
- A. E. Lindsay, A. R. Greenbaum and D. O’Hare, Anal. Chim. Acta, 2004, 511, 185 CrossRef.
- G. Mannaioni, A. Vannacci, C. Marzocca, A. M. Zorn, S. Peruzzi and F. Moroni, J. Toxicol. Clin. Toxicol., 2002, 40, 181 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef ; Win GX program set.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659–660 CrossRef CAS ; PARST program, release Nov. 1999.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–566 CrossRef CAS ; ORTEP 3 program.
- Mercury 1.1.2, Copyright CCDC 2001-2002, All rights reserved. For more information about this application, please contact User Support by e-mail at: support@ccdc.cam.ac.uk, or at the www site http://www.ccdc.cam.ac.uk/mercury/.
Footnotes |
| † Crystal data and experimental details for 3d. Empirical formula: C24H44N2Na2O12, formula weight, 598.6, crystal colour, colourless, cryst. dim. [mm3], 0.02 × 0.02 × 0.05, lattice parameters, a [Å], 21.1806 (15), c [Å], 5.6912 (6), volume [Å3], 2211.1 (3), crystal system, hexagonal, space group, P62, Z, 3, ρcalc [mg m−3], 1.349, µ [mm−1], 0.131, θrange [°], 1.92 to 24.98, coll. refl., 21426, indep. refls [Rint], 1441, data/restr/param, 1441/26/248, GOOF, 1.112, R indices [I > 2σ(I)], 0.0553, R indices (all data), 0.1364, Δρmin [e Å−3], −0.53, Δρmax [e Å−3], 0.74. CCDC reference number 255743. See http://dx.doi.org/10.1039/b504406e for crystallographic data in CIF or other electronic format. |
| ‡ In the test, a vial of the Photobacterium phosphoreum culture was reconstituted in 1 mL of solution and maintained at 3 °C in an incubator well on the analyzer. For each test, a serial dilution of the compounds was prepared in 2% brine. In addition, a series of brine solutions containing approximately 106 colony forming units of P. phosphoreum were prepared in glass cuvettes by pipetting 10 µL of the suspension into 500 µL of 2% brine. As a bacterial population control, these solutions were incubated in temperature controlled wells (6 °C) over 15 min and measured. The activity of compounds was recorded after 5 and 15 min of exposure. The results of the Microtox™ tests are expressed in terms of the EC50 value. |
| This journal is © The Royal Society of Chemistry 2005 |