Diversity in electrochemical oxidation of dihydroxybenzoic acids in the presence of acetylacetone. A green method for synthesis of new benzofuran derivatives†
Davood
Nematollahi
* and
Mohammad
Rafiee
Department of Chemistry, Faculty of Science, University of Bu-Ali-Sina, Hamadan, 65174, Iran. E-mail: nemat@basu.ac.ir; Fax: 0098-811-8272404; Tel: 0098-811-8271541
First published on 21st July 2005
Abstract
Electrochemical oxidation of diol derivatives of benzoic acid (1–3) have been studied in the presence of acetylacetone (4) as the nucleophile in aqueous solutions, using cyclic voltammetry and controlled-potential coulometry. The results indicate that the quinones derived from dihydroxybenzoic acids (1a–3a) participate in Michael addition reactions with acetylacetone (4) and via various mechanisms convert to the corresponding benzofurans (1d–3d). In this work, we derive various products with good yields based on electrochemical oxidation under controlled potential conditions in aqueous solutions, without toxic reagents and solvents at a carbon electrode in an undivided cell, using an environmentally friendly method.
Introduction
Because electrochemical oxidation very often parallels the cytochrome P450 catalyzed oxidation in liver microsomes, it was interesting to study the anodic oxidation of catechols in the presence of the β-diketone as CH-acidic nucleophiles. In addition, because of the pharmacological uses of benzofurans the syntheses and pharmacological properties of benzofuran derivatives have been extensively investigated.1–8 In this direction, recently, we have investigated the electrochemical oxidation of catechols in the presence of acethylacetone and dimedone as nucleophiles. The results indicate formation of benzofuran derivatives (Scheme 1).9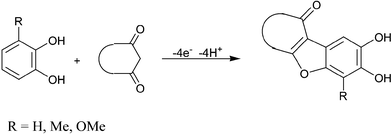 | ||
| Scheme 1 | ||
In this work electrochemical oxidation of some ortho and para dihydroxybenzoic acids (1–3) has been studied in aqueous solutions in the presence of acetylacetone (4) as a possible nucleophile. The results indicate different mechanisms for each case. The present work has led to the development of a facile and environmentally friendly reagentless electrochemical method for synthesis of some new benzofuran derivatives in aqueous solutions with high atomic economy and safe waste (sodium chloride and acetic acid), under ambient conditions and in an undivided cell using a graphite electrode.
Results and discussion
Electrochemical study of 3,4-dihydroxybenzoic acid (1)
The electrochemical study of 1 mM solution of 3,4-dihydroxybenzoic acid (1) in an aqueous sodium acetate solution (0.2 M) as supporting electrolyte, at a bare glassy carbon electrode has been performed using cyclic voltammetry (Fig. 1, curve a). The voltammogram shows one anodic (A1) and corresponding cathodic peak (C1), at 0.34 V and 0.08 V versus a standard calomel electrode (SCE), respectively, which correspond to the transformation of 3,4-dihydroxybenzoic acid (1) to o-benzoquinone (1a) and vice versa within a quasi-reversible two-electron process (Scheme 2, eqn. (1)). A peak current ratio (IClp/IAlp) of near unity, particularly during the repetitive recycling of potential, can be considered as a criterion for the stability of o-benzoquinone (1a) produced at the surface of the electrode under the experimental conditions. In other words, hydroxylation10 or dimerization11 reactions are too slow to be observed on the time scale of cyclic voltammetry. The oxidation of 3,4-dihydroxybenzoic acid (1) in the presence of acetylacetone (4) as a nucleophile was studied in some detail. Fig. 1, curve b shows the cyclic voltammogram obtained for a 1 mM solution of 1 in the presence of 1 mM acetylacetone (4). The voltammogram exhibits two anodic peaks (A1 and A2). The cathodic counterpart of the anodic peak A1 nearly disappears. Comparison of voltammograms b and d reveals that the peak A2 corresponds to the oxidation of acetylacetone (4) or acetylacetone linked to 3,4-dihydroxybenzoic acid (1). The multi-cyclic voltammetry of 1a in the presence of 4 shows that parallel to the shift of the A1 peak in a positive direction, a new peak (A0) appears as a shoulder at a less positive potential in the second cycle (Fig. 1, curve c). This new peak is related to electro-oxidation of intermediate 1b. The positive shift of the A1 peak in the presence of acetylacetone is due to the formation of a thin film of product at the surface of the electrode.9 In Fig. 1, curve d is the voltammogram of 4.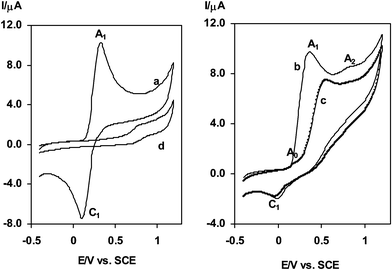 | ||
| Fig. 1 Cyclic voltammograms of 1 mM 3,4-dihydroxybenzoic acid: (a) in the absence of acetylacetone, (b and c) first and second scan in the presence of 1 mM acetylacetone and (d) 1 mM acetylacetone in the absence of 3,4-dihydroxybenzoic acid. Supporting electrolyte: sodium acetate solution (0.2 M). Scan rate: 100 mV s−1. t = 25 ± 1 °C. | ||
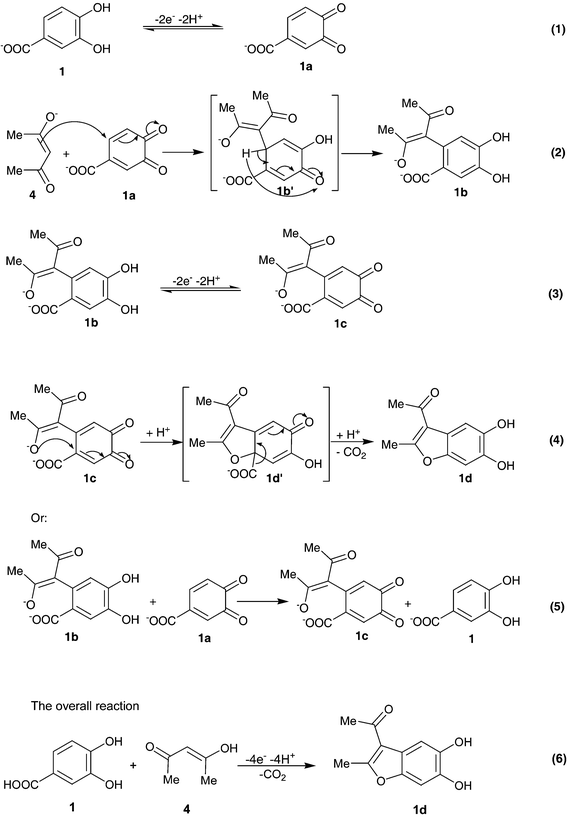 | ||
| Scheme 2 | ||
Furthermore, it is seen that proportional to the augmentation of potential sweep rate, the height of the C1 peak increases (Fig. 2, curves a–e). A plot of peak current ratio (IAlp/IClp) versus scan rate for a mixture of 3,4-dihydroxy benzoic acid (1) and acetylacetone (4) confirms the reactivity of 1 towards 4, appearing as an increase in the height of the cathodic peak C1 at higher scan rates (Fig. 2, curve f). On the other hand, the current function for the A1 peak, (IAlp/v1/2), decreases on increasing the scan rate and such a behavior is seen as indicative of an ECEC mechanism.12
 | ||
| Fig. 2 Typical cyclic voltammograms of 1 mM 3,4-dihydroxybenzoic acid in the presence of 1 mM acetylacetone at a glassy carbon electrode, in sodium acetate solution (0.2 M). Scan rates from (a) to (e) are: 25, 50, 100, 250 and 500 mV s−1, respectively. (f): Variation of peak current ratio (IAlp/IClp) versus scan rate. t = 25 ± 1 °C. | ||
Controlled-potential coulometry was performed in aqueous solution containing 0.5 mmol of 1 and 0.5 mmol of 4 at 0.4 V versus SCE. Cyclic voltammetric analysis carried out during the electrolysis shows the progressive formation of a new anodic (A0) peak, parallel to the disappearance of the A1 peak (Fig. 3). All anodic (A0, A1 and A2) and cathodic peaks disappear when the charge consumption becomes about 4e− per molecule of 1 (Fig. 3, inset). These observations allow us to propose the pathway in Scheme 2 for the electro-oxidation of 1 in the presence of 4.
 | ||
| Fig. 3 Cyclic voltammograms of 0.5 mmol 3,4-dihydroxybenzoic acid in the presence of 0.5 mmol acetylacetone, at a glassy carbon electrode during controlled potential coulometry at 0.4 V versus SCE. After consumption of: (a) 0, (b) 30, (c) 65, (d) 100, (e)140, (f) 170 and (g) 200 C. Inset: variation of peak current (IAlp) versus charge consumed. Supporting electrolyte: sodium acetate solution (0.2 M). Scan rate 100 mV s−1; t = 25 ± 1 °C. | ||
According to our results, it seems that the Michael addition reaction of anion enolate 4 to o-benzoquinone (1a) (eqn. (2)) is faster than other secondary reactions, leading to the intermediate (1b). The oxidation of this compound (1b) is easier than the oxidation of the parent-starting molecule (1) by virtue of the presence of an electron-donating group. The intramolecular reaction was performed via the 1,4-(Michael) addition reaction with an electro-decarboxylation reaction (eqn. (4)). Since the oxidation of formed dihydroxybenzofurane occurs at more positive potentials, the over-oxidation of 1d was circumvented during the controlled potential preparative reaction. As can be seen from the mechanism shown in Scheme 2, o-benzoquinone 1c can be generated through homogeneous oxidation (eqn. (5)). The synthesis of 1d has been performed using electrochemical oxidation of 3,4-dihydroxybenzoic acid (1) in the presence of acetylacetone (4) in aqueous sodium acetate, in an undivided cell at A1 peak potential (see Table 1).
Electrochemical study of 2,3-dihydroxybenzoic acid (2)
The electrochemical oxidation of 2,3-dihydroxybenzoic acid (2) in the presence of acetylacetone (4) as a nucleophile in sodium acetate solution (0.2 M) proceeds in a similar way to that of 1. Fig. 4 shows the voltammograms of 2 in the present and absence of 4. In this figure, curve a, is a cyclic voltammogram of 2 in the absence of 4. The anodic (A1) and corresponding cathodic peak (C1) correspond to the transformation of 2,3-dihydroxybenzoic acid (2) to the related o-benzoquinone and vice versa within a quasi-reversible two-electron process. The oxidation of 2,3-dihydroxybenzoic acid (2) in the presence of acetylacetone (4) was studied in some detail (Fig. 4, curve b). Under these conditions, the anodic peak A1 increases and the cathodic counterpart of A1 disappears. Other conditions are similar to those for the electro-oxidation of 3,4-dihydroxybenzoic acid (1) in the presence of acetylacetone (4). In this case, each molecule of 2 in the presence of 4 converts to 2d, via inter- and intramolecular Michael addition reactions with consumption of 4e−. The overall reaction mechanism for electro-oxidation of 2,3-dihydroxybenzoic acid (2) in the presence of acetylacetone (4) is presented in Scheme 3. | ||
| Fig. 4 Cyclic voltammograms of 1 mM 2,3-dihydroxybenzoic acid: (a) in the absence, (b) in the presence of 1 mM acetylacetone. Supporting electrolyte: sodium acetate solution (0.2 M). Scan rate: 25 mV s−1; t = 25 ± 1 °C. | ||
 | ||
| Scheme 3 | ||
The existence of a carboxylic group probably causes the o-benzoquinone (2a) derived from the oxidation of 2 to be attacked by acetylacetone (4) at the C-5 or C-6 positions to yield two types of products in each case. However, according to a previous report8a and considering the electron-withdrawing character of the carboxylic group, we suggest that o-benzoquinone 2a is attacked in the C-6 position by acetylacetone (4) leading to the formation of the products 2d. The electro-organic synthesis of 2d has been performed using oxidation of 2,3-dihydroxybenzoic acid (2) in the presence of acetylacetone (4) as described for 1d (see Table 1).
Electrochemical study of 2,5-dihydroxybenzoic acid (3)
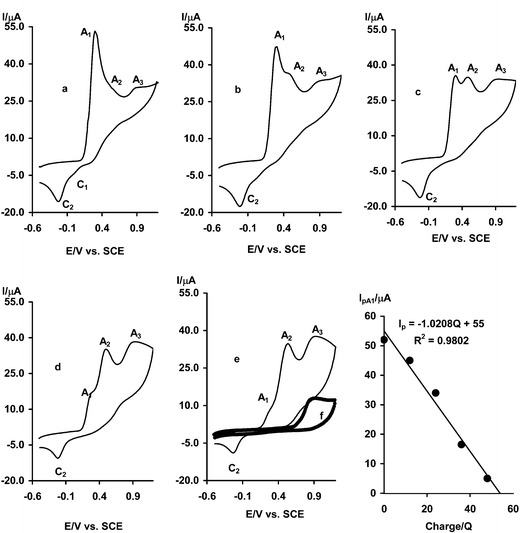
The electrochemical oxidation of 2,5-dihydroxybenzoic acid (3) in the presence of acetylacetone (4) in 0.2 M acetate solution has been performed using cyclic voltammetry. Fig. 5, curve a shows the cyclic voltammogram obtained for a 2 mM solution of 2,5-dihydroxybenzoic acid (3) in the presence of 2 mM acetylacetone (4). In comparison with cyclic voltammograms of 1 and 2 (Fig. 1, curve b and Fig. 4, curve b), p-benzoquinone (3a) derived from the oxidation of 3 has less reactivity towards the intermolecular Michael addition reaction, appearing as a decrease in peak current ratio (IAlp/IClp) (Fig. 5, curve b). Increasing the concentration of 4 and decreasing the scan rate caused an enhancement in the extent of chemical reaction during the time scale of cyclic voltammetry, appearing as an increase in peak current ratio (IAlp/IClp). This voltammogram shows a new anodic peak (A2) at higher potentials (0.51 V vs. SCE) and its corresponding cathodic peak (C2), which corresponds to the transformation of intermediate 3b to the related p-benzoquinone and vice versa (Fig. 5, curve b). The more positive Ep of peak A2 is related to relative stability of 3b towards oxidation due to extension of inter- and intramolecular hydrogen bonding. Also, this voltammogram shows another anodic (A3) in 0.92 V vs. SCE. | ||
| Fig. 5 Cyclic voltammograms of 2 mM 2,5-dihydroxybenzoic acid: (a) in the absence, (b) in the presence of 2 mM acetylacetone. Supporting electrolyte: sodium acetate solution (0.2 M). Scan rate: 50 mV s−1; t = 25 ± 1 °C. | ||
Controlled-potential coulometry was performed in aqueous solution containing 0.25 mmol of 3 and 0.25 mmol of 4 at 0.27 V versus SCE. Cyclic voltammetric analysis was carried out during the coulometry (Fig. 6). It was observed that, proportional to the advancement of coulometry, anodic peak A1 decreases, peak A2 at first increases and then remains constant and peak A3 nearly increases. Peak A1 disappears and peak A3 reaches a maximum value when the charge consumption becomes about 2e− per molecule of 3. These observations allow us to propose the pathway in Scheme 4 for the electro-oxidation of 3 in the presence of 4. In addition, cyclic voltammogram of the final product (after separation and purification) in acetate solution shows an irreversible anodic peak (A3). This peak, that is related to the irreversible oxidation of benzofuran 3d, indicates that there is no hydroquinone ring in the structure of 3d. Hydroquinone rings show a reversible or quasi-reversible behavior.
 | ||
| Fig. 6 Cyclic voltammograms of 0.25 mmol 2,5-dihydroxybenzoic acid in the presence of 0.25 mmol acetylacetone, at a glassy carbon electrode during controlled potential coulometry at 0.27 V versus SCE. After consumption of: (a) 0, (b) 12, (c) 24, (d) 36, and (e) 48 C. (f) Cyclic voltammogram saturated solution of final product after separation and purification. (g) Variation of peak current (IAlp) versus charge consumed. Supporting electrolyte: sodium acetate solution (0.2 M). Scan rate 100 mV s−1; t = 25 ± 1 °C. | ||
 | ||
| Scheme 4 | ||
The rate of the conversion of 3b to 3d is as low as that by the end of coulometry and both anodic peaks (A2 and A3) that are related to oxidation of 3b and 3d respectively will be observed (Fig. 6, curve e). However, during the preparative process in acidic media, 3b is rapidly and quantitatively converted to 3d (Fig. 6, curve f).
The existence of a carboxylic group probably causes the Michael acceptor 3a to be attacked by acetylacetone (4) at the C-3, C-4 or C-6 positions to yield three types of products in each case (Scheme 5). In this connection, the 1H NMR spectrum of obtained product indicates that the coupling constants, J, for the two aromatic peaks (6.74 and 7.54 ppm) are 8.8 Hz, that is, in agreement with the existence of two protons in the hydroquinone ring in the ortho positions.13 Therefore, according to the 1H NMR results we suggest that p-benzoquinone 3a is attacked in the C-6 position by 4 leading to the formation of 3d.
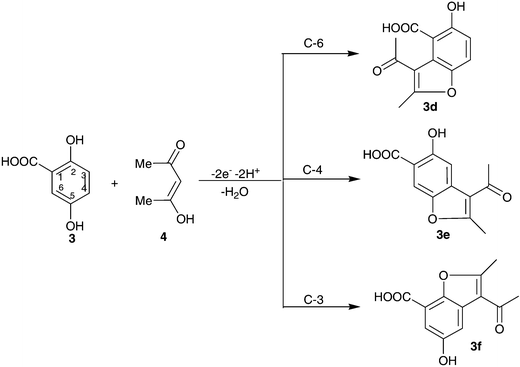 | ||
| Scheme 5 | ||
The synthesis of 3d has been performed using electrochemical oxidation of 2,5-dihydroxybenzoic acid (1) in the presence of acetylacetone (4) in aqueous sodium acetate, in an undivided cell at a potential less than the A1 peak potential (see Table 1).
Conclusion
The results of this work show that dihydroxybenzoic acids are oxidized in aqueous media to their respective quinones. The quinones are then attacked by the anion of acetylacetone via intermolecular Michael addition reaction. We observed diversity in their behavior in the steps that followed. In 3,4-dihydroxybenzoic acid (1), after the intermolecular Michael addition reaction and oxidation of the intermediate (1b) (Scheme 2, eqns. (2) and (3)), an intramolecular Michael addition reaction takes place that is accompanied by a decarboxylation reaction. We have recently reported the synthesis of this compound via electrooxidation of catechol in the presence of acetylacetone (4).9b In the case of 2,3-dihydroxybenzoic acid (2), the final product (2d) contains a carboxylic group and was obtained after a normal intramolecular Michael addition reaction as recently reported.9 Contrary to 1 and 2, in the case of 2,5-dihydroxybenzoic acid (3), the final product was obtained only after consumption of 2e− per molecule of 3 and a dehydration reaction. The overall reaction mechanisms for anodic oxidation of 3,4-dihydroxy benzoic acid (1), 2,3-dihydroxybenzoic acid (2) and 2,5-dihydroxybenzoic acid (3), in the presence of acetylacetone (4) is presented in Schemes (2), (3) and (4), respectively. These mechanisms show a good diversity in anodic oxidation of dihydroxybenzoic acids in the presence of acetylacetone (4).Experimental
Apparatus and reagents
Reaction equipments are described in an earlier paper.9b All chemicals (dihydroxybenzoic acid and acetylacetone) were reagent-grade materials, and sodium acetate was of proanalysis grade. These chemicals were used without further purification.Electroorganic synthesis of 1d–3d
In a typical procedure, 80 ml of sodium acetate solution (0.2 M) was pre-electrolyzed at the chosen potential (see Table 1), in an undivided cell; then 2 mmol of dihydroxybenzoic acid (1–3) and acetylacetone (4) (2 mmol) were added to the cell. The electrolysis was terminated when the decay of the current became more than 95%. The process was interrupted during the electrolysis and the graphite anode was washed in acetone in order to reactivate it. At the end of electrolysis, 20 ml 1 M hydrochloric acid was added to the solution and the cell was placed in a refrigerator overnight. The precipitated solid was collected by filtration and washed with water. After washing, products were characterized by: IR, 1H NMR, 13C NMR and MS. The isolated yields of 1d–3d after washing are reported in Table 1.Characterization of compounds: 3-Acetyl-5,6-dihydroxy-2-methylbenzofuran (1d) (C11H10O4): This compound was identified by comparison with an authentic sample (IR, 1H NMR, 13C NMR, MS, mp).9b3-Acetyl-5,6-dihydroxy-2-methylbenzofuran-4-carboxylic acid (2d) (C12H10O6): Mp 248–250 °C (dec.). IR(KBr): 3342, 1691, 1594, 1488, 1430, 1365, 1195, 1083, 906, 808, 595 cm−1. 1H NMR, δ (90 MHz DMSO d6): 2.07 (s, 3 H), 2.45 (s, 3 H), 7.46 (s, 1 H), 10.45 (broad). 13C NMR, δ (90 MHz DMSO d6): 13.2, 31.5, 102.1, 107.1, 116.6, 124.2, 146.8, 153.4, 156.7, 157.8, 170.1, 196.6. MS: m/z (relative intensity); 250(20), 232(62), 206(50), 191(35), 175(17), 146(55), 118(22), 90(43), 69(100), 43(72). 3-Acetyl-5-hydroxy-2-methylbenzofuran-4-carboxylic acid (3d) (C12H10O5) Mp: 198–200 °C (dec.). IR(KBr): 3080, 1680, 1630, 1615, 1572, 1426, 1202, 1168, 1060, 977,835, 705 cm−1. 1H NMR, δ (500 MHz DMSO d6): 2.29 (s, 3H), 2.39 (s, 3H), 6.84 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 11.0 (broad). 13C NMR, δ (500 MHz DMSO d6): 13.5, 32.0, 106.9, 114.2, 117.2, 120.9, 124.7, 147.2, 157.6, 158.0, 170.8, 197.2. MS: m/z (relative intensity); 234(46), 216(100), 201(25), 190(43), 175(80), 160(23), 146(18), 118(15), 89(20), 63(20), 43(69).
References
- M. G. Kadieva and E. T. Oganesyan, Chem. Heterocycl. Compd. (Eng. Transl.), 1997, 33, 1245 CAS
.
- G. R. Brown and A. J. Foubister, Eur. J. Med. Chim. Ther., 1992, 27, 723 Search PubMed
- A. H. Abdel-Rahman, A. M. Khalil and A. M. Hanna, Boll. Chim. Farm., 1997, 136, 262 Search PubMed
- E. L. Tolmach, A. B. Kudryavtsev, A. Ya. Zheltov and B. I. Stepanov, J. Org. Chem. USSR (Eng. Transl.), 1989, 25, 1594 Search PubMed
- S. Maeda, H. Masuda and T. Tokoroyama, Chem. Pharm. Bull., 1994, 42, 2536 CAS
- S. A. Zotova, T. M. Korneeva, V. I. Shvedov, N. I. Fadeeva and I. A. Leneva, Pharm. Chem. J. (Eng. Transl.), 1995, 29, 57 Search PubMed
- O. H. Hishmat, A. H. Abd el Rahman, N. M. A. El-Ebrashi, H. I. El-Diwani and A. I. El-Diwani, Indian J. Chem., Sect. B, 1983, 22, 3313
-
(a) S. M. Golabi and D. Nematollahi, J. Electroanal. Chem., 1997, 420, 127 CrossRef CAS
-
(a) D. Nematollahi, D. Habbibi, M. Rahmati and M. Rafiee, J. Org. Chem., 2004, 69, 2637 CrossRef CAS
-
(a) L. Papouchado, G. Petrie and R. N. Adams, J. Electroanal. Chem., 1972, 38, 389 CrossRef CAS
-
(a) D. I. Stum and S. N. Suslov, Biofizika, 1979, 21, 40 Search PubMed
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 2001, pp. 496–500 Search PubMed
-
W. Kemp, NMR in chemistry, Macmillan Education LTD, London, 1986, p. 63 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See http://dx.doi.org/10.1039/b503408f |
| This journal is © The Royal Society of Chemistry 2005 |