Polyethylene glycol and solutions of polyethylene glycol as green reaction media
Ji Chen†, Scott K. Spear, Jonathan G. Huddleston and Robin D. Rogers*
Center for Green Manufacturing and Department of Chemistry, The University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: rdrogers@bama.ua.edu
First published on 25th January 2005
Abstract
In this review, we examine the concept that aqueous biphasic reactive extraction (ABRE) can successfully integrate the solvent properties of polyethylene glycol (PEG) and its phase-transfer characteristics into a single efficient system which can additionally be manipulated to facilitate the separation of reactants and/or catalysts from products. We also suggest that the properties of these systems may recommend them as being relatively environmentally benign in comparison to the current use of organic solvents in extraction and in reactive extraction. In developing this concept, we review a number of the physical and chemical properties of PEG and aqueous solutions of PEG in the context of recent applications to chemical reaction engineering. Thus, we cover the interesting physical properties of PEG solutions in water, their unique solvent properties, and finally the metal cation coordination ability of PEG solutions. These properties are important in the application of low molecular weight liquid PEG as a solvent in chemical reactions; in the use of PEG as an alternative phase-transfer catalyst (PTC); and in the application of ABRE in the development of alternative pulping processes, catalytic chemistry, and enzymatic catalysis.
1. Introduction
Regulatory pressure is increasingly focusing on the use, manufacture, and disposal of organic solvents, and thus the development of nonhazardous alternatives (one of several goals of green chemistry and engineering) is vitally important for the continued and sustainable development of the chemical enterprise.1,2 There are many potential advantages to replacing volatile organic compounds (VOCs) with water or various types of aqueous solutions. The most obvious are low cost, reduced flammability, reduced toxicity, and reduced environmental risk as a result of discharges of the supporting phase. Water and aqueous-based solvent systems may represent an increasingly significant choice for the replacement of traditional solvents in synthetic chemistry. Other leading VOC solvent alternatives include supercritical fluids, ionic liquids, immobilized solvents, solventless conditions, and the use of fluorous solvents.1A recent special edition of Chemical Reviews and a recent American Chemical Society Symposium volume highlighted some aspects of the use of water and aqueous solutions in green chemistry,3,4 however, relatively few articles have focused on the use of aqueous PEG solutions and related materials in chemical reactions. This is despite the fact that, unlike several of the ‘neoteric solvents’ such as ionic liquids (ILs) where toxicity and environmental burden data are for the most part unknown, complete toxicity profiles are available for a range of polyethylene glycol (PEG) molecular weights and indeed, many are already approved for internal consumption by the US FDA.5,6
Table 1 lists a number of recent reviews of organic reactions conducted in water and aqueous solutions including the use of high temperature water, soluble polymer supports, micellar solutions, and PEG derivatives. In this context, PEG has been used as a solvent and phase-transfer catalyst (PTC) in organic synthesis.
| Medium and main reaction type | Ref. |
|---|---|
| High temperature water as reaction medium | 5,7 |
| Organometallic reactions performed in water | 8–11 |
| Reactions mediated by soluble polymers as catalysts and supports including PEG | 12,13 |
| Micellar solutions as catalysis and reaction media | 14–19 |
| PEG and its derivatives as PTC and solvents for organic synthesis | 20,21 |
A number of recent reviews have also covered PEG chemistry and its applications in biotechnology and medicine,22,23 PEG and PEG-supported catalysis,24 PEG-based aqueous biphasic systems (ABS) as alternative separation media,25 aqueous two-phase systems (ATPS) in bioconversion,26–28 and PEG and its derivatives as solvent and PTC in organic synthesis.20,21 However, none of these articles has focused on PEG solutions as alternative reaction media. Here we present a comprehensive review and analysis of the role of aqueous solutions of PEG in the development of alternative reaction media, an area of green chemistry which seems to have been rather overlooked in comparison to other solvent systems such as, for example sc-CO2 and ILs.4,29,30
Our interest in PEG solutions as green reaction media was spurred by several factors including; the present high interest in green separation chemistry,31–38 our long-term study of PEG–metal ion coordination,39–44 our studies of ABS solvent properties,45–49 and our application of aqueous biphasic reactive extraction (ABRE) in the development of alternative processes for wood pulping and green catalytic oxidation systems.50–57 In this review, we draw together five strands of the current literature: (1) the interesting physical properties of aqueous PEG solutions; (2) the unique solvent properties and cation coordination ability of PEG solutions; (3) the application of low molecular weight liquid PEG as a solvent in chemical reactions; (4) PEG as an alternative PTC; and (5) the application of ABRE in the development of green pulping processes, catalytic chemistry, and enzymatic catalysis.
2. Aqueous PEG solution properties
Polyethylene glycol, PEG: HO–(CH2CH2O)n–H, is available in a variety of molecular weights from 200 to tens of thousands. At room temperature, the water soluble and hygroscopic polymer is a colorless viscous liquid at molecular weight <600 and a waxy, white solid at molecular weights >800.58 The numerical designation of PEGs generally indicates the number average molecular weight (e.g., PEG-2000), although typically they are not monodisperse polymers. Liquid PEG is miscible with water in all proportions, and solid PEG is highly soluble in water, for example, PEG-2000 has a solubility of about 60% in water at 20 °C. Lower molecular weight liquid PEGs can be used as solvents in their own right with or without addition of water. These we define loosely here as low molecular weight PEG.PEG has a number of benign characteristics that underlie, for example, its application in bioseparations.59 PEG is on the FDA's GRAS list, (compounds Generally Recognized as Safe) and has been approved by the FDA for internal consumption.60,61 PEG is weakly, if at all, immunogenic, a factor which has enabled the development of PEG–protein conjugates as drugs.62–65 Aqueous solutions of PEG are biocompatible and are utilized in tissue culture media and for organ preservation.59
Unlike VOCs, low molecular weight liquid PEGs are nonvolatile. The vapor density for low molecular weight PEG is greater than 1 relative to air according to available MSDS data,66 and this is consistent with the industry standard for selection of alternative solvents to VOCs.1 PEG also has low flammability, and is biodegradable.
PEG has been found to be stable to acid, base, high temperature,50,56,57,67 O2, H2O2 high oxidation systems,68 and NaBH4 reduction systems,69,70 although partial oxidation of the PEG terminal –CH2OH group to –COOH may occur in such systems as H2O2–Na2WO4.56 In addition, PEG may be recovered from aqueous solution by extraction with a suitable solvent or by direct distillation of water or solvent.71
2.1. Phase separation
Aqueous solutions of PEG display a miscibility gap characterized by lower and upper critical solution temperatures (LCST and UCST, respectively) in which the polymer and water segregate into polymer-rich and polymer-poor phases.45 The physical basis of this behavior is dependent upon the amphipathic nature of the polymer chain with its hydrophobic methylene groups interspersed with ether groups which can compliment the hydrogen bonded structure of water.72–76 Hydration/dehydration of these groups promotes the observed behavior.Similar phase incompatibility may be found in a number of different polymers such as PEG–PPG co-polymers, poly-vinylpyrrolidone, poly-N-isopropylacrylamide, and in the clouding of non-ionic surfactants.25,59,77 Additionally, aqueous solutions of PEG phase separate in the presence of other, incompatible, but still water soluble polymers. The exact nature of phase separation here is incompletely understood, but appears to be an UCST phenomenon perhaps based on incompatible hydrogen bonded structures, or on an excluded volume mechanism.25,59,78
PEG-based biphasic systems are generally formed by the addition of PEG and other water soluble polymers such as Dextran, above critical concentrations, however, many other pairs of polymers show incompatibility (and not only in aqueous solution);25 the phase separation of Ficoll and Dextran is an important example.59,77,79 Alternatively, 1-butyl-3-methylimidazolium chloride may be combined with a kosmotropic salt such as K3PO4, above critical concentrations in aqueous solution resulting in two distinct aqueous phases.80 This is simply an extension of the temperature induced phase separation of PEG as a result of the lowering of the cloud point brought about by the salting-out effects of the salts.81 These effects follow the Hofmeister series.45
Thus, aqueous PEG solutions may display phase separation under controlled conditions. Such phase separated solutions are known as ABS (Aqueous Biphasic Systems) or alternatively, ATPS (Aqueous Two-Phase Systems), and have been exploited in bioseparation for nearly fifty years since their discovery by Albertsson.59
A typical phase diagram for the system, PEG-2000–NaHSO4 in 50% H2O2–H2O at room temperature is shown in Fig. 1. Mixture compositions lying to the left of the binodal curve are monophasic, whereas mixture compositions to the right of the binodal curve are biphasic. Tie lines (e.g., A–B in Fig. 1) connect mixture compositions to points on the binodal curve (the nodes A and B) representing the compositions of the two phases in equilibrium. All mixture compositions lying on the same tie line give identical compositions of the equilibrium phase. Only the relative volume of the phase changes, and this may be used as a means to manipulate recovery of products. The length of the tie line is called Tie Line Length (TLL).59 Increasing PEG or salt concentration results in longer TLL and increasing phase divergence. The phase divergence has been shown to be proportional to the chemical potential difference between the phases. Thus, solute preference for one phase over another increases with increasing phase divergence.45
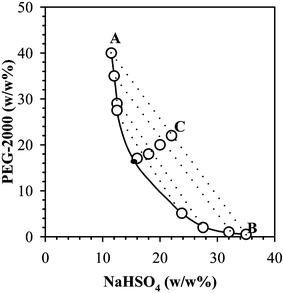 | ||
| Fig. 1 The phase diagram for PEG-2000–NaHSO4 in 50% H2O2 at room temperature used for catalytic oxidation of cyclic olefins.56,57 | ||
Differences in phase preference between species can be adjusted by controlling the ABS system composition. The fact that PEG, and other polymers, form biphasic systems, having tunable phase properties is an important advantage that allows the development of versatile reactive extraction systems; a property of PEG solutions which has yet to be fully exploited.56,57
2.2. Solution polarity
The poor solubility of organic reactants and their intermediates in water is the main limitation to the widespread application of water as a reaction solvent. Enhancement of the solubility of organic solutes in aqueous solutions, including the use of supercritical water, surfactants, co-solvents, and complexants,7,82–84 is an important aspect of the application of aqueous solutions as alternative solvents. For example, supercritical water is characterized by a reduced dielectric constant and a weaker hydrogen bonded network, which greatly increases the solubility of organic compounds.7,82 Eckert et al. have pointed out that this unusual combination of physical properties can provide environmentally benign separation and reaction processes.82Similarly, surfactants with polar and non-polar regions orient themselves into micelles with a hydrophobic solvent-like interior and a polar water-exposed surface. Organic solutes become localized in the interior or within the surface of the micelle, and this is believed to be responsible for the success of organic and enzyme catalyzed reactions in micellar media.14–16 A significant application of micellar catalysis lies in the field of homogeneous catalysis. In such systems, aqueous micellar solutions serve, not only as the reaction medium, but also to improve reaction selectivity and to facilitate catalyst recycling.9,17–19 In metal-mediated carbonyl additions, the allylmetal intermediate complexes are formed as a result of the action of a wide variety of intermolecular forces, and as a result can effectively “activate” the metal center of some organometallic reagents and enhance allylation and other organometallic reactions in water.11,85
The water soluble polymer PEG can be considered a co-solvent in water which leads to an apparent decrease of the aqueous solution polarity.77,84 The consequent increase in solubility of organic molecules has led to the application of PEG as a solvent and a PTC in organic synthesis.20,21
PEG, in aqueous solution, acting as a co-solvent, significantly changes many of the properties of water.83 Solutions of PEG in water may be viewed as monophasic, consisting of two homogeneously mixed components, or as biphasic, having large hydrated polymer molecules separated by regions of free water.77 Zaslavsky has made the argument that the latter viewpoint invites comparison of the resultant solution properties to the effects ascribed to vicinal water, i.e. water adjacent to solid interfaces, which has significantly different properties from pure water.77 In comparison to bulk water, vicinal water shows a decrease in density and dielectric relaxation, and an increase in viscosity and ionic conductance.77
Perhaps of even more significance to a discussion of PEG solutions as reaction media, is the effect of PEG on the polarity of aqueous solutions. Solution polarity is frequently measured by reference to the solvatochromatic properties of certain polarity sensitive dyes, in particular to Reichardt's betaine, from which is derived the ET scale of polarity.25 Zaslavsky found that the ET(30) values of PEG-6000–Dextran-500 were very close to, but somewhat less than those of pure water and the values decreased with increasing polymer concentration.86
We have found in the PEG-2000–K3PO4 system, that values of ET(30) are considerably lower than water at the critical point, and decrease in the PEG-rich phase with increasing phase divergence.46 However, for the lower salt-rich phase, the apparent polarity as measured by ET(30) increases with increasing TLL and distance from the critical point until it is virtually indistinguishable from that of water. Thus, there is an increasing difference in the polarity of the two phases as measured by this technique.
The polarity difference in PEG–Dextran and PEG–K3PO4 ABS with TLL is shown in Fig. 2. PEG–Dextran shows a small difference in polarity between the phases, whilst the greatest difference is found for PEG–K3PO4 ABS and this difference increases with TLL. In broad terms, the polarity of the PEG-rich phase could be described as being similar to a short chain alcohol when measured on the ET(30) scale.46
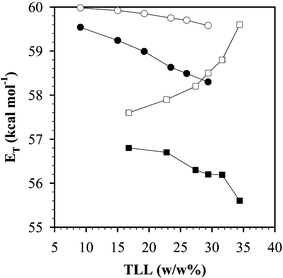 | ||
| Fig. 2 Solvent polarity scale for PEG-6000–Dextran-50086 and PEG-2000–K3PO446 ABS systems: (○) Dextran-rich bottom phase in PEG–Dextran ABS, (●) PEG-rich top phase in PEG–Dextran ABS, (□) K3PO4-rich bottom phase in PEG–K3PO4 ABS, (■) PEG-rich top phase in PEG–K3PO4 ABS. | ||
The relative affinity of the solvent medium for a non-polar CH2 group, ΔGCH2, has also been used to measure the solvent properties of different organic solvents, and various aqueous polymer solutions.46 This scale is not related to the polarity of the medium, but to the free energy of cavity formation or the cohesive energy density. The polarity of the separated top and bottom phases of a PEG-2000–K3PO4 ABS have been thoroughly investigated.48 The relationship between −ΔGCH2 and the degree of phase separation of the co-existing phases of the ABS, expressed in terms of the difference in concentration of the total number of ethylene oxide monomers (ΔEO) or the TLL between the phases, was investigated in various PEG–salt ABS differing in salt type (K3PO4, K2CO3, (NH4)2SO4, NaOH, Li2SO4, MnSO4, ZnSO4) and PEG molecular weight.48 No matter the salt type, the concentration, or the molecular weight of PEG, −ΔGCH2 was the same at the same degree of phase divergence (expressed as ΔEO, ΔPEG, or TLL). That is the free energy of transfer of a methylene group in all PEG–salt systems investigated was the same at the same degree of phase divergence. The difference in concentration of the polymers between the phases at a given temperature has been shown to be a measure of the chemical potential difference between the phases.45
Fig. 3 shows both the ET(30) and −ΔGCH2 relative solvent hydrophobicity scales as applied to PEG-based ABS and some organic solvents and surfactant solutions.46ET(30) is given for the organic phase (PEG in the case of ABS), and −ΔGCH2 represents the free energy of transfer from the aqueous to the organic phase. The ET(30) values of both PEG-6000–Dextran-500 and PEG-2000–K3PO4 aqueous polymer solutions are closer to that of pure water than most of the organic solvents shown in Fig. 3. However, it is possible to construct ABS, such as the PEG–K3PO4 system mentioned previously, having phase compositions such that −ΔGCH2 may range from zero to about 0.85 kcal mol−1. Not only does this mean that the hydrophobicity of the system is controllable, but in some cases the free energy of transfer may approach and exceed that of many organic solvents. It may thus be possible to achieve considerable solubility of some organic solutes in such systems, and this may allow certain organic reactions to be carried out in these relatively hydrophobic aqueous solutions.
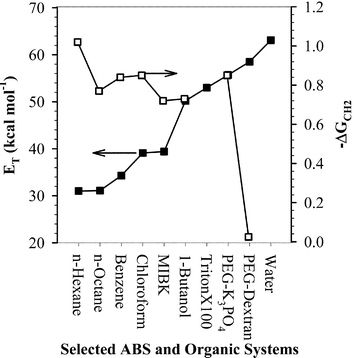 | ||
| Fig. 3 Relative solvent polarity ET (■) and −ΔGCH2 (□) for selected PEG-based ABS solutions and for some organic solvents. | ||
The molar transition energy, ET, however, taken from the wavelength of absorption of the long wavelength absorbance band of Reichardt's betaine dye,87 includes not only the effect of molecular dipole and induced dipole interactions, but also the ability of the solvent to interact with the betaine by hydrogen bond donation.87 Also, the free energy of transfer of a methylene group is only a measure of the energy associated with cavity formation, ignoring the contribution of molecular interactions between solute and solvent to the overall free energy of transfer for all solutes beyond simple alkyl chains.
Huddleston et al. have characterized the phases of selected polymer–polymer and polymer–salt systems in terms of their polarity using the Kamlett and Taft π* parameter (relative polarity/polarizability),46 and extended these studies to include α (hydrogen bond acidity) and β (hydrogen bond basicity)49 contributions to solute selectivity in these systems using a suite of solvatochromatic indicators. The parameters π* and β were found to show little difference between the two phases in PEG-2000–(NH4)2SO4 or K3PO4 ABS, but a big difference was found for the α value.49 At 37% w/w TLL, the α value between the PEG-2000-rich top phase and K3PO4 salt-rich phase could be as low as 0.3. The hydrogen bond donation ability was greatly depleted in the PEG-rich top phase. The solvatochromatic study of π*, β, and α lends support to later conclusions, derived from the application of linear free energy relationships (LSER),48 that polarity and solvent basicity play little part in solute distribution in ABS.
Other studies using solvatochromatic probes and the solvation model of Kamlett and Taft, have found that low molecular weight PEG, such as pure PEG-300, has a polarity value (π* = 0.94) greater than methanol (0.63), a hydrogen bond acceptor ability (β = 0.60) similar to methanol (0.62), and a hydrogen bond donor ability (α = 0.45) less than methanol (0.93).88 In the Hansen three component description of solubility behavior, the solubility parameters for tetraethylene glycol have been determined in terms of a dispersion component (δd2 = 16.6), polarity component (δp2 = 5.7) and a hydrogen bonding component (δh2 = 16.8) and these parameters were found to be similar to those of n-propanol.89
Linear solvent free-energy relationships (LSERs or LFERs) based on chemical equilibria and Gibbs energy relationships, have been applied to the investigation of the solvent properties of ABS.46,48 The general solvation equation of Abraham is usually given as in eqn. 1:
| log SP = c + rR2 + sπ2H + a∑α2H + b∑β2H + vVx | (1) |
LSERs are useful in the determination of the solute–solvent interactions that govern solute partitioning in ABS, and it is also possible to compare ABS to, for instance, aqueous micellar systems and traditional organic–aqueous biphasic systems.90 Such comparisons may greatly aid the selection of suitable solvent systems in a search for replacements for VOC systems. The solvent properties which were found to be of prime importance in driving solute partitioning in ABS, were solvent hydrogen bond acidity (donation ability) and the free energy of cavity formation.
2.3. PEG–metal ion complexes
PTCs are usually used to transport an aqueous reagent into an organic phase in an activated state, so that the reaction can proceed as a consequence of bringing the aqueous reagent and organic reagent together.67 PEG has the ability to serve as a PTC since the polyethylene oxide chains can form complexes with metal cations, similar to crown ethers.21 To maintain electroneutrality, such PEG–metal cation complexes must bring an equivalent anion into the organic phase, thus making the anion available for reaction with the organic reactants. Many factors affect phase catalytic activity, such as PEG molecular weight, chain end effects, and the nature of the associated cations and anions.Fig. 4 shows that the stability constants, log K, for PEG complexes with Na+ in ethanol (using a Na+ electrode with Ag+/AgCl as the reference electrode) depend on both the PEG molecular weight and the end-group substituents.91–93 The degree of complexation of solid alkali metal salts by PEG-400 is strongly dependent on both the salt anion and cation as shown in Fig. 5.94,95 For the Na+ and K+ salt–PEG complexes, commonly used in phase-transfer reactions, there is a greater dependency on the nature of the anion than the cation. The anions OH−, F−, HSO4−, and HCO3− were found (by conductimetric and refractive index measurements) to be ion paired with the PEG cation complex, and assumed to be relatively naked anions, while Cl−, Br−, I−, SCN−, NO3−, NO2−, and HF2− were assumed to be disassociated anions with significant solvation shells.95
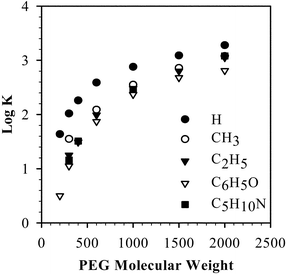 | ||
| Fig. 4 The stability constants of PEG–Na+ complexes with various PEG molecular weights and functional group substituents.91–93 PEG terminal groups were symmetrically substituted (see legend) and the molecular weight was set as that of the unsubstituted PEG. | ||
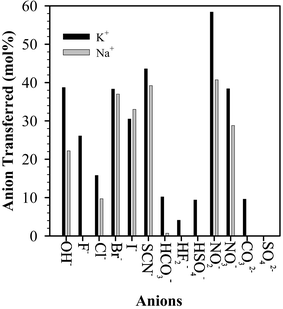 | ||
| Fig. 5 The metal ion (K+ and Na+) transfer ratio (mol%) from solid alkali metal salts of different anions to PEG-400.94,95 | ||
The binding constants of Na+ with PEG, with PEG monomethyl ethers (PEG-MME), with PEG dimethyl ethers (PEG-DME), and with crown ethers have been measured in anhydrous MeOH solutions.96 The binding constants (K) of one PEG chain containing 3–312 EO units with the sodium cation show a linear relationship with the number of binding sites available.96 It has also been proposed that for some alkoxylation reactions, several catalytic centers exist in a single PEG molecule.97 Dramatically reduced costs of PEG compared to crown ethers are a considerable incentive for investigating the properties of these “crown-like” PTCs.
PEG–cation complex structures have been analyzed by a variety of methods (Table 2) such as single crystal X-ray structure analyses, mass spectrometry, NMR, IR and Raman spectroscopy, powder X-ray diffraction, and electrochemistry. X-Ray analyses have shown that low molecular weight PEG (expressed as (EO)n) can usually form single crystal complexes with metal cations in acetonitrile, methanol, and their mixtures. For example, Sr2+ will typically organize 3–6 EO units into a pseudocyclic crown ether-like equatorial array. Thus (EO)3 or (EO)4 chains complex Sr2+ in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 fashion, whereas longer chains (EO)9–(EO)10 are able to helically wrap the Sr2+ ion.44 Because Sr2+ can be completely wrapped by (EO) units, the complex is given a hydrophobic exterior, effectively dehydrating the metal cation, and allowing Sr(EO)n2+ complexes to be transferred to an organic phase. This is the basis for the synergistic extraction of Sr2+ from aqueous solution using a mixture of cobalt dicarbollide and PEG.98
∶1 fashion, whereas longer chains (EO)9–(EO)10 are able to helically wrap the Sr2+ ion.44 Because Sr2+ can be completely wrapped by (EO) units, the complex is given a hydrophobic exterior, effectively dehydrating the metal cation, and allowing Sr(EO)n2+ complexes to be transferred to an organic phase. This is the basis for the synergistic extraction of Sr2+ from aqueous solution using a mixture of cobalt dicarbollide and PEG.98
Compared with crown ether complexation, the same PEG can coordinate different sizes of metal cations such as Sr2+ (1.12 Å)44 and Li+ (0.76 Å),114 thus, PEG presents a more flexible structure for metal cation complexation. Extraction by PEG polymer complexation also represents a considerable cost saving over crown ether complexation.
Pb2+ and (EO)5 can form Pb(EO)5 chelate rings, and the six oxygen donors arrange themselves in a nearly hexagonal equatorial plane, so that the anion can approach the two axial sites.41 Lanthanides have important applications in organic synthesis in protic solvents, and the crystal structures of dozens of LnX3 (X = Cl−, SCN−, NO3−) complexes formed with a range of small PEGs ((EO)3 to (EO)7) have been investigated by Rogers et al.42 These results showed that different EO chain lengths result in differences in the coordination sphere and can, in some cases, require additional inner-sphere ligands, such as an anion, water, or solvent molecules. These results have been the subject of extensive review.42
The complexation of metal ions with PEG has also been investigated by 1H NMR spectroscopy. There is a strong ion–dipole binding affinity with metals, and the preferred coordination numbers for Na+ and Ca2+ of 6 EO, and for Sr2+ of 7 EO, have emerged using the ion radius concept.102,103 Mass spectrometry has shown that the lowest energy structure Na+(EO)9 results from the Na+ ion being “solvated” by seven nearest neighbor atoms, and thus, the cation is completely encased by the (EO)9.99
IR and Raman spectroscopic studies of a PEG–Na+ complex in the context of the development of polymer electrolytes, (EO)nNaCF3SO3, have shown that free ions, ion pairs, and aggregates exist due to the interaction of Na+ with (EO)n and CF3SO3− at different concentrations.107 The electrochemical characterization of the Li+/Li redox electrode reaction in solid polymer electrolytes (SPEs) of PEG-10000–LiClO4 shows higher conductivity than polyethylene oxide (PEO) electrolytes due to the increase in the number of carriers and the facilitation of the formation of the conduction pathways.113 A powder X-ray diffraction study shows that Li+–EO3 forms isolated and helical EO chains with Li+ inside the helix,108 which is similar in conclusion to the results of X-ray crystallographic studies. In the case of Li+–EO6 complexation, double helical EO chains were shown to surround the Li+ cations, so that the anions were left outside the helical structure unpaired with Li+ ions.109 The conclusions were supported spectroscopically by FT-IR and Raman studies that showed that Li+ is completely surrounded by the two EO6 chains forming a cylindrical structure without significant anion coordination in the EO6∶LiAsF6 complex.105
PEGs certainly have the ability to form complexes with metal cations. This property may be of some importance in the application of PEGs as alternative PTCs and in ABRE processes. Crown ether metal ion complexation depends on the fixed size of the cavity in the center of the crown. However, fixed molecular weight PEGs show more flexible selectivity in the binding of different size metal cations due to changes in helical conformation. During the phase-transfer process, PEG may transfer metal cations from the aqueous phase to the organic phase by complexation and partition. The corresponding anion is also forced to transfer to maintain electroneutrality, thus the inorganic or organic solvated anion may be activated in the organic phase for participation in the chemical reaction.
3. Liquid PEG as reaction solvent
PEG and its aqueous solutions represent interesting solvent systems for solvent replacement, and may stand comparison to other currently favored systems such as ionic liquids, supercritical carbon dioxide, and micellar systems. Recently, Sheftel has reviewed the general toxicological action of PEG such as acute toxicity, the mean lethal doses of all molecular weights of PEG, short and long-term, reproductive toxicity, and the mutagenicity in vitro or vivo.115 Additionally, relevant laws or regulations governing use in the European Union and from the US FDA were also included. One important difference between using PEG and other ‘neoteric solvents’ is that all of the toxicological properties, the short and long-term hazards, and the biodegradability, etc., are established and known. For many of the other alternative solvent systems, lengthy investigations will be required to establish the necessary background safety and toxicological information.PEG is a hydrophilic polymer, easily soluble in water and many organic solvents including: toluene, dichloromethane, alcohol, and acetone, but it is not soluble in aliphatic hydrocarbons such as hexane, cyclohexane, or diethyl ether.58 The non-polar and hydrophobic hexane, slightly polar 2-octanone, and mildly polar heptanol, all showed some solubility in 70% PEG-300(aq), giving solutions of 0.01, 0.1, and 0.8 M, respectively.88 Solubilities of 1.7 M and 0.5 M were found for 2,3-dimethyl-1,3-butadiene in PEG-300 and its 90% PEG aqueous solution, and 4.5 M and 1.2 M for 1-bromobutane in these two media, respectively.88 This solubility enhancement has been applied to aqueous synthesis in both Diels–Alder and SN1 reactions.88,116,117
Of particular interest, is the high solubility of some salts in PEG-400, such as 1.8 M CH3COOK, 2.1 M KI, 1.1 M KNO3, 0.25 M KCN, and 0.16 M K2Cr2O7. This has enabled the use of this medium in some homogeneous oxidation and substitution reactions resulting in high yields.118 The solubility of these salts in PEG is comparable with their solubility in dimethylsulfoxide (DMSO). PEG-1500 used in a phenoxide allylation reaction was claimed to effectively enhance the solubility of the reactant allyl chloride in PEG aqueous solution.119
Low molecular weight liquid PEGs can be regarded as protic solvents with aprotic sites of binding constituted by the EO units. A few inorganic salts and many organic substrates are soluble in low molecular weight liquid PEG, and thus, they have been proposed as solvents for organic reactions.71,88,116,117 PEGs have been termed “host” solvents71 due to their ability to form complexes with metal cations as illustrated in Figs. 4 and 5.
Three main types of reactions have been investigated: substitution, oxidation, and reduction. However, application of liquid PEG is not limited to these reactions, polymerization of methyl methacrylate and styrene has been reported in PEG-400, and a higher rate of polymerization of methyl methacrylate was found than that in toluene, but the rate was slower for styrene in PEG-400 than that in xylene. Moreover, the easy removal of the copper catalyst following the reaction is comparable with the performance of other solvent alternatives such as 1-butyl-3-methylimidazolium hexafluorophosphate and fluorinated biphasic systems.120 Other types of liquid PEG have been used in substitution and reduction reactions such as high molecular weight PEGs including molecular weights of 900, 1000, 2000, and 3400 in conjunction with sc-CO2 above the melting point of the PEG.121–123 The similar small molecular weight liquid polypropylene glycols (PPG) 425 and 1000 have also found application in solvent substitution.88,116,117,124,125
3.1 Substitution reactions
Table 3 provides examples of substitution reactions performed using liquid PEG as the solvent. Potassium thioacetate in PEG-400 has been used as a nucleophilic reagent to substitute alkyl halides, tosylates, or mesylates, and the product yields were about 92–98%.127 The hydrolysis rate constant for the synthesis of 2-chloro-2-methylpropane in PEG-300–H2O has been compared to those found for acetic acid, methanol, ethanol, and acetone as co-solvent with H2O, and rate constants were found to increase by 1–3 orders of magnitude at high mass% PEG(aq) over those found for the above organic co-solvents. The Diels–Alder reaction rates of 2,3-dimethyl-1,3-butadiene with nitrosobenzene in PEG-300 showed a 3.3 fold increase compared to the same reactions in dichloromethane, and a 2.5 fold increase over that found in ethanol. 2,3-Dimethyl-1,3-butadiene in reaction with acrolein showed a 14 fold increase in rate constant in 70 mass% PEG-300(aq) over that in methanol.88,116 PPG-425 showed a similar enhancement of reaction rate, but this was lower than that of PEG-300.88,116| Substrate (RX) | Substitution (Y) | PEG | Product | Ref. |
|---|---|---|---|---|
| R-CH2Br | CH3COO−, I−, C6H5O−, CN− | PEG-400 | R–CH2Y | 126 |
| R = C6H5, C3H7, C9H19, C7H15 | 118 | |||
| R1-CHX-R2 | CH3COS− | PEG-400 | R1-CHY-R2 | 127 |
| R1 = C7H15, C9H19, C11H23, C10H21, C6H5 | ||||
| R2 = H, CH3 | ||||
| X = CH2C6H4SO2, CH2SO2, Cl, Br, I | ||||
| (CH3)3CCl | H2O | PEG-300 | (CH3)3COH | 88 |
(CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCl CHCl | PPG-425 | (CH3)2C(OH)CH3, | 117 | |
| (CH3)2CHCH2OH | ||||
| CH2CH(CH3)CH(CH3)CH2 | CH2CH-CHO | PEG-300 | OCH-C6H7-(CH3)2 | 88 |
| C6H5-NO | PPG-425 | C6H5-NOC4H4-(CH3)2 | ||
| R′X | RSO2− | PEG-400 | R-SO2-R′ + S2O2R′ | 128 |
| R′ = C6H5CH2Cl, BrCH2Br | PEG-400-C2H5 | |||
| R″-Br | ||||
| R″
= C2H5, n-C4H9, n-C8H17, CH2CHCH2 | ||||
| R1-(SO3-R2)n | F− | PEG-400 | R1-(F)n | 129 |
| R1 = n-C8H17, n-C6H13-CH, (CH3)3CCH2, C6H5(CH2)2 | ||||
| R2 = CH3, 4-CH3C6H4, 2-C10H10, 2,4,6-(CH3)3C6H5 | ||||
| ArNH2 | Cl−, Br−, I−, CN− | PEG-200-CH2Cl2 | ArY | 130 |
| Ar = C6H5, p-PhCOC6H4, 1-C10H9, p-ClC6H4, p-CH3OC6H4, | ||||
o-CH3OC6H4, m-CH3OC6H4, p-C6H5C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6H4 CC6H4 | ||||
| R-C6H4Br | C6H5-B(OH)2 | PEG-400 | R-C6H4-C6H5 | 131 |
| (C6H5)2CNCH2CO2PEG | R2X | PEG-3400 | (C6H5)2CNCHR2CO2PEG | 121 |
| R-C6H4-Br | CH2CHX | PEG-2000 | R-C6H4-CHCHX | 122 |
| R = Cl, OCH3, CH2O2 | X = C6H5, CO2C2H5, n-CH3(CH2)3O | (Pd(CH3OAc)2 catalyst) | ||
The reaction of sodium p-toluenesulfonate monohydrate with various alkyl halides in PEG-400 or its diethyl ether have been investigated, and it has been shown that sulfones can be obtained in good to excellent yields, while the alkylation of sodium p-toluenesulfinate and N, N′-dimethylformamide in methanol gives only moderate yields.128 The reaction of KF with the mesylates and tosylates of alcohols has been found to provide good yields of fluoro derivatives in PEG-400 compared to methanol and apolar crown ethers.129 The diazotization and Sandmeyer reactions of arylamines in PEG-CH2Cl2 were found to be effective for the preparation of halogenoarenes and cyanoarenes even in 10 mM dilute substrate. This is still comparable with these reactions in various organic solvents (e.g., xylene, DMSO, 18-crown-6, chloroform, and tetrahydrofuran (THF)), giving high yields only in high concentrations of substrate (≥73 mM)).130 The ordinary aqueous Sandmeyer reactions reported in Organic Synthesis gave only very poor yields.130
A Schiff base protected glycine modified by end functionalized PEG-3400 reacted readily with various electrophiles without the need for other organic solvents under microwave activation. PEG served as both polymer support and solvent in this reaction. The reaction was comparable with that in acetonitrile.121 The microwave-assisted Suzuki cross-coupling of arylboronic acids with aryl halides using PdCl2 as catalyst and KF as base in PEG-400 was found to offer ease of operation and the ability to recycle the catalyst. The recyclability of the PdCl2 and PEG-400 solvent by ether extraction makes the reaction more economic and thus, increases its viability for commercial exploitation.131
Molten liquid PEG-2000 at 80 °C was used as reaction solvent for Heck coupling reactions and gave a fast reaction rate and high yield.122 The reaction rate, yield, and regio- and stereoselectivities in this solvent system are comparable with the conventional organic solvents dimethyl formamide (DMF), DMSO, CH3CN, and the ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate. The recyclability of both PEG-2000 and Pd(OAc)2 can be achieved by simple ether extraction of the product, and the higher yield can be sustained even after four subsequent experiments.
Raston and co-workers have shown that PPG-425, HO–(CH(CH3)CH2)n–H, can be a solvent for aldehyde synthesis from 4-hydroxybenzaldehyde, K2CO3, and the corresponding bromo- or dibromoalkane at 60 °C.124 PPG-1000 was also successfully used in indium metal mediated synthesis of homoallylic amines.125 Yields and reaction times were better than and comparable with aqueous reactions and those employing traditional organic solvents. In addition, the product was easily isolated.
PPG is a viscous liquid with a negligible vapor pressure, stable under these reaction conditions, and is easily recovered. However, in contrast to PEG used as a solvent, most commercially available PPG, such as Dow PPGs from molecular weight 250 to 4000, are viscous liquids. Low molecular weight PPG-250 and 425 are in fact water soluble, but PPG shows an inverse temperature–solubility relationship, along with a rapid decrease in water solubility as the molecular weight increases. PPG-2000 has only 0.012% w/w solubility in water at 25 °C, and this may invite exploitation as a relatively more hydrophobic phase in comparison to PEG. These physical properties may limit the wide use of PPGs as reaction media.
3.2. Oxidation reactions
Oxidation reactions conducted in PEG-200 and PEG-400 are given in Table 4. Among the few oxidants examined, K2Cr2O7, which is relatively soluble in PEG-400, can oxidize benzyl bromide with good yields.71 This is similar to the reaction of Na2Cr2O7 in hexamethylphosphoramide (HMPA) and crown ether using the same substrate. Aerobic oxidations catalyzed by a polyoxometalate, H5PV2Mo10O40, in PEG-200 or 400 solvents showed wider application to alcohols, cyclic dienes, sulfides, and in the Wacker reaction.68 Only 5% weight loss during benzyl alcohol oxidation after three recycles was observed in a solvent recycling process due to PEG degradation, resulting from a 1.7% weight loss for each cycle. No oxidation of the terminal hydroxyl of PEG occurred.| Substrate | Oxidant | PEG | Product | Ref. |
|---|---|---|---|---|
| C6H5CH2Br | K2Cr2O7 | PEG-400 | C6H5CHO | 71 |
| R-CH2OH | Aerobic oxidation (catalyzed by H5PV2Mo10O40) | PEG-200 | R-CHO | 68 |
| R = R′-C6H4 | PEG-400 | |||
| CnH2n−4 (cyclic dienes) | Same as above | PEG-200 | CnH2n−6 | 68 |
| R-S-R (sulfide) | Same as above | PEG-200 | R-SO2-R (sulfoxides and sulfones) | 68 |
| CH3CHCH2 | Aerobic oxidation (catalyzed by H5PV2Mo10O40 and palladium) | PEG-200 | CH3COCH3 | 68 |
| R1CHCHR2 | N-Methylmorpholine (NMO) (catalyzed by OsO4) | PEG-400 | R1CH(OH)CH(OH)R2 | 132 |
| R1 = CnH2n+1, C6H5 | ||||
| R2 = H, CnH2n+1, C6H5 | ||||
The dihydroxylation of olefins has been conducted using PEG-400 as solvent and OsO4 as catalyst,132 and high yields of diols were achieved in a short time (2–3 h). The PEG-400 and OsO4 were easily reused by simple extraction of product diols using ether, and more than 90% yield was sustained even after 5 recycles. Additionally, the reaction was suitable for asymmetric dihydroxylation (Sharpless reaction) with high yield and good enantioselectivity.
3.3. Reduction reactions
Table 5 lists some important reduction reactions which have been demonstrated utilizing PEG as solvent. In PEG-400, carbonyl compounds could be reduced by NaBH4 more efficiently than by slow reaction in THF.71 It was also found that the reduction of alkyl and aryl esters to the corresponding alcohols by NaBH4 in PEG-400 was enhanced, where these compounds had been considered inert toward reduction in other organic solvents.134 The halide reduction by NaBH4 in PEG-400 is comparable to that in a variety of polar protic solvents such as DMSO, sulfone, HMPA, and DMF, and the acyl chloride reduction can effectively occur in PEG-400 as a substitute for the inert solvent dioxane.135 Lindlar's catalyst, Pd–CaCO3 poisoned with PbO, is found to partially reduce triple bonds in alkynes to cis-olefins in PEG-400,133 and PEG and the catalyst can be used 3–5 times without loss of activity or yield.| Substrate | Reductant | PEG | Product | Ref. |
|---|---|---|---|---|
| CH3COC6H13 | NaBH4 | PEG-400 | CH3CHOHC6H13 | 71 |
| R1-COO-R2 | NaBH4 | PEG-400 | R1-CH2OH | 134 |
| R1 = alkyl, aryl | ||||
| R2 = CH3, C2H5 | ||||
| R1-CHX-R2 | NaBH4 | PEG-400 | R1-CH-R2 | 135 |
| R1 = alkyl, aryl | ||||
| X = Cl, Br, I | ||||
| R2 = H, CH3, C4H9 | ||||
| R-COCl | NaBH4 | PEG-400 | R-COH | 135 |
| R = C15H31, C9H19, C6H5, p-BrC6H4 | ||||
| C6H5-CHCH2 | H2 | PEG-900 | C6H5-CH2CH3 | 123 |
The waxy solid PEG-900 at 155 bar pressure in sc-CO2 at 40 °C, showed liquid solvent characteristics, and was successfully used as the solvent for hydrogenation of styrene.123 The RhCl(PPh3)3-catalyzed hydrogenation of styrene to ethyl benzene in PEG-900 at 155 bar, 55 °C in sc-CO2 was conducted as a homogeneous catalysis reaction. Ethyl benzene was extracted into sc-CO2, and the catalyst-containing PEG phase was reused without loss.123
4. PEG as phase-transfer catalyst (PTC)
PEGs and their many derivatives have been extensively investigated as PTCs, and are used in many commercial processes to replace expensive, and environmentally-harmful PTCs.20,21,24,136,137 Of the commonly used PTCs, linear PEGs are much cheaper than analogous macrocyclic crown ethers and cryptands.138 PEGs are also more stable at high temperatures, up to 150–200 °C, and show higher stability to acidic and basic conditions than quaternary onium salts.674.1. Free phase-transfer catalysts
| R1 of substrate (R1OH) | Substrate (R2X) | PEG and solvent | Product | Ref. |
|---|---|---|---|---|
| a Furfuryl alcohol. | ||||
| CnH2n+1 | CnH2n+1X | PEG-350–2000 | R1-O-R2 | 139–141 |
| n = 10, 12 | n = 4, 8 | PEG-(C2H5)2 | ||
| X = Br, Cl, I | No solvent | |||
| C6H5, 4-Cl-C6H4, 3,5-(CH3)2-C6H3, | CnH2n+1X | PEG-400, 1500, 6000 | R1-O-R2 | 119 |
| 4-OH-C6H4, C6H5-C6H4 | CnH2nX | No solvent | 142–145 | |
| C6H5CH3X | ||||
| n = 1, 2, 3, 4, 8 | ||||
| X = Cl, Br, I | ||||
| OC4H4-CH2,a or | XRX | PEG-400, 600, 800–C6H6 or CH2Cl2 | (OC4H4CH2O)2R | 146 |
| Ar = CH3C6H4, Cl-C6H4, | X = Br, Cl | or | 147 | |
| NO2-C6H4, HO2CCH2-C6H4, | R = (CH2)n, C2H5OC2H5 | Ar-ORO-Ar | ||
| OHC-C6H4, C6H5-C6H4 | n = 1, 4, 5 | |||
| PEG-300, PPG-425, 1000 | RBr | PEG-300 | PEG-OR | 148 |
| R = CnH2n+1 | PPG-425, 1000 | or | ||
| n = 2, 4, 6 | KOH/no solvent | RO-PEG-OR | ||
| Substrate | Nucleophilic reagents | PEG/solvent and base | Product | Ref. |
|---|---|---|---|---|
| Cl-CH2CO2H | OH-C6H4-OH | PEG-600/CH3C6H5 | HO2CCH2-O-C6H4-O-CH2CO2H | 149 |
| p-ClOCCH2-O-C6H4-O-CH2COCl | R-C6H4-OH | PEG-400/CH2Cl2 | R-C6H4-OCCH2-O-C6H4-O-CH2CO-C6H4-R | 149 |
| m-(ClCOCH2O)2-C6H4 | ArOH | PEG-400/NaOH | (ArO2CCH2O)2-C6H4 | 150 |
| Ar = X-C6H5 | 151 | |||
| X = Cl, NO2, C6H5, CH3 | ||||
| C6H4-N2HC-R | ArOCH2COCl | PEG-400/ | C6H4-N2HC (COCH2OAr)R | 152 |
| R = H, C6H5OCH2, 2,4-Cl2C6H3OCH2, 2,4,5-Cl3C6H2OCH2 | Ar = C6H5, 4-ClC6H4, 4-CH3C6H4 | CH3CN, K2CO3 | ||
| p-Y-C6H4-N2+BF4− | CCl3Br | PEG-1000/ | p-Y-C6H4-Br | 153 |
| Y = Br, NO2 | CH3I | CH3COOK, CHCl3 | p-Y-C6H4-I | |
| p-Y-C6H4-N2+BF4− | PEG-1000/ | p-Y-C6H4-C6H5 | 153 | |
| Y = Br, NO2 | CH3COOK/C6H6 | |||
| C6H4-C2H2NHR1 | R2X | PEG-OCH3/ | C6H4-C2H2NR1R2 | 154 |
| R1 = H, C6H5 | R2 = CH3, C6H5, C12H25 | NaOH/C6H5CH3 | ||
| X = Br, I | ||||
| C6H5CH2Cl | KSCN | PEG w/o sc-CO2 | C6H5CH2SCN | 142,155 |
| p-CH3-C6H4-SO2Cl | MF | PEG-115–40000/ | p-CH3-C6H4-SO2F | 156 |
| M = Li, Na, K, Rb, Cs | CH3COCH3 | |||
| p-X-C6H4-Y | MOR | PEG-150–20000000/ | p-Y-C6H4-OR | 97 |
| X = Br, Cl | M = alkali metal | KOH | ||
| Y = Br, Cl, H | R = alkyl, aryl | |||
| 2-OC4H4-CO2H | C6H5-SO2Cl | PEG-400 | 2-C4H4O-CO2-SO2-C6H5 | 157 |
| R-CO-Cl | NH4SCN | PEG-400/CH2Cl2 | RCOSCN | 158 |
| R = Ar-C4H4O, 2-ClC6H4 | 159 | |||
| POCl3 | ArONa | PEG/CHCl3 | (ArO)3PO | 160 |
| Ar = RC6H4, 2,4-Cl2C6H3 | ||||
| R = CH3, C4H8, Cl, C6H5 | ||||
| C8H17Cl | NaCN | PEG/C10H22 | C8H17CN | 96 |
| CH2Br2 | R-C6H4-CO2K | PEG-600/CH3CN | R-C6H4-CO2-CH2-CO2-C6H4-R | 161 |
| C6H5-CH2Br | CH3CO2K | PEG/CH3CN | C6H5-CH2OCH3 | 162 |
PEG as PTC has been employed in a sc-CO2 solvent to convert benzyl chloride with potassium cyanide to form phenylacetonitrile, although the yield is lower than a similar reaction employing tetraheptylammonium cyanide. Nevertheless, this is a promising beginning for the development of a low cost, green reaction process.155 The effect of PEG molecular weight on catalytic effectiveness in the reaction of mono- and di-halobenzenes with alkoxide ions to form monoalkoxybenzenes has been examined.97 The yield of product was found to increase with primary, secondary, and tertiary alkoxide ions, and high molecular weight PEG was more effective than low molecular weight PEG. The synthesis of N-phenyl-N′-2-chlorobenzoyl-thiourea from 2-chlorobenzoyl chloride and ammonium thiocyanate using PEG-400 as PTC is more effective than with most quaternary ammonium salts and crown ethers.159
| Substrate | Oxidant or reductant/catalyst | PEG/solvent and base | Product | Ref. |
|---|---|---|---|---|
| C6H5-CH2OH | KOCl | PEG-6000/CH3COOC2H5 | C6H5-CHO | 142 |
| R-C6H4-CH2X | Air/Co2(CO)8 | PEG-400/2-CH3-C4H9OH | R-C6H4-CO | 163 |
| X = Cl, Br | ||||
| p-NO2-C6H4-CH3 | O2 | PEG-400/C6H5-CH3/KOH | p-NO2-C6H4-CO2H | 164 |
| RI | Air/CoCl2 | PEG-400/KCN/KOH | RCO2H | 165 |
| R = CH3-C6H4CH3(CH2)n | ||||
| C6H5-CH2-C6H5 | O2 | PEG-6000 | C6H5-CO-C6H5 | 166 |
| RCHCBr2 | Air/Pd(diphos)2 | PEG-400/NaOH | RCH2COOH | 167 |
| R = CH3C6H4, C6H4, CH3, CH3OC6H4, CH3CHCH | ||||
| C6H5-CCH | Mn(CO)3Br | PEG-400/NaOH | C6H5-C5H6O2 | 168 |
| R1COR2 | NaBH4 | PEG-400/C6H6 | R1CH(OH)R2 | 169 |
| R1 = C6H5, C6H5CH2 | 69 | |||
| R2 = CH3, C6H5 | ||||
| RCOY | PEG–Li(Na, K)BH4 | PEG-350 | RC(OH)Y | 70 |
| Y = H, R′, OR″, RCH2Br | RCH3 | |||
PEG with NaBH4 and PEG–NaBH4 complexes have been used in the reduction reactions of ketones and aldehydes.69,70,169 Free PEG can catalyze ketone reduction by NaBH4,169 while the PEG–NaBH4 derivative can selectively reduce aldehydes in the presence of ketones without concurrent reduction of the ketone group.69 Base-catalyzed autoxidation of picoline showed that PEG-6000 in benzene was more effective than the use of crown ethers or quaternary ammonium salts,166 and the reaction can replace previously used expensive aprotic polar solvents such as DMF, Me2SO, and HMPA. The conversion of vinyl dibromides to 1-bromoalkyne in PEG-400 has been shown to be superior to the same reaction using benzyltriethylammonium.167 The conversion of aldehydes to homologous acids by a simple two-step procedure using PEG is also considered more practical than the widely used conversion of p-anisaldehyde to p-methoxyphenylacetic acid.167 The conversion of alkylene to lactone by manganese carbonyl under PEG-400 has been shown to perform almost as well as the more expensive reaction employing benzyltriethylammonium chloride.168
PEG as PTC has also been investigated in the form of a liquid–gas phase reaction in the isomerization of allylbenzene by diffusion and adsorption.170 The results showed remarkably higher activity for dehydrohalogenation of 2-bromoethyl benzene than the use of benzyltriethylammonium and 18-crown-6.171
4.2. PEG-supported PTC
In addition to its own phase-transfer activity, PEG has also been employed as a polymer support for other PTCs. PEG has been modified with some typical PTCs such as crown ethers, ammonium salts, cryptands, and polypodands to enhance phase-transfer in two-phase reactions. 16-Crown-5, 19-crown-6, and 18-crown-6 ethers have been attached to PEG-3400 and PEG-6800 and demonstrated effective transfer of metal picrates into CH2Cl2 from H2O, which were able to catalyze the reaction of CH3COOK with benzyl bromide.162 The quantitative recovery of these crown–PEG polymers from CH2Cl2 and acetonitrile can be achieved by ether precipitation.The covalent attachment of quaternary ammonium salts to PEG-600 resulted in higher reaction rates for dehydrobromination of 2-bromoethylbenzene at 85 °C than those of PEG and quaternary salts alone.172 In the dehydrohalogenation of 4-bromo-1-chloroethylbenzene to 4-bromostyrene, the catalytic activity of PEG-600–ammonium salt depended on both the PEG end hydroxyl substitution and ammonium salt structure.172 The PEG-5000–quaternary ammonium salt complex was also easily recovered by precipitation using hexane, diethyl ether, and tert-butyl methyl ether, in which PEG is not soluble, and by filtration, without activity loss.12,173,174
PEG monoalkyl ether ((EO)n, n = 3, 4, 5) has been attached to hexachlorophosphazene to form cyclophosphazenic polypodands, and these compounds were found to have higher phase-transfer catalytic activity for nucleophilic substitution and alkylation reactions,175,176 especially when commercial Brij 30 (polyoxyethylene-4-lauryl ether) was used as the reactant. The reaction may be competitive with most commonly used PTCs because of the high catalytic activity and low price.176 The use of PEG of molecular weights from 2000 to 20000 as soluble polymer supports for catalysts and reagents has been reviewed by Janda and co-workers.12
5. Aqueous biphasic reactive extraction (ABRE)
The term aqueous biphasic reactive extraction (ABRE) has been used to describe the use of ABS as biphasic reaction media in which controlled partitioning of reactants, catalysts, and products, by choice of phase systems and manipulation of TLL, can be used to enable higher reaction yields, greater product selectivity, and simultaneous separation of reactants, products, and catalysts.50ABS have been used for separations primarily relating to biological solutes and particles for almost half a century.31,59,79,177,178 Recent extension of this technology has been directed toward application as green separation media25 including, separation of organic hydrocarbon species32,33,45 and metal ions.34–38 However, both PEG and ABS have been largely ignored as “green reaction media” until recent papers indicated their application as new solvents for delignification of cellulose,50–55 cyclic olefin oxidation,56,57 polyoxometalate catalyzed aerobic oxidation,68 SN1, Diels–Alder, and enzymatic reactions,88,116,117 and olefin catalytic oxidation to diols.132
ABRE has three main characteristics: (1) phase separation benefiting separation of reactants and products, and providing a reaction driving force; (2) the PEG-rich top phase in PEG–salt ABS has organic solvent-like properties as a medium for reaction; (3) ABS components, PEG and metal salts, may be included in the form of PTCs56,57 and metal-catalysts.50 Two types of ABRE have been noted. One takes the form of a three-phase reaction,56,57,179–181 and the other a two-phase reaction.50–55 The three-phase reaction results from adding organic reactants with or without corresponding supporting organic solvent into an ABS, thus, the system forms three phases initially as shown in Scheme 1. The three phases are usually composed of the light organic phase, and the typical PEG–salt ABS made of the PEG-rich top phase and salt-rich bottom phase. The two-phase reaction results from the chemical or enzyme catalyzed reaction supported only by the normal ABS biphases as demonstrated in Scheme 2.
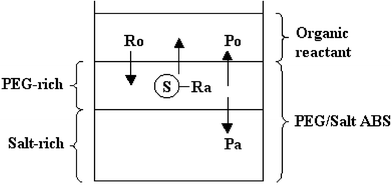 | ||
| Scheme 1 Three-phase ABRE: Ro = organic reactant; Ra = aqueous reactant; Po = organic product; Pa = aqueous product; S = PTC. | ||
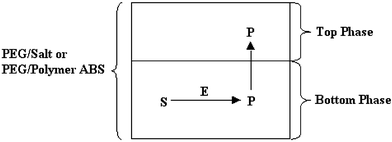 | ||
| Scheme 2 Typical enzyme hydrolysis reaction in ABS: S = substrate; E = enzyme; P = product. | ||
5.1. Three-phase reactions
In order to enhance reaction rate and yield, PEG–salt ABS were formed to increase the organic reactant solubility and PEG concentration in the PEG-rich top phase, which has been described as an ABRE process applied to catalytic oxidation,56,57 substitution, and isomerization reactions.179–181 The currently rather limited number of investigations of ABRE processes listed in Table 9, suggest that the role of ABS may be very important for increasing the reaction yield, however, the underlying mechanism is still largely unexplored. For the reactions shown in Scheme 1, ABS formation results in a PEG-rich top phase, which facilitates an increase in the solubility of some organic reactants.56,57,119 Secondly, phase separation is useful for reactant and product separation, and thirdly, alteration of the reactant, product or catalyst distribution may be used to manipulate the reaction rate. In three-phase reactions, many solvophilic and solvophobic reactants may display some solubility in and partition, at least to some extent, to the PEG-rich top phase and the reaction may occur in this phase.56,57,180| ABS and PEG | Organic phase | Product | Ref. |
|---|---|---|---|
| C4H9ONa + PEG + NaOH | C6H5-CH2Cl | C6H5-CH2-OC4H9 | 179 |
| (PEG-600, 3000) | (in C6H6 or C12H26) | 181–183 | |
| (Hex)4NBr + PEG + KOH | 2-C8H17Br | C8H16 | 184 |
| (PEG-200) | (in C12H26) | ||
| KOH + PEG | CH3O-C6H4-CH2-CHCH2 | CH3O-C6H4-CHCH-CH3 | 180 |
| (PEG-400–6000) | (in C6H5-CH3) | ||
| H2O2 + PEG + NaHSO4 | C6H10 | HOOC-(CH2)4-COOH | 56 |
| (PEG-600–20000) | C5H8 | HOOC-(CH2)3-COOH | 57 |
In the synthesis of n-butyl phenyl ether by three-phase catalysis,181 addition of NaOH resulted in a higher reaction rate than the addition of other kinds of salts such as NaCl, KBr and NaBr. PEG and OH− easily form ABS due to the more negative Gibbs free energy of hydration (ΔGhyd) of the hydroxide anion than that of Cl− and Br−. In the three-phase reaction, the mechanism shown in Scheme 1 is supposed to be that of organic reactant diffusion into the PEG-rich top aqueous phase56,57 (or PEG as PTC carrying aqueous reactant into the organic phase181), with subsequent reaction resulting in product precipitation (or dissolution) in the bottom phase, or alternatively, organic product partition into the organic phase.179 For the above reaction, in an ABS made up of PEG-3000, tetrahexylammonium bromide/KOH and dodecane as organic phase, the production rate of the three-phase system was seven times higher than a corresponding aqueous–organic two-phase system, however when toluene was added as the organic phase, no ABS was formed, because PEG-3000 is soluble in toluene. The yield in toluene was about 10% lower than that in dodecane under the same experimental conditions. The ether production rate and its selectivity are dependent on the initial concentration of n-butanol.181
Synergistic effects of PEG and (Hex)4NBr in the form of a third phase for dehydrohalogenation of 2-bromooctane, and butylphenyl ether have been studied.184 The amount of quaternary ammonium salt can be reduced to one-nineteenth of the original amount and easily recycled without any loss in catalytic activity in the 2nd and 3rd recycle.184
The mechanism of base-catalyzed three-phase reactions with PEG has been studied using the isomerization of allylanisole as a model reaction.180 The kinetically controlled reaction is successful only under three-phase conditions due to the phase separation of NaOH–PEG aqueous solutions. A potential advantage lies in the fact that PEG is more stable under the experimental conditions, under which quaternary ammonium salts undergo Hoffmann elimination and deactivation.
Recently, we have developed a new green catalytic oxidation of cyclic olefins (e.g., cyclohexene, cyclopentene, and 1,2,3,6-tetrahydrophthalic anhydride (THPA)) to dicarboxylic acid (e.g., adipic acid, glutaric acid, and 1,2,3,4-butanetetracarboxylic acid (BTCA)) in a PEG-2000–NaHSO4 50% H2O2 ABS and in the absence of organic solvent and specific PTC. Phase separation in PEG–NaHSO4 was found to be essential to enhance reaction yield.56,57 The ABS selection, and effect on adipic acid yield were carefully investigated, and in this ABRE process, the results showed that phase compositions, TLL, and PEG molecular weight can all affect the reaction yield. This model reaction demonstrated the potential “green” characteristics of the use of organic solvent-free, inexpensive, low toxicity PEG and NaHSO4 ABS components, combined with an ecologically benign and clean oxidant (50% H2O2).
5.2. Biphasic reactions
Hydrogel microspheres of methylacrylate bound Dextran polymer can be obtained from the polymerization of methylacrylate bound Dextran in PEG-10000–methacrylated Dextran (Dextran-40000 or 220000) ABS without using organic solvent.186 A 90% yield can be obtained within short times at relatively low concentrations of radical initiators potassium peroxydisulfate (KPS) and tetramethylethylenediamine (TEMED). Comparison of microspheres from ABS with a macroscopic hydrogel product with a system confined to one aqueous phase, showed a higher reaction rate. With the macroscopic hydrogel process in one phase, the KPS concentration decreased gradually with time, whereas for the microspheres in ABS, the KPS concentration remained constant due to the partition equilibrium between KPS in the PEG-rich top phase and the Dextran-rich bottom phase.
The enzyme hydrolysis and biomass bioconversions in ABS listed in Table 10, represent important applications of ABRE in bioengineering.26,28 Compared with one-phase reactions, several important characteristics are illustrated in Scheme 2. (1) The ABS must selectively distribute enzyme and substrate to one phase (the bottom phase was assumed in Scheme 2), while the desired product should be partitioned to the other phase (the top phase was assumed in Scheme 2). This can be achieved through differences in molecular weight or molecular structure. The un-hydrolyzed biomass, such as cellulose or starch, is macromolecular, while products, such as glucose or cellobiose, are much smaller. Transformation of substrate to product by an enzyme confined in the bottom phase can effectively enhance reaction by distribution of product to the top phase. Even if the low molecular weight product is evenly partitioned between the phases, the reaction can still be conducted by continuous removal of product from one phase. (2) Product inhibition can effectively be avoided by separation of enzyme and product into two different phases. (3) In some cases, product hydrolysis can be effectively prevented due to partition in the high concentration PEG phase.199
| Substrate | Enzyme | PEG and ABS | Product | Ref. |
|---|---|---|---|---|
| Cellulose (C6H10O5)n | Endo-β-glucanase | PEG-40000, 200000 | Glucose (C6H12O6) | 189–194 |
| Exo-β-glucanase | Dextran (MW = 1, 4, 11, 50, 200 × 104) | |||
| β-Glucosidase | ||||
| Starch or native starch | α-Amylase; | PEG-6000, 20000 | Maltose, glucose | 195–198 |
| Glucoamylase | Dextran (MW = 5–7 × 104) | |||
| Amyloglucosidase | PEO-PPO-2500 | |||
| MgSO4, (NH4)2SO4 | ||||
| p-Nitrophenyl α-mannoside | α-Mannosidase | PEG-8000 | Oligosaccharides | 199 |
| p-Nitrophenyl β-galactoside | Dextran-500000 | |||
| Bovine hemoglobin | Papain | PEG-6000 | Soluble peptides | 195 |
| Dextran (MW = 5–7 × 104) | ||||
| Ultra fine silica particles (Snowtex 30) | ||||
| N-Acetyl-L-methionine | Acylase | PEG-600 | Acetic acid and L-methionine | 200,201 |
| K3PO4 | ||||
| Penicillin G | Penicillin acylase | PEG-6000, 20000 | 6-Aminopenicillanic acid (6-APA) | 202–204 |
| Potassium salt | K3PO4 | |||
| Benzylpenicillin (BP) | ||||
| 7-amino-deacetoxicephalosporanic acid (7-ADCA) | Penicillin G acylase | PEG-400 | Cephalexin | 205,202 |
| Phenylglycine methyl ester (PGME) | MgSO4 | |||
| C21H30O5 (Hydrocortisone) | Bacterium | PEG | Prednisolone | 187 |
| Reppal-PES | ||||
In the bioconversion of cellulose to ethanol, the enzymes for hydrolysis, and enzyme recycling constitute the major portion of the process costs.189 Biphasic systems can be used in cellulose pretreatment, hydrolysis, and fermentation. Both the cellulose substrate and cellulolytic enzyme may be partitioned to the bottom phase, while the hydrolysis product, glucose and some soluble reducing sugars, will be partitioned to the top phase.189–192 Use of ultra filtration technology193 and an attrition bioreactor194 in combination with ABRE, have been found to enhance the reaction rate and improve the yield.193,194
Various crude starches have been hydrolyzed by the synergistic action of α-amylase and glucoamylase in PEG–Dextran and PEG–starch ABS.188 The main advantage in the use of starch is that the starch serves as one of the phase forming polymers, which markedly decreases the cost of the reaction.188 The enzyme hydrolysis of corn starch in ABS using α-amylase immobilized on ultra fine silica particles by covalent cross linking with glutaraldehyde has been studied,195 and the enzyme showed high activities. The immobilized enzyme was partitioned to the PEG-rich top phase in PEG–Dextran ABS, and the products were recovered from the bottom phase. Bovine Hb and papain immobilized on ultrafine silica particles by covalent crosslinking with glutaraldehyde have been studied in PEG-6000–Dextran-60000.195 The immobilized papain was totally partitioned to the top PEG-rich phase, and the soluble peptide product was found in the bottom phase.
Conversion of native waxy maize starch to glucose by α-amylase and glucoamylase has also been conducted in a PEG-20000–crude Dextran ABS in combination with membrane ultra filtration.196 A continuous stream of glucose could be produced, and PEG, Dextran, and the starch-degrading enzymes could be recycled. It was possible to cut the starch bioconversion time almost in half by employing a PEO-PPO-2500–MgSO4 ABS compared to a single phase process.197,198 (PEO-PPO-2500 is a random copolymer of ethylene oxide and propylene oxide with an average molecular weight of 2500.)
PEG-8000–Dextran-500000 ABS have also been applied to the synthesis of oligosaccharides by reverse action of Jack bean α-mannosidase.205 The reaction and whole yields were similar in two-phase systems and one-phase aqueous buffer systems, but the yield of product per unit of enzyme increased ten fold when using the ABS. This is likely to become a rapidly developing area of the application of ABRE systems since the availability of carbohydrate enzymes in high purity and modest cost has increased with the advent of the wide application of genetic manipulation (GM) techniques.206
Cephalexin has been synthesized in ABS by using penicillin G acylase (PGA) as a catalyst and 7-amino-deacetoxicephalosporanic acid (7-ADCA) and phenylglycine methyl ester (PGME) as substrates. A 60% yield of cephalexin was achieved in ABS compared to 21% in an entirely aqueous single phase reaction.202 The deacylation of penicillin G has been studied using penicillin acylase in a PEG–K3PO4 ABS. In this system, the cells partitioned to the bottom phase and the products to the top phase.203 Cephalexin synthesis from 7-ADCA and PGME catalyzed by PGA, which is covalently immobilized inside a glyoxyl-agarose porous support, can be conducted in PEG-600–(NH4)2SO4 ABS. The yield of 90% in the biphasic reaction was higher than that of 55% found in the monophasic reaction.199
Chirally selective enzymatic acylase hydrolysis of N-acetyl-L-methionine into acetic acid and L-methionine was carried out in PEG–K3PO4 ABS in liquid–liquid centrifugal partition chromatography (CPC), and the products and reactants were obtained separately in the same process.200,201
Enzyme-biocatalysts are often considered to be inherently green, clean, and nontoxic as opposed to traditional catalysts, which are often toxic metal compounds. Enzymatic syntheses are capable of providing high stereo- and regio-selectivities without using chemical protection-deprotection.207 ABS may have an important contribution to make in this area. ABS allow manipulation of product and reactant distributions allowing the possibility of separation and yield enhancements. ABS also provide a benign non-denaturing environment for enzymes in contrast to classical solvent media.
6. Conclusions
In recent years PEG aqueous solutions have been widely used in many different kinds of reaction systems. Their low-toxicity, low volatility, and biodegradability represent important environmentally benign characteristics, which are particularly attractive when combined with their relatively low cost as a bulk commodity chemical. In addition, aqueous PEG solutions may often substitute for expensive and often toxic PTCs. The developed state of knowledge with regard to the toxicological properties of PEG is of considerable current advantage compared to the paucity of knowledge for many other potential alternative solvent systems.The wide range of reactions conducted in pure liquid PEG demonstrates its resilience to degradation and the low occurrence of unwanted side reactions. The role of PEG as a cheap substitute PTC supports the idea that it can assist in promoting the molecular proximity of reactants and catalysts. ABRE combines many of the above advantages along with a measure of process integration and intensification in which chemical synthetic steps are combined with extractive steps. Aqueous solutions of polymers, and in particular PEG, can profoundly affect water structure, reducing cohesivity and some aspects of hydrogen bonding and thereby increasing the solubility of relatively less polar species.
The critical phase formation phenomena associated with PEG aqueous solutions are uniquely important in ABRE, since this brings together phases having different properties and which may be described as having different degrees of hydrophobicity. These phases are uniquely tunable and thus, taken together, ABRE systems can cover a large range of relative difference in chemical potential difference between the phases. In order to achieve this, ABRE systems need to be thought of as a continuum of self-similar systems ranging from Dextran–Ficoll and similar systems, through to PEG–Dextran, and ultimately, PEG–salt ABS. In all of these systems, the polymer molar concentration difference between the phases is proportional to the chemical potential difference engendered across the interface. Metal salts and metal-catalysts may be expected to benefit from solubility in the lower salt-rich phase and yet continued solubility in the more hydrophobic phase may provide for intimate contact with less polar species.
Phase separation can also provide a driving force for chemical reaction through the law of mass action by separation of reactants and products at the moment of formation. Phase separation in ABRE processes may also significantly aid in catalyst and product recovery by providing means of distribution to different phases. In turn, this can aid in the efficient use of reactants and the recycling of catalysts.
Solute distribution is controllable to a much greater extent than with conventional extraction systems. With careful design, this can result in enhanced reaction rates and improved yields in specific reactions. It is also possible to achieve the result that expensive and inefficient recycling and recovery steps can be minimized.
On the other hand, ABRE processes represent complex systems with a large number of variable reaction parameters, requiring optimization and analysis, such as temperature, pH, the addition of particular reactants, or the formation of products or soluble intermediates. All these factors will need to be carefully considered in the design of ABRE processes.
In the course of our recent studies on ABRE processes, we have identified two-types of ABRE process. The first is a three-phase system which includes insoluble liquid organic reactants. The second type is a biphasic process in which the reactants are completely soluble. In the former case, as for example in the oxidation of cyclic olefins in a PEG–NaHSO4–H2O2–H2O ABS, the solubility of the cyclic olefins in the PEG-rich phase is of prime importance. Successful design of the ABS, including selection of PEG molecular size and salt type, depends on an understanding of ABS phase behavior and phase polarity. Such reactions may be applicable in many organic synthetic processes.
Pure biphasic processes may be widely applicable where most components are more soluble. However, even here the existence of a third phase may be noted in some cases, for example a phase of residual solid wood pulp in alternative delignification processes for hardwood and softwood paper pulping in PEG–salt ABS. Such ABRE pulping processes were shown to compare favorably with Kraft type processes and with current organosolv practice. Another example may be given in the form of enzyme hydrolysis reactions, for example the degradation of cellulose in PEG/polymer ABS has shown these ABRE systems can lead to enhanced reaction rates, and cost and energy savings.
It may be anticipated, given the greater availability, higher purity, and reduced cost of enzymes resulting from the impact of rDNA technology, combined with the exquisite specificity of these enzymes and our increasing ability to engineer their properties, that the use of enzymatic synthesis in synthetic chemistry will greatly expand in the years to come. In this context, ABRE processes provide a compatibility with macromolecular stability unmatched by any other extractive solvent systems. Thus, the application of ABRE in the development of controlled enzymatic synthesis, for example of complex carbohydrates, is very promising.
It is to be hoped that this review will further stimulate interest in the extension of ABS applications to yet more novel catalytic processes and reactive extractions. The application of ABRE in synthetic organic chemistry represents both an opportunity and a challenge, and the wide availability of PEG at low cost may give such processes a promising future in green chemistry and green engineering.
Abbreviations
| ABRE | Aqueous biphasic reactive extraction |
| ABS | Aqueous biphasic systems |
| 7-ADCA | 7-Amino-deacetoxicephalosporanic acid |
| ATPS | Aqueous two-phase systems |
| BTCA | 1,2,3,4-Butanetetracarboxylic acid |
| CPC | Centrifugal partition chromatography |
| DMF | Dimethyl formamide |
| DMSO | Dimethyl sulfoxide |
| GM | Genetic manipulation |
| GRAS | Generally recognized as safe |
| HMPA | Hexamethylphosphoramide |
| IL | Ionic liquid |
| KPS | Potassium peroxydisulfate |
| LCST | Lower critical solution temperature |
| PEG | Polyethylene glycol |
| PGA | Penicillin G acylase |
| PGME | Phenylglycine methyl ester |
| PPG | Polypropylene glycol |
| PTC | Phase-transfer catalyst |
| sc-CO2 | Supercritical carbon dioxide |
| TEMED | Tetramethylethylenediamine |
| THF | Tetrahydrofuran |
| THPA | 1,2,3,6-Tetrahydrophthalic anhydride |
| TLL | Tie line length |
| UCST | Upper critical solution temperature |
| VOC | Volatile organic compound |
Acknowledgements
Our work with PEG is supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Research, US Department of Energy (Grant DE-FG02-96ER14673).References
- P. T. Anastas, in Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002, p. 1 Search PubMed.
- J. Sherman, B. Chin, P. D. Huibers, R. Garcia-Valls and T. A. Hatton, Health Perspect., 1998, 106(Suppl 1), 253 Search PubMed.
- L. R. Pratt, Chem. Rev., 2002, 102, 2625 CrossRef CAS.
- Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002 Search PubMed.
- N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725 CrossRef CAS.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef.
- P. E. Savage, Chem. Rev., 1999, 99, 603 CrossRef CAS.
- C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef.
- T. H. Chan, L. H. Li, Y. Yang and W. S. Lu, in Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002, p. 166 Search PubMed.
- C. J. Li, J. X. Haberman, C. C. K. Keh, X. H. Yi, Y. Meng, X. G. Hua, S. Venkatraman, W. C. Zhang, T. Nguyen, D. Wang, T. S. Huang and J. H. Zhang, in Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002, p. 178 Search PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325 CrossRef CAS.
- D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345 CrossRef CAS.
- S. Tascioglu, Tetrahedron, 1996, 52, 11113 CrossRef CAS.
- N. W. Fadnavis and A. Deshpande, Curr. Org. Chem., 2002, 6, 393 CAS.
- S. N. Shtykov, J. Anal. Chem., 2000, 55, 608 CrossRef CAS.
- J. H. M. Heijnen, V. G. De Bruijn, L. J. P. Van den Broeke and J. T. F. Keurentjes, in Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002, p. 191 Search PubMed.
- G. Oehme, I. Grassert, E. Paetzold, R. Meisel, K. Drexler and H. Fuhrmann, Coord. Chem. Rev., 1999, 185–186, 585 CrossRef CAS.
- G. Oehme, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2002, vol. 2, p. 835 Search PubMed.
- G. E. Totten and N. A. Clinton, J. Macromol. Sci. Rev. Macromol. Chem. Phys., 1988, C28, 293 Search PubMed.
- G. E. Totten, N. A. Clinton and P. L. Matlock, J. Macromol. Sci. Rev. Macromol. Chem. Phys., 1998, C38, 77 Search PubMed.
- Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications, ed. J. M. Harris, Plenum Press, New York, 1992 Search PubMed.
- Poly(ethylene glycol) Chemistry and Biological Applications, ed. J. M. Harris and S. Zalipsky, ACS Symposium Series 680, American Chemical Society, Washington, DC, 1997 Search PubMed.
- C. M. Starks, C. L. Liotta and M. Halpern, Phase-Transfer Catalysis: Fundamentals, Applications and Industrial Perspectives, Chapman & Hall, New York, 1994, p. 158 Search PubMed.
- J. G. Huddleston, H. D. Willauer, S. T. Griffin and R. D. Rogers, Ind. Eng. Chem. Res., 1999, 38, 2523 CrossRef CAS.
- F. Tjerneld and H. O. Johansson, Int. Rev. Cytol., 2000, 192, 137 Search PubMed.
- R. Hatti-Kaul, Mol. Biotechnol., 2001, 19, 269 Search PubMed.
- Methods in Biotechnology, Vol. 11, Aqueous Two-Phase systems, Methods and Protocols, ed. R. Hatti-Kaul, Human Press, NJ, 2000, p. 411 Search PubMed.
- Ionic Liquids, Industrial Applications to Green Chemistry, ed. R. D. Rogers and K. R. Seddon, ACS Symposium Series 818, American Chemical Society, Washington, DC, 2002 Search PubMed.
- Ionic Liquids as Green Solvents: Progress and Prospects, ed. R. D. Rogers and K. R. Seddon, ACS Symposium Series 856, American Chemical Society, Washington, DC, 2003 Search PubMed.
- R. D. Rogers and M. A. Eiteman, Aqueous Biphasic Separations: Biomolecules to Metal Ions, Plenum Press, New York, 1995 Search PubMed.
- J. G. Huddleston, H. D. Willauer, K. R. Boaz and R. D. Rogers, J. Chromatogr. B, 1998, 711, 237 CrossRef CAS.
- R. D. Rogers, H. D. Willauer, S. T. Griffin and J. G. Huddleston, J. Chromatogr., B: Biomed. Appl., 1998, 711, 255 CrossRef CAS.
- R. D. Rogers, A. H. Bond, C. B. Bauer, J. H. Zhang and S. T. Griffin, J. Chromatogr., B: Biomed. Appl., 1996, 680, 221 CrossRef CAS.
- R. D. Rogers, A. H. Bond, J. H. Zhang and C. B. Bauer, Appl. Radiat. Isot., 1996, 47, 497 CrossRef CAS.
- R. D. Rogers, A. H. Bond, C. B. Bauer, J. H. Zhang, S. D. Rein, R. R. Chomko and D. M. Roden, Solvent Extr. Ion Exch., 1995, 13, 689 CrossRef CAS.
- R. D. Rogers, A. H. Bond and C. B. Bauer, Sep. Sci. Technol., 1993, 28, 139 CrossRef CAS.
- R. D. Rogers, A. H. Bond and C. B. Bauer, Sep. Sci. Technol., 1995, 30, 1203 CrossRef CAS.
- R. D. Rogers, A. H. Bond and J. L. Wolff, J. Coord. Chem., 1993, 29, 187 CAS.
- R. D. Rogers, A. H. Bond, S. Aguinaga and A. Reyes, J. Am. Chem. Soc., 1992, 114, 2967 CrossRef CAS.
- R. D. Rogers, A. H. Bond and D. M. Roden, Inorg. Chem., 1996, 35, 6964 CrossRef CAS.
- R. D. Rogers, J. H. Zhang and C. B. Bauer, J. Alloys Compd., 1997, 249, 41 CrossRef CAS.
- R. D. Rogers, M. M. Benning, R. D. Etzenhouser and A. N. Rollins, J. Coord. Chem., 1992, 26, 299 CrossRef CAS.
- R. D. Rogers, M. L. Jezl and C. B. Bauer, Inorg. Chem., 1994, 33, 5682 CrossRef CAS.
- H. D. Willauer, J. G. Huddleston and R. D. Rogers, Ind. Eng. Chem. Res., 2002, 41, 1892 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer and R. D. Rogers, J. Chromatogr., B: Biomed. Appl., 2000, 743, 137 CrossRef CAS.
- H. D. Willauer, J. G. Huddleston, S. T. Griffin and R. D. Rogers, Sep. Sci. Technol., 1999, 34, 1069 CrossRef CAS.
- H. D. Willauer, J. G. Huddleston and R. D. Rogers, Ind. Eng. Chem. Res., 2002, 41, 2591 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer and R. D. Rogers, Phys. Chem. Chem. Phys., 2002, 4, 4065 RSC.
- Z. Guo, M. Li, H. D. Willauer, J. G. Huddleston, G. C. April and R. D. Rogers, Ind. Eng. Chem. Res., 2002, 41, 2535 CrossRef CAS.
- M. Li, H. D. Willauer, J. G. Huddleston and R. D. Rogers, Sep. Sci. Technol., 2001, 36, 835 CrossRef CAS.
- Z. Guo, J. G. Huddleston, R. D. Rogers and G. C. April, Ind. Eng. Chem. Res., 2003, 42, 248 CrossRef CAS.
- H. D. Willauer, J. G. Huddleston, M. Li and R. D. Rogers, J. Chromatogr., B: Biomed. Appl., 2000, 743, 127 CrossRef CAS.
- Z. Guo, G. C. April, M. Li, H. D. Willauer, J. G. Huddleston and R. D. Rogers, Chem. Eng. Commun., 2003, 190, 1155 Search PubMed.
- Z. Guo, Ph.D thesis, Assessment of Aqueous Biphasic Systems (ABS) as an Alternative Delignification Process, The University of Alabama, Tuscaloosa, 2002 Search PubMed.
- J. Chen, S. K. Spear, J. G. Huddleston, J. H. Holbrey, R. P. Swatloski and R. D. Rogers, Ind. Eng. Chem. Res., 2004, 43, 5358 CrossRef CAS.
- J. Chen, S. K. Spear, J. G. Huddleston, J. H. Holbrey and R. D. Rogers, J. Chromatogr., B: Biomed. Appl., 2004, 807, 145 CrossRef CAS.
- F. E. Bailey, Jr. and J. V. Koleske, Poly(Ethylene Oxide), Academic Press, New York, 1976 Search PubMed.
- P. Å. Albertsson, Partition of Cell Particles and Macromolecules, Wiley, New York, 3rd edn., 1986 Search PubMed.
- J. M. Harris, in Polyethylene Glycol Chemistry, Biotechnical and Biomedical Applications, ed. J. M. Harris, Plenum Press, New York and London, 1992, p. 7 Search PubMed.
- D. A. Herold, K. Keil and D. E. Bruns, Biochem. Pharmacol., 1989, 38, 73 CrossRef CAS.
- M. G. Peglation, Cancer Treat. Rev., 2002, 28, 13 CrossRef.
- D. Bhadra, S. Bhadra, P. Jain and N. K. Jain, Pharmazie, 2002, 57, 5 CAS.
- G. E. Francis, D. Fisher, C. Delgado, F. Marlik, A. Gardiner and D. Neale, Int. J. Hematol., 1998, 68, 1 CrossRef CAS.
- F. M. Veronese, P. Caliceti, O. Schiavon and M. Sergi, Adv. Drug Delivery Rev., 2002, 54, 587 CrossRef CAS.
- Carbowax PEG-200 MSDS of Canadian Center for Occupational Health and Safety (CCOHS), Record Number 2426725, Oct. 2002, http://www.safety.vanderbilt.edu/pdf/hcs_msds/carbowaxPEG200.pdf.
- S. D. Naik and L. K. Doraiswamy, AIChE J., 1998, 44, 612 CrossRef CAS.
- A. Haimov and R. Neumann, Chem. Commun., 2002, 876 RSC.
- J. R. Blanton, Synth. Commun., 1997, 27, 2093 CrossRef CAS.
- J. R. Blanton, React. Funct. Polym., 1997, 33, 61 CrossRef CAS.
- E. Santaniello, A. Manzocchi and P. Sozzani, Tetrahedron Lett., 1979, 20, 4581 CrossRef.
- R. Kjellander and E. Florin, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2053 RSC.
- R. Kjellander, J. Chem. Soc., Faraday Trans. 2, 1982, 77, 2025 RSC.
- M. Carlsson, D. Hallen and P. Linse, J. Chem. Soc., Faraday Trans., 1995, 91, 2081 RSC.
- G. Karlström, J. Phys. Chem., 1985, 89, 4962 CrossRef.
- H. O. Johansson, G. Karlstrom, F. Tjerneld and C. A. Haynes, J. Chromatogr., B: Biomed. Appl., 1998, 711, 3 CrossRef.
- B. Y. Zaslavsky, Aqueous Two-Phase Partitioning: Physical Chemistry and Bioanalytical Applications, Marcel Dekker, New York, 1995 Search PubMed.
- Y. Guan, T. E. Treffry and T. H. Lilley, J. Chromatogr., A, 1994, 668, 31 CrossRef CAS.
- H. Walter and G. Johansson, Aqueous Two-Phase Systems, Vol. 228, Methods in Enzymology Series, Academic Press, New York, 1994 Search PubMed.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632 CrossRef CAS.
- J. G. Huddleston, T. K. Looney, G. A. Broker, S. T. Griffin, S. K. Spear and R. D. Rogers, Ind. Eng. Chem. Res., 2003, 42, 6088 CrossRef CAS.
- C. A. Eckert, B. L. Knutson and P. G. Debenedetti, Nature, 1996, 383, 313 CrossRef CAS.
- S. H. Yalkowsky, Solubility and Solubilization in Aqueous Media, Oxford University Press, New York, 1999 Search PubMed.
- S. H. Yalkowsky and S. Banerjee, Aqueous Solubility: Methods of Estimation for Organic Compounds, Marcel Dekker, New York, 1992 Search PubMed.
- C. J. Li and T. H. Chan, Tetrahedron, 1999, 55, 11149 CrossRef CAS.
- B. Y. Zaslavsky, A. A. Borvskaya, N. D. Gulaeva and L. M. Miheeva, Biotechnol. Bioeng., 1992, 40, 1 CrossRef.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- N. F. Leininger, R. Clontz, J. L. Gainer and D. J. Kirwan, in Clean Solvents, Alternative Media for Chemical Reactions and Processing, ed. M. A. Abraham and L. Moens, ACS Symposium Series 819, American Chemical Society, Washington, DC, 2002; p. 208 Search PubMed.
- N. P. Cheremisinoff, Industrial Solvent Handbook, Marcel Dekker, New York, 2nd edn., 2003 Search PubMed.
- M. H. Abraham, A. M. Zissimos, J. G. Huddleston, H. D. Willauer, R. D. Rogers and W. E. Acree, Ind. Eng. Chem. Res., 2003, 42, 413 CrossRef CAS.
- L. Toke and G. T. Sazbo, Acta Chim. Acad. Sci. Hung., 1977, 93, 421 CAS.
- L. Toke, G. T. Sazbo and K. Somogyi-Werner, Acta Chim. Acad. Sci. Hung., 1979, 101, 47 CAS.
- G. T. Sazbo, K. Aranyosi and L. Toke, Acta Chim. Acad. Sci. Hung., 1982, 110, 215.
- R. Neumann and Y. Sasson, J. Mol. Catal., 1985, 31, 81 CrossRef CAS.
- R. Neumann, S. Dermeik and Y. Sasson, Isr. J. Chem., 1985, 26, 239 CAS.
- G. W. Gokel, D. M. Goli and R. A. Schultz, J. Org. Chem., 1983, 48, 2837 CrossRef CAS.
- R. Neumann and Y. Sasson, Tetrahedron, 1983, 39, 3437 CrossRef CAS.
- P. Selucky, J. Plesek, J. Rais, M. Kyrs and L. Kadlecova, J. Radioanal. Nucl. Chem., 1991, 149, 131 CAS.
- G. Von Helden, T. Wyttenbach and M. T. Bowers, Science, 1995, 267, 1483.
- G. Von Helden, T. Wyttenbach and M. T. Bowers, Int. J. Mass Spectrom. Ion Processes, 1995, 146–147, 349 CrossRef.
- T. Wyttenbach, G. Von Helden and M. T. Bowers, Int. J. Mass Spectrom. Ion Processes, 1997, 165–166, 377 CrossRef.
- S. Yanagida, K. Takahashi and M. Okahara, Bull. Chem. Soc. Jpn., 1978, 51, 1294 CAS.
- S. Yanagida, K. Takahashi and M. Okahara, Bull. Chem. Soc. Jpn., 1978, 51, 3111 CAS.
- L. J. M. Sawers, D. P. Tunstall and P. G. Bruce, Solid State Ionics, 1998, 107, 13 CrossRef CAS.
- C. P. Rhodes and R. Frech, Macromolecules, 2001, 34, 1365 CrossRef CAS.
- L. Ducasse, M. Dussauze, J. Grondin, J. C. Lassegues, C. Naudin and L. Servant, Phys. Chem. Chem. Phys., 2003, 5, 567 RSC.
- C. P. Rhodes and R. Frech, Solid State Ionics, 1999, 121, 91 CrossRef CAS.
- G. S. MacGlashan, Y. G. Andreev and P. G. Bruce, Nature, 1999, 398, 782.
- P. Lightfoot, M. A. Mehta and P. G. Bruce, Science, 1993, 262, 883 CrossRef CAS.
- P. Lightfoot, M. A. Mehta and P. G. Bruce, J. Mater. Chem., 1992, 2, 379 RSC.
- B. G. Bogdanov, M. Michailov and C. V. Uzov, J. Polym. Mater., 1991, 8, 193 Search PubMed.
- J. B. Thomson, P. Lightfoot and P. G. Bruce, Solid State Ionics, 1996, 85, 203 CrossRef.
- D. K. Cha and S. M. Park, J. Electroanal. Chem., 1998, 459, 135 CrossRef CAS.
- G. A. Broker, J. Chen, J. G. Huddleston, S. T. Griffin, J. Baldwin, J. D. Holbrey, S. K. Spear and R. D. Rogers, Structural Chemistry of Tetraethylene Glycol Complex of LiCl, in preparation Search PubMed.
- V. O. Sheftel, Indirect Food Additives and Polymers: Migration and Toxicology, Lewis Publishers, Inc., Boca Raton, FL, 2000, p. 1114 Search PubMed.
- N. F. Leininger, R. Clontz, J. L. Gainer and D. J. Kirwan, Chem. Eng. Commun., 2003, 190, 431 Search PubMed.
- N. F. Leininger, J. L. Gainer and D. J. Kirwan, AIChE J., 2004, 50, 511 CrossRef CAS.
- E. Santaniello, A. Manzocchi and P. Sozzani, Tetrahedron Lett., 1979, 20, 4581 CrossRef.
- M. L. Wang and K. R. Chang, Can. J. Chem. Eng., 1991, 69, 340 CAS.
- S. Perrier, H. Gemici and S. Li, Chem. Commun., 2004, 604 RSC.
- B. Sauvagnat, F. Lamaty, R. Lazaro and J. Martinez, Tetrahedron Lett., 2000, 41, 6371 CrossRef CAS.
- S. Chandrasekhar, Ch. Narsihmulu, S. S. Sultana and N. R. Reddy, Org. Lett., 2002, 4399 Search PubMed.
- D. J. Heldebrant and P. G. Jessop, J. Am. Chem. Soc., 2003, 125, 5600 CrossRef CAS.
- B. A. Roberts, G. W. V. Cave, C. L. Raston and J. L. Scott, Green Chem., 2001, 3, 280 RSC.
- P. C. Andrews, A. C. Peatt and C. L. Raston, Green Chem., 2004, 6, 119 RSC.
- E. Santaniello, in Crown Ethers and Phase Transfer Catalysis in Polymer Science, ed. L. J. Mathias and C. E. Carraher, Jr., Plenum Press, New York, 1984, p. 397 Search PubMed.
- P. Ferravoschi, A. Fiecchi, P. Grisenti, E. Santaniello and S. Trave, Synth. Commun., 1987, 17, 1569 CrossRef CAS.
- K. Sukata, Bull. Chem. Soc. Jpn., 1984, 57, 613 CAS.
- D. Badone, G. Jommi, R. Pagligrin and P. Tavecchia, Synthesis, 1987, 920 CrossRef CAS.
- N. Suzuki, T. Azuma, Y. Kaneko, Y. Izawa, H. Tomioka and T. Nomoto, J. Chem. Soc., Perkin Trans. 1, 1987, 645 RSC.
- V. V. Namboodiri and R. S. Varma, Green Chem., 2001, 3, 146 RSC.
- S. Chandrasekhar, Ch. Narsihmulu, S. S. Sultana and N. R. Reddy, Chem. Commun., 2003, 1716 RSC.
- S. Chandrasekhar, Ch. Narsihmulu, G. Chandrashekar and T. Shyamsunder, Tetrahedron Lett., 2004, 45, 2421 CrossRef CAS.
- E. Santaniello, P. Ferraboschi and P. Sozzani, J. Org. Chem., 1981, 46, 4584 CrossRef CAS.
- E. Santaniello, A. Fiecchi, A. Manzocchi and P. Ferraboschi, J. Org. Chem., 1983, 48, 3074 CrossRef CAS.
- Phase-Transfer Catalysis: Mechanisms and Syntheses, ed. M. E. Halpern, ACS Symposium Series 659, American Chemical Society, Washington, DC, 1997 Search PubMed.
- J. M. Harris, N. H. Hundley, T. G. Shannon and E. C. Struck, in Crown Ethers and Phase Transfer Catalysis in Polymer Science, ed. L. J. Mathias and C. E. Carraher, Jr., Plenum Press, New York, 1984, p. 371 Search PubMed.
- C. B. Dartt and M. E. Davis, Ind. Eng. Chem. Res., 1994, 33, 2887 CrossRef CAS.
- B. Abribat, Y. Le Bigot and A. Gaset, Synth. Commun., 1994, 24, 2091 CrossRef CAS.
- B. Abribat, Y. Le Bigot and A. Gaset, Tetrahedron, 1996, 52, 8245 CrossRef CAS.
- B. Abribat and Y. Le Bigot, Tetrahedron, 1997, 53, 2119 CrossRef CAS.
- D. Balasubramanian, P. Sukumar and R. Chandani, Tetrahedron Lett., 1979, 20, 3543 CrossRef.
- E. Angeletti, P. Tundo and P. Venturello, J. Chem. Soc., Perkin Trans. 1, 1982, 1137 RSC.
- M. L. Wang and K. R. Chang, J. Mol. Catal., 1991, 67, 147 CrossRef CAS.
- S. Slaoui, R. Le Goaller, J. L. Pierre and J. L. Luche, Tetrahedron Lett., 1982, 23, 1681 CrossRef CAS.
- J. Sun and C. G. Yan, Synth. Commun., 2002, 32, 1735 CrossRef CAS.
- M. M. Salunkhe, B. P. Kavitake, S. V. Patil and P. P. Wadgaonkar, J. Chem. Res. (S), 1995, 503 Search PubMed.
- B. Abribat, Y. Le Bigot and A. Gaset, Synth. Commun., 1994, 24, 1773 CrossRef CAS.
- T. B. Wei, J. C. Chen, X. C. Wang, Y. M. Zhang and L. L. Wang, Synth. Commun., 1996, 26, 1447 CrossRef CAS.
- X. C. Wang, Z. Li, L. M. Gao, T. B. Wei and J. C. Chen, Synth. Commun., 2000, 30, 2083 CrossRef CAS.
- X. C. Wang, T. B. Wei, J. C. Chen and J. R. Li, Synth. Commun., 1996, 26, 2765 CrossRef CAS.
- L. L. Wang, S. J. Lu, S. B. Li, Y. M. Zhang and T. B. Wei, Synth. Commun., 1998, 28, 1005 CrossRef CAS.
- R. A. Bartsch and I. W. Wang, Tetrahedron Lett., 1979, 20, 2503 CrossRef.
- R. S. Davidson, A. M. Patel, A. Safdar and D. Thornthwaite, Tetrahedron Lett., 1983, 24, 5907 CrossRef CAS.
- K. Chandler, C. W. Culp, D. R. Lamb, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 1998, 37, 3252 CrossRef CAS.
- E. N. Krylov and T. A. Buslaeva, Russ. J. Gen. Chem., 1997, 67, 106 CAS.
- T. B. Wei and Y. M. Zhang, Synth. Commun., 1999, 29, 2943 CrossRef CAS.
- T. B. Wei, J. C. Chen and Y. M. Zhang, Synth. Commun., 1995, 25, 1885 CrossRef CAS.
- Y. M. Zhang, T. B. Wei and L. L. Wang, Synth. Commun., 1997, 27, 751 CrossRef CAS.
- V. K. Krishnakumar, Synth. Commun., 1984, 14, 189 CrossRef CAS.
- B. P. Kavitake, M. M. Salunkhe and P. P. Wadgaonkar, Synth. Commun., 1997, 27, 1703 CrossRef CAS.
- J. M. Harris, N. H. Hundley, T. G. Shannon and E. C. Struck, J. Org. Chem., 1982, 47, 4789 CrossRef CAS.
- C. Zucchi, G. Pályi, V. Galamb, E. Sámpár-Szerencsés, L. Markó, P. Li and H. Alper, Organometallics, 1996, 15, 3222 CrossRef CAS.
- R. Neumann and Y. Sasson, Chem. Commun., 1985, 616 RSC.
- J. T. Lee and H. Alper, Organometallics, 1990, 9, 3064 CrossRef CAS.
- R. Neumann and Y. Sasson, J. Org. Chem., 1984, 49, 1282 CrossRef CAS.
- P. Li and H. Alpert, J. Org. Chem., 1986, 51, 4354 CrossRef CAS.
- J. X. Wang and H. Alpert, J. Org. Chem., 1986, 51, 273 CrossRef.
- B. G. Zupancic and M. Kokalj, Synth. Commun., 1982, 12, 881 CrossRef CAS.
- R. Neumann and Y. Sasson, J. Mol. Catal., 1985, 33, 201 CrossRef CAS.
- Y. Kimura and S. L. Regen, J. Org. Chem., 1983, 48, 195 CrossRef CAS.
- S. Grinberg and E. Shaubi, Tetrahedron, 1991, 47, 2895 CrossRef CAS.
- R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi and G. Tocco, Org. Lett., 2000, 2, 1737 CrossRef CAS.
- D. Albanese, M. Benaglia, D. Landini, A. Maia, V. Lupi and M. Penso, Ind. Eng. Chem. Res., 2002, 41, 4928 CrossRef CAS.
- M. P. Bullitta, E. Maccioni, L. Corda and G. Podda, J. Heterocycl. Chem., 1993, 30, 93 CrossRef CAS.
- A. Gobbi, D. Landini, A. Maia, G. Delogu and G. Podda, J. Org. Chem., 1994, 59, 5059 CrossRef CAS.
- J. G. Huddleston, A. Veide, K. Kohler, J. Flanagan, S. O. Enfors and A. Lyddiatt, Trends Biotechnol., 1991, 9, 381 CrossRef CAS.
- J. G. Huddleston and A. Lyddiatt, Appl. Biochem. Biotechnol., 1990, 26, 249 CrossRef CAS.
- H. C. Hsiao, S. M. Kao and H. S. Weng, Ind. Eng. Chem. Res., 2000, 39, 2772 CrossRef CAS.
- R. Neumann and Y. Sasson, J. Org. Chem., 1984, 49, 3448 CrossRef CAS.
- H. C. Hsiao and H. S. Weng, J. Chem. Technol. Biotechnol., 2001, 76, 959 CrossRef CAS.
- G. Jin, T. Ido and S. Goto, Catal. Today, 2001, 64, 279 CrossRef CAS.
- T. Ido, T. Yoshikawa, G. Jin and S. Goto, Kagaku Kogaku Ronbunshu, 2002, 28, 88 Search PubMed.
- G. Jin, T. Ido and S. Goto, J. Chem. Eng. Jpn., 1999, 32, 417 Search PubMed.
- T. J. McDonough, Tappi J., 1993, 76, 186 Search PubMed.
- R. J. H. Stenekes and W. E. Hennink, Polymer, 2000, 41, 5563 CrossRef CAS.
- R. Hatti-Kaul and B. Mattiasson, Appl. Microbiol. Biotechnol., 1986, 24, 259.
- A. Karakatsanis and M. Liakopoulou-Kyriakides, Starch/Stärke, 1998, 50, 349 Search PubMed.
- F. Tjerneld, I. Persson, P. Å. Albertsson and B. Hahn-Hägerdal, Biotechnol. Bioeng., 1985, 27, 1036 CrossRef CAS.
- F. Tjerneld, I. Persson, P. Å. Albertsson and B. Hahn-Hägerdal, Biotechnol. Bioeng., 1985, 27, 1044 CrossRef CAS.
- I. Persson, H. Stålbrand, F. Tjerneld and B. Hahn-Hägerdal, Appl. Biochem. Biotechnol., 1992, 27, 27 CAS.
- C. F. Mandenius, B. Nilsson, I. Persson and F. Tjerneld, Biotechnol. Bioeng., 1988, 31, 203 CrossRef CAS.
- F. Tjerneld, I. Persson, P. Å. Albertsson and B. Hahn-Hägerdal, Biotechnol. Bioeng. Symp., 1986, 15, 419 Search PubMed.
- F. Tjerneld, I. Persson and J. M. Lee, Biotechnol. Bioeng., 1991, 37, 876 CrossRef CAS.
- A. Kondo, T. Urabe and K. J. Higashitani, J. Ferment. Bioeng., 1994, 77, 700 Search PubMed.
- M. Larsson, V. Arasaratnam and B. Mattiasson, Biotechnol. Bioeng., 1989, 33, 758 CAS.
- M. Li, J. W. Kim and T. L. Peeples, Biochem. Eng. J., 2002, 11, 25 CrossRef CAS.
- M. Li, J. W. Kim and T. L. Peeples, J. Biotechnol., 2002, 93, 15 CrossRef CAS.
- O. H. Hernandez-Justiz, R. F. Fernandez-Lafuente, M. Terreni and J. M. Guisan, Biotechnol. Bioeng., 1998, 59, 73 CrossRef CAS.
- J. L. Den Hollander, B. I. Stribos, M. J. Van Buel, K. Ch. A. M. Luyben and L. A. M. Van der Wielen, J. Chromatogr., B: Biomed. Appl., 1998, 711, 223 CrossRef CAS.
- J. L. Den Hollander, Y. W. Wong, K. Ch. A. M. Luyben and L. A. M. Van der Wielen, Chem. Eng. Sci., 1999, 54, 3207 CrossRef CAS.
- D. Z. Wei, J. H. Zhu and X. J. Cao, Biochem. Eng. J., 2002, 11, 95 CrossRef CAS.
- L. C. Liao, C. S. Ho and W. T. Wu, Process Biochem., 1999, 34, 417 CrossRef CAS.
- E. Andersson, B. Mattiasson and B. Hahn-Hägerdal, Enzyme Microb. Technol., 1984, 6, 301 CrossRef CAS.
- T. J. Bartlett, R. A. Rastall, N. H. Rees, M. W. Adlard and C. Bucke, J. Chem. Technol. Biotechnol., 1992, 55, 73 CAS.
- T. K. Mong, H. K. Lee, S. G. Duron and C. H. Wong, Proc. Natl. Acad. Sci. USA, 2003, 100, 797 CrossRef CAS.
- A. Pandey, S. Sarfaraz and L. Iyengar, in Green Chemistry: Environment Friendly Alternatives, ed. R. Sanghi and M.M. Srivastava, Narosa Publishing House, New Delhi, India, 2003, p. 294 Search PubMed.
Footnote |
| † Current address: Key Lab. of Rare Earth Chemistry and Physics, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street 5625, Changchun, 130022, People’s Republic of China. |
| This journal is © The Royal Society of Chemistry 2005 |